

Abstract
Endoplasmic reticulum (ER) stress is associated with development and progression of fibrotic diseases, including idiopathic pulmonary fibrosis (IPF). ER stress was first implicated in the pathogenesis of IPF >15 years ago with the discovery of disease-causing mutations in surfactant protein C, which result in a misfolded gene product in type II alveolar epithelial cells (AECs). ER stress and the unfolded protein response (UPR) have been linked to lung fibrosis through regulation of AEC apoptosis, epithelial-mesenchymal transition, myofibroblast differentiation, and M2 macrophage polarization. Although progress has been made in understanding the causes and consequences of ER stress in IPF and a number of chronic fibrotic disorders, further studies are needed to identify key factors that induce ER stress in important cell types and define critical down-stream processes and effector molecules that mediate ER stress-related phenotypes. This review discusses potential causes of ER stress induction in the lungs and current evidence linking ER stress to fibrosis in the context of individual cell types: AECs, fibroblasts, and macrophages. As our understanding of the relationship between ER stress and lung fibrosis continues to evolve, future studies will examine new strategies to modulate UPR pathways for therapeutic benefit.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a severe form of interstitial lung disease characterized by progressive dyspnea, exercise intolerance, hypoxemia, and respiratory failure [1]. The pathological equivalent of IPF, known as usual interstitial pneumonia (UIP), is identified by the presence of patchy areas of fibrotic remodeling in the distal lung parenchyma with fibroblastic foci [1,2]. Collagen deposition occurs in association with accumulation of activated (myo) fibroblasts in areas subjacent to the epithelial surface, which can be composed of hyperplastic type II alveolar epithelial cells (AECs) or epithelium with bronchiolar appearance [3]. Collapse of remodeled alveoli creates focal areas of fibrosis and stretches adjacent lung parenchyma, resulting in micro-honeycombing and traction bronchiectasis, which are characteristic of this disorder. In addition to AECs and fibroblasts, inflammatory cells are thought to contribute to the development of fibrosis [4]. Specifically, macrophages with the M2 phenotype are found within parenchymal areas in IPF and secrete mediators that can impact fibrosis [4,5]. The current paradigm to explain IPF is that repetitive epithelial injury, along with genetic factors, results in persistent fibroblast activation and ongoing collagen and matrix deposition.
The endoplasmic reticulum (ER) is a specialized organelle responsible for maintaining protein homeostasis or “proteostasis”. The ER is primarily required for protein folding and performing quality control of proteins before they reach their intracellular or extracellular destinations. Any condition that perturbs protein processing can lead to accumulation of misfolded proteins in the ER – a condition termed ER stress. In response to ER stress, the cell launches a signaling cascade termed the unfolded protein response (UPR). The UPR initially aims to restore proteostasis, but may result in cell death under prolonged or severe ER stress [6]. Chronic ER stress is a key contributor to numerous diseases including neurodegeneration, diabetes, cancer, and metabolic diseases [7–10]. Current evidence points to a critical pathogenic role of ER stress/UPR in fibrosis in a number of organs, including kidney, heart, liver, gastrointestinal tract, and lung [8]. Potential mechanistic links between ER stress and lung fibrosis were first identified >15 years ago with the discovery that familial IPF can be caused by mutations in surfactant protein C (SFTPC), which is produced by type II AECs [11–14]. When mutant SFTPC constructs were expressed in lung epithelial cell lines, the result was accumulation of mutant protein in ER and induction of ER stress [15–20]. Subsequently, immunohistochemistry studies from our group [15] and others [21] showed that ER stress markers are common in the lungs of patients with both familial and sporadic IPF. This review will focus on progress in understanding the role of ER stress in lung fibrosis.
Overview of ER stress and the UPR
A cell produces up to 4 × 106 proteins every minute with the ER bearing the responsibility of folding and processing at least a third of those [22,23]. Functions of the ER include orchestrating biogenesis, folding, assembly, and trafficking of proteins, as well as degradation of defective proteins [24–27]. ER function is tightly regulated by a combination of factors including protein load, metabolic demands of the cell, chaperone efficiency, redox balance, and calcium homeostasis [28–31]. Perturbations in any of these factors can lead to ER stress and activation of the UPR.
The UPR pathway consists of three ER transmembrane proteins: 1) PKR-like endoplasmic reticulum kinase (PERK), 2) activating transcription factor 6 (ATF6), and 3) inositol requiring enzyme 1 α (IRE1α) [24,25,27]. In an unstressed cell, binding immunoglobulin protein (BiP), which is a chaperone that facilitates protein folding [26,27], remains bound to PERK, ATF6 and IRE1α, and maintains these sensors in an inactive state. In a stressed cell with accumulation of proteins in the ER, BiP is sequestered away from the sensors, resulting in their activation and initiation of the UPR, which is designed to restore cellular homeostasis through reduction in overall protein translation along with selective increases in expression of critical chaperone and redox proteins [32,33].
PERK
On sensing ER stress, PERK is activated and undergoes auto-phosphorylation and dimerization [34]. PERK then phosphorylates the α subunit of eukaryotic translation initiation factor 2α (eIF2α) at Ser51, which is the only known direct target of PERK [35]. Phosphorylation of eIF2α leads to overall inhibition of protein synthesis. Phospho-eIF2α, however, can selectively up-regulate activating transcription factor 4 (ATF4), which is a transcription factor that induces expression of a set of genes required for cellular homeostasis [35–39]. ATF4 can also activate expression of pro-apoptotic factors, including growth arrest and DNA damage-inducible protein 34 (GADD34) [36,40] and C/EBP homologous protein (CHOP) [41,42].
ATF6
After its release from BiP, ATF6 translocates to the Golgi where it is cleaved into amino terminal and carboxy terminal (cytosplasmic) domains by site1 and site2 proteases [43,44]. Cleaved cytoplasmic ATF6 then translocates to the nucleus where it activates transcription of several chaperones proteins, such as BiP, calreticulin, and protein disulphide isomerase (PDI) [45]. Activated ATF6 also induces the expression of X-box binding protein 1 (XBP1), a downstream transcription factor activated through the IRE1α pathway [46,47].
IRE1α
When released from chaperone binding, IRE1α undergoes oligomerization and auto-phosphorylation, resulting in activation of an RNase domain that cleaves XBP1 into an active form. Spliced XBP1 (XBP1s) then activates expression of target genes, including ER degradation enhancing α-mannosidase like protein (EDEM), which promotes protein degradation through the ER-associated degradation (ERAD) pathway [48]. In some settings, IRE1α can be activated to degrade a wider range of ER-localized mRNAs through a process known as IRE1α-dependent decay of mRNA (RIDD), thus affecting cell survival and other cellular phenotypes [49,50].
Factors influencing ER stress in IPF
In addition to genetically determined expression of mutant proteins, ER stress can be induced by: 1) exogenous factors that increase protein expression (e.g. viral infections) or produce reactive oxygen species (ROS) and/or impair redox balance in cells (e.g. cigarette smoke, inhaled particulates), and 2) intracellular processes that impair bioenergetics (e.g. hypoxia, aging), cause calcium shifts (e.g. mechanical stretch), or reduce clearance of misfolded proteins (e.g. proteasomal dysfunction, impaired autophagy) (Fig. 1).
Fig. 1.
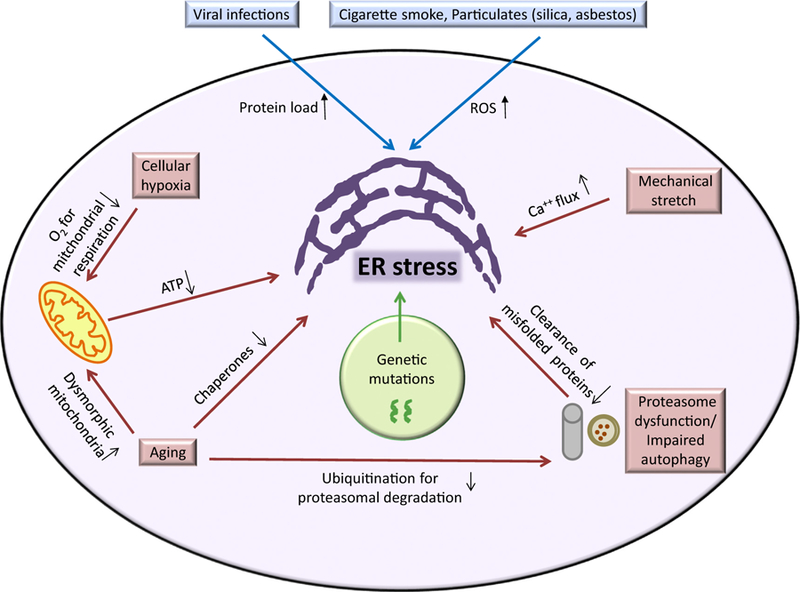
Potential causes of ER stress.
In IPF, several groups have detected herpesvirus antigens in lungs of patients by PCR and immunohistochemistry [51]. Our group has reported concomitant expression of herpesvirus antigens and ER stress markers in AECs in IPF patients [15] as well as in asymptomatic relatives of patients with familial IPF [52], thereby suggesting a role of herpesvirus-induced ER stress in lung fibrosis. Also, increased expression of a repertoire of hypoxia-inducible genes has been reported in IPF lungs [53,54] and in single cell analysis of AECs in IPF [55] supporting the existence of cellular hypoxia, which can induce ER stress [56].
Aging represents another ER stress-influencing factor in patients with IPF, whose median age of diagnosis is 66 years [57–59]. Aging can significantly impair ER function through reduction in chaperone production, increase in dysmorphic mitochondria (impacting bioenergetics), and inefficient proteasomal degradation of proteins [60–63]. Torres-Gonzalez et al. reported increased ER stress markers in type II AECs in old mice compared to young mice in an experimental model of lung fibrosis [64], thus supporting a connection between ER stress and aging. In addition to direct effects of aging, impairment in ER quality control mechanism (e.g. ERAD, autophagy) can contribute to ER stress [65,66]. In this regard, staining for microtubule-associated protein 1A/1B-light chain 3-II (LC3-II), which is reflective of autophagy, was shown to be absent in type II AECs in fibrotic lesions while expression was prominent in non-fibrotic areas [66].
Regulation of effector pathways by ER stress
ER stress and UPR signaling have been linked to fibrosis through apoptotic cell death, activation/ differentiation of fibroblasts, epithelial-mesenchymal transition (EMT), and activation or polarization of inflammatory responses [6,8,67].
Epithelial cell apoptosis is a key component of fibrotic diseases in a number of organs, including the lungs [3,6]. Prolonged or excessive ER stress can induce apoptosis in epithelial cells through several UPR-dependent downstream mechanisms, including induction of CHOP, activation of the ER bound caspase (caspase-4 in humans, caspase-12 in mice), or activation of c-Jun NH2 terminal kinase (JNK) [24–26,31]. Of these three pathways, apoptosis through induction of CHOP is best studied. CHOP is a member of the C/EBP family of transcription factors. It functions both as an inhibitor of C/EBP transcriptional activity and an inducer of gene targets through interactions with alternative enhancer sites [68,69]. CHOP regulates the apoptotic pathway through activation of pro-apoptotic genes and downregulation of anti-apoptotic genes, such as B-cell lymphoma 2 (BCL2) [70]. In most reports, CHOP is primarily induced downstream of PERK/ATF4, but all 3 arms of the UPR can participate in CHOP induction [71]. ER stress can also cause apoptosis through UPR-independent cell death mechanisms. For example, excessive ER stress releases calcium into the cytosol, which can trigger apoptosis through Bcl2-associated X protein (Bax) and Bcl2 homologous antagonist killer (Bak) [72].
In addition to effects on cell survival, ER stress can affect cell differentiation. In this regard, XBP1 contributes to plasma cell differentiation [73]. In the context of fibrosis, ER stress has been shown to affect differentiation of fibroblasts to myofibroblasts, which are an activated subset of fibroblasts common in fibrotic diseases that produce large amounts of collagen and other extracellular matrix components. For example, fibroblasts treated with ER stress-inducing agents are more susceptible to myofibroblast differentiation induced by TGFβ [74,75]. In epithelial cells, ER stress has been shown to induce EMT, resulting in a cellular phenotype that may directly participate in fibrotic remodeling [19,20,76]. In addition to in vitro studies showing ER stress-induced EMT, treatment of mice with cyclosporine, which can induce ER stress in renal tubular epithelial cells, has been shown to cause EMT through both TFGβ dependent and independent mechanisms [76,77].
Inflammation is linked to fibrosis in a number of organs, including lungs, and ER stress and the UPR pathway have been shown to regulate inflammatory signaling pathways, including nuclear factor kappa B (NF-κB) and activator protein-1 (AP-1) [78]. In inflammatory bowel disease, ER stress has been observed in colonic mucosa and the IRE1α pathway has been shown to regulate disease progression [79]. Accumulation of mutant muc2 (mucin in intestinal goblet cells) results in ER stress and inflammation in the large intestine with increased expression of interleukin-1β (IL-1β), tumor necrosis factor α (TNFα), and interferon-γ [80]. In bone marrow derived macrophages subjected to LPS stimulation, ER stress is able to drive production and processing of pro–IL-1β [81].
Macrophages can be polarized into M1 (proinflammatory) or M2 (pro-fibrotic) subsets based on microenvironmental stimuli [82]. Based on available data, ER stress may be able to skew macrophage populations towards either a M1 or a M2 phenotype depending on the pathophysiological context. In obesity, IRE1α deficiency has been reported to favor the M2 macrophage phenotype [83] and CHOP deficiency can increase both M1 and M2 macrophages [84]. However, in diabetes, pulmonary fibrosis, and allergic airway inflammation, ER stress has been reported to skew towards M2 polarization through JNK or CHOP dependent mechanisms [85–87].
Pro-fibrotic effects of ER stress in individual cell types in lung fibrosis
Pro-fibrotic effects of ER stress may be transduced through several different cell types in the lungs, including alveolar epithelial cells, fibroblasts, and macrophages (Fig. 2). The impact of ER stress on pro-fibrotic characteristics of each of these cell types is discussed below.
Fig. 2.
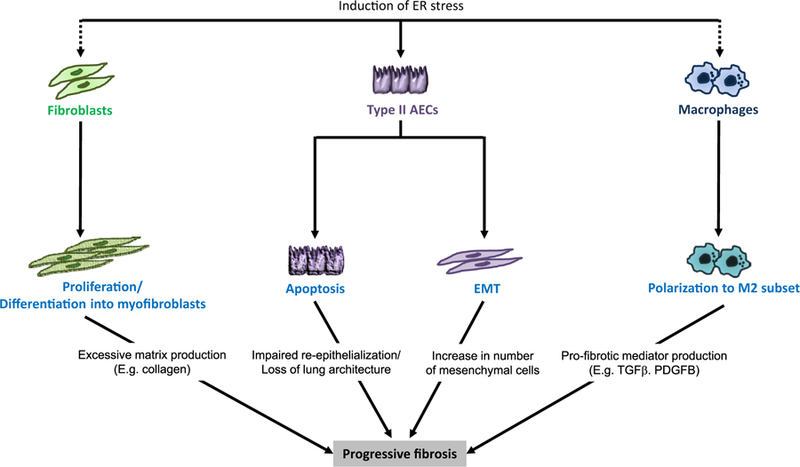
Pro-fibrotic effects of ER stress in different cell types in lung fibrosis.
Alveolar epithelial cells
In lungs of IPF patients, ER stress markers have been identified primarily in type II AECs [15,21]. By immunohistochemistry, we identified increased expression of BiP, EDEM, and XBP1 in AECs from patients with both familial and sporadic IPF [15]. In addition, Korfei et al. reported increased expression of ATF4, ATF6, and CHOP in AECs from lung sections obtained from IPF patients, along with evidence of apoptosis by immunostaining for cleaved caspase 3 [21].
In type II AEC lines, over-expression of mutant SFPTC genes (either exon 4 deleted or L188Q SFTPC) results in ER stress and increased apoptosis [15,17,18]. Constitutive expression of exon 4 deleted SFTPC in mice resulted in fetal lethality with disrupted lung morphogenesis and defective surfactant protein processing [88]. We generated doxycycline inducible transgenic mice in which mutant L188Q SFTPC is expressed in type II AECs [89]. When this transgene was induced in type II AECs or when the ER-stress inducing agent tunicamycin was administered through direct intratracheal instillation into the lung, ER stress was observed in type II AECs; however, fibrosis did not develop and AEC apoptosis was not identified. In contrast, treatment with low dose bleomycin resulted in a substantial increase in fibrosis with greater AEC apoptosis than treatment with bleomycin alone [89]. Together, these data indicate that ER stress in AECs, at least at the levels achievable in these models, is not sufficient to induce lung fibrosis. Rather, ER stress appears to sensitize to development of fibrosis following epithelial injury.
The mechanisms by which ER stress regulates survival/apoptosis of AECs are incompletely understood and may be context dependent. In cultured type II AECs over-expressing mutant forms of SFTPC, Mulegeta et al. identified caspase 4/12 as an important mediator of apoptosis [18]. Similarly, activation of caspase-12 and cleaved caspase-3 was detected in lungs of mice expressing L188Q SFTPC in type II AECs following bleomycin treatment [89]. CHOP has also been implicated in ER stress-dependent AEC apoptosis [21], although current studies show inconsistent effects of CHOP deletion on lung fibrosis. Tanaka et al. [90] reported that CHOP deficient mice have reduced AEC apoptosis and lung fibrosis after bleomycin treatment. In contrast, Ayaub et al. [91] found that CHOP deficient mice had higher mortality and increased fibrosis compared to wild-type controls following bleomycin treatment.
Several other mechanisms have been implicated in ER stress-induced AEC apoptosis, including regulation of ER Ca2+. Calmodulin-dependent kinase II (CAMKII) is an important regulator of ER Ca2+ and can modulate apoptosis [92]. Inhibition of CAMKII using a transgenic mice with AEC specific expression of the CaMKII inhibitor peptide AC3-I resulted in decreased intracellular Ca2+, reduced CHOP, decreased AEC apoptosis, and protection from fibrosis following bleomycin treatment [92]. In addition, Bueno M. et al. [62] showed that the mitochondrial protective factor PTEN-induced putative kinase 1 (PINK1) is decreased in IPF lungs. In experimental models, ER stress induction resulted in decreased PINK1 in AECs, along with altered bioenergetics and increased AEC apoptosis [62]. PINK1 loss can also induce ER stress, indicating the possibility of a feedback mechanism linking ER stress, mitochondrial dysfunction, and fibrotic remodeling [93].
In addition to AEC apoptosis, another possible mechanistic link between ER stress and fibrosis comes from studies in which ER stress-inducing agents or expression of mutant SFTPC were shown to induce EMT in AECs with increased expression of mesenchymal markers such as α smooth muscle actin (αSMA), vimentin, N-Cadherin and reduction of epithelial markers like E-Cadherin, and zona occludens-1 (ZO-1) [19,20]. Further studies suggested the IRE1α/XBP1 pathway is critical for regulating EMT in type II AECs [19]. Along these lines, induction of ER stress due to expression of mutant SFTPC (exon4 deletion) in A549 cells increased collagen production and secretion [94].
Fibroblasts
Myofibroblast differentiation is involved in wound healing and is characteristic of fibrotic diseases since these activated myofibroblasts produce increased amounts of collagen and matrix components [95]. In fibroblasts from IPF patients, pharmacological inhibition of ER stress through 4-phenylbutyric acid (4-PBA) treatment reduced TGFβ1-induced myofibroblast differentiation, αSMA expression, and collagen production [74]. Knockdown of the ER chaperone calreticulin using siRNA in mouse and human IPF fibroblasts reduced TGFβ1-induced collagen and fibronectin production [96]. Similarly, treatment of fibroblasts with an IRE1α inhibitor reduced collagen 1α2 and fibronectin expression and reduced autophagy in IPF lung fibroblasts following TGFβ treatment [75]. In addition, ER stress regulation of PI3K/AKT signaling has been implicated in fibroblast proliferation and differentiation [97]. Together, current data implicate ER stress in regulating myofibroblast differentiation in the lungs; however, whether this is a critical pathogenic mechanism in IPF remains to be determined.
Macrophages
Macrophages can contribute to lung fibrosis through secretion of pro-fibrotic mediators (e.g. TGFβ), chemokines, and matrix metalloproteases [4,98,99]. In the lungs, ER stress has been reported in macrophages obtained by bronchoalveolar lavage from asbestosis patients and alveolar macrophages from mice with asbestos-induced lung fibrosis [100]. In IPF, Yao et al. reported expression of CHOP in M2 macrophages in IPF patients [86].
Several groups have shown that ER stress can affect macrophage phenotypes; however, current studies are inconsistent regarding the impact of ER stress on M2 macrophage polarization. In mouse obesity models, genetic deficiency of IRE1α or CHOP resulted in increased M2 macrophages [83,84]. On the other hand, several studies have shown that induction of ER stress skews towards M2 polarization. Specifically, ER stress-induced JNK activation has been shown to induce M2 polarization in mouse peritoneal macrophages [85]. In lung macrophages, CHOP has been reported to induce M2 polarization in the bleomycin model of pulmonary fibrosis and a model of allergic airway inflammation [86,87]. Similarly, treatment of murine bone marrow-derived macrophages with PBA was reported to prevent palmitate-induced M2 polarization [101]. Although available studies suggest that the effects of ER stress on macrophage polarization depend on the disease context, ER stress in lung macrophages appears to favor the M2 phenotype.
In addition to effects on macrophage polarization, ER stress may impact fibrosis by inducing macrophage apoptosis, thereby abrogating the effects of M2 polarization. Ayaub et al. showed that CHOP expression induced macrophage apoptosis and protected from bleomycin-induced lung fibrosis [91]. Similarly, silica has been shown to induce alveolar macrophages apoptosis via ER stress in vitro [102]. Taken together, these data reveal a complex interplay between ER stress and macrophage phenotype. Further studies are needed to clarify whether the net effect of ER stress in macrophages is pathogenic or protective in different models of lung fibrosis.
Potential therapeutic approaches
There is substantial interest in targeting ER stress and UPR in many diseases; however, blocking the UPR should be approached with caution because of the adaptive and homeostatic nature of this system, as evidenced by prenatal or neonatal lethality of PERK, ATF6, IRE1, or XBP1 knockout mice [6,103–107]. Therefore, targeting primary UPR components could result in cellular toxicity rather than reduction of pathogenic effects. Despite these concerns, a small molecule inhibitor has been reported to allosterically modulate the RNAase activity of IRE1α oligomers (but not dimers) leading to cell survival under ER stress [108], thus suggesting that fine-tuned and nuanced targeting of upstream UPR mediators might prove beneficial. Downstream or terminal UPR effectors (e.g. CHOP) are not necessarily crucial for maintaining homeostasis in normal physiological conditions, as evidenced by CHOP knockout mice being perfectly viable [109]. Therefore, a productive strategy could be to target ER stress-downstream effectors that regulate cell survival and fibrotic remodeling.
An alternative approach to reduce ER stress could be to improve protein processing by improving chaperone functions through pharmacological agents. For example, the chemical chaperones 4-PBA and taurohyodeoxycholic acid (TUDCA) have been shown to have beneficial effects in mouse models of fibrotic diseases [90,110–114]; however, determination of the usefulness of these chaperones in IPF requires further work. Since ER stress and the integrated UPR require interplay of numerous intracellular factors, moving forward it would be important to take this biological complexity into account while choosing and targeting “druggable” nodes.
Future directions
Although current evidence suggests that ER stress-induced apoptosis in type II AECs is an important determinant of fibrosis susceptibility, further studies will be needed to delineate other potential pro-fibrotic effects of ER stress in these cells. Senescence biomarkers p16 and p21 have been observed in epithelial cells in IPF [115,116]. Therefore, the connection between ER stress and senescence, which has been reported in some cultured cells [117,118], represents an important area for future investigations. Secondly, in addition to ER stress caused by specific factors in individual cells, non-cell autonomous functions of UPR (transmissible ER stress) is another interesting area for future investigations. Recently, transmission of ER stress from one cell type to another has been reported in several settings, including diet-induced obesity and diabetes [119], viral myocarditis [120], and cancer [121–124]. Finally, while a combination of in vivo and in vitro studies have revealed signaling pathways involved in mediating the pro-fibrotic effects of ER stress, moving forward it will be critical to determine which of these pathways are predominantly dysfunctional in IPF patients. With upcoming technological advances, studies aimed at defining the genomic, transcriptomic, and proteomic signature of single cells isolated from IPF patients will likely fill knowledge gaps pertaining to the relative importance of different arms of the UPR and downstream pro-fibrotic pathways in this disease. A nuanced understanding of causes and mechanisms of ER stress in different cell types will hopefully better inform clinical studies aimed at targeting this important disease-mofifying pathway.
Conclusions
A variety of studies implicate ER stress as an important factor in progression of a variety of chronic, fibrotic diseases, including IPF. In the lung, ER stress appears to primarily localize in AECs, where this pathway has been linked to increased AEC apoptosis, as well as other pro-fibrotic phenotypic characteristics. In addition, ER stress in other cell types, including fibroblasts and macrophages, can affect pro-fibrotic responses by these cells. Further studies are required to define the upstream factors that induce ER stress in IPF and identify the effector molecules that mediate ER stress-related phenotypes. This information will facilitate new strategies to modulate these pathways for therapeutic benefit.
Acknowledgments
Supported by: American Heart Association Pre-doctoral Fellowship (15PRE25600007) (AB), P01 HL092870 (TSB), The Department of Veterans Affairs (TSB)
References
- [1].Mora AL, Rojas M, Pardo A, Selman M, Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease, Nat. Rev. Drug Discov 16 (11) (2017) 755–772. [DOI] [PubMed] [Google Scholar]
- [2].Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE Jr., Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ, An official ATS/ ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management, Am. J. Respir. Crit. Care Med 183 (6) (2011) 788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Selman M, Pardo A, Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers, Proc. Am. Thorac. Soc 3 (4) (2006) 364–372. [DOI] [PubMed] [Google Scholar]
- [4].Byrne AJ, Maher TM, Lloyd CM, Pulmonary macrophages: a new therapeutic pathway in Fibrosing lung disease? Trends Mol. Med 22 (4) (2016) 303–316. [DOI] [PubMed] [Google Scholar]
- [5].Byrne AJ, Mathie SA, Gregory LG, Lloyd CM, Pulmonary macrophages: key players in the innate defence of the airways, Thorax 70 (12) (2015) 1189–1196. [DOI] [PubMed] [Google Scholar]
- [6].Tanjore H, Blackwell TS, Lawson WE, Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis, Am. J. Phys. Lung Cell. Mol. Phys 302 (8) (2012) L721–L729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lenna S, Trojanowska M, The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis, Curr. Opin. Rheumatol 24 (6) (2012) 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tanjore H, Lawson WE, Blackwell TS, Endoplasmic reticulum stress as a pro-fibrotic stimulus, Biochim. Biophys. Acta 1832 (7) (2013) 940–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Oakes SA, Papa FR, The role of endoplasmic reticulum stress in human pathology, Annu. Rev. Pathol 10 (2015) 173–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang S, Kaufman RJ, The impact of the unfolded protein response on human disease, J. Cell Biol 197 (7) (2012) 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kabore AF, Wang WJ, Russo SJ, Beers MF, Biosynthesis of surfactant protein C: characterization of aggresome formation by EGFP chimeras containing propeptide mutants lacking conserved cysteine residues, J. Cell Sci 114 (Pt 2) (2001) 293–302. [DOI] [PubMed] [Google Scholar]
- [12].Maitra M, Wang Y, Gerard RD, Mendelson CR, Garcia CK, Surfactant protein A2 mutations associated with pulmonary fibrosis lead to protein instability and endoplasmic reticulum stress, J. Biol. Chem 285 (29) (2010) 22103–22113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nogee LM, Dunbar AE III, Wert SE, Askin F, Hamvas A, Whitsett JA, A mutation in the surfactant protein C gene associated with familial interstitial lung disease, N. Engl. J. Med 344 (8) (2001) 573–579. [DOI] [PubMed] [Google Scholar]
- [14].Thomas AQ, Lane K, Phillips III J, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, Roberts R, Haines J, Stahlman M, Loyd JE, Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred, Am. J. Respir. Crit. Care Med 165 (9) (2002) 1322–1328. [DOI] [PubMed] [Google Scholar]
- [15].Lawson WE, Crossno PF, Polosukhin VV, Roldan J, Cheng DS, Lane KB, Blackwell TR, Xu C, Markin C, Ware LB, Miller GG, Loyd JE, Blackwell TS, Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection, Am. J. Phys. Lung Cell. Mol. Phys 294 (6) (2008) L1119–L1126. [DOI] [PubMed] [Google Scholar]
- [16].Maguire JA, Mulugeta S, Beers MF, Endoplasmic reticulum stress induced by surfactant protein C BRICHOS mutants promotes proinflammatory signaling by epithelial cells, Am. J. Respir. Cell Mol. Biol 44 (3) (2011) 404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mulugeta S, Nguyen V, Russo SJ, Muniswamy M, Beers MF, A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation, Am. J. Respir. Cell Mol. Biol 32 (6) (2005) 521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mulugeta S, Maguire JA, Newitt JL, Russo SJ, Kotorashvili A, Beers MF, Misfolded BRICHOS SP-C mutant proteins induce apoptosis via caspase-4- and cytochrome c-related mechanisms, Am. J. Phys. Lung Cell. Mol. Phys 293 (3) (2007) L720–L729. [DOI] [PubMed] [Google Scholar]
- [19].Tanjore H, Cheng DS, Degryse AL, Zoz DF, Abdolrasulnia R, Lawson WE, Blackwell TS, Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress, J. Biol. Chem 286 (35) (2011) 30972–30980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhong Q, Zhou B, Ann DK, Minoo P, Liu Y, Banfalvi A, Krishnaveni MS, Dubourd M, Demaio L, Willis BC, Kim KJ, duBois RM, Crandall ED, Beers MF, Borok Z, Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein, Am. J. Respir. Cell Mol. Biol 45 (3) (2011) 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, Weaver TE, Guenther A, Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 178 (8) (2008) 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, Bennink JR, Yewdell JW, Quantitating protein synthesis, degradation, and endogenous antigen processing, Immunity 18 (3) (2003) 343–354. [DOI] [PubMed] [Google Scholar]
- [23].Guerriero CJ, Brodsky JL, The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology, Physiol. Rev 92 (2) (2012) 537–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Frakes AE, Dillin A, The UPR(ER): sensor and coordinator of organismal homeostasis, Mol. Cell 66 (6) (2017) 761–771. [DOI] [PubMed] [Google Scholar]
- [25].Hetz C, Saxena S, ER stress and the unfolded protein response in neurodegeneration, Nat. Rev. Neurol 13 (8) (2017) 477–491. [DOI] [PubMed] [Google Scholar]
- [26].Oyadomari S, Mori M, Roles of CHOP//GADD153 in endoplasmic reticulum stress, Cell Death Differ 11 (4) (2003) 381–389. [DOI] [PubMed] [Google Scholar]
- [27].Wouters BG, Koritzinsky M, Hypoxia signalling through mTOR and the unfolded protein response in cancer, Nat. Rev. Cancer 8 (11) (2008) 851–864. [DOI] [PubMed] [Google Scholar]
- [28].Oslowski CM, Urano F, Measuring ER stress and the unfolded protein response using mammalian tissue culture system, Methods Enzymol 490 (2011) 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Mandl J, Meszaros T, Banhegyi G, Csala M, Minireview: endoplasmic reticulum stress: control in protein, lipid, and signal homeostasis, Mol. Endocrinol 27 (3) (2013) 384–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Fu S, Watkins SM, Hotamisligil GS, The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling, Cell Metab 15 (5) (2012) 623–634. [DOI] [PubMed] [Google Scholar]
- [31].Ma Y, Hendershot LM, ER chaperone functions during normal and stress conditions, J. Chem. Neuroanat 28 (1–2) (2004) 51–65. [DOI] [PubMed] [Google Scholar]
- [32].Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D, Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response, Nat. Cell Biol 2 (6) (2000) 326–332. [DOI] [PubMed] [Google Scholar]
- [33].Schroder M, Kaufman RJ, The mammalian unfolded protein response, Annu. Rev. Biochem 74 (2005) 739–789. [DOI] [PubMed] [Google Scholar]
- [34].Liu CY, Schroder M, Kaufman RJ, Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum, J. Biol. Chem 275 (32) (2000) 24881–24885. [DOI] [PubMed] [Google Scholar]
- [35].Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG, Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation, Mol. Cell. Biol 21 (15) (2001) 5018–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Brush MH, Weiser DC, Shenolikar S, Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2, Mol. Cell. Biol 23 (4) (2003) 1292–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D, Perk is essential for translational regulation and cell survival during the unfolded protein response, Mol. Cell 5 (5) (2000) 897–904. [DOI] [PubMed] [Google Scholar]
- [38].Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D, An integrated stress response regulates amino acid metabolism and resistance to oxidative stress, Mol. Cell 11 (3) (2003) 619–633. [DOI] [PubMed] [Google Scholar]
- [39].Ma Y, Hendershot LM, Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress, J. Biol. Chem 278 (37) (2003) 34864–34873. [DOI] [PubMed] [Google Scholar]
- [40].Novoa I, Zeng H, Harding HP, Ron D, Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2+, J. Cell Biol 153 (5) (2001) 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fawcett TW, Martindale JL, Guyton KZ, Hai T, Holbrook NJ, Complexes containing activating transcription factor (ATF)/cAMP-responsive-element-binding protein (CREB) interact with the CCAAT/enhancer-binding protein (C/EBP)-ATF composite site to regulate Gadd153 expression during the stress response, Biochem. J 339 (Pt 1) (1999) 135–141. [PMC free article] [PubMed] [Google Scholar]
- [42].Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D, Regulated translation initiation controls stress-induced gene expression in mammalian cells, Mol. Cell 6 (5) (2000) 1099–1108. [DOI] [PubMed] [Google Scholar]
- [43].Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL, ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs, Mol. Cell 6 (6) (2000) 1355–1364. [DOI] [PubMed] [Google Scholar]
- [44].Shen J, Prywes R, Dependence of site-2 protease cleavage of ATF6 on prior site-1 protease digestion is determined by the size of the luminal domain of ATF6, J. Biol. Chem 279 (41) (2004) 43046–43051. [DOI] [PubMed] [Google Scholar]
- [45].Vekich JA, Belmont PJ, Thuerauf DJ, Glembotski CC, Protein disulfide isomerase-associated 6 is an ATF6-inducible ER stress response protein that protects cardiac myocytes from ischemia/reperfusion-mediated cell death, J. Mol. Cell. Cardiol 53 (2) (2012) 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee K, Tirasophon W, Shen X, Michalak M, Prywes R, Okada T, Yoshida H, Mori K, Kaufman RJ, IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response, Genes Dev 16 (4) (2002) 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K, XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor, Cell 107 (7) (2001) 881–891. [DOI] [PubMed] [Google Scholar]
- [48].Back SH, Lee K, Vink E, Kaufman RJ, Cytoplasmic IRE1alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress, J. Biol. Chem 281 (27) (2006) 18691–18706. [DOI] [PubMed] [Google Scholar]
- [49].Hollien J, Weissman JS, Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response, Science 313 (5783) (2006) 104–107. [DOI] [PubMed] [Google Scholar]
- [50].Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS, Regulated Ire1-dependent decay of messenger RNAs in mammalian cells, J. Cell Biol 186 (3) (2009) 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Moore BB, Moore TA, Viruses in idiopathic pulmonary fibrosis. Etiology and exacerbation, Ann. Am. Thorac. Soc 12 (Suppl. 2) (2015) S186–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kropski JA, Pritchett JM, Zoz DF, Crossno PF, Markin C, Garnett ET, Degryse AL, Mitchell DB, Polosukhin VV, Rickman OB, Choi L, Cheng DS, McConaha ME, Jones BR, Gleaves LA, McMahon FB, Worrell JA, Solus JF, Ware LB, Lee JW, Massion PP, Zaynagetdinov R, White ES, Kurtis JD, Johnson JE, Groshong SD, Lancaster LH, Young LR, Steele MP, Phillips Iii JA, Cogan JD, Loyd JE, Lawson WE, Blackwell TS, Extensive phenotyping of individuals at risk for familial interstitial pneumonia reveals clues to the pathogenesis of interstitial lung disease, Am. J. Respir. Crit. Care Med 191 (4) (2015) 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, Tsiambas E, Karameris A, Bouros D, Aidinis V, Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis, Am. J. Respir. Crit. Care Med 176 (11) (2007) 1108–1119. [DOI] [PubMed] [Google Scholar]
- [54].Kusko RL, Brothers JF 2nd, Tedrow J, Pandit K, Huleihel L, Perdomo C, Liu G, Juan-Guardela B, Kass D, Zhang S, Lenburg M, Martinez F, Quackenbush J, Sciurba F, Limper A, Geraci M, Yang I, Schwartz DA, Beane J, Spira A, Kaminski N, Integrated genomics reveals convergent transcriptomic networks underlying chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 194 (8) (2016) 948–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xi Y, Kim T, Brumwell AN, Driver IH, Wei Y, Tan V, Jackson JR, Xu J, Lee DK, Gotts JE, Matthay MA, Shannon JM, Chapman HA, Vaughan AE, Local lung hypoxia determines epithelial fate decisions during alveolar regeneration, Nat. Cell Biol 19 (8) (2017) 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fels DR, Koumenis C, The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth, Cancer Biol. Ther 5 (7) (2006) 723–728. [DOI] [PubMed] [Google Scholar]
- [57].Ley B, Collard HR, King TE Jr., Clinical course and prediction of survival in idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 183 (4) (2011) 431–440. [DOI] [PubMed] [Google Scholar]
- [58].American Thoracic Society, Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS), Am. J. Respir. Crit. Care Med 161 (2 Pt 1) (2000) 646–664. [DOI] [PubMed] [Google Scholar]
- [59].Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G, Incidence and prevalence of idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 174 (7) (2006) 810–816. [DOI] [PubMed] [Google Scholar]
- [60].Martinez G, Duran-Aniotz C, Cabral-Miranda F, Vivar JP, Hetz C, Endoplasmic reticulum proteostasis impairment in aging, Aging Cell 16 (4) (2017) 615–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bratic A, Larsson NG, The role of mitochondria in aging, J. Clin. Invest 123 (3) (2013) 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bueno M, Lai YC, Romero Y, Brands J, St CM, Kamga Croix C, Corey C, Herazo-Maya JD, Sembrat J, Lee JS, Duncan SR, Rojas M, Shiva S, Chu CT, Mora AL, PINK1 deficiency impairs mitochondrial homeostasis and promotes lung fibrosis, J. Clin. Invest 125 (2) (2015) 521–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Saez I, Vilchez D, The mechanistic links between proteasome activity, aging and age-related diseases, Curr. Genomics 15 (1) (2014) 38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Torres-Gonztílez E, Bueno M, Tanaka A, Krug LT, Cheng DS, Polosukhin VV, Sorescu D, Lawson WE, Blackwell TS, Rojas M, Mora AL, Role of endoplasmic reticulum stress in age-related susceptibility to lung fibrosis, Am. J. Respir. Cell Mol. Biol 46 (6) (2012) 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ruggiano A, Foresti O, Carvalho P, Quality control: ER-associated degradation: protein quality control and beyond, J. Cell Biol 204 (6) (2014) 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K, Insufficient autophagy in idiopathic pulmonary fibrosis, Am. J. Phys. Lung Cell. Mol. Phys 304 (1) (2013) L56–69. [DOI] [PubMed] [Google Scholar]
- [67].Zhang L, Wang Y, Pandupuspitasari NS, Wu G, Xiang X, Gong Q, Xiong W, Wang CY, Yang P, Ren B, Endoplasmic reticulum stress, a new wrestler, The Pathogenesis of Idiopathic Pulmonary Fibrosis, Am J Transl. Res, 9(2), 2017, pp. 722–735. [PMC free article] [PubMed] [Google Scholar]
- [68].Cassel TN, Nord M, C/EBP transcription factors in the lung epithelium, Am. J. Physiol. Lung Cell. Mol. Physiol 285 (4) (2003) L773–L781. [DOI] [PubMed] [Google Scholar]
- [69].Lekstrom-Himes J, Xanthopoulos KG, Biological role of the CCAAT/enhancer-binding protein family of transcription factors, J. Biol. Chem 273 (44) (1998) 28545–28548. [DOI] [PubMed] [Google Scholar]
- [70].McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ, Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state, Mol. Cell. Biol 21 (4) (2001) 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ma Y, Brewer JW, Diehl JA, Hendershot LM, Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response, J. Mol. Biol 318 (5) (2002) 1351–1365. [DOI] [PubMed] [Google Scholar]
- [72].Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ, BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis, Science 300 (5616) (2003) 135–139. [DOI] [PubMed] [Google Scholar]
- [73].Reimold AM, Iwakoshi NN, Manis J, Vallabhajosyula P, Szomolanyi-Tsuda E, Gravallese EM, Friend D, Grusby MJ, Alt F, Glimcher LH, Plasma cell differentiation requires the transcription factor XBP-1, Nature 412 (6844) (2001) 300–307. [DOI] [PubMed] [Google Scholar]
- [74].Baek HA, Kim DS, Park HS, Jang KY, Kang MJ, Lee DG, Moon WS, Chae HJ, Chung MJ, Involvement of endoplasmic reticulum stress in myofibroblastic differentiation of lung fibroblasts, Am. J. Respir. Cell Mol. Biol 46 (6) (2011) 731–739 (2011–0121OC) [DOI] [PubMed] [Google Scholar]
- [75].Ghavami S, Yeganeh B, Zeki AA, Shojaei S, Kenyon NJ, Ott S, Samali A, Patterson J, Alizadeh J, Moghadam AR, Dixon IMC, Unruh H, Knight DA, Post M, Klonisch T, Halayko AJ, Autophagy and the unfolded protein response promote pro-fibrotic effects of TGFbeta1 in human lung fibroblasts, Am. J. Phys. Lung Cell. Mol. Phys 314 (0) (2017) L493–L504 (ajplung). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].McMorrow T, Gaffney MM, Slattery C, Campbell E, Ryan MP, Cyclosporine A induced epithelial-mesenchymal transition in human renal proximal tubular epithelial cells, Nephrol. Dial. Transplant 20 (10) (2005) 2215–2225. [DOI] [PubMed] [Google Scholar]
- [77].Pallet N, Bouvier N, Bendjallabah A, Rabant M, Flinois JP, Hertig A, Legendre C, Beaune P, Thervet E, Anglicheau D, Cyclosporine-induced endoplasmic reticulum stress triggers tubular phenotypic changes and death, Am. J. Transplant 8 (11) (2008) 2283–2296. [DOI] [PubMed] [Google Scholar]
- [78].Hotamisligil GS, Endoplasmic reticulum stress and the inflammatory basis of metabolic disease, Cell 140 (6) (2010) 900–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, Blumberg RS, XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease, Cell 134 (5) (2008) 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, Thornton DJ, Png CW, Crockford TL, Cornall RJ, Adams R, Kato M, Nelms KA, Hong NA, Florin TH, Goodnow CC, McGuckin MA, Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis, PLoS Med 5 (3) (2008), e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Shenderov K, Riteau N, Yip R, Mayer-Barber KD, Oland S, Hieny S, Fitzgerald P, Oberst A, Dillon CP, Green DR, Cerundolo V, Sher A, Cutting edge: endoplasmic reticulum stress licenses macrophages to produce mature IL-1beta in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway, J. Immunol 192 (5) (2014) 2029–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Braga TT, Agudelo JS, Camara NO, Macrophages during the fibrotic process: M2 as friend and foe, Front. Immunol 6 (2015) 602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Shan B, Wang X, Wu Y, Xu C, Xia Z, Dai J, Shao M, Zhao F, He S, Yang L, Zhang M, Nan F, Li J, Liu J, Liu J, Jia W, Qiu Y, Song B, Han JJ, Rui L, Duan SZ, Liu Y, The metabolic ER stress sensor IRE1alpha suppresses alternative activation of macrophages and impairs energy expenditure in obesity, Nat. Immunol 18 (5) (2017) 519–529. [DOI] [PubMed] [Google Scholar]
- [84].Grant R, Nguyen KY, Ravussin A, Albarado D, Youm YH, Dixit VD, Inactivation of C/ebp homologous protein-driven immune-metabolic interactions exacerbate obesity and adipose tissue leukocytosis, J. Biol. Chem 289 (20) (2014) 14045–14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Oh J, Riek AE, Weng S, Petty M, Kim D, Colonna M, Cella M, Bernal-Mizrachi C, Endoplasmic reticulum stress controls M2 macrophage differentiation and foam cell formation, J. Biol. Chem 287 (15) (2012) 11629–11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Yao Y, Wang Y, Zhang Z, He L, Zhu J, Zhang M, He X, Cheng Z, Ao Q, Cao Y, Yang P, Su Y, Zhao J, Zhang S, Yu Q, Ning Q, Xiang X, Xiong W, Wang CY, Xu Y, Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production, Mol. Ther 24 (5) (2016) 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Wang Y, Zhu J, Zhang L, Zhang Z, He L, Mou Y, Deng Y, Cao Y, Yang P, Su Y, Zhao J, Zhang S, Yu Q, Hu J, Chen Z, Ning Q, Xiang X, Xu Y, Wang CY, Xiong W, Role of C/EBP homologous protein and endoplasmic reticulum stress in asthma exacerbation by regulating the IL-4/signal transducer and activator of transcription 6/ transcription factor EC/IL-4 receptor alpha positive feedback loop in M2 macrophages, J. Allergy Clin. Immunol 140 (6) (2017) 1550–1561 e8. [DOI] [PubMed] [Google Scholar]
- [88].Bridges JP, Wert SE, Nogee LM, Weaver TE, Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice, J. Biol. Chem 278 (52) (2003) 52739–52746. [DOI] [PubMed] [Google Scholar]
- [89].Lawson WE, Cheng DS, Degryse AL, Tanjore H, Polosukhin VV, Xu XC, Newcomb DC, Jones BR, Roldan J, Lane KB, Morrisey EE, Beers MF, Yull FE, Blackwell TS, Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs, Proc. Natl. Acad. Sci. U. S. A 108 (26) (2011) 10562–10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Tanaka Y, Ishitsuka Y, Hayasaka M, Yamada Y, Miyata K, Endo M, Kondo Y, Moriuchi H, Irikura M, Tanaka KI, Mizushima T, Oike Y, Irie T, The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice, Pharmacol. Res 99 (Supplement C) (2015) 52–62. [DOI] [PubMed] [Google Scholar]
- [91].Ayaub EA, Kolb PS, Mohammed-Ali Z, Tat V, Murphy J, Bellaye PS, Shimbori C, Boivin FJ, Lai R, Lynn EG, Lhotak S, Bridgewater D, Kolb MR, Inman MD, Dickhout JG, Austin RC, Ask K, GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis, J. Pathol 239 (4) (2016) 411–425. [DOI] [PubMed] [Google Scholar]
- [92].Winters CJ, Koval O, Murthy S, Allamargot C, Sebag SC, Paschke JD, Jaffer OA, Carter AB, Grumbach IM, CaMKII inhibition in type II pneumocytes protects from bleomycin-induced pulmonary fibrosis by preventing Ca(2+)-dependent apoptosis, Am. J. Phys. Lung Cell. Mol. Phys 310 (1) (2016) L86–L94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Celardo I, Costa AC, Lehmann S, Jones C, Wood N, Mencacci NE, Mallucci GR, Loh SH, Martins LM, Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease, Cell Death Dis 7 (6) (2016), e2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Nakajima F, Aratani S, Fujita H, Yagishita N, Ichinose S, Makita K, Setoguchi Y, Nakajima T, Synoviolin inhibitor LS-102 reduces endoplasmic reticulum stress-induced collagen secretion in an in vitro model of stress-related interstitial pneumonia, Int. J. Mol. Med 35 (1) (2015) 110–116. [DOI] [PubMed] [Google Scholar]
- [95].Klingberg F, Hinz B, White ES, The myofibroblast matrix: implications for tissue repair and fibrosis, J. Pathol 229 (2) (2013) 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zimmerman KA, Graham LV, Pallero MA, Murphy-Ullrich JE, Calreticulin regulates transforming growth factor−+¦-stimulated extracellular matrix production, J. Biol. Chem 288 (20) (2013) 14584–14598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, Hung SC, Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis, 7 ed., 2017. [DOI] [PMC free article] [PubMed]
- [98].Young LR, Gulleman PM, Short CW, Tanjore H, Sherrill T, Qi A, McBride AP, Zaynagetdinov R, Benjamin JT, Lawson WE, Novitskiy SV, Blackwell TS, Epithelial-macrophage interactions determine pulmonary fibrosis susceptibility in Hermansky–Pudlak syndrome, JCI Insight 1 (17) (2016), e88947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Young LR, Borchers MT, Allen HL, Gibbons RS, McCormack FX, Lung-restricted macrophage activation in the pearl mouse model of Hermansky–Pudlak syndrome, J. Immunol 176 (7) (2006) 4361–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Ryan AJ, Larson-Casey JL, He C, Murthy S, Carter AB, Asbestos-induced disruption of calcium homeostasis induces endoplasmic reticulum stress in macrophages, J. Biol. Chem 289 (48) (2014) 33391–33403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Xiu F, Diao L, Qi P, Catapano M, Jeschke MG, Palmitate differentially regulates the polarization of differentiating and differentiated macrophages, Immunology 147 (1) (2016) 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Hu YB, Wu X, Qin XF, Wang L, Pan PH, Role of endoplasmic reticulum stress in silica-induced apoptosis in RAW264.7 cells, Biomed. Environ. Sci 30 (8) (2017) 591–600. [DOI] [PubMed] [Google Scholar]
- [103].Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D, Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival, Mol. Cell 7 (6) (2001) 1153–1163. [DOI] [PubMed] [Google Scholar]
- [104].Lee AH, Chu GC, Iwakoshi NN, Glimcher LH, XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands, EMBO J 24 (24) (2005) 4368–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Reimold AM, Etkin A, Clauss I, Perkins A, Friend DS, Zhang J, Horton HF, Scott A, Orkin SH, Byrne MC, Grusby MJ, Glimcher LH, An essential role in liver development for transcription factor XBP-1, Genes Dev 14 (2) (2000) 152–157. [PMC free article] [PubMed] [Google Scholar]
- [106].Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ, The unfolded protein response sensor IRE1alpha is required at 2 distinct steps in B cell lymphopoiesis, J. Clin. Invest 115 (2) (2005) 268–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ, ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress, Dev. Cell 13 (3) (2007) 351–364. [DOI] [PubMed] [Google Scholar]
- [108].Ghosh R, Wang L, Wang ES, Perera BG, Igbaria A, Morita S, Prado K, Thamsen M, Caswell D, Macias H, Weiberth KF, Gliedt MJ, Alavi MV, Hari SB, Mitra AK, Bhhatarai B, Schurer SC, Snapp EL, Gould DB, German MS, Backes BJ, Maly DJ, Oakes SA, Papa FR, Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress, Cell 158 (3) (2014) 534–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D, CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum, Genes Dev 12 (7) (1998) 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Choi SW, Ryu OH, Choi SJ, Song IS, Bleyer AJ, Hart TC, Mutant tamm-horsfall glycoprotein accumulation in endoplasmic reticulum induces apoptosis reversed by colchicine and sodium 4-phenylbutyrate, J. Am. Soc. Nephrol 16 (10) (2005) 3006–3014. [DOI] [PubMed] [Google Scholar]
- [111].Liu SH, Yang CC, Chan DC, Wu CT, Chen LP, Huang JW, Hung KY, Chiang CK, Chemical chaperon 4-phenylbutyrate protects against the endoplasmic reticulum stress-mediated renal fibrosis in vivo and in vitro, Oncotarget 7 (16) (2016) 22116–22127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rani S, Sreenivasaiah PK, Kim JO, Lee MY, Kang WS, Kim YS, Ahn Y, Park WJ, Cho C, Kim DH, Tauroursodeoxycholic acid (TUDCA) attenuates pressure overload-induced cardiac remodeling by reducing endoplasmic reticulum stress, PLoS One 12 (4) (2017), e0176071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, Hung SC, Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis, Sci. Rep 7 (1) (2017), 14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Cao SS, Zimmermann EM, Chuang BM, Song B, Nwokoye A, Wilkinson JE, Eaton KA, Kaufman RJ, The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice, Gastroenterology 144 (5) (2013) 989–1000 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, Hara N, P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis, Am. J. Respir. Crit. Care Med 154 (2 Pt 1) (1996) 477–483. [DOI] [PubMed] [Google Scholar]
- [116].Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, Oberg AL, Birch J, Salmonowicz H, Zhu Y, Mazula DL, Brooks RW, Fuhrmann-Stroissnigg H, Pirtskhalava T, Prakash YS, Tchkonia T, Robbins PD, Aubry MC, Passos JF, Kirkland JL, Tschumperlin DJ, Kita H, LeBrasseur NK, Cellular senescence mediates fibrotic pulmonary disease, Nat. Commun 8 (2017), 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, Pointer JN, Gruber SB, Su LD, Nikiforov MA, Kaufman RJ, Bastian BC, Soengas MS, Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway, Nat. Cell Biol 8 (10) (2006) 1053–1063. [DOI] [PubMed] [Google Scholar]
- [118].Matos L, Gouveia AM, Almeida H, ER stress response in human cellular models of senescence, J. Gerontol. A Biol. Sci. Med. Sci 70 (8) (2015) 924–935. [DOI] [PubMed] [Google Scholar]
- [119].Williams KW, Liu T, Kong X, Fukuda M, Deng Y, Berglund ED, Deng Z, Gao Y, Liu T, Sohn JW, Jia L, Fujikawa T, Kohno D, Scott MM, Lee S, Lee CE, Sun K, Chang Y, Scherer PE, Elmquist JK, Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis, Cell Metab 20 (3) (2014) 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Zhang H, Yue Y, Sun T, Wu X, Xiong S, Transmissible endoplasmic reticulum stress from myocardiocytes to macrophages is pivotal for the pathogenesis of CVB3-induced viral myocarditis, Sci. Rep 7 (2017), 42162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Mahadevan NR, Zanetti M, Tumor stress inside out: cell-extrinsic effects of the unfolded protein response in tumor cells modulate the immunological landscape of the tumor microenvironment, J. Immunol 187 (9) (2011) 4403–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Mahadevan NR, Rodvold J, Sepulveda H, Rossi S, Drew AF, Zanetti M, Transmission of endoplasmic reticulum stress and pro-inflammation from tumor cells to myeloid cells, Proc. Natl. Acad. Sci. U. S. A 108 (16) (2011) 6561–6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M, Cell-extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8(+) T cell priming, PLoS One 7 (12) (2012), e51845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR, Willert K, Lin JH, Zanetti M, Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells, Sci. Signal 10 (482) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]