

Summary
The bone marrow (BM) microenvironment (niche) plays important roles in supporting normal/abnormal haematopoiesis. We investigated the interaction between leukaemic mesenchymal niche and haematopoietic stem and progenitor cells (HSPCs) using the model of Fanconi anaemia (FA), a genetic disorder characterized by BM failure and leukaemia. Healthy donor HSPCs co-cultured on mesenchymal stromal cells (MSCs) derived from FA patients with acute myeloid leukaemia (AML) exhibited higher human engraftment and myeloid expansion in Non-obese diabetic severe combined immunodeficiency IL-2γ−/−/SGM3 recipients. Untargeted metabolomics analysis revealed the progressively elevated prostaglandins (PGs) in the MSCs of FA patients with myelodysplastic syndromes (MDS) and AML. Reduced secretion of PGs subsequent to inflammatory cyclooxygenase 2 (COX2) inhibition ameliorated HSPC/myeloid expansion. Transcriptome analysis demonstrated dysregulation of genes involved in the NR4A family of transcription factors (TFs) and WNT/β-catenin signalling pathway in FA-AML-MSC-co-cultured-CD34+ cells. COX2 inhibition led to significantly decreased NR4A TFs and WNT signalling genes expression. Mechanistically, NR4A1 and NR4A2 synergistically activate the CTNNB1 gene promoter. Knocking down CTNNB1 or NR4A1 in AML-MSC-co-cultured-CD34+ cells increased leukaemia-reactive T-effector cells production and rescued anti-leukaemia immunity. Together, these findings suggest that specific interactions between leukaemic mesenchymal niche and HSPCs orchestrate a novel COX2/PG-NR4A/WNT signalling axis, connecting inflammation, cellular metabolism and cancer immunity.
Keywords: haematopoietic stem cell transplantation, mesenchymal stromal cells, prostaglandins, nuclear hormone transcription factors, WNT signalling
Haematopoietic stem cell transplantation (HSCT) is a potential curative therapy for patients with high-risk haematological malignancies (van den Brink & Burakoff, 2002; Nikiforow & Ritz, 2016), including Fanconi anaemia (FA), a cancer-prone disease associated with bone marrow (BM) failure and leukaemia (Bagby & Alter, 2006; Dong et al, 2015). During the last several decades, there have been major advances in FA HSCT due to significant improvement in patient and donor selection (human leucocyte antigen [HLA] typing), conditioning regimens, graft manipulation methods, graft-versus-host disease (GVHD) prophylaxis and supportive care (de la Fuente et al, 2003; Dalle, 2008; MacMillan et al, 2011). However, overall survival rates of FA patients after HSCT are still below 30% and the eradication of residual leukaemia stem cells (LSCs), which often contributes to relapse (van den Brink & Burak-off, 2002), remains a major challenge.
Haematopoietic stem cells (HSCs) and LSCs are supported by BM stromal cells and other additional factors, which form the BM microenvironment (niche), to preserve their shared characteristics, including quiescence, multipotency and self-renewal (Wilson & Trumpp, 2006). Recent studies have revealed that disordered niche function could contribute to disease. For example, several studies show that altered signalling in the BM stroma/niche can result in leukaemia initiation or progression (Walkley et al, 2007; Raaij-makers et al, 2010; Zhang et al, 2012; Morrison & Scadden, 2014). Conversely, leukaemic cells can reprogramme the BM niche in favour of leukaemic proliferation while compromising normal haematopoiesis (Kode et al, 2014). To date, only a few studies have described the functional characteristics of FA BM mesenchymal stromal cells (MSCs; Smyth et al, 2009; Schepers et al, 2013; Fontaine et al, 2016), all of which implicate a role of the FA BM niche in the pathophysiology of FA BM failure. However, the role of the FA BM niche in the maintenance of the residue LSCs and leukaemia relapse after HSCT has not been defined.
Prostaglandins (PGs) are lipid compounds of the eicosanoid family, which play major roles in inflammation and immune response (Smyth et al, 2009) and are an important component of the mesenchymal secretome (Fontaine et al, 2016). Synthesized by the pro-inflammatory cyclooxygenases COX1 and COX2 through oxidation of the derivative arachidonic acid, major PGs include PGE2, PGG2, PGH2 and PGI2 (Smith et al, 2000). Among them, PGE2 has been subjected to intensive investigation for its role in HSC expansion and engraftment in the context of HSCT (Durand & Zon, 2010; Hoggatt & Pelus, 2010). In the context of cancer, the key inflammatory enzyme for the biosynthesis of PGs, COX2 has long been known to be deregulated in many cancers (Gallo et al, 2002; Wu et al, 2010; Janakiram & Rao, 2014; Sicking et al, 2014). Inhibition of COX2 using either non-steroidal anti-inflammatory drugs (NSAIDs) or specific COX2 inhibitors has been shown to be beneficial for the treatment of a variety of solid tumours (Ranger, 2014). It is also been shown that various oncogenes can induce expression of COX2 in haematopoietic cells and clinical human leukaemias uniformly express COX2 in circulating blasts (Bernard et al, 2008). COX2 selective inhibitors reduce the burden of haematological malignancies (Lilly et al, 2004; Ramon et al, 2013); however, the underlying mechanisms remain to be elucidated.
In the present study, we investigated the interaction between leukaemic mesenchymal niche and haematopoietic stem and progenitor cells (HSPCs) using ex vivo co-culture and in vivo xenotransplant, and identified a novel COX2/PG-NR4A/WNT signalling axis as a crucial regulator of leukaemic mesenchymal niche-HSPC interaction. Our results suggest that specific interaction between the leukaemic mesenchymal niche and HSPCs orchestrates this novel immunometabolic axis, connecting inflammation, cellular metabolism and cancer immunity, and that targeting this regulatory axis might be beneficial for developing innovative therapeutic strategies for leukaemia and other haematological malignancies.
Materials and methods
Mice
Non-obese diabetic severe combined immunodeficiency (NOD/SCID)/rL2γ−/−/SGM3 (NSGS) mice, which express transgenic cDNAs encoding human stem cell factor (SCF, also termed KITLG), granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin 3 (IL3) (Wunderlich et al, 2010) were purchased from Jackson Laboratory and housed in Transgenic Animal Facility at West Virginia University (WVU). NSGS mice at age of 6–8 weeks were used as recipients for BM transplantation (BMT). All experimental procedures conducted in this study were approved by the Institutional Animal Care and Use Committee of West Virginia University.
Human bone marrow stromal cell (hBMSC) culture and treatment
Human MSC culture protocol was adapted and modified from a previously described method (Mirsaidi et al, 2017). Briefly, cell cultures were maintained at 37°C, in 5% CO2 and 98% humidity in normal growth medium consisting of Dulbecco’s modified eagle medium (DMEM-low glucose, with GlutaMAX; ThermoFisher Scientific, Reinach, Switzerland), supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA), penicillin/streptomycin (50 units/ml; 50 μg/ml). All MSCs were used at passage 3, which displayed the characteristic MSC surface phenotype (CD45−HLA−DR−CD105+CD73+CD90+CD44+CD146+).
To inhibit PG biosynthesis, MSCs derived from FA patients with acute myeloid leukaemia (FA-AML) were pre-treated with COX2 inhibitor (COX2i; celecoxib, 10 μmol/l) for 2 h (Rezavand et al, 2013) followed by co-culture with normal BM CD34+ cells for 2 weeks. Cell culture media containing the specified treatment agents was regularly replenished every 3–4 days throughout the course of the experiment.
Bone marrow haematopoietic stem progenitor cell (HSPC)-MSC coculture
1–2 × 106 BM CD34+ cells from healthy donors (HD) were seeded on 70% confluent MSCs derived from different sources and cultured in serum-free medium containing 100 ng/ml each of SCF, FLT3 ligand, thrombopoietin (TPO, also termed THPO) and granulocyte colony-stimulating factor (G-CSF) for 2 weeks, followed by flow cytometry, colony-forming unit cell (CFU-C) assay or BMT.
NSGS BMT and in vivo treatment
5 × 105 normal BM CD34+ cells from the co-culture on MSCs of HD, FA patients with cytopenias but no cancer (FA-BMF) or FA-AML were transplanted into sublethally-irradiated (250 cGy) NSGS mice. Recipients were intraperitoneally (i.p.) injected with the GVHD inhibitor, OKT3 (10 μg/kg; BioXCell, West Lebanon, NH, USA) at 24 h and 1 week after transplant (Wunderlich et al, 2014). Total human engraftment (hCD45+), HSPCs (CD34+) and myeloid (CD33+)/lymphoid (CD19+) cells of the recipients were analysed by flow cytometry.
For in vivo treatment, NSGS mice transplanted with HD MSC co-cultured HSPCs were subjected to i.p. injection of a long-acting agonist of PGE2, 16–16 dimethyl-PGE2 (dmPGE2; 10 μg/kg body weight; Cayman Chemical, Ann Arbor, MI, USA; Goessling et al, 2009) or vehicle (Neat oil) followed by flow cytometry analysis for donor-derived chimera or different lineages at 12 weeks post-transplant.
Flow cytometry analysis
Bone marrow cells isolated from NSGS recipients were subjected to flow cytometry analysis using antibodies for human CD45 for total human engraftment, CD34 for human progenitor frequency, and CD33 and CD19 for myeloid or lymphoid lineage, respectively (BD Biosciences, San Jose, CA, USA). The frequency of regulatory T cells (Treg) or effector T cells (Teff) were determined by staining BM cells from the indicated experimental animals with antibodies against CD4, CD25, FOXP3 or CD3, CD8 and CCR7 (BD Biosciences).
Metabolite profiling
Metabolites were extracted as previously described (Amarachintha et al, 2015). Briefly, MSCs were washed with ice cold Dulbecco’s PBS twice to remove any culture media. Cells were collected into 300-μl LC/MS-grade H2O containing 1 mmol/l HEPES and 1 mmol/l EDTA (pH 7·2). Samples were vortexed for 30 s and incubated for 1–2 min in boiling water and subsequently in liquid nitrogen for 1 min. Samples were then thawed on ice and normalized based on the protein content. 2 ml of −20°C metabolite extraction solution containing methanol, acetonitrile and H2O at a ratio of 2:2:1 was added to each sample and vortexed for 1 min. Samples were then incubated at 4°C for 30 min followed by centrifugation at 1500 g for 10 min. The supernatants (~2 ml total) were pooled in a high performance liquid chromatography vial (Catalogue number 27115-U; Sigma-Aldrich) and dried under forced nitrogen at room temperature before reconstituted for liquid chromatography - quadrupole - time of flight - mass spectrometry (LC-Q-TOF-MS) analysis. All pure standards were purchased from Sigma-Aldrich. Samples were resuspended in 50 μl of 50:50 water/acetonitrile solutions for mass spectrometry analysis. Untargeted metabolomics was performed on the MSC extract to identify which metabolite levels are altered in MSCs derived from FA-MDS or FA-AML patients compared to those from healthy donor MSCs. Samples analysed at Scripps Center for Metabolomics and Mass Spectrometry (La Jolla, CA, USA). LC-Q-TOF-MS detected hundreds of peaks with unique m/z ratio and retention time in MSCs. Each peak, termed a metabolomics feature, is characterized on the basis of its accurate mass, retention time and tandem mass spectral fragmentation pattern by using the METLIN metabolite database (https://metilin.scripps.edu). The data was then analysed with the bioinformatics program XCMS online (Amarachintha et al, 2015), widely used XCMS software that is freely available at https://xcmsonline.scripps.edu/landing_page.php?pgcontent=mainPage.
Prostaglandins assessment
Prostaglandin levels in the indicated MSCs were measured by enzyme-linked immunosorbent assay (ELISA; Cayman Chemical, Ann Arbor, MI, USA) as previously described (Rozenberg et al, 2016). Cells were seeded in 6-well plates and allowed to reach 90% confluence and then treated with 6MV-X-ray in growth medium without supplements for the indicated times. In each condition, PGs were determined in supernatants by ELISA following manufacturer directions using BioTEK ELX808 Absorbance Microplate Readers (BioTEK, Winooski, VT, USA). Each condition was evaluated for PGs by averaging a minimum of two optical density measurements. Measured ELISA test value from supernatant of MSC for standard results were taken as the value for the 10 × 104 per MSCs.
Reporter gene assays
Approximately 106 HEK 293 cells were co-transfected with the indicated combinations of a CTNNB1 reporter construct containing −591 to +147 of the proximal CTNNB1 promoter (Zhou et al, 2008) and NR4A TFs, and plated on a 6-well plate. The cells were harvested 12 h after transfection, washed with PBS, and luciferase activity was assessed using the dual luciferase assay reporter kit (Promega, Madison, WI, USA).
Cytotoxic T lymphocyte (CTL) cytotoxicity assay
A semi-automated mini-cytotoxicity assay was used to determine specific lysis as previously described (Molldrem et al, 2000). Effector cells (peripheral blood mononuclear cells; PBMC or sorted CTLs) were prepared in doubling dilutions from 2·5 × 103 to 20 × 103 cells/well and were plated in 40 μl, 60-well Terasaki trays (Robbins Scientific, Sunnyvale, CA, USA) with six replicates per dilution. Target cells (BM cells from HLA-mismatched AML patients) at a concentration of 2 × 106 cells/ml were stained with 1 μg/ml of Calsein-AM (Molecular Probes, Eugene, OR, USA) for 60 min at 37°C. After washing three times in culture medium plus 10% AB serum, target cells were re-suspended at 105 cells/ml. Wells with target cells alone and medium alone were used for maximum (max) and minimum (min) fluorescence emission, respectively. After a 4-h incubation at 37°C in 5% CO2, 5 μl FluoroQuench (One Lambda, Canoga Park, CA, USA) was added to each well and the trays were centrifuged for 1 min at 60 g before measurement of fluorescence (excitation at 485 nm, emission measured at 530 nm) using an automated CytoFluor II plate reader (PerSeptive Biosystems, Foster City, CA, USA). A decrease in the fluorescence emission is proportional to the degree of lysis of target cells, once the released dye was quenched by the haemoglobin contained in the FluoroQuench reagent. The percentage of lysis was calculated as follows:
Statistical analysis
Paired or unpaired Student’s t-test was used for two-group comparisons. Survival data were plotted by the Kaplan–Meier curve method and analysed by the log-rank test. Values of P < 0·05 were considered statistically significant. Results are presented as mean ± SD. Detailed methods are included in Appendix S1.
Results
FA-AML MSCs promote HSPC and myeloid expansion of normal BM CD34+ cells
To investigate the haematopoietic supportive function of FA MSCs in the context of disease progression, we co-cultured normal BM CD34+ cells on MSCs derived from HD, FA-BMF, or FA-AML, and then analysed the proliferation of the co-cultured HSPCs. We found that total CD34+ progeny and CFU-C were significantly expanded on all three groups of MSCs after 2 weeks of co-culture (Table S1). These results suggest that the ability of FA MSCs to expand normal CD34+ ex vivo is not compromised. Furthermore, FA-AML showed the greatest ability to expand the normal human BM HSPCs.
We next determined the effect of FA MSCs on the in vivo repopulating ability of the co-cultured HSPCs by transplanting the co-cultured cells into sublethally-irradiated NSGS mice, which express transgenic cDNAs encoding human SCF, GM-CSF and IL3 (Wunderlich et al, 2010). We have successfully used these “humanized” mice to establish the FA AML xeno-transplant model (Du et al, 2011). Analysis of human engraftment at 12 weeks post-transplant revealed that the recipients transplanted with cells co-cultured on FA-BMF MSCs gave nearly the same total (hCD45+) and CD34+ engraftment as those receiving cells co-cultured on HD MSCs (Fig 1). This suggests that the ability of these FA-BMF MSCs to support haematopoietic repopulation in vivo is not compromised. Significantly, the recipients of HSPCs co-cultured on the FA-AML MSCs not only exhibited much higher total (hCD45+) and CD34+ engraftment than the other two groups, but also showed characteristics of myeloid expansion at the expense of the lymphoid lineage (Fig 1A). The observed HSPC/myeloid expansion promoted by the FA-AML MSCs may be an essential initiating event in FA leukaemogenesis, which progresses exclusively to myeloid leukaemia (Bagby & Alter, 2006; Dong et al, 2015), as the expansion can enlarge the pool of transformable cells.
Fig 1.
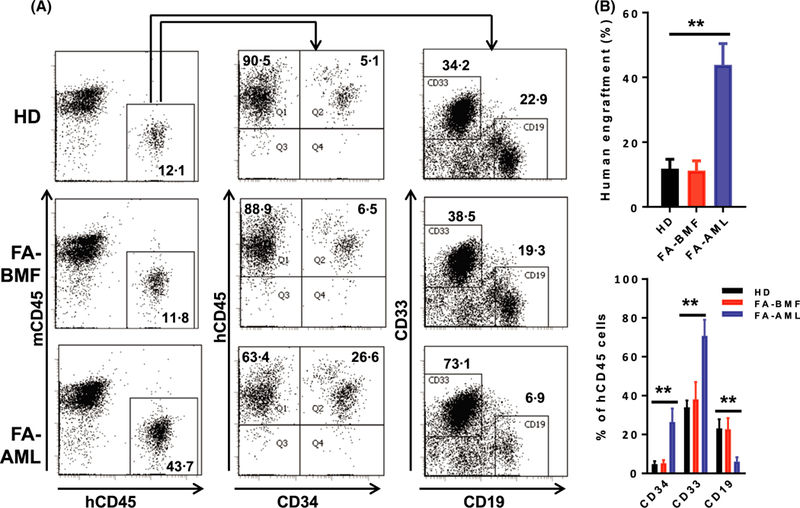
NSGS mice transplanted with normal cells co-cultured on FA-AML MSCs exhibit haematopoietic stem and progenitor cells and myeloid expansion. 5 × 105 normal bone marrow (BM) cells co-cultured on mesenchymal stromal cells (MSCs) from healthy donors (HD), Fanconi anaemia (FA) patients with cytopenias but no cancer (FA-BMF) or FA patients with acute myeloid leukaemia (FA-AML) (3–5 BM samples for each group) were transplanted into sublethally-irradiated NSGS mice. Recipients were intraperitoneally (i.p.) injected with the GVHD inhibitor OKT3 (10 μg/kg) at 24 h and 1 week after transplant. BM cells were subjected to flow cytometry analysis for human cell content at 12 weeks post-transplant. (A) Representative flow cytometry plots of total human engraftment (hCD45+; left), CD34+ (middle) and myeloid (CD33+)/lymphoid (CD19+) cells. (B) Quantification of human cell content by flow cytometry analysis depicted in (A). Results are means ± SD of three independent experiments (n = 9–12 for each group). **P < 0·01. [Colour figure can be viewed at wileyonlinelibrary.com]
Over-production of PGs in FA-AML MSCs is correlated with HSPC/myeloid expansion in NSGS recipients
To identify the molecular mechanism underlying the effect of the FA MSCs on the fate of co-cultured HSPCs, we turned our attention to metabolic components, as metabolites produced by MSCs in the BM niche are vital to normal HSC functions and play crucial roles in leukaemic transformation (Raaijmakers et al, 2010; Morrison & Scadden, 2014). We performed untargeted metabolic profiling to analyse the metabolome of BM-derived MSCs from HD and FA patients at the disease stages of BM failure (FA-BMF), MDS (FA-MDS) and AML (FA-AML), by LC-Q-TOF-MS) (Schultz et al, 2013). This global metabolic platform identified 934–2252 metabolites upregulated and 728–1091 metabolites downregulated in FA MSCs compared to HD MSCs (Fig 2A, B). Among the de-regulated metabolites, PGs, which are an important component of the inflammatory mesenchymal secretome (Fontaine et al, 2016), were the only metabolites that are progressively elevated in FA-MDS and FA-AML MSCs (Fig 2C; data not shown). Furthermore, using PG-specific ELISA kits, we found that intracellular levels of several PGs, namely PGD2, PGE1 and PGE2, were indeed increased in the MSCs from FA-BMF, FA-MDS and FA-AML patients compared to those from HD (Fig 2D).
Fig 2.
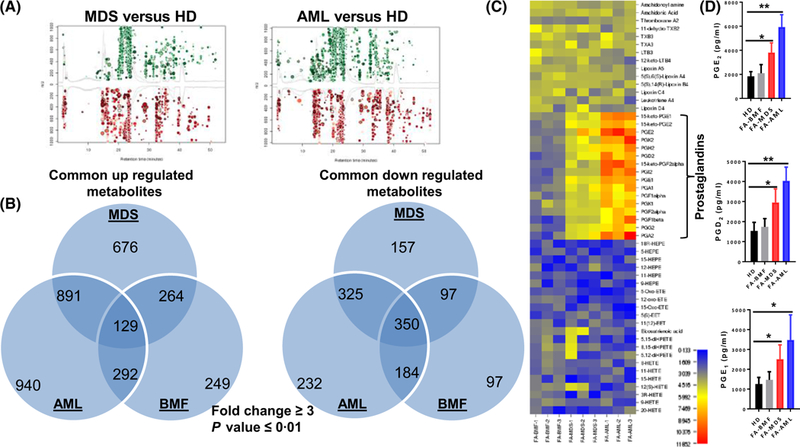
Elevated levels of prostaglandins (PGs) in FA-MDS and FA-AML MSCs. Liquid chromatography-mass spectroscopy -based untargeted metabolomics analysis of mesenchymal stromal cells (MSCs) from healthy donors (HD), Fanconi anaemia (FA) patients with cytopenias but no cancer (FA-BMF), FA patients with myelodysplastic syndrome (FA-MDS) or FA patients with acute myeloid leukaemia (FA-AML). (A) Cloud plot presentation of metabolite features of FA-MDS versus HD MSCs and FA-AML versus HD MSCs with fold change ≥3 and P ≤ 0·01. The statistical significance of the fold change was calculated by a Welch t test with unequal variances. Upregulated features (features that have a positive fold change) are graphed above the x-axis in green while downregulated features (features that have a negative fold change) are graphed below the x-axis in red. The x-axis represents retention time. The y-axis represents mass-to-charge (m/z) ratio. Features with higher fold change have larger radii. Features with lower P-value have higher colour intensity. (B) Venn diagram demonstrating the separate and overlapping metabolite features in BMF, MDS and AML MSCs compared to HD MSCs showing both upregulated and downregulated metabolites with fold change ≥3 and P value ≤0·01. (C) The levels of the metabolites in the Arachidonic acid metabolism pathway are shown. (D) Elevated level of the indicated PGs in MSCs analysed by enzyme-linked immunosorbent assay. Results are means ± SD of three independent experiments (n = 9 for each group). *P < 0·05; **P < 0·01.
To determine whether the effects of FA-AML MSCs on HSPC/myeloid expansion in the transplanted recipients were indeed mediated by these MSC-derived PGs, we employed two approaches: (i) blocking PG synthesis by a COX2i, celecoxib (Rezavand et al, 2013); (ii) treating the transplanted recipients with a long-acting agonist of PGE2, 16–16 dimethyl PGE2 (dmPGE2; Goessling et al, 2009) (Fig 3A). We first confirmed that the levels of PGD2 and PGE2 in the celecoxib-treated FA-AML MSCs were significantly reduced (Fig 3B). Strikingly, celecoxib treatment almost completely abolished the otherwise HSPC/myeloid expansion observed in FA-AML MSC-co-cultured cells (Fig 3C). In the second experiment, we found that while it did not completely recapitulate the effect of the FA-AML MSCs, dmPGE2 treatment induced HSPC/myeloid expansion in NSGS recipients (Fig 3D). Together, these studies identify the inflammatory COX2-PG secretome as a potential mediator of the effects of the FA-AML MSCs on HSPC/myeloid expansion.
Fig 3.
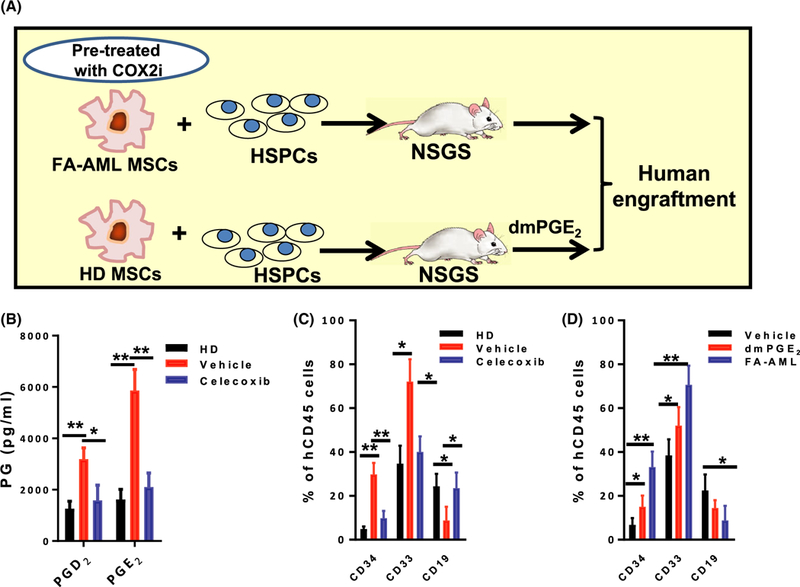
The AML-MSC COX2-PG secretome promotes HSPC/myeloid expansion in humanized recipients. (A) Schematic representation of the experimental design. Reduction of prostaglandin (PG) biosynthesis by COX2 inhibitor (COX2i) in mesenchymal stromal cells (MSCs) from Fanconi anaemia (FA) patients with acute myeloid leukaemia (FA-AML). FA-AML MSCs were pre-treated with COX2i (celecoxib, 10 μmol/l) for 2 h followed by co-culture with normal bone marrow (BM) CD34+ cells for two weeks. The progenies were then transplanted into sublethally-irradiated NSGS recipients. In another set of experiments, normal BM CD34+ cells were co-cultured on healthy donor (HD) MSCs for 2 weeks followed by BM transplantation into NSGS mice. The recipients were then injected with 16–16 dimethyl-PGE2 (dmPGE2; 10 μg/kg body weight) twice per day for 7 days starting at 24 h after transplantation. (B) COX2i celecoxib reduces PG production in FA-AML MSCs. FA-AML MSCs were pre-treated with COX2i (COX2i; celecoxib, 10 μmol/l) or vehicle (5% dimethyl sulphoxide) for 2 h. The levels of the indicated PGs in MSCs were measured by enzyme-linked immunosorbent assay. Results are means ± SD of three independent experiments (n = 9 for each group). (C) COX2i celecoxib prevents haematopoietic stem and progenitor cell (HSPC)/myeloid expansion of FA-AML MSC-co-cultured cells. Co-cultured cells described in (B) were transplant into sublethally-irradiated NSGS mice (5 × 105 cells/mouse; n = 6). Total human engraftment (hCD45+), HSPCs (CD34+) and myeloid (CD33+)/lymphoid (CD19+) cells were analysed by flow cytometry eight weeks post-transplant. Results are means ± SD of three independent experiments (n = 9–12 for each group). (D) dmPGE2 treatment induces HSPC/myeloid expansion in NSGS recipients. The NSGS recipient mice transplanted with HD MSC co-cultured cells were subjected to daily i.p. injection of dmPGE2 (10 μg/kg body weight) or vehicle (Neat oil) twice per day for 7 days starting at 24 h post-transplant. Total human engraftment (hCD45+), HSPCs (CD34+) and myeloid (CD33+)/lymphoid (CD19+) cells of the recipients were analysed by flow cytometry. Results are means ± SD of three independent experiments (n = 9–12 for each group). *P < 0·05, **P <0·01. [Colour figure can be viewed at wileyonlinelibrary.com]
The link between AML-MSC COX2-PG secretome and NR4A-Treg signalling
To identify the signalling pathways that specifically mediate the effect of the leukaemic mesenchymal COX2-PG secretome, we performed two sets of gene-expression profiling (GEP) by RNA-seq analysis. GEP 1 used co-cultured cells on HD, FA-BMF or FA-AML MSCs and GEP 2 used cells co-cultured on FA-AML MSCs treated with or without COX2i, or on HD MSCs stably expressing COX2 (Figure S1A). We noted that treatment of FA-AML MSCs with the COX2i, celecoxib, reduced several PGs to the levels comparable to those in HD MSCs (Fig 3B), and that HD MSCs transfected with COX2 not only expressed much higher levels of both PTGS2 mRNA (Figure S1B) and COX2 protein (Figure S1C) but also increased PGE2 biosynthesis (Figure S1D) than those transfected with the empty vector (Figure S1C). Pathway analysis identified 10 top pathways that were shared by both sets of GEP, with signalling receptor activity, WNT signalling and regulatory T cell differentiation pathways most significantly affected (Figure S1E). Specifically, genes involved in those pathways include the NR4A nuclear hormone TF family (NR4A2 [NURR1], NR4A3 [NOR1], NR4A1 [NUR77], PRL, MMP13, EGR1, JUNB, PLK2; Ramirez-Herrick et al, 2011), WNT/β-catenin signalling (LEF1, SOX9, WNT5A, FZD9, FHL2, DLG2, CTNNB1, PTGS2, CCND1, MMP9; Hovanes et al, 2001) and Treg function (FOXP3, CTLA4, LGALS3, IL2RA / ISG20 [CD25], CCR7, TNFRSF18 [GITR], TIAF1, IL2, CYSLTR1, LTB4R; Marson et al, 2007; Zheng et al, 2007) pathways (Figure S1E, F). These three pathways are known to be involved in leukaemogenesis (Nishikawa & Sakaguchi, 2010; Mohan et al, 2012), and functionally related to PGs, particularly PGE2 (Smith et al, 2000; Goessling et al, 2009; Smyth et al, 2009; Durand & Zon, 2010; Hoggatt & Pelus, 2010; Fontaine et al, 2016).
Given that inflammatory PGs can induce the expression of the NR4A TFs (Ramirez-Herrick et al, 2011), we further examined whether there was a potential link between the AML-MSC COX2-PG secretome and NR4A signalling. We found that reduced secretion of PGs by mesenchymal inhibition of COX2 was correlated with decreased expression of the NR4A TFs and Treg genes (FOXP3 and CTLA4) in the AML MSC-cocultured CD34+ cells (Fig 4A–C). Conversely, pre-treatment with dmPGE2 increased expression of the NF4A TFs and Treg genes in HD MSC-cocultured CD34+ cells but had no further effect on FA-AML MSC co-cultured CD34+ cells (Fig 4D, E). These results provide initial evidence linking the AML-MSC COX2-PG secretome to NR4A signalling.
Fig 4.
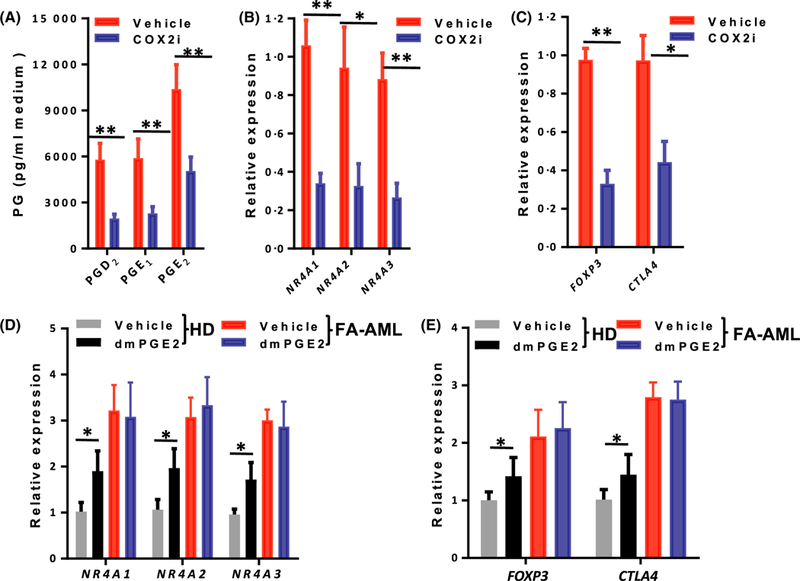
The link between AML-MSC COX2-PG secretome and NR4A-Treg signalling. (A) Mesenchymal inhibition of COX2 reduces the secretion of prostaglandins (PGs). Mesenchymal stromal cells (MSCs) from Fanconi anaemia (FA) patients with acute myeloid leukaemia (FA-AML) were pre-treated with COX2 inhibitor (celecoxib, 10 μmol/l) or vehicle (5% dimethyl sulphoxide) for 2 h, and the levels of the indicated PGs in the culture medium were measured by enzyme-linked immunosorbent assay. Results are means ± SD of three independent experiments (n = 9 for each group). (B) Mesenchymal inhibition of COX2 reduces the expression of the NR4A transcription factors (TFs) in co-cultured CD34+ cells. The MSCs in (A) were co-cultured with normal CD34+ cells for 2 weeks and expression of the three NR4A TF genes was determined by real-time polymerase chain reaction (PCR). Results are means ± SD of three independent experiments (n = 9 for each group). (C) Mesenchymal inhibition of COX2 reduces the expression of Treg genes in co-cultured CD34+ cells. The MSCs in (A) were co-cultured with normal CD34+ cells for 2 weeks and expression of FOXP3 and CTLA4 was determined by real-time PCR. The normal CD34+ cells were from 12 healthy donors. Results are means ± SD of three independent experiments (n = 9 for each group). (D) 16–16 dimethyl-PGE2 (dmPGE2) treatment increases the expression of the NR4A TF genes in co-cultured CD34+ cells. MSCs derived from HD were pre-treated with dmPGE2 followed by co-culture with normal CD34+ cells for 2 weeks. The expression of the three NR4A TF genes was determined by real-time PCR. Results are means ± SD of three independent experiments (n = 9 for each group). (E) dmPGE2 treatment increases the expression of Treg genes in co-cultured CD34+ cells. Co-cultured normal CD34+ cells described in (D) were subjected to real-time PCR analysis for FOXP3 and CTLA4. The normal CD34+ cells were from 12 healthy donors. Samples were normalized to the level of GAPDH mRNA. Results are means ± SD of three independent experiments (n = 9 for each group). *P < 0·05, **P < 0·01. [Colour figure can be viewed at wileyonlinelibrary.com]
Crosstalk between NR4A and WNT signalling in the regulation of anti-leukaemia T-effector cells
The canonical WNT pathway involves post-translational mechanisms that prevent proteasomal degradation of the proto-oncogene β-catenin (CTNNB1) and allow it to bind to the TCF-LEF family of TFs for transactivation of the WNT target genes (Behrens et al, 1996). Our gene-profiling studies showed that CTNNB1 mRNA was significantly increased in CD34+ cells co-cultured on FA AML-derived MSCs (Figure S1). Given that NR4A TFs regulate CTNNB1 transcription (Rajalin & Aarnisalo, 2011; Smith et al, 2011; Han & Cao, 2012), we thus determined whether the AML-MSC COX2-PG secretome could induce crosstalk between NR4A TFs and WNT/β-catenin signalling in the AML MSC-co-cultured HSPCs. CTNNB1 promoter activity was measured using a CTNNB1-luciferease reporter (−591 to +147; Nollet et al, 1996), in response to ectopic expression of the NR4A TFs. HEK293 cells transfected with the CTNNB1-luci-ferease reporter showed marginal luciferase activity induced by ectopic expression of individual NR4A TFs but a synergistic enhancement of the reporter activity when both NR4A1 and NR4A2 (but not NR4A3) expression vectors were present (Fig 5A), suggesting that NR4A1 and NR4A2 were acting in concert to activate the CTNNB1 promoter. By using a WNT GFP reporter assay (Fuerer & Nusse, 2010; Sertorio et al, 2016), we confirmed the significantly enhanced WNT activation in CD34+ cells co-cultured with FA-AML MSCs compared to those co-cultured with HD-MSCs (Fig 5B).
Fig 5.
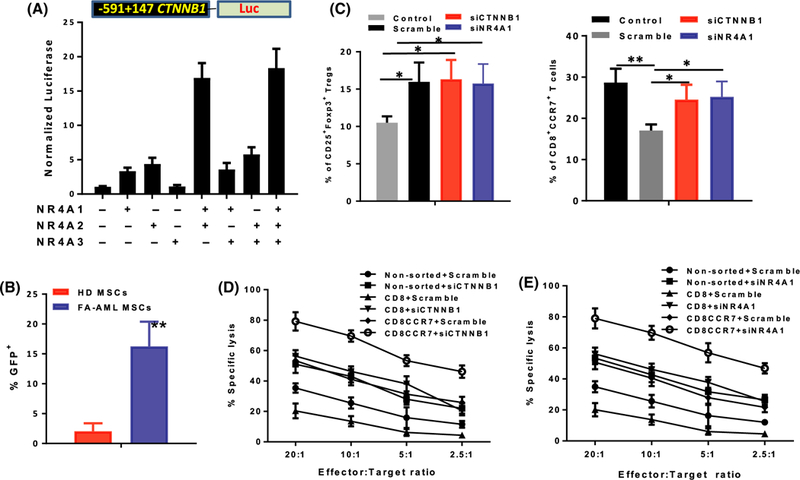
Crosstalk between NR4A and WNT signalling in regulation of anti-leukaemia T effector cells. (A) NR4A1 and NR4A2 act synergistically to enhance CTNNB1 reporter activity. HEK293 cells expressing a CTNNB1 reporter construct containing −591 to +147 of the proximal CTNNB1 promoter were co-transfected with the indicated combinations of the NR4A transcription factors (TFs). Twenty-four hours after transfection, cells were analysed for luciferase activity. Results are means ± SD of three independent experiments (n = 9 for each group). (B) Enhanced WNT activation in CD34+ cells co-cultured with mesenchymal stromal cells (MSCs) from Fanconi anaemia (FA) patients with acute myeloid leukaemia (FA-AML). CD34+ cells were transduced with 7TGC-eGFP WNT reporter lentivirus followed by co-culture with HD-MSC or FA-AML-MSC for two weeks. Percentage of GFP+ cells were determined by Flow cytometry. Results are means ± SD of three independent experiments (n = 9 for each group). (C) CTNNB1 or NR4A1 knockdown reduces CD8+CCR7+ T cells. Normal CD34+ cells were transfected with 25 nmol/l siGenome SMARTpool siRNAs for CTNNB1, NR4A1 or non-targeted Scramble. After 48 h, cells were co-cultured with FA-AML MSCs for 2 weeks and subjected to by flow cytometry analysis for different T-cell subsets. The graphs show quantification of the indicated T-cell subsets in gated CD3CD4 T cells (CD25+FOXP3+; left) or gated CD3CD8 T cells (CD8+CCR7+; right). Results are means ± SD of three independent experiments (n = 9 for each group). (D, E) CTNNB1 or NR4A1 knockdown rescues anti-leukaemia immunity. CD8+ or CD8+CCR7+ cells were sorted by FACS. The indicated T-cell subsets or non-sorted cells expressing Scramble shRNA or shRNA targeting CTNNB1 (D) or NR4A1 (E) were then plated with bone marrow cells from HLA-mismatched AML patients in a 4 h cytotoxicity assay at Effector:Target (E:T) ratios from 20:1 to 2–5:1. Data were pooled from two separate experiments, and six replicate wells were used for each dilution of effector cells. Data are displayed as mean specific lysis ± SD. Results are means ± SD of three independent experiments (n = 9 for each group). *P < 0·05, **P < 0·01. [Colour figure can be viewed at wileyonlinelibrary.com]
To test the functional consequence of upregulated WNT/β-catenin signalling, we reduced CTNNB1 expression by siRNA in the AML MSC-cocultured CD34+ cells (Figure S2A) and determined the effect of CTNNB1 knockdown on regulation of anti-leukaemia T effector cells. Consistent with upregulated FOXP3 (Fig 4), the AML MSC-cocultured HSPCs favoured CD25+FOXP+ Treg differentiation (Fig 5C, left). Knock-down of CTNNB1 had no effect on the frequency of Treg cells (Fig 5C, left). Strikingly, knocking down CTNNB1 significantly increased the production of CD8+CCR7+ T effector cells (Fig 5C, right), a population known to play a major role in anti-leukaemia immunity (Nowyhed et al, 2015). Next, we performed a semi-automated mini-cytotoxicity assay (Marson et al, 2007) to determine specific lysis of HLA-mismatched leukaemia target cells. The results show that the sorted co-cultured CD8+CCR7+ cells from a healthy donor (HLA-2·2+) exhibited greater lysis of blast cells taken from an AML patient (HLA-A2·1+) than the unsorted co-cultured cells or total CD8+ cells (Fig 5D). Marginal lysis of the AML cells was observed for the CD8+CCR7− T cells (data not shown). Furthermore, knocking down CTNNB1 markedly enhanced the capacity of the sorted CD8+CCR7+ cells or total CD8+ cells to lyse the leukaemia cells (Fig 5D).
Similar results were obtained in NR4A1 knockdown experiments (Fig 5C, E), in which knocking down NR4A1 (Figure S2B) significantly reduced the expression of the WNT signalling gene CTNNB1, significantly increased CD8+CCR7+ population without affecting Treg cell frequency (Fig 5C) and markedly enhanced the cytotoxic capacity of the sorted CD8+CCR7+ cells or total CD8+ cells on the HLA-mismatched leukaemia cells (Fig 5E). It is noteworthy that knocking down NR4A1 also significantly reduced the expression of the WNT signalling gene CTNNB1, suggesting that the NR4A TFs may regulate β-catenin at the transcription level. Taken together, our results suggest that the upregulated NR4A-WNT signalling axis may act to attenuate anti-leukaemia immunity by blocking the production of leukaemia-reactive CD8 cytotoxic T lymphocytes.
Discussion
Acute myeloid leukaemia has a 5-year survival rate of 25% (Georgi et al, 2016). Clinical evidence continues to support the need to identify novel targets and therapeutics for the treatment of this deadly disease. Experimental evidence has clearly shown that interaction of the leukaemic cells with the tumour microenvironment (TME) contributes to de novo drug resistance and, probably, failure to eliminate minimal residual disease (MRD; Ossenkoppele & Schuurhuis, 2013). However, the impact of TME-induced immune response in mediating MRD following standard of care treatment and BMT is currently less clear. Here we investigated the regulation of the interaction between leukaemic mesenchymal niche and normal HSPCs and identified a novel COX2/PG-NR4A/WNT signalling axis, connecting inflammation, cellular metabolism and cancer immunity. There are several findings that highlight the significance of our study: (i) The recipients transplanted with normal HSPCs co-cultured on the FA-AML MSCs exhibited HSPC and myeloid expansion; (ii) PGs were progressively elevated in FA-MDS and FA-AML MSCs; (iii) Treatment with the COX2i, celecoxib, reduced PGs produced by FA-AML MSCs and abolished HSPC/myeloid expansion in FA-AML MSC-co-cultured cells; (iv) Gene-expression profiling identified NR4A/WNT/Treg signalling pathways mediating the effect of the AML-MSC COX2-PG secretome; (v) Reduced secretion of PGs by mesenchymal inhibition of COX2 led to decreased expression of NR4A TFs and the Treg genes, FOXP3 and CTLA4, in the AML-MSC-cocultured CD34+ cells; (vi) The upregulated NR4A-WNT/β-catenin signalling acted to attenuate anti-leukaemic immunity by upregulating Tregs and blocking the production of leukaemia-reactive CD8 cytotoxic T lymphocytes.
One intriguing finding of the present study is our observation that the ability of FA-BMF-derived MSCs to support in vitro HSPC proliferation and in vivo haematopoietic repopulation is not compromised. Specifically, we showed that total normal human CD34+ and CFU-C were significantly increased on all three groups of MSCs, with FA-AML derived MSCs showing the greatest ability to support expansion of normal human BM HSPCs (Table S1). This is in striking difference with previous studies describing phenotypic haematopoiesis-supportive defects of MSCs from Fancc; Fancg double KO, Fanca−/− and Fancc−/− mice in an in vitro co-culture system in which HSPCs from wild type mice were seeded on these FA MSCs (Zhou et al, 2017; Xu et al, 2018). It is noteworthy that the results of these in vitro studies in FA mouse models have not been confirmed in vivo. The discrepancy in the results from the in vitro co-culture studies may be due to several factors. First, the haematopoietic supportive property may be different between mice and humans. Second, the co-culture conditions are different: the mouse studies used cytokine-free MSC medium; whereas our human studies used serum-free medium containing SCF, FLT3 ligand, TPO and G-CSF. Finally, there generally is a phenotypic variety among patients, which may contribute functional differences between mice and humans in particular in vitro experiments.
Mesenchymal PGs have been shown to be important components of the stromal secretome (Fontaine et al, 2016), and play major roles in inflammation and immune response (Schepers et al, 2013). Among them, PGE2 is known to play critical roles in HSC expansion and engraftment in the context of HSCT (Durand & Zon, 2010; Hoggatt & Pelus, 2010). By unbiased metabolomics analysis, here we identified PGs as the only metabolites that were progressively elevated during FA disease progression (Fig 2). Co-culture of CD34+ cells from HD on high PG-producing FA-AML MSCs caused HPSC and myeloid expansion in transplanted recipients. Reduction of PGs subsequent to pharmacological and genetic inhibition of the inflammatory COX2 in AML MSCs ameliorated HSPC and myeloid expansion in transplanted recipients (Fig 3). These results are in line with previous studies, that clinical human leukaemia uniformly expresses COX2 in circulating blasts (Bernard et al, 2008) and COX2 selective inhibitors have shown potential as immunotherapeutics for haematological malignancies (Lilly et al, 2004; Ramon et al, 2013). Therefore, targeting the COX2-PG signalling axis may present an effective strategy for the treatment of many haematological malignancies.
Members of the orphan nuclear hormone receptor NR4A subfamily, composed of closely related molecules NR4A1 (NUR77), NR4A2 (NURR1) and NR4A3 (NOR1), are early response genes induced by a variety of stimuli including growth factors, inflammation, cytokines, peptide hormones and cellular stress (Maxwell & Muscat, 2006). Their expression is highly enriched in Treg cells (Lin et al, 2007; Moran et al, 2011), which play an important role in the establishment and maintenance of immune tolerance after allogeneic HSCT (Rezvani et al, 2006; Rieger et al, 2006). Recent studies have shown that the NR4A family of TFs transactivate expression of FOXP3 and are critical for the generation of Tregs (Bandukwala & Rao, 2013; Sekiya et al, 2013; Michonneau et al, 2016). Consistent with these observations, our transcriptomic studies showed dysregulated genes involved in NR4A nuclear hormone TF family and Treg function in FA-AML MSC co-cultured CD34+ cells. Consistently, reduced biosynthesis of PGs by mesenchymal inhibition of COX2 decreased expression of the NR4A TFs and the Treg genes, FOXP3 and CTLA4, in the AML MSC-cocultured CD34+ cells (Fig 4). It is in this context that our results provide the first line of evidence linking the AML-MSC COX2-PG secretome to the NR4A-Treg signalling axis and thus support a function role of altered NR4A signalling in Treg biology.
The remarkable activity of donor T cells against malignant cells in the context of allogeneic HSCT is arguably the most potent clinical immunotherapy for cancer (van den Brink et al, 2002). In fact, the success of HSCT relies on the donor CTL-mediated graft-versus-leukaemia (GVL) effect (Michonneau et al, 2016). Therefore, induction of functional CTLs is one of the major goals for stem cell therapy. Recent studies have demonstrated an essential role of the WNT signalling for CTL manipulation in autoimmune diseases and immune therapy for certain cancers (Jeannet et al, 2010; Xiao et al, 2013). Our transcriptomic studies showed dysregulated expression of genes involved in NR4A TFs and WNT pathways, which is associated with expansion of Tregs and a reduction of the leukaemia-reactive CD8+CCR7+ pool. Mechanistically, we showed that NR4A1 and NR4A2 acted in concert to activate the CTNNB1 promoter (Fig 5). Although further studies are needed to elucidate the exact mechanism, our findings that knocking down CTNNB1 or NR4A1 significantly increased production of the leukaemia-reactive CD8+CCR7+ population (Nowyhed et al, 2015), and markedly enhanced the capacity of the sorted CD8+CCR7+ cells to lyse leukaemia cells (Fig 5D, E), shed new light on understanding the role of leukaemic niche factors in regulating donor anti-leukaemia immunity in HSCT.
Defining niche components and the molecular mechanisms by which these factors work to regulate haematopoiesis provides opportunities to understand how disordered niche function could contribute to disease, and reveal potential therapies for patients with leukaemia and other haematological malignancies (Schepers et al, 2013). Our present findings identify the specific interactions between leukaemic mesenchymal niche and donor HSPCs to orchestrate a novel COX2/PG/NR4A/WNT signalling axis, connecting inflammation, cellular metabolism and cancer immunity, and contribute to better understanding of the underlying mechanisms in HSCT-associated disease relapse.
Supplementary Material
Acknowledgement
This work was supported by the West Virginia University (WVU) Health Science Center (HSC) and School of Pharmacy (SOP) startup funds, a NIH/NIGMS CoBRE award (P20GM121322), a NIH/NIGMS WV CTSI Award (U54 GM104942), a Leukemia Research Foundation (LRF) Award and an American Cancer Society Institutional Research Grant at WVU. The bone marrow samples from Fanconi anaemia patients were received through the Fanconi Anemia Comprehensive Care Center, which administers a Fanconi Anemia Cell Repository at Cincinnati Children’s Research Foundation.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Supporting Information
Additional supporting information may be found online in the Supporting Information section at the end of the article.
References
- Amarachintha S, Sertorio M, Wilson A, Li X & Pang Q (2015) Fanconi anemia mesenchymal stromal cells-derived glycerophospholipids skew hematopoietic stem cell differentiation through toll-like receptor signaling. Stem Cells, 33, 3382–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagby GC & Alter BP (2006) Fanconi anemia. Seminars in Hematology, 43, 147–156. [DOI] [PubMed] [Google Scholar]
- Bandukwala HS & Rao A (2013) ‘Nurr’ishing Treg cells: Nr4a transcription factors control Fox-p3 expression. Nature Immunology, 14, 201–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R & Birchmeier W (1996) Functional interaction of beta-catenin with the transcription factor LEF-1. Nature, 382, 638–642. [DOI] [PubMed] [Google Scholar]
- Bernard MP, Bancos S, Sime PJ & Phipps RP (2008) Targeting cyclooxygenase-2 in hematological malignancies: rationale and promise. Current Pharmaceutical Design, 14, 2051–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink MR & Burakoff SJ (2002) Cytolytic pathways in haematopoietic stem-cell transplantation. Nature Review of Immunology, 2, 273–281. [DOI] [PubMed] [Google Scholar]
- Dalle JH (2008) HSCT for Fanconi anemia in children: factors that influence early and late results. Bone Marrow Transplantation, 42, S51–S53. [DOI] [PubMed] [Google Scholar]
- Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H & Vasiliou V (2015) Update of the human and mouse Fanconi anemia genes. Human Genomics, 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Li X, Sipple J & Pang Q (2011) Overexpression of IL-3R alpha on CD34+CD38 — stem cells defines leukemia-initiating cells in FA AML. Blood, 117, 4243–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand EM & Zon LI (2010) Newly emerging roles for prostaglandin E2 regulation of hematopoiesis and hematopoietic stem cell engraftment. Current Opinion in Hematology, 17, 308–312. [DOI] [PubMed] [Google Scholar]
- Fontaine MJ, Shih H, Schäfer R & Pittenger MF (2016) Unraveling the mesenchymal stromal cells’ paracrine immunomodulatory effects. Transfusion Medicine Review, 30, 37–43. [DOI] [PubMed] [Google Scholar]
- de la Fuente J, Reiss S, McCloy M, Vulliamy T, Roberts IA, Rahemtulla A & Dokal I (2003) Non-TBI stem cell transplantation protocol for Fanconi anaemia using HLA compatible sibling and unrelated donors. Bone Marrow Transplantation, 32, 653–656. [DOI] [PubMed] [Google Scholar]
- Fuerer C & Nusse R (2010) Lentiviral vectors to probe and manipulate the Wnt signaling pathway. PLoS ONE, 5, e9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo O, Masini E, Bianchi B, Bruschini L, Paglierani M & Franchi A (2002) Prognostic significance of cyclooxygenase-2 pathway and angiogenesis in head and neck squamous cell carcinoma. Human Pathology, 33, 708–714. [DOI] [PubMed] [Google Scholar]
- Georgi JA, Taube F, Kramer M, Herold S, Schaefer-Eckart K, Schmitz N, Eberlein C, Stasik S, Aulitzky WE, Kraemer A, Roesler W, Haenel M, Einsele H, Baldus CD, Middeke JM, Platzbecker U, Roellig C, Serve H, Berdel WE, Ehninger G, Bornhaeuser M, Schetelig J & Thiede C (2016) Differences between CEBPA bZIP and TAD mutations and their effect on outcome-an analysis in 4578 patients with acute myeloid leukemia. Blood, 128, 283. [Google Scholar]
- Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT & Zon LI (2009) Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell, 136, 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han YF & Cao GW (2012) Role of nuclear receptor NR4A2 in gastrointestinal inflammation and cancers. World Journal of Gastroenterology, 18, 6865–6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggatt J & Pelus LM (2010) Eicosanoid regulation of hematopoiesis and hematopoietic stem and progenitor trafficking. Leukemia, 24, 1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, Holcombe RF & Waterman ML (2001) Beta-catenin-sensitive isoforms of lymphoid enhancer factor- 1 are selectively expressed in colon cancer. Nature Genetics, 28, 53–57. [DOI] [PubMed] [Google Scholar]
- Janakiram NB & Rao CV (2014) The role of inflammation in colon cancer. Advances in Experimental Medicine and Biology, 816, 25–52. [DOI] [PubMed] [Google Scholar]
- Jeannet G, Boudousquié C, Gardiol N, Kang J, Huelsken J & Held W (2010) Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proceedings of the National Academy of Science of the United States of America, 107, 9777–9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, Brum A, Park D, Galili N, Mukherjee S, Teruya-Feldstein J, Raza A, Rabadan R, Berman E & Kousteni S (2014) Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature, 506, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly MB, Drapiza L, Sheth M, Zemskova M, Bashkirova S & Morris J (2004) Expression of cyclooxygenase-2 (COX-2) in human leukemias and hematopoietic cells. Blood, 104, 4336. [Google Scholar]
- Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB & Chatila TA (2007) Regulatory T cell development in the absence of functional Foxp3. Nature Immunology, 8, 359–368. [DOI] [PubMed] [Google Scholar]
- MacMillan ML, Hughes MR, Agarwal S & Daley GQ (2011) Cellular therapy for fanconi anemia: the past, present, and future. Biology of Blood Marrow Transplantation, 17, S109–S114. [DOI] [PubMed] [Google Scholar]
- Marson A, Kretschmer K, Frampton GM, Jacobsen ES, Polansky JK, MacIsaac KD, Levine SS, Fraenkel E, von Boehmer H & Young RA (2007) Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature, 445, 931–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell MA & Muscat GE (2006) The NR4A subgroup: immediate early response genes with pleiotropic physiological roles. Nuclear Receptor Signaling, 4, e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michonneau D, Sagoo P, Breart B, Garcia Z, Celli S & Bousso P (2016) The PD-1 axis enforces an anatomical segregation of CTL activity that creates tumor niches after allogeneic hematopoietic stem cell transplantation. Immunity, 44, 143–154. [DOI] [PubMed] [Google Scholar]
- Mirsaidi A, Tiaden AN & Richards PJ (2017) Prostaglandin E2 inhibits matrix mineralization by human bone marrow stromal cell-derived osteoblasts via Epac-dependent cAMP signaling. Scientific Reports, 7, 2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan HM, Aherne CM, Rogers AC, Baird AW, Winter DC & Murphy EP (2012) Molecular pathways: the role of NR4A orphan nuclear receptors in cancer. Clinical Cancer Research, 18, 3223–3228. [DOI] [PubMed] [Google Scholar]
- Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE & Davis MM (2000) Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nature Medicine, 6, 1018–1023. [DOI] [PubMed] [Google Scholar]
- Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J & Hogquist KA (2011) T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. Journal of Experimental Medicine, 208, 1279–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ & Scadden DT (2014) The bone marrow niche for haematopoietic stem cells. Nature, 505, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforow S & Ritz J (2016) Dramatic expansion of HSCs: new possibilities for HSC transplants? Cell Stem Cell, 18, 10–12. [DOI] [PubMed] [Google Scholar]
- Nishikawa H & Sakaguchi S (2010) Regulatory T cells in tumor immunity. International Journal of Cancer, 127, 759–767. [DOI] [PubMed] [Google Scholar]
- Nollet F, Berx G, Molemans F & van Roy F (1996) Genomic organization of the human beta- catenin gene (CTNNB1). Genomics, 32, 413–424. [DOI] [PubMed] [Google Scholar]
- Nowyhed HN, Huynh TR, Thomas GD, Blatchley A & Hedrick CC (2015) Cutting edge: the orphan nuclear receptor Nr4a1 regulates CD8+ T cell expansion and effector function through direct repression of Irf4. Journal of Immunology, 195, 3515–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossenkoppele G & Schuurhuis GJ (2013) MRD in AML: time for redefinition of CR? Blood, 121, 2166–2168. [DOI] [PubMed] [Google Scholar]
- Raaijmakers MH, Mukherjee S, Guo S, Zhang S, Kobayashi T, Schoonmaker JA, Ebert BL, Al-Shahrour F, Hasserjian RP, Scadden EO, Aung Z, Matza M, Merkenschlager M, Lin C, Rommens JM & Scadden DT (2010) Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature, 464, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalin AM & Aarnisalo P (2011) Cross-talk between NR4A orphan nuclear receptors and β-catenin signaling pathway in osteoblasts. Archives of Biochemistry and Biophysics, 509,44–51. [DOI] [PubMed] [Google Scholar]
- Ramirez-Herrick AM, Mullican SE, Sheehan AM & Conneely OM (2011) Reduced NR4A gene dosage leads to mixed myelodysplastic/ myeloproliferative neoplasms in mice. Blood, 117, 2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon S, Woeller CF & Phipps RP (2013) The influence of Cox-2 and bioactive lipids on hematological cancers. Current Angiogenesis, 2, 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger GS (2014) Current concepts in colorectal cancer prevention with cyclooxygenase inhibitors. Anticancer Research, 34, 6277–6282. [PubMed] [Google Scholar]
- Rezavand N, Khazaei M, Oliapanah E, Nikzad H & Khazaei MR (2013) Low doses of celecoxib stimulate human endometrium growth in A three-dimensional culture model. International Journal of Fertility & Sterility, 7, 7–12. [PMC free article] [PubMed] [Google Scholar]
- Rezvani K, Mielke S, Ahmadzadeh M, Kilical Y, Savani BN, Zeilah J, Keyvanfar K, Montero A, Hensel N, Kurlander R & Barrett AJ (2006) High donor FOXP3-positive regulatory T-cell (Treg) content is associated with a low risk of GVHD following HLA-matched allogeneic SCT. Blood, 108, 1291–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger K, Loddenkemper C, Maul J, Fietz T, Wolff D, Terpe H, Steiner B, Berg E, Miehlke S, Bornhäuser M, Schneider T, Zeitz M, Stein H, Thiel E, Duchmann R & Uharek L (2006) Mucosal FOXP3+ regulatory T cells are numerically deficient in acute and chronic GvHD. Blood, 107, 1717–1723. [DOI] [PubMed] [Google Scholar]
- Rozenberg A, Rezk A, Boivin MN, Darlington PJ, Nyirenda M, Li R, Jalili F, Winer R, Artsy EA, Uccelli A, Reese JS, Planchon SM, Cohen JA & Bar-Or A (2016) Human mesenchymal stem cells impact Th17 and Th1 responses through a prostaglandin E2 and myeloid-dependent mechanism. Stem Cells Translational Medicine, 5, 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC & Passegué E (2013) Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell, 13, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz AW, Wang J, Zhu ZJ, Johnson CH, Patti GJ & Siuzdak G (2013) Liquid chromatography quadrupole time-of-flight characterization of metabolites guided by the METLIN database. Nature Protocols, 8, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya T, Kashiwagi I, Yoshida R, Fukaya T, Morita R, Kimura A, Ichinose H, Metzger D, Chambon P & Yoshimura A (2013) Nr4a receptors are essential for thymic regulatory T cell development and immune homeostasis. Nature Immunology, 14, 230–237. [DOI] [PubMed] [Google Scholar]
- Sertorio M, Amarachintha S, Wilson A & Pang Q (2016) Loss of Fancc impairs antibody-secreting cell differentiation in mice through deregulating the Wnt signaling pathway. The Journal of Immunology, 196, 2986–2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicking I, Rommens K, Battista MJ, Böahm D, Gebhard S, Lebrecht A, Cotarelo C, Hoffmann G, Hengstler JG & Schmidt M (2014) Prognostic influence of cyclooxygenase-2 protein and mRNA expression in node-negative breast cancer patients. BMC Cancer, 14, 952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith WL, DeWitt DL & Garavito RM (2000) Cyclooxygenases: structural, cellular, and molecular biology. Annual Review of Biochemistry, 69, 145–182. [DOI] [PubMed] [Google Scholar]
- Smith AG, Lim W, Pearen M, Muscat GE & Sturm RA (2011) Regulation of NR4A nuclear receptor expression by oncogenic BRAF in melanoma cells. Pigment Cell Melanoma Research, 24, 551–563. [DOI] [PubMed] [Google Scholar]
- Smyth EM, Grosser T, Wang M, Yu Y & FitzGerald GA (2009) Prostanoids in health and disease. Journal of Lipid Research, 50, S423–S428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT & Purton LE (2007) A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor γ deficiency. Cell, 129, 1097–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A & Trumpp A (2006) Bone-marrow haematopoietic-stem-cell niches. Nature Review of Immunology, 6, 93–106. [DOI] [PubMed] [Google Scholar]
- Wu WK, Sung JJ, Lee CW, Yu J & Cho CH (2010) Cyclooxygenase-2 in tumorigenesis of gastrointestinal cancers: an update on the molecular mechanisms. Cancer Letter, 295, 7–16. [DOI] [PubMed] [Google Scholar]
- Wunderlich M, Chou FS, Link KA, Mizukawa B, Perry RL, Carroll M & Mulloy JC (2010) AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia, 24, 1785–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Brooks RA, Panchal R, Rhyasen GW, Danet-Desnoyers G & Mulloy JC (2014) OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood, 123, e134–e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Sun Z, Smyth K & Li L (2013) Wnt signaling inhibits CTL memory programming. Molecular Immunology, 56, 423–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Li X, Cole A, Sherman Z & Du W (2018) Reduced Cdc42 activity compromises hematopoiesis-supportive function of Fanconi anemia mesenchymal stromal cells. Stem Cells, 36, 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, Hair A, Holyoake TL, Huettner C, & Bhatia R (2012) Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell, 21, 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA & Rudensky AY (2007) Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature, 445, 936–940. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Du W, Koretsky T, Bagby GC & Pang Q (2008) TAT-mediated intracellular delivery of NPM-derived peptide induces apoptosis in leukemic cells and suppresses leukemo-genesis in mice. Blood, 112, 2474–2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, He Y, Xing W, Zhang P, Shi H, Chen S, Shi J, Bai J, Rhodes SD, Zhang F, Yuan J, Yang X, Zhu X, Li Y, Hanenberg H, Xu M, Robertson KA, Yuan W, Nalepa G, Cheng T, Clapp DW & Yang FC (2017) An abnormal bone marrow microenvironment contributes to hematopoietic dysfunction in Fanconi anemia. Haematologica, 102, 1017–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.