

SUMMARY
Most genes mutated in microcephaly patients are expressed ubiquitously, and yet the brain is the only major organ compromised in most patients. Why the phenotype remains brain specific is poorly understood. In this study, we used in vitro differentiation of human embryonic stem cells to monitor the effect of a point mutation in kinetochore null protein 1 (KNL1;CASC5), identified in microcephaly patients, during in vitro brain development. We found that neural progenitors bearing a patient mutation showed reduced KNL1 levels, aneuploidy, and an abrogated spindle assembly checkpoint. By contrast, no reduction of KNL1 levels or abnormalities was observed in fibroblasts and neural crest cells. We established that the KNL1 patient mutation generates an exonic splicing silencer site, which mainly affects neural progenitors because of their higher levels of splicing proteins. Our results provide insight into the brain-specific phenomenon, consistent with microcephaly being the only major phenotype of patients bearing KNL1 mutation.
Graphical Abstract
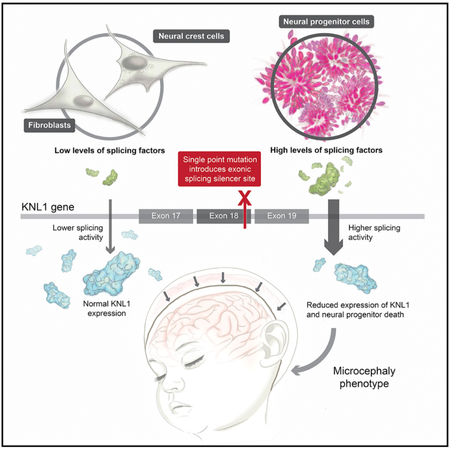
In Brief
Using 3D neural spheroids, Javed et al. investigate a mutation in KNL1 that causes microcephaly. Their study shows that, despite ubiquitous mutant KNL1 expression, KNL1 mRNA processing is affected only in neural precursors due to difference in splicing protein levels, offering insights into why the phenotype remains brain specific in patients.
INTRODUCTION
One approach for investigating recent human brain evolution is to study brain size regulator genes (Cox et al., 2006). Many such genes have been recognized because mutations in their sequence were identified in microcephalic patients (Faheem et al., 2015). Mutations in genes linked to autosomal recessive primary microcephaly result in a head circumference similar to that of early hominids, suggesting their involvement in brain evolution (Ponting and Jackson, 2005; Shi et al., 2017). These genes encode proteins that localize to the cell-division machinery and likely play important roles in this process. In the case of most mutations, it has been speculated that changes to the centrosome or spindle apparatus influence many processes such as cell survival and cell division, reducing the number of neural progenitors and, over the course of development, the total number of neurons (Thornton and Woods, 2009; Woods et al., 2005).
Individuals with microcephaly usually display body height and weight similar to that of the normal population, suggesting a brain-specific effect of the mutation (Woods et al., 2005). However, kinetochore null protein 1 (KNL1), a known microcephaly gene, is ubiquitously expressed (Hayette et al., 2000) and is essential for kinetochore assembly, chromosome segregation, and spindle assembly checkpoint (SAC) signaling (Bolanos-Garcia et al., 2011; Cheeseman et al., 2004, 2006, 2008; Kiyomitsu et al., 2007), suggesting that it is relevant for all dividing cell types. Thus, it is unclear why the KNL1 mutant phenotype remains brain specific instead of generally compromising organismal growth and viability or predisposing an individual to tumor formation due to increased aneuploidy.
Homozygous point mutations in KNL1 have been identified in microcephaly patients (Genin et al., 2012; Jamieson et al., 1999; Saadi et al., 2016; Szczepanski et al., 2016). The function of KNL1 during neurogenesis has never been studied, though RNA sequencing of human neocortex at 13–16 gestational weeks showed that KNL1 is upregulated in the ventricular zone, the brain layer with active neural progenitor proliferation, and downregulated in the cortical plate (Fietz et al., 2012). In addition, the publicly available human brain expression Brain-Span website showed that KNL1 expression is highest at the 9th gestational week, at the onset of neurogenesis, and decreases after birth, suggesting a role of KNL1 during brain development (Shi et al., 2017).
Here, we studied the role of KNL1 in brain development by introducing a point mutation identified in patients with microcephaly into human embryonic stem cells (hESCs) (Genin et al., 2012). We observed that mutant neural progenitors, derived from this hESC line, presented reduced levels of KNL1, aneuploidy, a decreased proliferation rate, increased cell death, and an abrogated spindle assembly checkpoint. Furthermore, when cultured in a 3D neural spheroid system, the overall size was reduced due to the depletion of neural progenitors in favor of premature differentiation. As opposed to neural progenitors, mutant fibroblasts and neural crest cells, derived from the same parental stem cell lines, did not present a reduction of KNL1 levels, cell growth, or genomic integrity. We revealed that the KNL1 point mutation disrupts an exonic splicing enhancer site and generates an exonic splicing silencer site. The newly generated exonic splicing silencer site is recognized by the inhibitory splicing protein heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), which is highly expressed in neural progenitors, leading to a cell-specific phenotype. This phenotype has not been previously reported and could provide a new paradigm for understanding microcephaly.
RESULTS
Neural Progenitors Bearing a KNL1 Point Mutation Have Reduced KNL1 Expression and Protein Levels
To assess the molecular mechanism underlying KNL1 function and its relevance in microcephaly, we designed a CRISPR/Cas9 targeting strategy in hESCs to recreate one of the point mutations identified in individuals with microcephaly (Genin et al., 2012). The homozygous missense coding-variant changes guanine to adenine at position 6125 in exon 18 of the KNL1 gene (also referred to as CASC5) (c.6125G > A; p.M2041I), which encodes an amino acid located at the KNL1 C terminus in the region proposed to interact with ZW10 Interacting Kinetochore Protein (ZWINT-1), a kinetochore protein that mediates kinetochore assembly (Kiyomitsu et al., 2011) (Figure 1A). The homozygous hESC clones were further validated by Sanger sequencing (Figure 1B). Each subsequent experiment was performed on one non-targeted wild-type clone, two independent targeted wild-type clones, and three independent targeted patient mutation clones derived from a single CRISPR targeting experiment performed on the hESC WIBR3 background (Lengner et al., 2010). Using the same strategy, an additional induced pluripotent stem cell (iPSC) line was generated and further characterized (Figure S1).
Figure 1. Neural Progenitors Bearing KNL1 Patient Point Mutation Have a Newly Formed Exonic Splicing Silencer Site and a Reduced Level of KNL1.

(A) Cas9 nuclease is targeted to genomic KNL1 (green) and mediates a double-stranded break 3 bp upstream of the 5′-NGG motif (arrow). Cas9-gRNA-single strand oligonucleotides (ssODN) co-electroporation was performed to allow the insertion of the homozygous mutation in the genome (red).
(B) Sanger sequencing on targeted hESC clones, confirming a homozygous patient point mutation.
(C) Immunostaining using antibodies against NESTIN (red) and PAX6 (green) on neural progenitors at 10 days post-differentiation (scale bar, 25 μm).
(D) Non-quantitative touchdown PCR strategy to amplify the region flanking the point mutation (exon 18) from cDNA and the resulting agarose gel electrophoresis from wild-type and KNL1c.6125G > A neural progenitors.
(E) Real-time qPCR analysis of KNL1 expression in neural progenitors, normalized to GAPDH. Welch’s t test (two-tailed) was applied.
(F) Western blot analysis of KNL1, HNRNPA1, SRSF7, SRSF10, and Vinculin in wild-type and KNL1c.6125G > A neural progenitors.
(G) Quantification of KNL1 protein levels, normalized to Vinculin in wild-type and KNL1c.6125G > A neural progenitors. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
(H) The point mutation generated a new ESS sites based on (1, blue) an algorithm from Sironi et al. (2004), (2, orange) an algorithm from Zhang and Chasin (2004), and (3, green) human splicing finder (HSF) matrices.
(I) RNA immunoprecipitation assay, using SRSF7, SRSF10, and HNRNPA1 antibody, followed by real-time qPCR to detect KNL1 RNA in wild-type and KNL1c.6125G > A neural progenitors.
One non-targeted wild-type clone, two wild-type clones, and three patient mutation clones derived from the same CRISPR-Cas9 targeting are plotted in each graph. Results are mean ± SEM. *p < 0.05, ***p < 0.001.
It has been reported that in patients fibroblasts and lymphoblastoid cell lines, the c.6125G > A point mutation disrupts an exon splicing enhancer (ESE) site, leading to the partial skipping of exon 18 (Genin et al., 2012). This is predicted to result in a premature stop codon in exon 19. Since the ESE sites are not the only motif recognized by the splicing machinery, the full form of the KNL1 transcript is still present in patient cells. Surprisingly, a prior study that used patient-derived material found that, despite the presence of the truncated transcript, patient fibroblasts and lymphoblastoids did not display altered KNL1 total protein levels (Genin et al., 2012). The mechanisms underlying this truncated mRNA transcript/protein discrepancy remain unexplored.
To investigate phenotypes that may represent in vitro correlates of microcephaly, we differentiated the isogenic parental and KNL1 mutant cell lines into neural progenitors using a SMAD-inhibition protocol (Muffat et al., 2016). At 10 days, the majority of cells were PAX6+/NESTIN+, confirming their neural progenitor identity (Figure 1C). Based on a non-quantitative touchdown PCR strategy, we detected a truncated form of KNL1 mRNA present in the neural progenitors containing the point mutation KNL1c.6125G > A (Figure 1D). To test the possibility that the truncated mRNA was degraded through nonsense-mediated decay, KNL1 mRNA and protein expression were monitored. KNL1c.6125G > A neural progenitors also displayed a statistically significant decrease in KNL1 mRNA (Figure 1E) and protein expression (Figures 1F and 1G), consistent with nonsense-mediated decay occurring as a result of the presence of the premature stop codon.
ESE sites are not the only splicing regulatory element present on exons, and it is possible that the engineered mutation interferes with other sites such as exonic splicing silencing (ESS) sites. Contrary to ESE sites, which promote the inclusion of an exon in the mRNA transcript by recruiting serine arginine (SR) splicing protein, ESS sites stimulate the exclusion of the exon and are recognized by inhibitory splicing factor such as hetereogenous nuclear ribonucleoproteins (hnRNPs) (Wang and Burge, 2008). We used the Human Splicing Finder bioinformatics tool, which combines 12 different algorithms to calculate effects of the point mutation on other splicing regulatory elements (Desmet et al., 2009). We found that the point mutation not only disrupted an ESE site but also generated an ESS site (Figure 1H). The Human Splicing Finder bioinformatics tool projected that the disrupted ESE site is most likely recognized by the exon enhancer Serine/arginine-Rich Splicing Factor 7 (SRSF7) and Serine/arginine-Rich Splicing Factor 10 (SRSF10), and that the newly generated ESS site mediates the interaction with the exon silencer protein HNRNPA1. We first evaluated the expression of these splicing factors in isogenic neural progenitors populations and confirmed that they are expressed at similar levels between the two genotypes (Figure 1F). To further investigate the interaction of KNL1 with SRSF7, SRSF10 and HNRNPA1 splicing protein in wild-type and KNL1c.6125G > A neural progenitors, we performed an RNA immunoprecipitation assay followed by a real-time qPCR and showed that the interaction between KNL1 mRNA and SRSF7/10 decreased, whereas its interaction with HNRNPA1 increased significantly in KNL1c.6125G > A neural progenitors (Figure 1H).
These data validate the predicted binding activity and confirm the disruption of an ESE site coinciding with the generation of an ESS site in KNL1c.6125G > A neural progenitors, which might lead to the reduction of KNL1. The decreased level of KNL1 described here in KNL1c.6125G > A neural progenitors reflects a much stronger difference from the one previously reported in patient fibroblasts and lymphoblastoid cells (Genin et al., 2012) and may reflect neural-specific differences in KNL1 mRNA processing.
KNL1c.6125G > A Neural progenitors Have Reduced Cell Growth, Altered Cell-Cycle Phases, and Higher Cell-Death Rate
It has been shown that KNL1 plays a role in cell division in human cells (Caldas and DeLuca, 2014; Cheeseman et al., 2008; Kiyomitsu et al., 2007). We first investigated the impact of the KNL1 point mutation on cell growth in hESCs-derived neural progenitors. KNL1c.6125G > A neural progenitors showed reduced growth rate when compared with isogenic wild-type controls as measured by an ATP assay (Figure 2A). Since both reduced proliferation rate and increased cell death can contribute to reduced growth, we measured cell-cycle distribution in neural progenitors labeled with propidium iodide using fluorescence-activated cell sorting. Consistent with altered cell division, we found a significant increase in the proportion of KNL1 mutant cells in G2/M based on fluorescence-activated cell sorting (FACS) analysis, with 9% ± 7% of wild-type neural progenitors (Figure 2B), but 18% ± 7% of KNL1c-6125G > A mutant progenitors in G2/M phase (Figures 2C and 2D). To test whether the duration of mitosis is increased in KNL1c.6125G > A neural progenitors, we performed live imaging analysis, which revealed a modest but statistically significant increase in the time required to complete mitosis time from 25 ± 1 min in wild-type cells to 32 ± 1 min (n = 33) in KNL1c.6125G > A neural progenitors (Figure 2E). Because this difference is insufficient to explain the marked increase in G2/M cells in the FACS analysis, we performed further live imaging and found that KNL1c.6125G > A neural progenitors were prone to failed cell division, leading to aborted cycles (Figure 2F). An apoptosis assay, which measures caspase-3/7 released by dying cells, showed significantly more apoptotic cells in KNL1c.6125G > A neural progenitors (Figure 2G). Thus, our data indicate that an increase in mitotic duration, cytokinesis failure, and cell death is associated with reduced cell growth of the KNL1c.6125G > A neural progenitors, reflecting the complexity of the mutant phenotype.
Figure 2. Reduced Cell Growth and Increased Cell Death Are Detected in KNL1c.6125G > A Neural Progenitors.

(A) ATP assay, represented in relative light units, on a 2D adherent neural progenitor culture; day 0 represents the plating day. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
(B and C) FACS analysis showing cell-cycle distribution of wild-type (B) and KNL1c.6125G > A (C). Propidium iodide (PI)-labeled DNA from neural progenitors. This approach displayed G1/G0, S, and G2/M cell-cycle phases based on the quantized fluorescence of labeled DNA.
(D) Quantification of wild-type and KNL1c.6125G > A neural progenitors in the G2/M phase. Welch’s t test (two-tailed) was applied.
(E) Quantification of the mitosis time in wild-type and KNL1c.6125G > A neural progenitors using live imaging. Each point represents a wild-type or KNL1c.6125G > A neural progenitors, from three different wild-type and KNL1c.6125G > A neural progenitors clones.
(F) Quantification of successful cell division in wild-type and KNL1c.6125G > A neural progenitors using live imaging. Welch’s t test (two-tailed) was applied.
(G) Apoptosis assay in wild-type and KNL1c.6125G > A neural progenitors, based on caspase-3/7 activity. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
One non-targeted wild-type clone, two wild-type clones, and three patient mutation clones derived from the same CRISPR-Cas9 targeting are plotted in each graph, except for Figure 1E. Results are mean ± SEM. **p < 0.01, ****p < 0.0001.
KNL1c.6125G > A Neural progenitors Are Susceptible to Aneuploidy and Have an Abrogated Spindle Assembly Checkpoint
One safeguard of genomic integrity is the spindle assembly checkpoint, which ensures accurate chromosome segregation by preventing anaphase initiation before proper kinetochore attachment. Unattached kinetochores trigger the activation of the spindle assembly checkpoint, sending a “wait” signal to delay anaphase onset. It has been shown that KNL1 provides the platform for assembly of the spindle assembly checkpoint machinery in human cells (Caldas and DeLuca, 2014). Using immunostaining, we found that KNL1 is localized at centromeres (ACA) in dividing phospho-Histone 3 (pH3) positive neural progenitors (Figure 3A), consistent with observations in other cell types (Cheeseman et al., 2008; Genin et al., 2012; Kiyomitsu et al., 2007). In contrast, we observed a decreased KNL1 signal at kinetochores in KNL1c.6125G > A neural progenitors as compared to wild-type cells (Figure 3B), in agreement with the mRNA and protein expression data (Figures 1G and 1H). Furthermore, ZWINT-1, which is essential for KNL1 stabilization and interacts directly with KNL1 (Kiyomitsu et al., 2011), also displayed reduced kinetochore localization in mutant neural progenitors (n = 15, Figures 3C and 3D), consistent with reduced KNL1 levels.
Figure 3. Aneuploidy and Abrogated Spindle Assembly Checkpoint Is Observed in KNL1c.6125G > A Neural Progenitors.

(A) Immunostaining using antibodies against KNL1 (green) and centromere (ACA; red), a kinetochore marker, on neural progenitors (scale bar, 10 μm).
(B) Quantification of KNL1 fluorescence intensity at the kinetochore, based on the ratio of KNL1 integrated fluorescence intensity to the fluorescence intensity of ACA at the kinetochore. Welch’s t test (two-tailed) was applied.
(C) Immunostaining using antibodies against ZWINT-1 (green) and ACA (red), a kinetochore marker, on neural progenitors (scale bar, 5 μm).
(D) Quantification of ZWINT-1 fluorescence intensity at the kinetochore, based on the ratio of ZWINT-1 integrated fluorescence intensity to the fluorescence intensity of ACA at the kinetochore. Welch’s t test (two-tailed) was applied.
(E) Quantification of aneuploid cells in wild-type and KNL1c.6125G > A hESCs, after subsequent passages. Welch’s t test (two-tailed) was applied.
(F) Karyotype analysis of wild-type and KNL1c.6125G > A neural progenitors.
(G) Quantification of aneuploid cells in wild-type and KNL1c.6125G > A neural progenitors, after subsequent passages. Welch’s t test (two-tailed) was applied.
(H) Quantification of wild-type and KNL1c.6125G > A neural progenitors arrested in mitosis, in the presence of nocodazole. Welch’s t test (two-tailed) was applied.
One non-targeted wild-type clone, two wild-type clones and three patient mutation clones derived from the same CRISPR-Cas9 targeting are plotted in each graph. Results are mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001.
Based on the reduction of KNL1 expression, we tested the genome integrity of KNL1c.6125G > A hESC and neural progenitors by karyotype analysis.
Normal karyotype was found in wild-type hESC, KNL1c.6125G > A hESC, and wild-type neural progenitors, regardless of passage number (Figures 3E–3G). In contrast, 40% ± 5% of KNL1 mutant progenitors displayed chromosomal aberrations in early (P4) and late (P7) passages (Figures 3F and 3G). To test the presence of proper spindle assembly checkpoint function in the KNL1 mutant progenitors, we treated cells with the microtubule-depolymerizing drug nocodazole, which decouples kinetochores and microtubules. While high-dose nocodazole induced mitotic arrest and enriched wild-type neural progenitors in the G2/M phase cell population, mutant cells were not as susceptible to the drug with fewer cells found in mitotic arrest (Figure 3H) suggesting a defective spindle assembly checkpoint in mutant cells.
KNL1c.6125G > A Differentiating Neuro-Glial Cultures Show Premature Differentiation at the Expense of Neural Progenitor Proliferation
To recapitulate aspects of cortical development and study disease-relevant phenotypes, we investigated the expression of markers for neural progenitors (PAX6), neurons (MAP2), and astrocytes (GFAP) in a two-dimensional neuro-glial culture over 8 weeks of differentiation. Real-time PCR showed a decrease in PAX6 and an increase in MAP2 and GFAP overtime, mimicking the time course of neocortical development in vitro. Next, we compared the expression of PAX6, MAP2, and GFAP between wild-type and KNL1c.6125G > A neuro-glial cultures. Mutant neuro-glial cultures consistently displayed a reduced level of PAX6 as compared to wild-type cultures, and significantly higher expression of MAP2 and GFAP was observed at 4 and 6 weeks (Figures 4B and 4C), suggesting premature differentiation in KNL1 mutant cells. To confirm this finding, we sorted cells based on cell-surface markers to quantify the portion of neural progenitors, neurons, and astrocytes present in the differentiating cultures using the following set of markers: CD184+/CD271−/CD44−/CD24+, CD184−/CD15low/CD44−/CD24+, and CD184+/CD44+. The validity of this FACS-based approach was confirmed using real-time PCR, substantiating the specific identity of each group of purified cells (Figure S2). As early as 2 weeks post-differentiation, fewer neural progenitors and more astrocytes and neurons were present in the mutant as compared to in wild-type cultures (Figures 4D–4F). Consistent with these data, immuno-staining showed that, compared to their isogenic controls, mutant cultures had higher levels of the early neuronal (TUJ1) and astrocyte (S100β) markers at 4 weeks and had higher levels of the mature neuronal (MAP2) and the astrocyte (GFAP) markers at 6 weeks (Figures 4G and 4H).
Figure 4. KNL1c.6125G > A Differentiating Neuro-Glial Cultures Differentiate Prematurely, at the Expense of Neural Stem Cell Proliferation.

(A–C) Time-course analysis of neural progenitors marker PAX6 (A), neuronal marker MAP2 (B), and glial marker GFAP (C), using real-time qPCR in a wild-type and KNL1c.6125G > A neuro-glial differentiating culture. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
(D–F) Time-course analysis of neural progenitors (D), neurons (E), and astrocytes (F), using quantification based on cell-surface markers of wild-type and KNL1c.6125G > A neuro-glial differentiating culture. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
(G) Immunostaining using antibodies against S100b and Tuj1 in wild-type and KNL1c.6125G > A in a neuro-glial culture, at 4 weeks of differentiation (scale bar, 50 μm).
(H) Immunostaining using antibodies against GFAP and MAP2 in wild-type and KNL1c.6125G > A in a neuro-glial culture, at 6 weeks of differentiation (scale bar, 50 μm).
(I) FACS-based quantification of EdU+TUJ1+/EdU+ cells following 1-day post EdU pulse-chase assay in 2-week-old neuro-glial differentiating culture. Welch’s t test (two-tailed) was applied.
(J) FACS-based quantification of EdU+S100β+/EdU+ cells following 1-day post EdU pulse-chase assay in 2-week-old neuro-glial differentiating culture. Welch’s t test (two-tailed) was applied.
(K) Quantification of the mitosis time in wild-type and KNL1c.6125G > A 1-week differentiating culture using live imaging, in the presence and absence of nocodazole. Each point represents a dividing wild-type or KNL1c.6125G > A cell, from three different wild-type and KNL1c.6125G > A clones. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
(L) Quantification of EdU+PAX6+/EdU+ in wild-type and KNL1c.6125G > A 1-week differentiating culture following a pre-incubation with EdU for 1 hr and a 90-min nocodazole treatment. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
One non-targeted wild-type clone, two wild-type clones and three patient mutation clones derived from the same CRISPR-Cas9 targeting are plotted in each graph. Results are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
To assess the fate of mutant neural progenitors in differentiating cultures, we performed an EdU pulse-chase assay followed by TUJ1 and S100β staining. At 1 day post EdU pulse, the majority of the EdU+ population was TUJ1-/S100β− in the wild-type culture, indicating an undifferentiated status. However, a different distribution was noted in the mutant culture, with significantly higher proportion of EdU+/TUJ1+ and EdU+/S100β+ populations, indicating that they have adopted neuronal or astrocytic identity. Therefore, KNL1 mutation depletes the pool of neural progenitors in favor of premature differentiation (Figures 4I and 4J).
To test whether an increase in mitosis time of neural progenitors is sufficient to explain the premature differentiation phenotype, we performed live imaging analysis following a nocodazole treatment on wild-type and mutant 1-week-old differentiating cultures. We quantified the EdU+/PAX6+ population in nocodazole-treated wild-type culture and non-treated mutant culture, which had a similar mitosis time (Figure 4K), and observed a significant lower population of EdU+/PAX6+ in the non-treated mutant culture, compared to nocodazole-treated wild-type culture. Therefore, increase in mitosis time is not solely responsible for the premature phenotype observed in mutant differentiating culture (Figure 4L).
KNL1c.6125G > A Three-Dimensional Neural Spheroids Reproduce the Human Microcephaly Phenotype
To explore the function of KNL1 in a 3D and more complex culture system, we generated cerebral organoids (Lancaster et al., 2013). As mutant hESCs display an altered cell cycle that would affect cell growth before neural differentiation (Figure S3), we initiated the 3D cultures from early neural progenitors and generated neural spheroids (Muffat et al., 2018). As shown in Figures 5A–5C, real-time PCR revealed that neural progenitor marker PAX6 was highly expressed at the start of the culture and decreased over time, while neuronal marker such as MAP2 and the astrocyte marker GFAP increased at later times of differentiation. However, in contrast to wild-type cultures and consistent with the 2D cultures (Figure 4), KNL1 mutant neural spheroids showed lower PAX6 expression early and a higher level of MAP2 and GFAP at 4 weeks indicating a higher fraction of differentiated cells. At 8 weeks, wild-type cultures had a higher number of differentiated cells, which could be explained by the larger number of neural progenitors generated initially (Figures 5A–5C). Similarly, immunohistochemistry on neural spheroids sections revealed that early KNL1 mutant cultures contained fewer PAX6+ cells than wild-type cultures and a higher fraction of TUJ1+ and S100β+ at later time points (Figure 5D).
Figure 5. KNL1c.6125G > A Three-Dimensional Neural Spheroids Are Also Prone to Premature Differentiation and Are Smaller.

(A–C) Time-course analysis of neural progenitors marker PAX6 (A), neuronal marker MAP2 (B), glial marker GFAP (C) using real-time qPCR in wild-type and KNL1c.6125g > A neural spheroids.
(D) Immunostaining using antibodies against PAX6/NESTIN at 2 weeks, S100β/TUJ1 at 4 and 6 weeks, and SOX9/NEUN at 10 weeks in wild-type and KNL1c.6125G > A neural spheroids sections (scale bar, 200 μm).
(E) Wild-type and KNL1c.6125G > A neural spheroids at 10 weeks (scale bar, 500 μm).
(F) Area measurement of wild-type and KNL1c.6125G > A neural spheroids in a time course of 10 weeks.
One non-targeted wild-type clone, two wild-type clones, and three patient mutation clones derived from the same CRISPR-Cas9 targeting are plotted in each graph. Results are mean ± SEM. *p < 0.05, ***p < 0.001, ****p < 0.0001.
Consistent with the proliferation defect of neural progenitors, the KNL1 mutant neural spheroids were reduced in size compared to wild-type as early as at 2 weeks of culture (Figure 5E). At 10 weeks, wild-type neural spheroids had grown to 2.5 mm in diameter, whereas mutant neural spheroids were 30% smaller. Unlike cerebral organoids, neural spheroids do not present fluid-filled ventricle-like regions, which exclude differences in internal cavities as a confounding factor (Figure 5D). The results in Figure 5 support the conclusion that the KNL1c.6125G > A mutation causes reduced proliferation of neural progenitors and premature differentiation, consistent with the microcephaly phenotype caused by the KNL1 mutation.
Reduced KNL1 Expression Causes Premature Differentiation
One possible explanation for the phenotypes seen in neural progenitors and differentiating cultures is the reduced level of KNL1 expression. To test this hypothesis, we infected hESCs with a lentivirus expressing the KNL1 short hairpin RNA (shRNA). Stable clones of hESCs expressing KNL1 shRNA were differentiated into neural progenitors (Figure 6A), and the reduction in KNL1 was confirmed by western blotting (Figure 6B). Human ESCs carrying KNL1 shRNA were differentiated into neural progenitors and used to generate neural spheroids. Figure 6C shows that the resulting neural spheroids were smaller than wild-type and similar in size as KNL1c.6125G > A neural spheroids and showed lower PAX6 expression than wild-type neural spheroids (Figures 6D–6F), suggesting fewer cells with a neural progenitor identity. At 4 weeks, MAP2 (Figure 6E) and GFAP (Figure 6F) expression was higher in spheroids carrying KNL1 shRNA than in wild-type spheroids, consistent with a higher number of cells with differentiated identity (Figures 6D–6F). These data suggest that global reduction in KNL1 expression, independent of the mutated protein sequence, is sufficient to cause premature differentiation and reduced size.
Figure 6. KNL1c.6125G > A Three-Dimensional Neural Spheroids Size Reduction and Premature Differentiation Is Mimicked by Knockdown and Rescue Experiments.

(A) Schematic overview of the generation of stable cell lines with shRNA and overexpressed KNL1.
(B) Western blot analysis and quantification of KNL1 expression of wild-type, KNL1c.6125G > A, wild-type shRNA, and KNL1c.6125G > A +KNL1 cDNA neural progenitors, 10 days post-differentiation. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
(C) Area measurement of wild-type, KNL1c.6125G > A, wild-type shRNA, and KNL1c.6125G > A +KNL1 cDNA neural spheroids in a time course at 10 weeks. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
(D–F) Time-course analysis of neural progenitors marker PAX6 (D), neuronal marker MAP2 (E), glial markers GFAP (F) using real-time qPCR in wild-type, KNL1c.6125G > A, wild-type shRNA, and KNL1c.6125G > A +KNL1 neural spheroids. ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test.
One non-targeted wild-type clone, two wild-type clones, and three patient mutation clones derived from the same CRISPR-Cas9 targeting are plotted in each graph. Results are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We also tested whether the ectopic overexpression of KNL1 in mutant cells is sufficient to rescue the size reduction. We infected KNL1c.6125G > A hESC with a lentivirus expressing the KNL1 cDNA and generated stable clones (Figure 6A). Western blot analysis showed that the level of KNL1 in KNL1c.6125G > A neural progenitors bearing KNL1 cDNA was similar to that of wild-type neural progenitors (Figure 6B). KNL1c.6125G > A neural progenitors containing KNL1 cDNA were then used to generate neural spheroids, which reached the same size as that of wild-type neural spheroids (Figure 6C). Real-time PCR showed similar expressions of PAX6 (Figure 6D), MAP2 (Figure 6E), and GFAP (Figure 6F) in KNL1c.6125G > A neural spheroids containing KNL1 cDNA compared to wild-type neural spheroids, demonstrating that wild-type KNL1 expression is sufficient to rescue the size difference and prevent premature differentiation. Our data suggest that KNL1 is essential for balancing cell proliferation and differentiation and identify KNL1 as a potential regulator of brain size.
KNL1c.6125G > A Fibroblasts and Neural Crest Cells Show Wild-Type Levels of KNL1 and Cell Growth, Due to a Lower Level of HNRNPA1 Protein
As KNL1 is expressed in all tissues, it is surprising that only the brain is impacted by its mutation. To investigate the role of the KNL1 mutation in cell types other than neural progenitors, isogenic mutant and wild-type hESC cells were differentiated into fibroblasts and neural crest cells. Each cell type was characterized by real-time PCR and immunostaining (Figure S4). It has been previously reported that KNL1 mutant fibroblasts and lymphoblastoids expressed a truncated form of the KNL1 transcript (Genin et al., 2012; Saadi et al., 2016; Szczepanski et al., 2016). To investigate the presence of KNL1 truncated mRNA, we performed a non-real-time qPCR on wild-type and mutant cells and found that the truncated KNL1 mRNA was present in all KNL1c.6125G > A cell types (Figures 7A and 7E). Additionally, we tested whether these cell types had altered KNL1 mRNA or protein levels. Surprisingly, KNL1 transcript was expressed similarly in all wild-type and KNL1c.6125G > A cell types except in neural progenitors (Figures 7B, 7C, 7F, and 7G). These results are consistent with those of a previous study of patient fibroblasts in which KNL1 transcript expression was not impacted.
Figure 7. Wild-Type and KNL1c.6125G > A Fibroblasts and Neural Crest Cells Do Not Present Any Difference in KNL1 Level or Cell Growth.

(A) Non-quantitative agarose gel electrophoresis of KNL1 cDNA, flanking the point mutation in neural crest cells.
(B) Real-time qPCR analysis of KNL1 expression, normalized to GAPDH, in neural crest cells. Welch’s t test (two-tailed) was applied.
(C) Western blot analysis and quantification of KNL1 expression of wild-type and KNL1c.6125G > A neural crest cells. Welch’s t test (two-tailed) was applied.
(D) ATP assay on wild-type and KNL1c.6125G > A neural crest cells; day 0 represents the plating day.
(E) Non-quantitative agarose gel electrophoresis of KNL1 cDNA, flanking the point mutation in fibroblasts.
(F) Real-time qPCR analysis of KNL1 expression, normalized to GAPDH, in n fibroblasts. Welch’s t test (two-tailed) was applied.
(G) Western blot analysis and quantification of KNL1 expression of wild-type and KNL1c.6125G > A fibroblasts. Welch’s t test (two-tailed) was applied.
(H) ATP assay on wild-type and KNL1c.6125G > A neural crest cells; day 0 represents the plating day.
(I) Western blot analysis of HNRNPA1 protein level in wild-type and KNL1c.6125G > A neural progenitors, neural crest cells, and fibroblasts.
(J) Western blot analysis of KNL1, HNRNPA1, and Vinculin protein levels and quantification of KNL1 expression of wild-type, KNL1c.6125G > A, and wild-type neural progenitors transfected with HNRNPA1 siRNA and KNL1c.6125G > A neural progenitors transfected with hnRNPA1 siRNA, 48 hr post-transfection.
(K) Western blot analysis of KNL1, HNRNPA1, and Vinculin protein levels and quantification of KNL1 expression of wild-type, KNL1c.6125G > A, and wild-type neural crest cells infected with HNRNPA1 cDNA and KNL1c.6125G > A neural crest cells infected with HNRNPA1 cDNA, 48 hr post-transfection.
(L) Western blot analysis of KNL1, HNRNPA1, and Vinculin protein levels and quantification of KNL1 expression of wild-type, KNL1c.6125G > A, and wild-type fibroblasts infected with HNRNPA1 cDNA and KNL1c.6125G > A fibroblasts infected with HNRNPA1 cDNA, 48 hr post-transfection.
ANOVA was performed followed by a post hoc group comparisons using a Bonferroni test for (D), (H), and (J)–(L). One non-targeted wild-type clone, two wild-type clones, and three patient mutation clones derived from the same CRISPR-Cas9 targeting are plotted in each graph. Results are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
The data in Figure 7 indicate that the KNL1 RNA was expressed in KNL1c.6125G > A fibroblasts and neural crest cells with total KNL1 RNA at similar levels to those in wild-type cells. Importantly, using an ATP assay, we show that KNL1 mutant fibroblasts and neural crest cells had similar growth rate as their isogenic wild-type controls (Figures 7D and 7H) and were karyo-typically normal (Figure S4). However, KNL1 knockdown in wild-type fibroblasts and neural crest cells led to a reduction of cell growth, as observed in mutant neural progenitors, highlighting the importance of KNL1 expression in modulating cell growth (Figure S5). Despite the same proliferation rate of neural progenitors and neural crest cells, only mutant neural progenitors (NPCs) growth was impacted, which highlight that the reduced cell growth phenotype in neural progenitors is cell type specific and not inherent to the cell proliferation rate.
It has been proposed that the expression of exon enhancer and silencer proteins affect the inclusion or exclusion of an exon, and that their differential expression may cause an exon to be included in one cell type and excluded in other cell type (Badr et al., 2016; Mayeda et al., 1993). We decided to focus on the HNRNPA1 protein, as its interaction with KNL1 is the most impacted in KNL1c.6125G > A neural progenitors, and monitored its protein expression in neural progenitors, neural crest cells, and fibroblasts. Western blot analysis revealed that HNRNPA1 is highly expressed in neural progenitors, which could explain the difference in KNL1 mRNA processing seen specifically in neural progenitors (Figure 7I). To confirm the implication of HNRNPA1 in the reduction of KNL1 expression in KNL1c.6125G > A neural progenitors, we knocked down HNRNPA1 and detected an increase of KNL1 protein level in KNL1c.6125G > A neural progenitors (Figure 7J). Moreover, KNL1 protein levels were reduced in KNL1c.6125G > A fibroblasts and neural crest cells upon the overexpression of HNRNPA1 (Figures 7K and 7L), confirming the implication of this splicing protein in the cell type-specific difference of KNL1 mRNA processing. Cell growths have been monitored in fibroblasts and neural crest cells overexpressing HNRNPA1 as well as in HNRNPA1 knockdown neural progenitors; however, due to the essential role of HNRNPA1 in mRNA processing, metabolism, and transport (Jean-Philippe et al., 2013), we observed a reduction of cell growth in all conditions (Figure S5).
In summary, our data show that the KNL1c.6125G > A point mutation generates a truncated mRNA form in fibroblasts and neural crest cells similar to what is seen in neural progenitors. However, overall KNL1 expression, cell growth, and genomic integrity were only impacted in neural progenitors but not in the other cell types, suggesting neural-specific differences in KNL1 mRNA processing. It has been speculated previously that only some cell types such as neural progenitors might express truncated or no KNL1 protein and thus would display a mutant phenotype (Genin et al., 2012). Here, we demonstrate that KNL1c.6125G > A neural progenitors express a reduced level of KNL1 while other cell types express normal levels due to difference in splicing protein expressions.
DISCUSSION
In this study, we engineered a hESC line containing a KNL1c.6125G > A patient-specific mutation and, using 2D and 3D culture systems, tested its consequences by differentiating ESCs into neural progenitors and differentiated cell types. We found that KNL1c.6125G > A mutant neural progenitor cells displayed impaired proliferation and were prone to premature differentiation. Furthermore, the cells developed rapidly an aneuploid karyotype. In contrast to neural progenitors, mutant fibroblasts and mutant neural crest cells had a normal karyotype and showed no growth defect. Thus, our results are consistent with microcephaly being the only major phenotype of the KNL1 mutation by demonstrating that, although KNL1 is ubiquitously expressed, the mutation affects only neural progenitors.
Why the brain is the only organ affected in microcephaly patients remains an open question. Although the clinical features of microcephaly have historically been defined as small head circumference, mental retardation, and normal height and weight, recent studies have questioned this definition as microcephaly patients often present with other cortical malformations or primordial dwarfism (Morris-Rosendahl and Kaindl, 2015; Woods et al., 2005). Thus, brain size may not be the only phenotype of microcephaly gene defects, but it is certainly the most consistent one. Here, we showed that a KNL1 point mutation leads to a reduction in neural progenitors growth but not in fibroblasts and neural crest cells. RNA splicing protein levels recognizing ESE/ESS sites differ between cell types resulting in decreased expression of KNL1 in neural progenitors but not in the other cell types. KNL1 is, however, not the only gene with a mutation that affects splicing sites as many other gene mutations identified in microcephaly patients cause altered splicing (Farooq et al., 2016; Hashmi et al., 2016; Kakar et al., 2012; Yang et al., 2012; Yu et al., 2010). Mutations affecting the splicing protein recognition sequence might impact tissues differently because of differential expression of its specific binding splicing proteins. This adds a layer of complexity to the understanding of microcephaly and may help understand the wide phenotype heterogeneity seen in patients. The brain has been described as the tissue with the highest level of alternative splicing events and the largest number of splicing factor genes, which could partly explain the brain-specific phenotype (Barbosa-Morais et al., 2012). Though our data are consistent with the HNRNPA1-KNL1 interaction explaining the neural precursor phenotype, further functional studies are needed to assess whether this splicing factor is sufficient to account for the brain-specific phenotype.
In addition to the KNL1c.6125G > A mutation, several other KNL1 mutations have been described. A point mutation in intron 24 has been shown to result in skipping of exon 25 and a downstream premature stop codon (Szczepanski et al., 2016). This mutation, KNL1c.6673–19T > A, caused reduced KNL1 protein expression accompanied by mitotic arrest and an altered DNA damage response in primary fibroblasts and lymphoblastoid cell lines (Szczepanski et al., 2016). As KNL1c.6673–19T > A fibroblasts also showed reduced KNL1 in this patient, combined with severe microcephaly (head circumference reduced by −13 to −17 SDs), we would not expect a brain-specific phenotype. In comparison, the patient carrying the KNL1c.6125G > A was reported to have a reduced head circumference of −3 to −8 SDs suggesting a milder phenotype than in the intron 24 mutation (Szczepanski et al., 2016).
Given the central and essential role of KNL1 in mediating proper chromosome segregation, Spindle Assembly Checkpoint signaling, and directing error correction of improper kinetochore-microtubule attachments, it is surprising that the KNL1c.6125G > A point mutation in KNL1 leads to a brain-specific phenotype, whereas another point mutation (KNL1c.6673–19T > A) affects lymphoblastoid and fibroblast cells. RNA splicing proteins involved in the recognition of ESE sites differ from those that recognize intron junctions (Cartegni et al., 2002). It is thus possible that proteins involved in recognizing intron junctions are ubiquitously present, while those mediating ESE identifications are more restricted to particular cell types. In addition, the KNL1 intron mutation results in the deletion of a region interacting with MIS12, which mediates the kinetochore-centromere connection (Kline et al., 2006). Immunofluorescence of KNL1c.6673–19T > A revealed a partially mislocalized KNL1, which could represent the abnormal truncated protein and have a deleterious effect (Szczepanski et al., 2016). In KNL1c.6125G > A hESCs and neural progenitors, we were unable to show that the truncated mRNA was translated into a protein. More studies are needed to understand the implications of the RNA splicing component and the truncated KNL1 proteins resulting from the defective splicing.
One of the predominant microcephaly models suggests that abnormal spindle orientation leads to an imbalance between proliferative and neurogenic division in favor of the latter, depleting neural stem cells and favoring the production of neurons (Konno et al., 2008). However, in vivo mouse studies showed that spindle orientation is not necessarily linked to a small brain (Konno et al., 2008). What could explain the switch between proliferation/differentiation in mutant KNL1 neural progenitors? Although many studies have investigated the role of the centrosome in microcephaly and linked it to cellular processes such as polarity or cell-cycle length, little is known about the function of KNL1 during neurogenesis. KNL1 mRNA is mostly detected at the onset of neurogenesis in the highly proliferative ventricular zone, which suggests that KNL1 plays a crucial role in regulating cell-cycle progression of neural progenitors. However, it remains to be determined whether KNL1 also has kinetochore-independent functions. While a reduction in KNL1 levels is anticipated to have diverse effects on kinetochore function, chromosome segregation, and mitotic progression, further studies are necessary to investigate the molecular actors and pathways involved in its brain-specific roles and provide insights into the mechanisms that trigger premature differentiation.
KNL1 shows distinct sequence divergence between modern humans and Neanderthals, and a set of amino acid changes has been identified that might contribute to recent evolutionary change in the human brain (Prüfer et al., 2014; Shi et al., 2017). It would be interesting to investigate whether these modifications interfere with KNL1 expression or mimic evolutionary changes. Much remains to be learned about KNL1 function and, more broadly, about genes identified in patients with microcephaly. Due to the large number of proteins involved in cell division, it is highly likely that more microcephaly genes will be discovered and the roles of the affected proteins will be clarified, improving our understanding of human brain development.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rudolf Jaenisch (jaenisch@wi.mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human embryonic stem cell culture
hESC WIBR3 cells were previously described (Muffat et al., 2016) and cultured at 5% CO2 on C-inactivated mouse embryonic fibroblasts (MEFs) in the following medium: DMEM/F12 (Invitrogen), 15% fetal bovine serum (Hyclone), 5% knockout serum replacement (Invitrogen), 1% non-essential amino acids (Invitrogen), 1mM glutamine (Invitrogen), 0.1mM b-mercaptoethanol (Sigma) and 4ng/ml bFGF (R&D Systems). Cultures were passaged with 1mg/ml collagenase type IV (Thermo), 20% knockout serum replacement (Invitrogen) or manually every 5-7 days. WIBR3 cell line was derived from a female healthy donor. All lines were maintained for over 10 passages, tested for mycoplasma negativity and sent for karyotype analysis. Cytogenetic analysis of karyotype will ensure the integrity of the chromosomes in each line.
Induced pluripotent stem cell culture
Human iPS cell line iPS-wt5 was previously described (Muffat et al., 2016) and cultured in medium containing DMEM/F12 (Thermo), 20% knockout serum replacement (Thermo), 1% non-essential amino acids (Thermo), 1mM Glutamax (Thermo), 0.1mM β-mercaptoethanol (Sigma) and 12ng/ml bFGF (Thermo). iPS-wt5 was reprogrammed by the Li-Huei Tsai Lab from a healthy male donor using non-integrative Sendai virus (OSKML). This line is routinely tested for mycoplasma negativity.
Statement of compliance with IRBs
All experiments involving cells from human donors and animals were performed in compliance with Whitehead Institute IRB.
METHOD DETAILS
CRISPR/Cas9 gRNA design
20 bp annealed oligonucleotides (gRNA) specific to the exon 18 of KNL1 was cloned in the BbsI restriction site of a plasmid carrying the CAS9 nuclease (modified px330 vector containing a CMV-EGFP cassette). The gRNA sequence was 5′- TCAAATAAAGATAGAT GAGA-3′, which contains a BclI restriction site.
To insert the point mutation, a 153 bp oligonucleotide has been designed were the PAM motif has been modified to insert the point mutation found in patients. The oligonucleotide sequence was 5′- G TCT ATA ATC TTT TGT ATT TAG AAT GAG AGG GAG AAA CTT CAA ATA AAG ATA GAT GAG ATa GAT AAA ATA CTT AAG AAG ATC GAT AAC TGt CTC ACT GAG ATG GAA ACA GGT AAA GTA TTT TAA ATA CTT TTC CAA AAG AAA ATT TAA TTT TA-3′.
CRISPR/Cas9-mediated genome editing
Twenty-four hours before the electroporation, hESC medium was complemented with 10 μM ROCK inhibitor Y27632 (10mM, Stemgent). hESC were first separated from MEFs using collagenase type IV and then dissociated using accutase (Stem Cell Technologies) to form a single cell suspension. Ten million cells were co-electroporated with 30ug of the vector px330-KNL1 gRNA and 30 μg of the single strand oligo. After 48 hr, cells were single dissociated again, GFP positive cell were sorted using FACS and plated at a clonal density in hESC medium containing ROCK inhibitor Y27632 for the first 24 hr. Clones were picked manually after 15-20 days and expanded. Genomic DNA was extracted and PCR fragment was generated with the following primers: 5′- GCCTGTTTGTGATCAT GATAGAGC-3′ and 5′- AGTGAGGCAGTTATCGATCTTCTT –3–. Clones were screened for the destruction of the BclI restriction site and promising homozygote clones were further validated by Sanger sequencing. One non-targeted wild-type clone, two wild-type clones and three patient mutation clones were selected for further characterization.
Lentivirus cloning, production, and transduction
KNL1 shRNA TRCN0000145591 was clone in the site AgeI/EcoRI of a pLKO plasmid (Addgene 10878). KNL1 (Clone Id: 100069116, Dharmacon) was cloned into pLEX 307 (Addgene 41392) using the Gateway cloning strategy. KNL1 cDNA was then modified to contain either the point mutation or a stop codon on exon 19. HNRNPA1 overexpression was performed using pLX-TRC304 backbone (Clone Accession: BC002355, Clone ID: ccsbBroad304_00765, Thermo).
HEK293T cells were transfected with pLKO-KNL1-shRNA, pLEX307-KNL1, pLEX307-KNL1 point mutation or pLEX307-KNL1 stop codon lentivirus constructs, with second-generation packaging plasmids using X-tremeGENE 9 (Sigma). Culture medium was changed 12-24 hr, collected 72 hr post-transfection, filtered through 0.45 μm and concentrated via ultracentrifugation. To transduce hESCs with lentivirus, hESCs cultured on MEFs were first dissociated from MEFs using collagenase type IV, and further dissociated using accutase to generate single cells. hESCs were plated onto matrigel-coated dishes and fed with mTeSR medium containing 10 μM ROCK inhibitor Y27632. The lentivirus was added right after passaging. hESCs were subsequently passaged as single cells at clonal density in plate coated with MEFs-feeders. Single clones were manually picked and expanded after 15-20 days. Expression of lentiviral-transgenes was evaluated using quantitative RT-PCR and immuno-blotting.
Differentiation to fibroblasts and maintenance
hESC were separated from MEFs using collagenase IV and plated onto a low-attachment plate in the following MEF medium: DMEM/F12 (Invitrogen), 15% fetal bovine serum (Hyclone), 1% non-essential amino acids (Invitrogen), 1 mM glutamine (Invitrogen). Every 2 days, colonies were collected by gravity separation in a 15 mL tube and the supernatant was discarded. At day 6, the colonies were transferred to gelatin-coated plates and passaged when confluence was reached. Fibroblast identity was confirmed using quantitative RT-PCR and immunofluorescence.
Differentiation to neural crest cells and maintenance
hESC were differentiated to neural crest cells based on a previously published protocol (Cohen et al., 2016). Briefly, neural induction was obtained by using 10 μM SB431542 (Stemgent) and 500 nM LDN193189 (Stemgent) for 3 days followed by an 8-day differentiation using 3 μM CHIR99021 (Stemgent). Neural crest cells were then maintained in N2B27 supplemented with bFGF (20 ng/mL) and EGF (20 ng/mL).
Differentiation to neural progenitors and maintenance
Four million cells from a hESC single cell suspension were filtered through a 40μm mesh and plated onto matrigel-coated dishes. Cells were cultured directly in NGD medium, complemented with 10 μM ROCK inhibitor Y27632 for the first 24 hr, with the addition of dorsomorphin (2.5 μM, Stemgent), bFGF (10 ng/mL) and insulin (10 ng/mL, Sigma Aldrich) for 10 days until super-confluent. Cells were subsequently passaged 1:1 using accutase and plated onto matrigel-coated dishes. Neural progenitors were expanded and maintained in NGD medium with 10ng/ml bFGF, and additional 10 ng/mL insulin. Cytogenetic analyses were performed at passage 4, 7 and 10.
Differentiation into neurons and astrocytes
To initiate differentiation of neural progenitors into neurons and glia, FGF was removed and insulin concentration was reduced from the NGD medium (5 μg/mL). Differentiating cultures were dissociated, at different time point, up to 6 weeks, in the presence of 0.05% DNase I and Accutase for 30 min at 37C with gentle agitation. Cells were then re-suspended in chilled HBSS/0.1% BSA and filtered through a 40 μM mesh before being centrifuged through a cushion of 4% BSA to remove cellular debris. The subsequent cell suspension was plated on PEI-coated plate or used for FACS sorting.
Differentiation to neural spheroids and maintenance
Neural progenitors were dissociated using accutase, 30,000 neural progenitors at passage 1 were plated into a well of an ultra-low-attachment 96-well plate (Corning) to form single EBs. NGD medium was complemented with ROCK inhibitor Y27632 (50 μM) for the first 24 hr. EBs were maintained in 96-well plates for 2 weeks and transferred to an orbital shaker (Unimax-1010, Heidolph Brinkmann) rotating continuously at 150 rpm.
ATP and Apoptosis assay
Cell growth and cell death rate was measured using an ATP assay (CellTiter-Glo Luminescent kit) or apoptosis assay (ApoTox-Glo Triplex Assay Kit), according to the manufacturer’s instructions (Promega). Signal intensity was read on a Tecan microplate reader. hESC, neural progenitors and fibroblasts were plated in matrigel-coated 96-well plates (black wall, clear bottom, Falcon) at 20,000 cells per well. ATP and apoptosis assay were performed on day 0 and day 3.
Cell cycle
Samples were fixed in 70% ethanol overnight, washed with PBS, treated with FxCycle PI/RNase Staining (Life Technologies) solution for 20 min and analyzed by FACS. For phospho-histone-H3 analysis, fixed cells were washed 2X in PBS-BSA 5%-Triton 0.1%, incubated with phospho-histone-H3 antibody (Cell Signaling Technology) for 2hours. After 2X wash with PBS-BSA5%-Triton0.1%, samples were incubated with Alexa secondary antibodies (Life Technologies), for two more hr. The samples were then washed 3X in PBS before analysis.
Nocodazole treatment
To induce mitotic arrest, cells were incubated in a high concentration of nocodazole (100 ng/ml) in NGD medium for 10 h. Floating cells were collected and combined with attached cells, detached using accutase. Cells were centrifuged, washed with PBS twice and fixed in 70% ethanol overnight.
To monitor the impact of mitosis time, cells were incubated with 0.1 μM nocodazole for 90 min and washed twice with NGD medium.
EdU click-it assay
EdU (10 μM, Life Technologies) was added on neural progenitors and differentiated culture (2 weeks) for 6 hr, after which cells were either collected immediately using accutase, or washed with PBS and fed fresh medium until collection day (24 hr later). EdU click-it assay was performed on fixed single cell suspension, per manufacture’s instruction (Life Technologies), followed by immuno-staining and FACS sorting.
Quantification of neural progenitors, neurons, and astrocytes, based on cell surface markers using FACS
Single cell suspension of differentiated culture was resuspended in NGD complemented with 0.5% BSA and 5 mM EDTA at a concentration of 10 million cells/ml. Cells were stained with fluorochrome-conjugated antibodies for 20–30 min at room temperature. CD184/CD271/CD44/CD24/CD15 were used to stain differentiated culture, at different time point, as per the manufacturer instructions. Neural progenitors, neurons, and astrocytes were CD184+/CD271−/CD44−/CD24+, CD184−/CD15low/CD44−/CD24+ and CD184+/CD44+, respectively.
Histology and imaging
Cells and tissues were fixed with 4% (w/v) paraformaldehyde in PBS. 5 μm paraffin sections were prepared from neural spheroids. Following membrane permeabilization with PBS containing 0.3% triton, cells and tissue sections were blocked with 3% normal donkey serum. Primary antibodies were against S100β, TUJ1, MAP2, GFAP, SOX9, NEUN, NESTIN, PAX6, and visualized by secondary antibodies conjugated with Alexa 488, 568, 594, 647 (Life Technologies), followed by counter-staining with DAPI). Fluorescent images of immuno-staining were captured on a Zeiss LSM-700 confocal microscope with the Axioxx 40X NA 1.5 (Zeiss) objective.
Live imaging was acquired either using brightfield or 405 laser line (pre-incubation with 100 ng/ml Hoechst33342 for 30 min), for 12 hr. Images were acquired every 5 min with a 50ms exposure. The incubation chamber was set to 37°C with 5% CO2.
For protein fluorescence intensity quantification, images were collected using a 63X/1.4NA oil objective on an AxioVert 200M inverted microscope (Zeiss) with a CSU-22 Spinning Disk Confocal scan head (Yokogawa). Kinetochore fluorescence intensity measurements were performed using the Fiji software. Background signal was calculated from control and mutant cytoplasm and subtracted from the integrated fluorescence intensity using the averaged cytoplasm pixel value. KNL1 kinetochore localization was estimated on maximal intensity projection images by calculating the ratio of KNL1 integrated fluorescence intensity to the fluorescence intensity of ACA at a single kinetochore.
RNA extraction, reverse transcription and quantitative PCR
Cells and tissues were homogenized in RLT+ buffer and total RNA was extracted using the RNeasy kit (QIAGEN) following manufacturer’s instructions. Total RNA concentrations were measured using NanoDrop ND-1000 spectrophotometer. RNA was reverse transcribed into cDNA using Superscript III reverse transcriptase (Invitrogen) with random hexamer primers. To reveal the truncated form a touchdown PCR program has been used (Genin et al., 2012) with the following primers: forward 5′- GAGCTGAAGGCCTTTG GAATTTA –3′ and reverse 5′- CCAAATTCTTAGTTTCTGAGCTGAC –3′. Transcript abundance was determined by quantitative PCR using SYBR Green PCR mix (Applied Biosystems), with primer pairs against KNL1, PAX6, NESTIN, KI67, S100β, TUJ1, GFAP, MAP2 and GAPDH. Raw Ct values were normalized to GAPDH.
RNA immunoprecipitation
Wild-type and KNL1c.6125G > A neural progenitors were harvested using a nuclear isolation buffer (1.28M sucrose, 40mM Tris-HCl pH 7.5, 20 mM MgCl2 and 4% Triton X-100) for 20 min on ice, followed by a centrifugation at 2500 g for 15 min. The nuclear pellets were sheared in a RIP buffer containing 150mM KCl, 25 mM Tris pH7.4, 5mM EDTA, 0.5 mM DTT, 0.5% of NP40,100 U/mL RNase inhibitor and protease inhibitors. Debris were removed by centrifugation and the supernatant was used to perform an immunoprecipitation using anti-SRSF7 (A303-773, Bethyl), anti-SRSF10 (sc-101132, Santa Cruz Biotechnology) or anti-HNRNPA1 (sc-32301, Santa Cruz Biotechnology) antibody, which were precoupled with protein A/G beads. Four washed were completed to remove the unbound material using the RIP buffer and the complexes were further treated with 20 units of RNase-free DNase I and a solution of 0.1% sodium dodecyl sulfate and 0.5 mg/ml Proteinase K. The RNA was purified using TRIzol RNA extraction kit. RIP results were normalized using relative levels of GAPDH mRNA in each IP sample and the relative KNL1 mRNA fold represents enrichment of each transcript over control IgG-IP sample.
Protein purification and immuno-blotting
Total protein was extracted from cells and tissues using RIPA lysis buffer (Millipore) complemented with protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail 2 and 3 (Sigma). Total protein from the supernatant was measured using BCA protein assay (Pierce). Primary antibodies were against KNL1 (Abcam), SRSF7 (Bethyl), SRSF10 (Santa Cruz Biotechnology) or anti-HNRNPA1 (Santa Cruz Biotechnology) and Vinculin (Abcam) and visualized with HRP-conjugated secondary antibodies, followed by LumiGlo Chemiluminescent Kit (KPL), according manufacturer’s instructions. Quantification of band intensity was performed using ImageJ. Values were normalized to Vinculin.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data values were presented as mean ± SEM. Welch’s t tests (two-tailed) were applied to data with two groups. ANOVA analyses were used for comparisons of data with greater than two groups. Post hoc group comparisons were performed with Bonferroni test. A value of p < 0.05 was considered significant. Experiments were always performed on one non-targeted wild-type clone, two independent wild-type clones and three independent patient mutation clones derived from a single CRISPR targeting experiment. Statistical details are also indicated in the legend of each figure.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-PAX6 | Covance | Cat#PRB-278P; RRID:AB_291612 |
| Mouse monoclonal anti-NESTIN | Millipore | Cat#MAB5326; RRID:AB_2251134 |
| Rabbit polyclonal anti-S100β | Dako | Cat#Z0311; RRID:AB_10013383 |
| Rabbit monoclonal anti-TUJ1 | Covance | Cat#MRB-435P; RRID:AB_663339 |
| Rabbit polyclonal anti-GFAP | Dako | Cat#z0334; RRID:AB_10013382 |
| Chicken polyclonal anti-MAP2 | Encor Biotechnology | Cat#CPCA-MAP2; RRID:AB_2138173 |
| Rabbit polyclonal anti-SOX9 | Millipore | Cat#AB5535; RRID:AB_2239761 |
| Mouse monoclonal anti-NEUN | Millipore | Cat#MAB377; RRID:AB_2298772 |
| Mouse monoclonal anti-KI67 | Dako | Cat#M7240; RRID:AB_2142367 |
| Mouse monoclonal anti-Phospho-HISTONE H3 | Cell Signaling Technology | Cat#9706; RRID:AB_331748 |
| Rabbit polyclonal anti-KNL1 (CASC5) | Abcam | Cat#ab70537; RRID:AB_1209410 |
| Rabbit polyclonal anti-SRSF7 | Bethyl | Cat#A303-773A; RRID:AB_11218390 |
| Mouse monoclonal anti-SRSF10 | Santa Cruz Biotechnology | Cat#SC-101132; RRID:AB_1123037 |
| Mouse monoclonal anti-HNRNPA1 | Santa Cruz Biotechnology | Cat#SC-32301; RRID:AB_627729 |
| Mouse monoclonal anti-VINCULIN | Abcam | Cat#ab129002; RRID:AB_11144129 |
| Mouse monoclonal anti-CD184 | BD Biosciences | Cat# 555976; RRID:AB_398616 |
| Mouse monoclonal anti-CD271 | BD Biosciences | Cat# 562123; RRID:AB_10926195 |
| Mouse monoclonal anti-CD15 | BD Biosciences | Cat# 562980 |
| Mouse monoclonal anti-CD24 | BD Biosciences | Cat# 555427; RRID:AB_395821 |
| Mouse monoclonal anti-CD44 | BD Biosciences | Cat# 555479; RRID:AB_395871 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nocodazole | Sigma-Aldrich | Cat#SML1665; CAS: 31430-18-9 |
| Critical Commercial Assays | ||
| CellTiter-Glo Luminescent kit | Promega | Cat#G7570 |
| ApoTox-Glo Triplex Assay Kit | Promega | Cat# G6320 |
| Click-iT Plus EdU Alexa Fluor 488 Imaging Kit | Life Technologies | Cat#C10637 |
| FxCycle PI/RNase Staining Solution | Thermo Fisher | Cat# F10797 |
| Experimental Models: Cell Lines | ||
| WIBR3 hESCs | Lengner et al., 2010 | N/A |
| WIBR3 KNL1 mutant hESCs | This paper | N/A |
| iPSC-wt5 (LHT22) | Li-Huei Tsai lab | N/A |
| Oligonucleotides | ||
| KNL1 shRNA targeting sequence (TRCN0000145591): CCACGAAATCTATTGGCTAAT | Dharmacon | Cat# RHS3979-201855275 |
| KNL1 gRNA: TCAAATAAAGATAGATGAGA | This paper | N/A |
| KNL1 ssODN: G TCT ATA ATC TTT TGT ATT TAG AAT GAG AGG GAG AAA CTT CAA ATA AAG ATA GAT GAG ATa GAT AAA ATA CTT AAG AAG ATC GAT AAC TGt CTC ACT GAG ATG GAA ACA GGT AAA GTA TTT TAA ATA CTT TTC CAA AAG AAA ATT TAA TTT TA | This paper | N/A |
| KNL1 gDNA amplification: Forward: GCCTGTTTGTGATCATG ATAGAGC; Reverse: AGTGAGGCAGTTATCGATCTTCTT | This paper | N/A |
| KNL1 qPCR: Forward: GAGCTGAAGGCCTTTGGAATTTA; Reverse: CCAAATTCTTAGTTTCTGAGCTGAC | This paper | N/A |
| SMARTpool: ON-TARGETplus HNRNPA1 siRNA - 5 nmol | Thermo Fisher | Cat# L-008221-00-0005 |
| SMARTpool: ON-TARGETplus SRSF10 siRNA - 5 nmol | Thermo Fisher | Cat# L-012914-02-0005 |
| SMARTpool: ON-TARGETplus SRSF7 siRNA - 5 nmol | Thermo Fisher | Cat# L-015909-00-0005 |
| Recombinant DNA | ||
| Plasmid: pLKO | Moffat et al., 2006 | Addgene 10878 |
| Plasmid: pLEX307 | Not published | Addgene 41392 |
| Plasmid: KNL1 cDNA - BC043597 - Clone Id: 100069116 | Dharmacon | MHS1010-202699116 |
| Software and Algorithms | ||
| Human Splicing Finder | Desmet et al., 2009 | http://www.umd.be/HSF3/HSF.shtml |
Highlights.
KNL1c.6673–19T > A patient mutation disrupts a splicing enhancer site
Mutant cells differentiate prematurely into neurons and astrocytes
The mutation affects splicing and mimics microcephaly in 3D neural spheroids
Though expressed ubiquitously, the KNL1 mutant causes only microcephaly
ACKNOWLEDGMENTS
We would like to thank Li-Huei Tsai for providing the iPS-wt5. The authors thank Dongdong Fu, Raaji Alagappan, and Carrie Garrett-Engele for technical support. Confocal microscopy was performed at the Keck Facility, with the precious help of Wendy Salmon. We thank members of the Jaenisch and Cheeseman laboratories for discussions. Fluorescent Activated Cell Scanning was performed with the expertise of the Whitehead FACS facility. We also thank Lisa Nip, for helping with the graphical abstract. A.O.J. was supported by the BIF fellowship. Y.L. was supported by a Simons Postdoctoral Fellowship, an International Rett Syndrome Foundation Postdoctoral Fellowship, and a NARSAD Young Investigator Grant. J.M. was supported by the European Leukodystrophy Association and a NARSAD Young Investigator Grant. R.J. was supported by NIH grants HD 045022, R37-CA084198, and 1U19AI131135-01.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online athttps://doi.org/10.1016/j.celrep.2018.09.032.
DECLARATION OF INTERESTS
R.J. is a cofounder of Fate Therapeutics, Fulcrum Therapeutics, and Omega Therapeutics.
REFERENCES
- Badr E, ElHefnawi M, and Heath LS (2016). Computational identification of tissue-specific splicing regulatory elements in human genes from RNA-seq data. PLoS ONE 11, e0166978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. (2012). The evolutionary landscape of alternative splicing in vertebrate species. Science 338, 1587–1593. [DOI] [PubMed] [Google Scholar]
- Bolanos-Garcia VM, Lischetti T, Matak-Vinković D, Cota E, Simpson PJ, Chirgadze DY, Spring DR, Robinson CV, Nilsson J, and Blundell TL (2011). Structure of a Blinkin-BUBR1 complex reveals an interaction crucial for kinetochore-mitotic checkpoint regulation via an unanticipated binding Site. Structure 19, 1691–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldas GV, and DeLuca JG (2014). KNL1: Bringing order to the kinetochore. Chromosoma 123, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, and Krainer AR (2002). Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet 3, 285–298. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Niessen S, Anderson S, Hyndman F, Yates JR 3rd, Oegema K, and Desai A (2004). A conserved protein network controls assembly of the outer kinetochore and its ability to sustain tension. Genes Dev 18, 2255–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Chappie JS, Wilson-Kubalek EM, and Desai A (2006). The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell 127, 983–997. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Hori T, Fukagawa T, and Desai A (2008). KNL1 and the CENP-H/I/K complex coordinately direct kinetochore assembly in vertebrates. Mol. Biol. Cell 19, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen MA, Wert KJ, Goldmann J, Markoulaki S, Buganim Y, Fu D, and Jaenisch R (2016). Human neural crest cells contribute to coat pigmentation in interspecies chimeras after in utero injection into mouse embryos. Proc. Natl. Acad. Sci. USA 113, 1570–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Jackson AP, Bond J, and Woods CG (2006). What primary microcephaly can tell us about brain growth. Trends Mol. Med 12, 358–366. [DOI] [PubMed] [Google Scholar]
- Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, and Béroud C (2009). Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, Pushparaj P, Ahmed F, Algahtani HA, Al-Qahtani MH, and Saleh Jamal H (2015). Molecular genetics of human primary microcephaly: An overview. BMC Med. Genomics 8 (Suppl 1), S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooq M, Fatima A, Mang Y, Hansen L, Kjaer KW, Baig SM, Larsen LA, and Tommerup N (2016). A novel splice site mutation in CEP135 is associated with primary microcephaly in a Pakistani family. J. Hum. Genet 61, 271–273. [DOI] [PubMed] [Google Scholar]
- Fietz SA, Lachmann R, Brandl H, Kircher M, Samusik N, Schröder R, Lakshmanaperumal N, Henry I, Vogt J, Riehn A, et al. (2012). Transcriptomes of germinal zones of human and mouse fetal neocortex suggest a role of extracellular matrix in progenitor self-renewal. Proc. Natl. Acad. Sci. USA 109, 11836–11841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genin A, Desir J, Lambert N, Biervliet M, Van Der Aa N, Pierquin G, Killian A, Tosi M, Urbina M, Lefort A, et al. (2012). Kinetochore KMN network gene CASC5 mutated in primary microcephaly. Hum. Mol. Genet 21, 5306–5317. [DOI] [PubMed] [Google Scholar]
- Hashmi JA, Al-Harbi KM, Ramzan K, Albalawi AM, Mehmood A, Samman MI, and Basit S (2016). A novel splice-site mutation in the ASPM gene underlies autosomal recessive primary microcephaly. Ann. Saudi Med 36, 391–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayette S, Tigaud I, Vanier A, Martel S, Corbo L, Charrin C, Beillard E, Deleage G, Magaud JP, and Rimokh R (2000). AF15q14, a novel partner gene fused to the MLL gene in an acute myeloid leukaemia with a t(11;15)(q23;q14). Oncogene 19, 4446–4450. [DOI] [PubMed] [Google Scholar]
- Jamieson CR, Govaerts C, and Abramowicz MJ (1999). Primary autosomal recessive microcephaly: Homozygosity mapping of MCPH4 to chromosome 15. Am. J. Hum. Genet 65, 1465–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean-Philippe J, Paz S, and Caputi M (2013). hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci 14, 18999–19024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakar N, Goebel I, Daud S, Nünberg G, Agha N, Ahmad A, Nünberg P, Kubisch C, Ahmad J, and Borck G (2012). A homozygous splice site mutation in TRAPPC9 causes intellectual disability and microcephaly. Eur. J. Med. Genet 55, 727–731. [DOI] [PubMed] [Google Scholar]
- Kiyomitsu T, Obuse C, and Yanagida M (2007). Human Blinkin/AF15q14 is required for chromosome alignment and the mitotic checkpoint through direct interaction with Bub1 and BubR1. Dev. Cell 13, 663–676. [DOI] [PubMed] [Google Scholar]
- Kiyomitsu T, Murakami H, and Yanagida M (2011). Protein interaction domain mapping of human kinetochore protein Blinkin reveals a consensus motif for binding of spindle assembly checkpoint proteins Bub1 and BubR1. Mol. Cell. Biol. 31, 998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline SL, Cheeseman IM, Hori T, Fukagawa T, and Desai A (2006). The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J. Cell Biol 173, 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno D, Shioi G, Shitamukai A, Mori A, Kiyonari H, Miyata T, and Matsuzaki F (2008). Neuroepithelial progenitors undergo LGN-dependent planar divisions to maintain self-renewability during mammalian neurogenesis. Nat. Cell Biol 10, 93–101. [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin C-A, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengner CJ, Gimelbrant AA, Erwin JA, Cheng AW, Guenther MG, Welstead GG, Alagappan R, Frampton GM, Xu P, Muffat J, et al. (2010). Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell 141, 872–883. [DOI] [PubMed] [Google Scholar]
- Mayeda A, Helfman DM, and Krainer AR (1993). Modulation of exon skipping and inclusion by heterogeneous nuclear ribonucleoprotein A1 and pre-mRNA splicing factor SF2/ASF. Mol. Cell. Biol 13, 2993–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. [DOI] [PubMed] [Google Scholar]
- Morris-Rosendahl DJ, and Kaindl AM (2015). What next-generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH). Mol. Cell. Probes 29, 271–281. [DOI] [PubMed] [Google Scholar]
- Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai L-H, Aubourg P, Ransohoff RM, and Jaenisch R (2016). Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat. Med 22, 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muffat J, Li Y, Omer A, Durbin A, Bosch I, Bakiasi G, Richards E, Meyer A, Gehrke L, and Jaenisch R (2018). Human induced pluripotent stem cell-derived glial cells and neural progenitors display divergent responses to Zika and dengue infections. Proc. Natl. Acad. Sci. USA 115, 7117–7122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting C, and Jackson AP (2005). Evolution of primary microcephaly genes and the enlargement of primate brains. Curr. Opin. Genet. Dev. 15, 241–248. [DOI] [PubMed] [Google Scholar]
- Prüfer K, Racimo F, Patterson N, Jay F, Sankararaman S, Sawyer S, Heinze A, Renaud G, Sudmant PH, de Filippo C, et al. (2014). The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 505, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saadi A, Verny F, Siquier-Pernet K, Bole-Feysot C, Nitschke P, Munnich A, Abada-Dendib M, Chaouch M, Abramowicz M, and Colleaux L (2016). Refining the phenotype associated with CASC5 mutation. Neurogenetics 17, 71–78. [DOI] [PubMed] [Google Scholar]
- Shi L, Hu E, Wang Z, Liu J, Li J, Li M, Chen H, Yu C, Jiang T, and Su B(2017). Regional selection of the brain size regulating gene CASC5 provides new insight into human brain evolution. Hum. Genet 136, 193–204. [DOI] [PubMed] [Google Scholar]
- Sironi M, Menozzi G, Riva L, Cagliani R, Comi GP, Bresolin N, Giorda R, and Pozzoli U (2004). Silencer elements as possible inhibitors of pseudoexon splicing. Nucleic Acids Res 32, 1783–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepanski S, Hussain MS, Sur I, Altmüller J, Thiele H, Abdullah U, Waseem SS, Moawia A, Nünberg G, Noegel AA, et al. (2016). A novel homozygous splicing mutation of CASC5 causes primary microcephaly in a large Pakistani family. Hum. Genet 135, 157–170. [DOI] [PubMed] [Google Scholar]
- Thornton GK, and Woods CG (2009). Primary microcephaly: Do all roads lead to Rome? Trends Genet 25, 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, and Burge CB (2008). Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 14, 802–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods CG, Bond J, and Enard W (2005). Autosomal recessive primary microcephaly (MCPH): A review of clinical, molecular, and evolutionary findings. Am. J. Hum. Genet 76, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J, et al. (2012). Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151, 1097–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu TW, Mochida GH, Tischfield DJ, Sgaier SK, Flores-Sarnat L, Sergi CM, Topçu M, McDonald MT, Barry BJ, Felie JM, et al. (2010). Mutations in WDR62, encoding a centrosome-associated protein, cause microcephaly with simplified gyri and abnormal cortical architecture. Nat. Genet 42, 1015–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XH-F, and Chasin LA (2004). Computational definition of sequence motifs governing constitutive exon splicing. Genes Dev 18, 1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.