

Abstract
Purpose of the review
Barth syndrome is an X-linked disease characterised by defective remodelling of phospholipid side chains in mitochondrial membranes. Major features include neutropenia, dilated cardiomyopathy, motor delay and proximal myopathy, feeding problems and constitutional growth delay. We conducted this review of neutropenia in Barth syndrome to aid in the diagnosis of this disease, and to improve understanding of both the consequences of neutropenia and the benefits of treatment with granulocyte colony-stimulating factor (G-CSF).
Recent findings
In 88 patients with Barth syndrome, neutropenia, i.e. at least one count below 1.5 × 109/L, was detected in 74 (84%) and 44% had severe chronic neutropenia, with multiple counts below 0.5 × 109/L. The pattern of neutropenia varied between intermittent and unpredictable, chronic and severe, or cyclical with mathematically regular oscillations. Monocytosis, i.e., monocytes >1.0 × 109/L, was observed at least once in 64 of 85 (75%) patients. Granulocyte colony stimulating factor (G-CSF) was administered to 39 of 88 subjects (44%). Weekly average G-CSF doses ranged from 0.12 to 10.92 mcg/kg/day (mean 1.16 mcg/kg/day, median 1.16 mcg/kg/day). Antibiotic prophylaxis was additionally employed in 21 of 26 neutropenic patients. Pre-treatment bone marrow evaluations predominantly showed reduced myeloid maturation which normalised on G-CSF therapy in seven of 13 examined. Consistent clinical improvement, with reduced signs and symptoms of infections, was observed in response to prophylactic G-CSF ± prophylactic antibiotics. However, despite G-CSF and antibiotics, one adult patient died with multiple infections related to indwelling medical devices and gastrostomy site infection after 15.5 years on G-CSF and a paediatric patient required gastrostomy removal for recurrent abdominal wall cellulitis.
Keywords: Granulocytes, Monocytes, Macrophages; Barth Syndrome; Granulocyte Colony-Stimulating Factor; Neutropenia
Summary:
Barth syndrome should be considered in any male with neutropenia accompanied by any of the characteristic features of this syndrome. Prophylaxis with G-CSF ± antibiotics prevents serious bacterial infections in the more severe neutropenic patients although infections remain a threat even in patients who are very compliant with therapy, especially in those with indwelling devices.
Background
Peter Barth first reported the syndrome which carries his name in 1983 as an X-linked triad of dilated cardiomyopathy (DCM), skeletal myopathy and neutropenia with high mortality. He described three affected male infants from a large Dutch family in which 17 boys had died at between three days and 31 months of age due to either neutropenic bacterial sepsis or cardiac failure.[1] He subsequently reported cyclic neutropenia and monocytosis.[2] The phenotype of Barth syndrome (BTHS) has expanded steadily and now includes developmental motor and constitutional growth delay, organic aciduria, cardiac endocardial fibroelastosis (EFE) and left ventricular non-compaction (LVNC), ventricular arrhythmia, fetal cardiomyopathy/fetal loss, feeding problems, hypoglycemia, lactic acidosis and characteristic facial features (reviewed in [3]).
Mutations in TAZ (Xq28) as the cause for BTHS were first described in 1996.[4] TAZ encodes an evolutionarily highly conserved acyltransferase, deficiency of which results in defective remodeling of inner mitochondrial membrane phospholipids, especially that of cardiolipin (CL). The aetiology of neutropenia in this disease remains the subject of debate; researchers postulated that mitochondrial abnormalities could lead to enhanced apoptosis of myeloid precursors and neutrophils, although the only published clinical study did not demonstrate shortened cell survival despite avid binding of annexin-V to BTHS blood neutrophils.[5] However, TAZ knockdown experiments suggest accelerated apoptosis of myeloid progenitor cells due to increased dissipation of mitochondrial membrane potential, aberrant release of cytochrome c and activation of caspase-3.[6] Neutrophil functions, such as directed motility, phagocytosis and killing, appear to be unimpaired.[1, 5]
BTHS is still rarely diagnosed, with approximately 200 living males worldwide currently known to the Barth Syndrome Foundation, although there is evidence for substantial under-diagnosis.[3] To aid in the understanding and diagnosis of this disease, particularly with reference to patterns of neutropenia and associated infection risk, we reviewed clinical data, routine blood counts and responses to granulocyte colony stimulating factor (G-CSF) therapy from 88 affected boys.
METHODS
Patients
Patient data were obtained from the Barth Syndrome Registry, NHS Specialised Services Barth Syndrome Service, Royal Hospital for Children, Bristol, UK, Department of Pediatrics, University of Florida Health Center, Gainesville FL and the Severe Chronic Neutropenia International Registry (SCNIR), University of Washington, Seattle WA. Patients or their parents or guardians gave consent for these studies as approved by the respective review boards. Retrospective clinical data were sourced from a combination of hospital notes and parental questionnaires.
BTHS was diagnosed through a combination of characteristic clinical features (predominantly cardiomyopathy and myopathy) and detection of excessive 3-MGC in urine. All were confirmed as having causative mutation of the TAZ gene (Barth Syndrome Registry data). Clinical data on illness and infections were gathered through chart reviews, patient questionnaires and annual reports of patients in the SCNIR. Complete blood counts (CBC) were examined from three sources: (a) the biennial Barth Syndrome Foundation International Scientific, Medical and Family conferences, (b) the clinics of the UK national Barth Syndrome Service or (c) patients’ local hospital clinics. Seventeen neutropenic males underwent a bone marrow examination.
Mathematical Analysis
The Lomb periodogram was used to test for statistical significance periodicity in blood cell counts.[7–9]
RESULTS
Clinical Characteristics
The frequency of common disease characteristics is shown in Table 1. Of those patients showing DCM, 68% manifested symptoms of cardiomyopathy in the first year of life.
Table 1:
Disease characteristics of patients
| Frequency within cohort | |
|---|---|
| Dilated cardiomyopathy | 72/75 (96.0%) |
| Neutropenia | 74/88 (84.1%) |
| Feeding problems / Growth delay | 56/75 (74.7%) |
| Skeletal myopathy | 54/75 (72.0%) |
| Motor delay | 52/75 (69.3%) |
| Bacterial infections | 47/68 (69.1%) |
| Mouth ulcers | 43/75 (57.3%) |
| Fatigue | 40/75 (53.3%) |
The frequency of common diseases characteristics based on patient, family and clinician observations
Incidence and Severity of Neutropenia
Absolute neutrophil counts (ANC) were recorded in all 88 patients at first visit to one of the BTHS clinics. The first recorded CBC of 51 (58%) patients revealed an ANC ≤ 1.5 × 109/L. Follow-up records revealed that 74 of 88 subjects (84%) had at least one ANC < l.5 × 109/L, 28 of whom (38%) had a mean ANC < 1.5 × 109/L.
Multiple CBCs (two or more counts) were available in 79 of the 88 (90%) patients, 44% of whom met the criteria for severe chronic neutropenia (SCN), with multiple ANCs < 0.5 × 109/L over greater than three months. Table 2 shows the data on neutropenia frequency in more detail.
Table 2:
Frequency of Neutropenia in 88 Barth Patients
| Total Study Group (88 patients) | |
| ≥ 2 CBC readings available | 79 (90%) |
| > 10 CBC readings available | 52 (59%) |
| Meeting criteria for CNa | 49 (56%) |
| Meeting criteria for SCNb | 39 (44%) |
| Never treated with G-CSF | 15 (29%) |
| Data from 52 patients with > 10 CBC | |
| ≥ 1 ANC reading <1.5 × 109/L | 49 (94%) |
| ≥ 10 ANC readings <1.5 × 109/L | 40 (77%) |
| ≥ 10 ANC readings <0.5 × 109/L | 18 (35%) |
Chronic Neutropenia (CN) and
Severe Chronic Neutropenia (SCN) were defined as multiple ANCs of < 1.5 × 109/L respectively over greater than three months
Fifty-two patients had more than 10 CBCs; 15 of 52 never received G-CSF. The distribution of ANCs in these 15 patients is shown in Figure 1A. Despite not receiving G-CSF, some showed wide amplitude of ANC, e.g. UPN1 with a range of 0.05–9.24 × 109/L in 63 counts (median 1.74).
Figure 1. A. Range of ANC in 15 individual patients with more than 10 individual CBC readings who never received G-CSF B. Differences between CBC readings within one family.
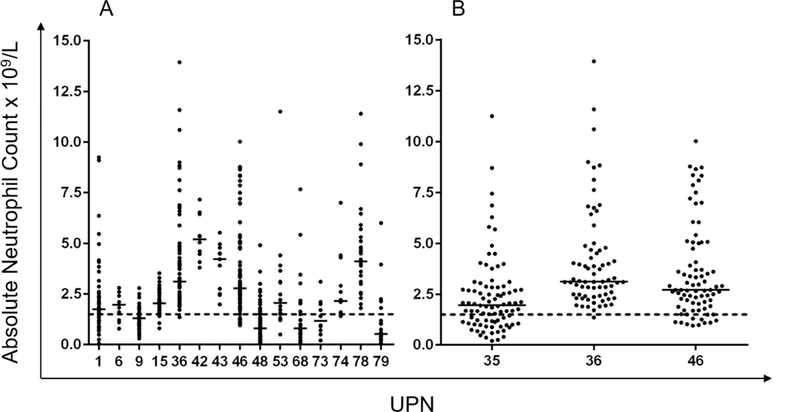
Details are shown of ANCs from CBCs taken principally from times of cardiac review, clinic visits and inpatient admissions. The dotted line indicates the threshold for neutropenia (ANC <1.5 × 109/L).
UPN35 and 36 are brothers, and UPN46 a maternal cousin with BTHS. The earliest DCM occurred in UPN36 who has only been minimally neutropenic (at 1.35 × 109/L) on one of 72 CBC tests. By contrast his brother has significant proximal myopathy but only mild DCM, yet has intermittent neutropenia down to 0.2 × 109/L. Their cousin (UPN46) had a minimum ANC of 0.95 × 109/L from 84 observations taken over 16 years; he has never received G-CSF or had a significant bacterial infection despite disease of sufficient severity to require cardiac transplantation at 3.5 years of age.
Fourteen of the 88 patients (16%) were not neutropenic on any of their CBCs (1–33 ANCs performed over 0–12.3 years). Mean ANCs in the 14 non-neutropenic patients ranged from 1.95–5.28 × 109/L. Twelve of these 14 (86%) non-neutropenic BTHS patients exhibited severe DCM. One other boy (UPN36) who presented with severe DCM was only mildly neutropenic once (ANC 1.35 × 109/L) from 72 CBCs taken over 9.58 years.
Monocytosis
Monocytosis (defined as an absolute monocyte count of greater than 1.0 × 109/L) was frequent, recorded at some stage in 64/85 patients (75%) with available monocyte counts. Twenty-three of the 64 patients (36%) had absolute monocyte counts of >3.0 × 109/L on at least one occasion, with the highest monocyte count being 9.69 × 109/L. Fifty of 85 patients (59%) had a monocyte count greater than 1.0 × 109/L at the time of neutropenia (ANC< 1.5 × 109/L); 13 of the 50 patients (26%) had a monocyte count greater than 3.0 × 109/L at the time of neutropenia. Seven of the 14 non-neutropenic patients (50%) also had monocyte counts greater than 1.0 × 109/L; one patient had two counts >3.0 × 109/L.
Difference in incidence and severity of neutropenia occurred among affected family members (Figure 1B, UPN35, 36 and 46). ANCs in members of this family did not correlate with their severity of cardiomyopathy, proximal myopathy or other symptoms. The severity of neutropenia sometimes changed with time. For example UPN9 (who has never received G-CSF) had a neutrophil count less than 1.0 × 109/L on 5 of 10 counts (median 0.95 × 109/L, range 0.3–1.76 × 109/L) performed serially at age 2–5 months, 1 of 8 counts (median 1.25 × 109/L, range 0.7–2.5 × 109/L ) performed serially at 4.75 years, yet none of 17 counts (median 1.3 × 109/L, range 1.1–1.9 × 109/L) taken over 6 weeks at 9.5 years and 4 of 21 counts at 12.5 years (median 1.28 × 109/L, range 0.45 – 2.99 × 109/L).
Rhythmic Fluctuations & Cyclic Neutropenia
Twenty-seven patients had serial blood counts before G-CSF and eight of the 27 had at least 21 counts, giving an adequate period of blood count profiling for assessment of cyclic neutropenia. Seven boys appeared to have oscillation of blood neutrophils, four of whom are shown in Figure 2A, C & D and Figure 3A.
Figure 2. Examples of extensive neutrophil profiles in BTHS patients.
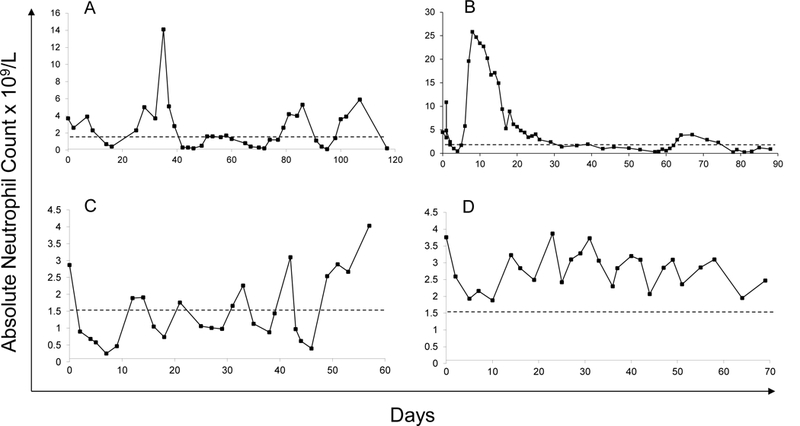
(A and B) UPN37 and 80 respectively did not satisfy mathematical criteria for cycling. (A) UPN37 appeared to cycle at an average of 25 days (range 23–28 days). His G-CSF response is shown in Figure 4A. (B) UPN48 showed spontaneous variability between an ANC of zero and 5.3 × 109/L over a period of 61 days; Unpredictable variation in this manner is the most common pattern seen in BTHS patients. (C and D) Two patients (UPN35 & 36 respectively) exhibited periods of rhythmic “microcycling” over periods averaging 8–9 days either around (in C) or above (in D) the threshold of neutropenia.
Figure 3. G-CSF responses.
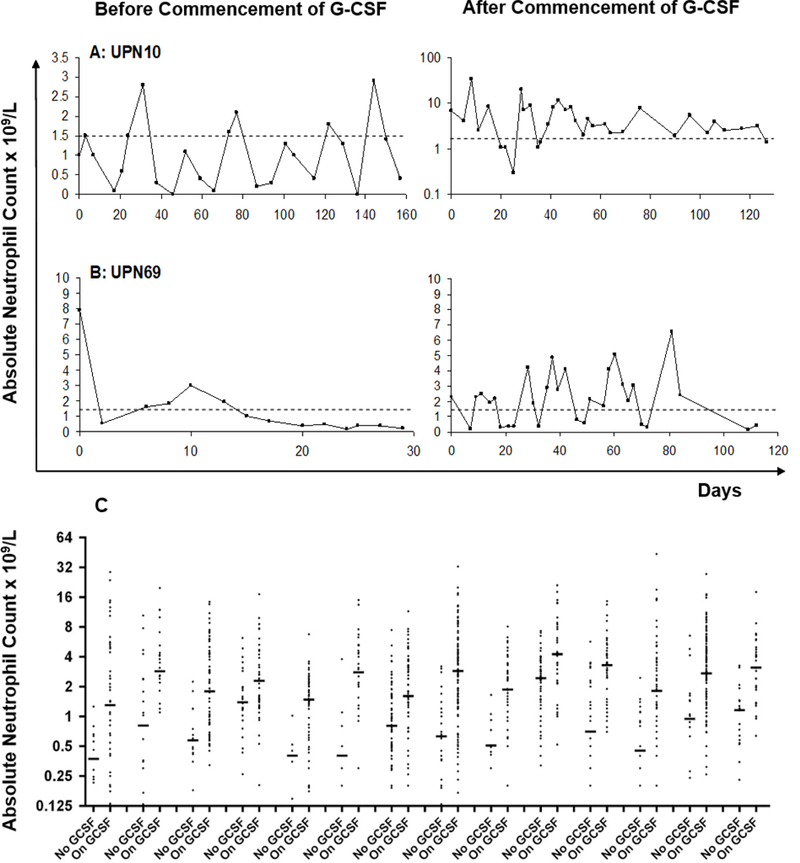
(A) Neutropenia resolved after treatment with G-CSF in UPN10, the only patient who satisfied strict mathematical criteria for cyclical neutropenia, with an average periodicity of 23.3 days. Doses of G-CSF were steadily titrated down from 5 to 0.5 mcg/kg/day in order to keep ANC above 1.5 × 109/L (note logarithmic scale with ANC ranging up to 33.9 × 109/L). (B) By contrast, UPN69 continues to show irregular periods of neutropenia after commencement of G-CSF at 3mcg/kg/dose three times weekly. (C) Effect of G-CSF on range of ANC in 14 other patients (UPN 5, 8, 13, 19, 20, 23, 34, 38, 39, 40, 41, 44, 58 and 67) with the largest numbers of on- and off-therapy CBC.
Only one boy (UPN10, Figure 3A) satisfied strict mathematical criteria for cyclic neutropenia. His cycle length ranged from 21 to 29 days with a mean of 23.3 days. It was not clear what effect G-CSF had on cycle duration due to experimentation with dosage and frequency of drug following initiation. In UPN37 (Figure 2A) aphthous ulceration occurred at nadirs of ANC during the period of study with an average cycle length of 25 days (range 23–28), although this patient did not satisfy mathematical criteria for cyclical neutropenia. Serial counts in two brothers suggested cycling at shorter time intervals, i.e., 10 days (range 8–13) and 9 days (6–13), respectively (UPN35, Figure 2C) and above (UPN36, Figure 2D), but these patterns were not apparent on repeat profiles on either boy performed three years later (data not shown).
Bone Marrow Characteristics
Bone marrow results were available from 19 tests performed in 17 patients. Six patients were tested before commencing G-CSF. The remaining aspirates comprised surveillance tests taken to exclude development of myelodysplasia or leukaemia secondary to G-CSF therapy.
The aspirate or biopsy was reported to be hypocellular in three patients; the remainder had normal or had only a modest reduction in cellularity. One patient had mild red cell dysplastic change after four years of G-CSF therapy. None of the marrows obtained on G-CSF showed an increase in marrow myeloblasts and no clonal cytogenetic change was detected.
Decreased myeloid maturation was reported for five of six patients prior to G-CSF therapy. The common abnormality was a relative paucity of mature neutrophils in the marrow. One patient was reported to have an arrest at the metamyelocyte stage; the others primarily had reduced numbers of mature neutrophils in the marrow. None of the aspirates were described as showing promyelocyte arrest.
Results were available on 13 patients investigated during G-CSF therapy. Seven marrows showed normalisation of myeloid differentiation, the remaining marrows still showing left-shifted myelopoiesis. Marrow cellularity increased in two of three patients who had marrows performed both before and on treatment with G-CSF. Increased or toxic granulation was observed in two patients.
Increased eosinophils or eosinophil precursors were reported for two of six patients prior to G-CSF therapy and six of 13 patients on therapy, one of whom had increased eosinophils prior to receiving G-CSF.
Responses to G-CSF
G-CSF was administered to 39 of 88 subjects (44%). Responses to G-CSF are illustrated in Figure 3. Some patients were treated just to cover periods of infection or mouth ulceration but in most G-CSF was given on a dosing schedule varying from daily to weekly, the most common regimes being three times weekly or alternate daily. For 17 patients receiving G-CSF on whom detailed data were available, doses averaged over a week ranged from 0.12 mcg/kg/day to 10.92 mcg/kg/day (mean 1.160 mcg/kg/day, median 1.160 mcg/kg/day). Each of these treated patients had previously exhibited severe symptomatic neutropenia. For this group of 17 patients, the median absolute neutrophil count pre-G-CSF therapy was 0.47 × 109/L and mean 0.57 × 109/L (range 0.00–10.45 × 109/L) compared with median ANC of 2.28 × 109/L, mean ANC of 3.93 × 109/L (range 0.00–33.89 × 109/L) during G-CSF therapy (p< 0.001, paired two tailed T test).
Determining a dose of G-CSF that alleviated symptoms without producing excessively high counts was a problem in some patients due to the wide amplitude of ANC prior to treatment. Figure 4A and B show two examples of patients with marked responses to G-CSF. Careful titration of G-CSF dose (exemplified by UPN10, Figure 3A) and alternate daily or thrice weekly dosing prevented excessive rises in neutrophil counts in most patients.
Figure 4. Exuberant responses to G-CSF.
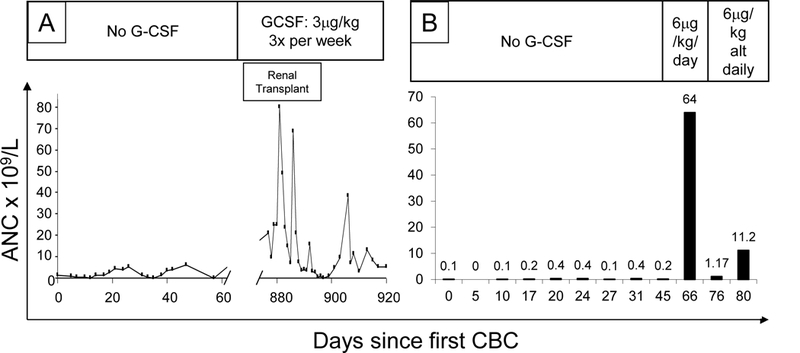
UPN37 showed a range of ANC of 0.1 to 5.9 × 109/L in the year before commencing G-CSF at 3mcg/kg/dose thrice weekly. His ANC ranged from 0.2 to 79.9 in the 6 weeks afterwards, although renal transplantation (required as a consequence of previous Haemophilus sepsis) may have contributed to these dramatic changes. By contrast, no events occurred in UPN51 (shown in B), whose ANC rose to 64 × 109/L, from a maximum ANC of 0.6 × 109/L in nine CBCs taken at regular intervals over the previous two months, after G-CSF was introduced at 10mg/kg/day; this necessitated switching to alternate daily dosing.
Infection Risk
A detailed review for significant bacterial infections was conducted in 35 patients under the longitudinal care of the UK NHS Barth Syndrome Service because all retrospective notes could be accessed in this group. Infections prior to introduction of G-CSF therapy included Streptococcal bacteremia/septicemia (2 patients, one resulting in acute tubular necrosis), Haemophilus septicemia (one patient, resulting in renal failure requiring renal transplantation), osteomyelitis (2 patients), septic arthritis (1), soft tissue abscesses (3), cellulitis (5), balanitis (2) lobar consolidation/pneumonia (4), gingivitis (2), severe recurrent aphthous ulceration (2), urinary tract infections (1) and secondary infection of an underlying keratosis pilaris rash (1). A further UK patient who predeceased this study had neutropenia and skin sepsis; he developed severe neurological handicap secondary to streptococcal septicemia. The overall registry data also identified further instances of streptococcal septicemia (1), pneumococcal bacteremia (1), Haemophilus septicemia (1) and Pseudomonas cellulitis (1) and four unspecified episodes of bacteremia/septicemia.
26 of these 35 UK (74%) patients had one or more counts in the severe neutropenic range, 14 (40%) having an ANC of zero on occasions. Of these 26 patients with severe neutropenia, 14 (54%) had experienced severe bacterial infections thought to be secondary to their underlying neutropenia.
Only five patients in the entire group had significant instances of suspected/proven bacterial infection after institution of G-CSF ± antibiotic prophylaxis. One patient died of sepsis after suffering recurrent staphylococcal infections, Candidaemia (C. parapsillosis) and Enterobacter cloacae infection associated with multiple indwelling central venous catheters, an implantable cardioverter defibrillator (ICD) and extensive granulation tissue which formed around long-term gastrostomies. He also required omphalectomy due to long-term umbilical inflammation / infection. All infections occurred despite consistent prevention of neutropenia by G-CSF therapy and death occurred after 15.5 years of G-CSF therapy; there was no evidence of myelodysplastic or malignant change on longitudinal bone marrow monitoring. Four other patients experienced cellulitis either following trauma such as cat scratches or insect bites (3 patients) or associated with poor compliance with G-CSF (1). One of these patients also had recurrent localised cellulitis at his gastrostomy site, which necessitated closure of the gastrostomy, despite consistent G-CSF therapy.
From self-reported data on 68 patients in the Barth Syndrome Registry, other more minor infections were seen as follow: ear infections (32%), sinusitis (18%) and urinary tract infections (11%). Thirty-two percent of parents reported that their children had been admitted to hospital on one or more occasions for investigation or treatment of febrile episodes or proven infections.
Discussion
Originally BTHS was reported as a composite X-linked syndrome of DCM, skeletal myopathy, neutropenia and growth delay. More recent data emphasise that motor delay, infections, mouth ulcers, fatigue and feeding problems (sometimes requiring tube or gastrostomy feeding) are also common features (see Table 1). However, tremendous phenotypic diversity and inter-patient variability is apparent (reviewed in detail by Clarke et al [3]).
This phenotypic diversity applies profoundly to neutropenia which this series shows can vary from being persistent and severe, through widely variable and unpredictable (the commonest pattern) to never being neutropenic. No genotype/phenotype correlation has been demonstrated for any aspect of this disease and this is perhaps not surprising when ANCs can vary widely even in males within a single family (as shown in Figure 1B). It should be noted that one member of this family (UPN36, ANC shown in Figure 1B) was minimally neutropenic just once (at 1.35 × 109/L, average ANC 4.08) from 72 CBCs taken at between 13 months and 11 years of age despite an otherwise severe disease phenotype.
Rigaud et al report similar findings in a French cohort in terms of initial ANC, percentage of patients with at least one neutropenic count and those with a median ANC <0.5 × 109/L. [10] However, these authors highlighted a correlation between severity of neutropenia at diagnosis and prognosis; all four patients in that study with an initial ANC ≤ 0.5 × 109/L died by the age of three years. By contrast 22 patients in our cohort had an initial ANC in the same range but only one death occurred (at 23 years, unrelated to infection). It seems likely that wider use of G-CSF in the patients reported in this series compared to the French series (44% versus 27%) contributed to this discrepancy.
BTHS neutropenia has often been described as cyclical. Our analysis of this was hampered by absence of adequately detailed profiles, i.e., blood counts taken thrice weekly for 3–6 weeks, in many patients. However, by stringent mathematic analysis, we were able to show definitive cycling behaviour - with an average periodicity of 23.3 days - in only one boy (UPN10, Figure 3A). Several other profiles also showed rhythmic variation (e.g. that of UPN37 in Figure 2A), which might be easy to confuse with the disease cyclic neutropenia. Several boys with BTHS also showed an intermittent pattern of severe mouth ulceration and gingivitis when neutropenic, which could add to this confusion. Similarly, none of the patients described by Rigaud et al exhibited cyclic neutropenia.[10] It is therefore more accurate to describe BTHS patients as having variable neutropenia.
Important differential diagnoses include cyclic, severe congenital and idiopathic neutropenia. Diagnostic confusion between severe congenital neutropenia (SCN) and BTHS could occur although only rarely; for example, one patient had nine out of 10 ANC below 0.2 × 109/L over a one-year period. Further features in common between SCN and BTHS include monocytosis, myelocyte arrest and eosinophilia or increased numbers of eosinophil precursors on blood / marrow examination.[1, 11, 12]
The cases that we have described stress the serious risks of infection in these children. Further evidence is provided by the family originally described by Peter Barth [1] and an 18% fatality rate due to infection in the French national experience: two deaths from septic shock and two cardiac deaths during unexplained high fever.[10] However, it is intriguing that there were no cases of Clostridial infections in this series (even though some children had severe persistent neutropenia) and comparatively few cases of deep tissue cellulitis. Gauging the risk of infection is also not easy due to the highly unpredictable nature of the neutropenia. As one example, UPN37 had mean neutrophil counts of 2.5, 2.17, 2.73, 2.06, 1.59 and 2.21 × 109/L in consecutive periods of two years yet developed recurrent aphthous ulceration at the nadirs of counts (at 23–28 day intervals, see Figure 2A) in only the last of these periods. This deterioration culminated in Haemophilus pneumonia and septicemia, renal failure (requiring paternal allograft), gut ischemia and Aspergillus peritonitis after peritoneal dialysis catheter insertion. His subsequent G-CSF responses are shown in Figure 4A.
G-CSF usage in Barth syndrome was first reported in 1995.[13] Thirty-nine of the 88 (44%) patients in the current series have received G-CSF, ranging in frequency from daily to weekly but most typically on a three times per week or daily basis. With averaged doses from 0.12 mcg/kg/day to 10.92 mcg/kg/day (mean 1.16 mcg/kg/day), many boys/men remained intermittently neutropenic (94% of evaluable patients, representative examples can be seen in Figure 3C). However, these patients were free of significant infections following introduction of G-CSF ± antibiotic prophylaxis in most cases. The notable exception was a man with longstanding severe BTHS manifestations requiring gastrostomy feeding for many years, intravenous feeding requiring central venous catheters, and placement of an ICD to prevent ventricular arrhythmia. He developed severe granulation reactions at gastrostomy sites, hypertrophic scars at surgical sites and infections related to central venous catheters and an ICD despite good control of neutrophil counts by G-CSF over a prolonged period. It is unclear whether some of these were unusual manifestations of BTHS or inanition due to prolonged illness.
Four other boys developed cellulitis: related to nail biting and non-compliance with G-CSF in one patient and suspected cellulitis/reactions of debatable relevance to neutropenia following insect bites and cat scratches in two others. The fourth boy experienced problems with insect bites and recurrent cellulitis around a gastrostomy which eventfully necessitated closure. Some patients reported alleviation of aphthous ulceration and reduction in malaise/fatigue although this is hard to evaluate. The French experience included two episodes of severe infection (including one episode of septic shock) whilst patients were receiving G-CSF therapy.[10] These infections underpin the rationale for additional use of antibiotic prophylaxis in a majority of neutropenic UK patients within this series.
Malignancy has been reported in only two boys with BTHS; single cases of juvenile myelomonocytic leukaemia (JMML, personal communication, Dr AB de Sousa) and post-cardiac transplant EBV negative T-cell non-Hodgkin lymphoma[14] respectively, neither child having received G-CSF. No other cases of myelodysplasia or leukemia have been reported or are currently known through the Barth Syndrome Registry or SCNIR. Nevertheless, we consider it wise to perform intermittent bone marrow assessments whilst experience of this treatment for BTHS remains limited.
Historically, BTHS has been regarded as extremely rare, with only 450 affected cases ever proven by genetic analysis worldwide. However, with improved case ascertainment and better diagnostic tests now available, a frequency as high as one in 140,000 has been suggested.[3] US Pediatric Cardiomyopathy Registry data showed that 3–5% of all males with cardiomyopathy had BTHS as their underlying diagnosis (Dr. Jeffrey Towbin, personal communication). A 2017 Chinese study, using next generation sequencing to investigate males and females with primary cardiomyopathy in a systematic fashion, showed that 6.5% of males with LVNC ± DCM had pathogenic TAZ mutations. This would imply that most pediatric hematologists working alongside specialist cardiac units should expect to meet one or more cases in their career. Considering this, there is surprisingly little haematological literature on BTHS.[5, 6, 10, 12, 15, 16, 17, 18, 19]
Historical testing for BTHS often relied on identification of 3-methylglutaconic aciduria but this is now recognised to be highly unreliable, with many patients lacking pathological aciduria at initial presentation.[18, 19, 20, 21, 22, 23, 24, 25, 26] A “gold standard” biochemical test for BTHS is however now available: the ratio of L4-cardiolipin (CL) to its closely related precursor monolysocardiolipin (MLCL) (termed the MLCL/CL ratio). This test appears to have 100% specificity and sensitivity and can be performed on a blood sample sent by routine post.[27, 28] An intermediate form of BTHS has been described recently.[29, 30] This is characterised by less severe CL deficiency but still an abnormal ratio due to high levels of MLCL. Interestingly this appears to be associated with almost complete absence of neutropenia which may hold clues to elucidating pathogenesis if confirmed in more patients. Definitive diagnosis can also be performed by identifying a causative mutation in the TAZ gene and TAZ sequencing should be included in next generation sequencing / clinical exome analyses designed to investigate idiopathic neutropenia.
Conclusions
We recommend that BTHS should be considered within the differential diagnosis of any boy with unexplained neutropenia - whether chronic severe, cyclical or intermittent in nature. This is most important if neutropenia is accompanied by motor delay, myopathy/cardiomyopathy, feeding problems, failure to thrive or suspected mitochondrial disease, although presentation with isolated neutropenia can occur (because DCM is either absent or subclinical or has completely resolved). Particular attention should be paid to boys who lack mutation of ELANE or the glucose-6-phosphatase, catalytic subunit 3 (G6PC3) gene (where structural heart abnormalities are also present).[31] Based on the clinical data summarized in the report, we recommend that neutropenic Barth syndrome males who have recurrent fevers and infections should be treated approximately 3 days per week with G-CSF in doses sufficient to normalize their ANCs. Prophylactic antibiotics, e.g., low dose daily penicillin V or trimethoprim-sulfamethoxazole may be helpful.
Key Points.
Barth syndrome is a rare sex-link genetic disorder associated with neutropenia that may be chronic, intermittent or cyclic and accompanied by infections of varying severity.
The susceptibility to infections is due only in part to neutropenia; congestive heart failure, nutritional deficiencies and the need for feeding tubes add to the risk of infections.
Neutropenia in Barth responds to long-term treatment with granulocyte colony stimulating factor (G-CSF), usually in low dose given subcutaneously daily or every other day.
In some patient prophylactic antibiotics may help to prevent serious infections.
Acknowledgements
DCD is a consultant and receives research support from Amgen, a biopharmaceutical company that produces and markets G-CSF. MCM receives research support from the Natural Sciences and Engineering Research Council of Canada.
CGS and DCD conceived the study, collated and analysed data, provided clinical care to patients and wrote the initial draft. All other authors co-authored the paper and edited the manuscript. In addition: SJG prepared the figures and analysed data, CS and MKM collated data via the Barth Syndrome Registry, BV and HO collated additional UK and Swiss data respectively, MKD, VMB and KRM organised data collection within the Barth Syndrome community, MCM provided mathematical analysis of cycling behaviour and AAB organised, collated and analysed data.
The authors dedicate this paper to the memory of Will McCurdy whose large body of neutrophil data stimulated wider use of G-CSF in this condition, better understanding of ANC variability and the development of this study. We also thank the many patients, families and clinicians who have subsequently collated and contributed blood count data to the Barth Syndrome Registry of the Barth Syndrome Foundation (www.barthsyndrome.org) and the Severe Chronic Neutropenia International Registry, Dr Leo Hamilton for provision of blood count data, and Dr Richard Kelley for comments on our manuscript. We are indebted to the COGENT Trust for supporting Sarah Groves and the early work which made this study possible, and for National Health Service (NHS) Specialised Services for supporting Professor Colin Steward and the development of a coordinated multidisciplinary UK service for BTHS.
Funding:
The COGENT Trust (private charity) supported SG who prepared figures and analysed data. National Health Service (NHS) Specialised Services supported Professor Steward’s clinical work on Barth Syndrome. The SCNIR (DCD & AAB) is supported by an NIH grant, R24AI049393, and by Amgen.
Footnotes
DECLARATIONS
Ethics approval and consent to participate
Informed consent was obtained from all patients. The Institutional Review Boards at the University of Washington, Seattle, Washington approved the SCNIR to collect data from consented patients (IRB application number 00000996). The Institutional Review Boards at the University of Florida approved the BSF to collect data from consented patients (IRB protocol number 186–2006).
References
- 1.Barth PG, Scholte HR, Berden JA, Vanderkleivanmoorsel JM, Luythouwen IEM, Vantveerkorthof ET, et al. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J Neurol Sci. 1983;62(1–3):327–355. [DOI] [PubMed] [Google Scholar]
- 2.Barth PG, Wanders RJA, Vreken P. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) - MIM 302060. J Pediatr. 1999;135(3):273–276. [DOI] [PubMed] [Google Scholar]
- 3.Clarke SL, Bowron A, Gonzalez IL, et al. Barth syndrome. Orphanet J Rare Dis. 2013;8(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bione S, Dadamo P, Maestrini E, et al. A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat Genet. 1996;12(4):385–389. [DOI] [PubMed] [Google Scholar]
- 5.Kuijpers TW, Maianski NA, Tool ATJ, et al. Neutrophils in Barth syndrome (BTHS) avidly bind annexin-V in the absence of apoptosis. Blood. 2004;103(10):3915–3923. [DOI] [PubMed] [Google Scholar]
- 6.Makaryan V, Kulik W, Vaz FM, et al. The cellular and molecular mechanisms for neutropenia in Barth syndrome. Eur J Haematol. 2012;88(3):195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lomb NR. Least-squares frequency analysis of unequally spaced data. Astrophys Space Sci. 1976;39:447–462. [Google Scholar]
- 8.Haurie C, Dale DC, Mackey MC. Occurrence of periodic oscillations in the differential blood counts of congenital, idiopathic, and cyclical neutropenic patients before and during treatment with G-CSF. Exp Hematol. 1999;27(3):401–409. [DOI] [PubMed] [Google Scholar]
- 9.Scargle JD. Studies in astronomical time series analysis. II - Statistical aspects of spectral analysis of unevenly spaced data. Astrophys J. 1982;263(Part 1):835–853. [Google Scholar]
- 10.Rigaud C, Lebre AS, Touraine R, et al. Natural history of Barth syndrome: a national cohort study of 22 patients. Orphanet J Rare Dis. 2013;8:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barth PG, Valianpour F, Bowen VM, et al. X-linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update. Am J Med Genet A. 2004;126A(4):349–354. [DOI] [PubMed] [Google Scholar]
- 12.Vernon HJ, Sandlers Y, McClellan R, Kelley RI. Clinical laboratory studies in Barth Syndrome. Mol Genet Metab. 2014;112(2):143–147. [DOI] [PubMed] [Google Scholar]
- 13.Cox GF, Pulsipher M, Rothenberg M, et al. Correction of neutropenia in Barth syndrome by G-CSF. Am J Hum Genet. 1995;57(4):1015–1015. [Google Scholar]
- 14.Ronghe MD, Foot ABM, Martin R, et al. Non-Epstein-Barr virus-associated T-cell lymphoma following cardiac transplantation for Barth syndrome. Acta Paediatr. 2001;90(5):584–586. [PubMed] [Google Scholar]
- 15.McCanta AC, Chang AC, Weiner K. Cardiomyopathy in a child with neutropenia and motor delay. Curr Opin Pediatr. 2008;20(5):605–7. [DOI] [PubMed] [Google Scholar]
- 16.Folsi V, Miglietti N, Lombardi A, et al. Cardiomyopathy in a male patient with neutropenia and growth delay. Ital J Pediatr. 2014;40:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Raam BJ, Kuijpers TW. Mitochondrial defects lie at the basis of neutropenia in Barth syndrome. Curr Opin Hematol. 2009;16(1):14–19. [DOI] [PubMed] [Google Scholar]
- 18.Woiewodski L, Ezon D, Cooper J, Feingold B. Barth Syndrome with late-onset cardiomyopathy: a missed opportunity for diagnosis. J Pediatr. 2017;183:196–198.***This report point to the challenge of diagnosis of Barth syndrome.
- 19.Wang J, Guo Y, Huang M, et al. Identification of TAZ mutations in pediatric patients with cardiomyopathy by targeted next-generation sequencing in a Chinese cohort. Orphanet J Rare Dis. 2017;12(1):26.**This recent report points out the world-wide distribution of this rare disease.
- 20.Gedeon AK, Wilson MJ, Colley AC, et al. X linked fatal infantile cardiomyopathy maps to Xq28 and is possibly allelic to Barth syndrome. J Med Genet. 1995;32(5):383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bleyl SB, Mumford BR, Brown-Harrison MC, et al. Xq28-linked noncompaction of the left ventricular myocardium: prenatal diagnosis and pathologic analysis of affected individuals. Am J Med Genet. 1997;72(3):257–65. [PubMed] [Google Scholar]
- 22.Schmidt MR, Birkebaek N, Gonzalez I, Sunde L. Barth syndrome without 3-methylglutaconic aciduria. Acta Paediatr. 2004;93(3):419–21. [DOI] [PubMed] [Google Scholar]
- 23.Brady AN, Shehata BM, Fernhoff PM. X-linked fetal cardiomyopathy caused by a novel mutation in the TAZ gene. Prenat Diagn. 2006;26(5):462–5. [DOI] [PubMed] [Google Scholar]
- 24.Marziliano N, Mannarino S, Nespoli L, et al. Barth syndrome associated with compound hemizygosity and heterozygosity of the TAZ and LDB3 genes. Am J Med Genet A. 2007;143A(9):907–15. [DOI] [PubMed] [Google Scholar]
- 25.Takeda A, Sudo A, Yamada M, et al. Barth syndrome diagnosed in the subclinical stage of heart failure based on the presence of lipid storage myopathy and isolated noncompaction of the ventricular myocardium. Eur J Pediatr. 2011;170(11):1481–4. [DOI] [PubMed] [Google Scholar]
- 26.Thiels C, Fleger M, Huemer M, et al. Atypical clinical presentations of TAZ mutations: an underdiagnosed cause of growth retardation? JIMD Rep. 2016;29:89–93.** Another report pointing out the challenge of recognizing Barth diagnosis.
- 27.Kulik W, van Lenthe H, Stet FS, et al. Bloodspot assay using HPLC-tandem mass spectrometry for detection of Barth syndrome. Clin Chem. 2008;54(2):371–378. [DOI] [PubMed] [Google Scholar]
- 28.Bowron A, Frost R, Powers VE, et al. Diagnosis of Barth syndrome using a novel LC-MS/MS method for leukocyte cardiolipin analysis. J Inherit Metab Dis. 2013;36(5):741–746. [DOI] [PubMed] [Google Scholar]
- 29.Bowron A, Honeychurch J, Williams M, et al. Barth syndrome without tetralinoleoyl cardiolipin deficiency: a possible ameliorated phenotype. J Inherit Metab Dis. 2015;38(2):279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson WR, DeCroes B, McClellan R, et al. New targets for monitoring and therapy in Barth syndrome. Genet Med 2016;18(10):1001–10. [DOI] [PubMed] [Google Scholar]
- 31.Boztug K, Appaswamy G, Ashikov A, et al. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360(1):32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]