

Introduction
It has been known for decades that the immune system can be spontaneously activated against melanoma. The presence of tumor infiltrating lymphocytes (TIL) in tumor deposits is a positive prognostic factor [1]. Cancer vaccination includes approaches to generate, amplify, or skew antitumor immunity. To accomplish this goal, tested approaches involve administration of tumor antigens, antigen presenting cells (APCs) or other immune modulators, or direct modulation of the tumor. Because the success of checkpoint blockade can depend in part on an existing antitumor response, cancer vaccination may play an important role in future combination therapies. In this review, we discuss a variety of melanoma vaccine approaches and methods to determine the biological impact of vaccination.
Historical perspective
Active immunotherapies are not a novel concept. Over a century ago, William B. Coley injected live streptococci into sarcoma patients to promote erysipelas and induce immune system-mediated tumor rejection. While therapies such as these have shown limited clinical benefit and high degree of systemic toxicity, they did usher in an era of cancer immunotherapy [2]. Some of the first melanoma cancer vaccine trials occurred in the 1970s. These included testing tumor lysate injections and pathogen adjuvants like Bacillus Calmette-Guerin (BCG) [3,4]; vaccinia virus oncolysate [5] or Corynebacterium parvum [6]. The results were promising, with some patients displaying clinical responses that were often transient. In the 1980s, there were studies focused on allogeneic melanoma cells [7], and in the early 1990s, gangloside (GM2, GD3)-based vaccines with adjuvants and immunogenic conjugates [8]. Cytokines were also tested at this time, both as systemically delivered agents (IL-2, IFNa) or as cytokine-secreting genetically modified tumor cell vaccines (especially GM-CSF) [9,10]. During the 1990s, the identification of the tumor antigens expressed by melanoma tumors which were recognized by the cytotoxic T cells (CTL) infiltrating tumors were beginning to be published which had a major impact on the field [11-13].
The most common approach to cancer vaccination in the last two decades involves immunization with shared tumor antigens expressed by many different patients’ tumors. The earliest tumor associated antigens (TAAs) identified were proteins that were overexpressed in tumor cells but minimally expressed in untransformed normal tissues [11,12,14,15]. TAAs were also identified after cloning the genes that encoded proteins that included epitopes recognized by tumor reactive TILs. Other types of TAA tested in melanoma include cancer testis antigens and mutated antigens (tumor specific or private antigens).
Melanoma antigens
Melanoma antigens can be segregated into four categories: overexpressed antigens, cancer testis antigens, mutated oncogenes, and patient-specific mutated neoantigens [16]. Overexpressed antigens include melanoma lineage antigens such as MART-1/Melan-A, tyrosinase, and gp100 (expressed in >90% of melanoma tumors [17]). All have shown efficacy as targets in vitro and in murine models, and have been tested in clinical trials with some objective tumors) clinical responses [18-20]. A potential explanation for the limited clinical activity seen when targeting such TAAs is that the highest avidity T cells specific to these normal “self” antigens may have been deleted or exhausted by chronic antigen stimulation, leaving only less effective, lower avidity T cells to be activated.
Cancer testis antigens are expressed in a proportion of most tumor tissue types and in germ cells that, because of their physiologic location, are generally ignored by the immune system. Such antigens include the large MAGE-A (expressed in 9-51% of melanomas [17]), MAGE-B, and MAGE-C families, and NY-ESO-1 (expressed in 45%of malanomas [17]). These antigens have been tested in human clinical trials and implicated in therapeutic responses [21].
Mutated oncogenes have been known for decades, and commonly occurring shared mutations in the RAS family of oncogenes (NRAS is mutated in 15-25% of melanomas [22-23]) have been identified. However, earlier it was thought unlikely that such mutations would be present in a processed, presented, and immunogenic MHC restricted epitope in a characterized MHC class I or II molecules, making shared mutations seemingly impossible to target by vaccination in a clinical trial [24]. Neoantigens are those antigens that arise from random somatic mutations in individual tumors [25]. This group of antigens will be discussed in greater detail below.
Cancer vaccine platforms
Most cancer vaccines are designed to activate tumor specific CD8+ CTL because studies in mice reproducibly support the key therapeutic role played by these cells. The most common vaccination strategies used have been based on MHC class I restricted peptide epitopes from TAAs. These have been delivered in a variety of adjuvant formulations (including cytokines and toll-like receptor (TLR) ligands) to promote in vivo presentation by endogenous APC. Peptide based vaccines take advantage of the existing data on MHC class I peptide binding motifs for the most common HLA types, and the algorithms which can screen protein amino acid sequences for peptide epitopes derived from TAAs. Data from animal models support the potential for such vaccines to have a substantial therapeutic effect [26-28].
Peptide-based strategies
Peptides formulated in adjuvants (such as Montanide, which is analogous to incomplete Freund’s adjuvant (IFA)), with or without cytokines, such as GM-CSF and interferon. (IFN.), or TLR agonists, have shown clinical benefit (partial responses, complete responses, and durable disease stabilization) in small and large scale clinical trials [29-32]. In smaller trials, peptides loaded onto APCs, such as dendritic cells, have also resulted in positive immune and clinical effects [33-35].
As peptide-based vaccines are tested, optimal adjuvants and formulations of these vaccines are still being identified. Clinical trials of peptide-based vaccines were recently reviewed [36]. A benefit of peptide-based approaches is that 9 to 10 amino acid long peptides are simple and inexpensive to manufacture. Large scale manufacture is possible and the peptides are stable when stored and shipped. While enthusiasm for peptide vaccines remains high due to their immunogenicity, data demonstrating clinical efficacy of vaccine peptide-specific T cells remains rare.
Because of HLA restriction, those who do not express common HLA types cannot be treated with this type of vaccine. In addition, the usual MHC class I binding short peptides do not activate CD4+ helper T cells, which may limit the functionality of CD8+ cytotoxic T cells. This problem has been overcome by the addition of non-tumor specific help (inclusion of keyhole limpet hemocyanin (KLH), tetanus, or (pan-DR binding synthetic helper (PADRE) peptides), although data are limited over the nature of the “help” provided by heterologous helper peptides. Shared melanoma antigen helper peptides have been tested in several trials, with data showing improved survival in vaccinated patients [16].
Another strategy that has shown significant clinical efficacy in the setting of cervical cancer is the use of synthetic peptides that are long enough to include multiple MHC class I and II epitopes [38,39]. These 23-45 amino acid long peptides, delivered subcutaneously, have been shown to be especially effective, possibly because they utilize a more efficient processing and presentation pathway, which leads to superior T cell activation [40]. We have observed in vitro that a 16 a.a. long MAGE-A6-derived peptide, MAGE-A6-172-187, that has been reported to be promiscuously presented by multiple HLA-DR alleles, can also induce HLA-A2-restricted CD8+ T cell responses against the MAGE-A6-176-185 epitope [16,41]. These results indicate that long peptides derived from melanoma antigens could also be implemented in future melanoma vaccine modalities.
Multiple peptides can be given at the same time, targeting several T cell clones and antigens at once [42-44]. A trial combining pre-vaccine cyclophosphamide with multiple peptides and GM-CSF showed that improved survival was associated with antigenic breadth of response and reduced suppressive circulating regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) [44]. The Slingluff group has previously generated a cocktail of 6 MHC class II-restricted peptides (6MHP; derived from MAGE, MART-1, gp100, and tyrosinase (TAAs)) which has been tested in two phase I/II trials. Survival outcomes of patients treated with the 6MHP vaccine are superior to those of matched institutional controls [46,47]. Currently, the vaccine is being tested in a new phase I/II study in combination with ipilimumab (NCT02385669).
To enhance the efficacy of cancer vaccines, “wild type” tumor antigen-derived epitopes can be modified in order to activate cross-reactive T-cell clones, resulting in activation of higher avidity T cell clones capable of superior tumor recognition and killing. One or two amino acid substitutions, normally within the predicted epitope anchor residues, have been shown to lead to higher binding affinity that resulted in induction of higher avidity, tumor-specific T cells [47,48]. The design of heteroclitic peptides can be difficult and labor-intensive. Recently, Cristian Capasso et al. have developed The Epitope Discovery and Improvement System (EDIS), an automated algorithm-driven platform to speed up the design of heteroclitic peptides [49].
Another potential strategy to enhance tumor antigen-derived epitope immunogenicity is to utilize highly homologous and cross-reactive “mimicking” peptides derived from proteins found in common microbial pathogens to which many individuals have pre-existing immunity. We have shown that a peptide derived from the Mycoplasma penetrans HF-2 permease protein, HF-2-216-229, shares a high degree of structural and functional homology with the aforementioned MAGE-A6-172-187 epitope. The functional avidities of CD4+ and CD8+ T cells primed with HF-2-216-229 are 100 and 1000x greater than those of CD4+ and CD8+ T cells primed with MAGE-A6172-187, respectively. Consequently, HF-2-216-229-stimulated T cells are superior at recognizing APCs pulsed with MAGE-A6-172-187 or recombinant MAGE-A6, as well as HLA-matched MAGE-A6+ melanoma cell lines [16,41].
APC based strategies
Many types of APCs have been investigated, including peripheral blood mononuclear cells (PBMC), activated B cells, and, more commonly, dendritic cells (DC). DC are a heterogeneous population of APCs that can efficiently take up antigens and sample their environment. They then process and present these antigens to CD4+ and CD8+ T cells and incorporate immune response modulating cues (including the secretion of cytokines such as interleukin 12 (IL-12) p70, which skews the immune response towards type 1 to modulate the type of response. A type 1 response involves IFN., IL-2, and tumor necrosis factor (TNF) and it promotes the activation of cytotoxic CD8+ T cells. Several recent reviews summarize the history, biology, and clinical application of these cells [50-52].
Clinical trials of autologous DC vaccines involve individualized patient vaccination approaches and single clinical trial arms. It is difficult to compare trials and draw firm conclusions about the efficacy or different approaches [53-55]. Natural CD1c+ DC as well as DC generated from monocytes (Figure 1) [56-58] and CD34+ progenitor cells have been tested with various antigen formats, including complex tumor lysates that contain normal, TAA and tumor specific antigens, or synthetic MHC class I restricted peptides. Vaccines have been injected into the blood, skin (subcutaneously or intradermally), and lymph nodes. The early lessons learned were that DC vaccines are safe, feasible and immunogenic and can promote clinically significant tumor regression in 4.2-7.1% of patients [35,59-65].
Figure 1. Basic monocyte-derived DC-based immunization protocol.
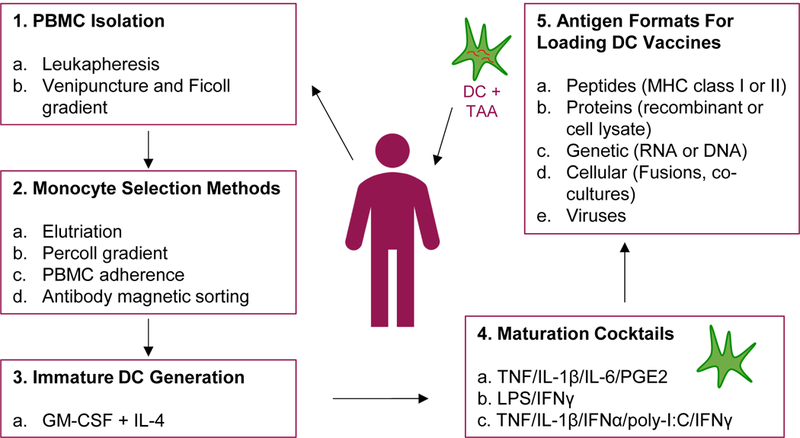
DC-based vaccines are generated by the selection of CD14+ monocytes from isolated PBMC. Monocytes are cultured with GM-CSF and IL-4, often for 5 days, to obtain immature DC. Immature DC are matured using various maturation cocktails as previously described by Kaka et al. (2009), Vujanovic et al. (2010), and Kalinski et al. (2005). After 24-48 hr maturation, matured DC are delivered to patients. DC are loaded with antigens either before, during, or after maturation
One therapeutic cancer vaccine—Sipuleucel-T manufactured by Dendreon—was approved by the Food and Drug Administration in 2010. It consists of autologous APC loaded with the TAA prostatic acid phosphatase plus GM-CSF and is approved for metastatic prostate cancer on the basis of a 4.1 month improvement in overall survival seen in data from large scale phase III clinical trials [66].
Tumor based strategies
Early cancer vaccine studies found that mice could be immunized with tumor cells that were killed and engineered to express immune stimulatory cytokines [67,68] including GM-CSF. The data supporting syngeneic, autologous, or allogeneic tumor cells transfected to express high amounts of GM-CSF supported clinical testing, with some immune and clinical responses [69-73]. Other strategies that use the personalized approach of harnessing autologous tumor antigens include using tumor lysates to load APCs ex vivo and fusion of tumor cells and autologous APCs. Immunity to undefined tumor lysates and foreign helper proteins has been demonstrated in some cases [74-76]. Autologous tumor cells can also be used to load APCs (autologous or derived from allogeneic cell lines) with tumor genomic DNA [77]. This allows uncharacterized mutated gene products specific to the tumor to be processed and presented for immune activation (Figure 1).
Oncolytic virus-based strategies
As mentioned above, the inclusion of pathogens in cancer vaccines can greatly increase immune stimulation in the context of presenting tumor antigens. Pathogens have complex arrays of molecules that can trigger multiple immune activation pathways. Oncolytic viruses have two anti-tumor mechanisms. First, they can infect and replicate inside the tumor cell, which directly mediates tumor lysis and further release of new viral particles into the tumor surrounding, which leads to additive tumor destruction. Second, tumor lysis leads to the release of tumor antigens, danger-associated molecular patterns, cytokines and chemokines into the tumor microenvironment, which leads to induction and/or enhancement of anti-tumor adaptive immunity [78].
Herpesviruses have been used as oncolytic viruses for cancer vaccination. A promising strategy has tested GM-CSF as an adjuvant or APC growth factor engineered into replicating herpesvirus vectors. One such vector, T-VEC (Talimogene Laherparepvec), was successfully tested in patients with melanoma in a randomized phase III trial [79]. The trial found an objective response rate of 26% and a complete clinical response in 11% of patients with stage IIIB-IV melanoma [80]. This virus was FDA approved in 2015 for these unresectable patients and has been shown to result in increased tumor PD-L1 expression and CD8+ T cell infiltration [81].
Neoantigen-based Cancer Vaccines
Personalized immunogenic cancer vaccines loaded with neoantigens, or tumor specific antigens (TSA), have now been tested in the clinic. Data suggests neoantigen cancer vaccines may potentially generate a stronger anti-tumor immune response, compared to classical DC based cancer vaccines loaded with TAAs[82]. Neoantigens are different from TAAs because they are absent from normal tissues and are completely specific to the cancer cells[83]. These antigens have been reported to originate from viral proteins, post-translational modifications, and somatic mutations[84]. Moreover, research studies have revealed that neoepitopes can either be classified as “shared” or “personalized”. Shared neoepitopes, or mutated oncogenes, are antigens that are found in a specific cancer type, where personalized neoepitopes, otherwise referred to as patient specific mutated neoantigens, are specific to an individual [85]. Examples of shared neoepitopes include BRAF, NRAS, and p53 mutations. Approximately 50% of melanomas harbor BRAF mutations at position V600 of the protein chain, the majority of which involve the substitution of glutamic acid for valine (V600E), resulting in constitutively active BRAF [86]. As stated above, NRAS mutations are found in approximately 15-25% of melanomas and occur at codons 12,13, and 61 [22,23]. It has been reported that p53 mutations are present in approximately 19% of melanomas. Additionally, wildtype p53 is reported to not function properly in melanomas. The literature indicates that wildtype p53 in melanomas fails to act as a tumor suppressor [87,88].
Neoantigen cancer vaccines are identified by the use of whole exome sequencing to identify mutations expressed in RNA from a tumor biopsy, compared to healthy tissues. Each mutation is screened using one of many algorithms to predict MHC binding and tested for immunogenicity (Figure 2). The mutations predicted to be processed and presented in the patients’ MHC molecules sufficiently have the best anti-tumor immune response are used to generate personalized DC cancer vaccines[89-92]. The cost of manufacturing a personalized, single neoantigen vaccine is estimated to be $60,000, which includes the sequencing and analysis of tumor and matched healthy tissue DNA samples [93,94].
Figure 2. Checkpoint + neoantigen vaccine combination therapies.
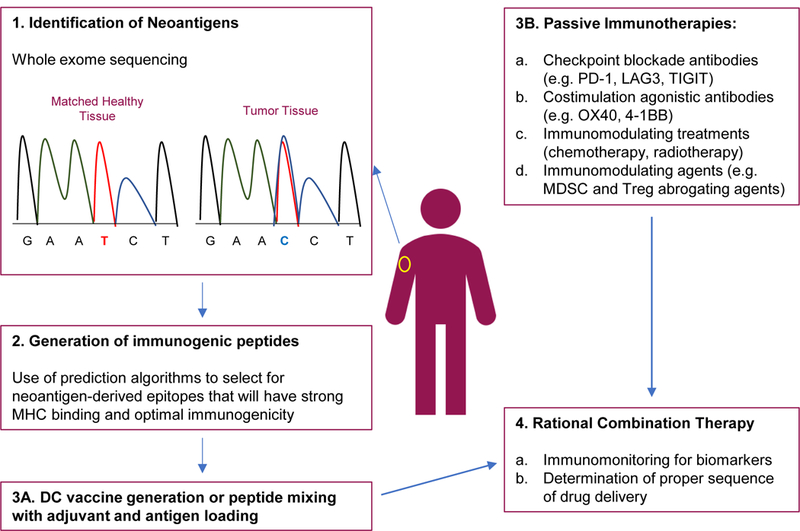
1-3A Neoantigen based vaccines are generated by the identification of neoepitopes through whole exome sequencing of tumor and matched donor tissues. The neoepitopes are tested for immunogenicity and strong MHC binding through the use of computational algorithms and in vitro testing. Selected neoantigens are then used for vaccine production. 3B. Other immunotherapies are in the clinic for the treatment of melanoma. These include numerous checkpoint inhibitors and agonistic therapies, standard chemotherapy and radiation, and immunomodulating agents. 4. Combinations of neoantigen vaccines and other immunotherapies might lead to an optimal anti-tumor immune response.
Murine studies have indicated the immunogenicity of neoantigens since the early 1990s. In 1916, Ernest Tyzzer reported the “acquisition of new immunogenic characteristics by cancer cells” [95]. Then, in 1943, Gross et al. showed mice were immune to re-challenge with tumor cells [96], suggesting that tumors could be immunogenic. In the early 2000s, Castle et al. and Kreitzer et al. each independently showed that nonsynonymous mutations are immunogenic and can produce CD4+ helper T cell responses [97,98]. Additionally, Castle et al. showed that mutated peptides can also be recognized by cytotoxic T lymphocytes in a B16F10 melanoma mouse, generating CD8+ T cell immune responses [97].
The pre-clinical data from murine studies indicate the use of neoantigen cancer vaccines in human clinical trials might lead to superior clinical response rates. Ott et al. were one of the first groups to propose a phase I study of a neoantigen cancer vaccine on the patient’s own melanoma [90]. In the study, Ott et al. designed a vaccine that targeted approximately 20 predicted personal tumor neoantigen peptides. The results from the study revealed the vaccine aided in the generation of CD4+ and CD8+ T cell responses specific to various predicted neoantigens. Additionally, four out of six patients had no reoccurrence of the tumor at 25 months after vaccination [90]. Importantly, the two patients that had progressed achieved complete tumor regression after subsequent treatment with anti-PD-1. After anti-PD-1 treatment, it was reported that these patients had an expansion in neo-antigen-specific T cell responses [90]. Additionally, it has recently been shown that neoantigen cancer vaccines also can lead to determinant spreading, resulting in a more diverse immune response. Carreno et al. reported that a DC based neoantigen vaccine not only resulted in neoantigen specific T cell responses, but also resulted in new human leukocyte antigen (HLA) class I - neoantigens post vaccination in late stage melanoma patients [89].
The above studies were done based on the generation of peptides in adjuvant or peptide-pulsed DC cancer vaccines (strategy summarized in Figure 2). Recently, investigators have also been generating personalized RNA mutanome vaccines for the treatment of advanced melanoma [92]. Sahin et al. were the first group to generate this type of vaccine for advanced melanoma. Non-synonymous mutations were detected in 13 late staged melanoma patients; five mutations were selected per vaccine, per patient. All of the patients generated neoantigen-specific T cell responses and 2 patients had an increase in neoantigen specific infiltrating T cells post vaccination [92]. They also reported that 1 patient achieved a complete response to the vacation when used in combination with anti-PD-1 [92]. Arguably, the above research studies suggest neoantigen cancer vaccines are capable of generating a potent and clinically effective immune response. Yet, neoantigen cancer vaccines may generate the best immune response when used in combination with checkpoint blockade treatments (anti-PD-1 and anti-CTLA-4).
Single and combination checkpoint blockade therapies have been very successful in a large subset of patients with advanced melanoma. Research studies have shown that ant-CTLA-4 treatment can broaden CD8+ T cell responses in advanced melanoma patients [99-101]. For example, Kvistborg et al. observed a significant expansion of melanoma-specific T cell responses post-treatment with anti-CTLA-4 in 40 melanoma patients [97]. Even though checkpoint blockade treatments have shown promise in the clinic, clinical data has suggested that only approximately 20-40% of patients respond to anti-PD-1/anti-CTLA-4, and the ones that do respond already have a mounted anti-tumor immune response [102].
In advanced melanoma patients, checkpoint blockade treatments have been reported to enhance overall survival in responding patients [100,103,104]. However, the molecular mechanisms correlating with clinical benefit from these therapies are largely unknown. Whole exome sequencing of melanoma tumors (pre and post anti-CTLA-4 treatment) and matched normal tissue have indicated that patients who clinically benefit from anti-CTLA-4 treatment have a high mutational load [99-101]. Therefore, these patients likely have a high neoantigen load that can be recognized by the immune system. Snyder et al. examined the tumor neoantigen landscape in late stage melanoma patients [100]. Using computational methods, Snyder et al. identified neoantigens in tumors from patients with clinical benefit to anti-CTLA-4; importantly, these neoantigens were absent in tumors from non-responding patients. Presence of these neoepitopes were not only associated with clinical benefit, but also were associated with overall survival. Follow-up in-vitro studies revealed that some of the predicted neoantigens were able to efficiently prime an activated T cell response [100]. These studies suggest that checkpoint blockade treatment, in combination with neoantigen cancer vaccines, might result in an optimal anti-tumor immune response that correlates with clinical benefit (Figure 2).
Research is now exploring combination therapies of vaccines and checkpoint inhibitors in the clinic (Figure 2). A new phase 1B trial for advanced melanoma, bladder, and lung cancer, is exploring the safety and therapeutic benefit of a new neoantigen vaccine (NEO-PV-01) in combination with ipilimumab (NCT02950766) or nivolumab (NCT02897765). The vaccine includes up to 20 distinct neoantigen peptides [105].
In all, neoantigen cancer vaccines have shown promising results in early clinical trials. Clinical data has indicated that these vaccines are capable of generating anti-tumor immune responses and determinant spreading. Moreover, predicted neoantigens have been shown to correlate with clinical outcome, and when used in combination with checkpoint blockade treatments, a subset of patients have experienced a complete response.
An area for further study is the stability of neoantigen-specific T cell responses. A recent study has shown neoantigen expression can be lost from the tumor cell population overtime [106]. The loss of neoantigen expression on tumor cells correlated with neoantigen-specific T cell responses, therefore, tumor resistance may occur in a personalized vaccine. To avoid such resistance, a broad neoantigen landscape and induction of determinant spreading should be considered for vaccine development, which might lead to improved clinical outcomes for advanced melanoma patients.
Determinant spreading
Although antigen choice is critical for generating antitumor immunity, the spread of the immune response from one antigen to another antigen expressed in the same tissue (“determinant spreading” [107] or “epitope spreading”) has been linked to superior clinical outcome [33,108-113] in multiple tumor types and vaccination settings. The phenomenon of in vivo cross presentation of tumor derived antigens released in one wave of T cell attack to promote subsequent waves of anti-tumor T cells directed against different antigens may be an important mechanism for tumor rejection [111]. A vaccine that targets shared antigens may set the stage for subsequent rounds of immunity to mutated neoantigens. The most important role of vaccines containing TAAs may be to induce determinant spread to tumor specific antigens that activate higher avidity T cells, which more effectively mediate tumor rejection. The in vivo mechanism of cross priming may also result in autoimmunity [114], which has been found to be a biomarker of clinical response to interferon in patients with melanoma [115].
Other recent promising vaccine strategies
Adjuvant and agonistic antibody immunization
Sagiv-Barfi, I. et al have shown in in multiple murine models that intratumoral (in situ) vaccination with CpG-enriched oligodeoxynucleotide (CpG; a Toll-like receptor 9 ligand) and agonistic anti-OX40 antibody triggers a potent systemic anti-tumor T cell immune response that mediates rejection of distant tumors and enhanced long-term survival of the animals [116]. This strategy is currently being tested in patients with low-grade B-cell non-Hodgkin lymphomas in a phase I trial (NCT03410901).
Oncolytic vectors in combination with checkpoint blockade
TBI-1401 (HF10) is a spontaneously mutated HSV-1 vector that has attenuated neuroinvasiveness. It has previously been tested in a small phase I study in patients with recurrent breast cancer, where tumor shrinkage was reported [117]. HF10 is currently being tested for efficacy and safety with repeated administration of intratumoral injections in combination with intravenous infusions ipilimumab in patients with melanoma (NCT03153085).
mRNA vaccines
The use of mRNA vaccines has shown significant potential as a vaccine modality due to its innate immunogenic properties. mRNA is a non-infectious, non-integrating platform, that gets degraded by normal cellular processes. It is an easily modifiable, stable and highly translatable vaccine platform [118]. BioNTech have demonstrated that DCs can be effectively targeted in vivo using intravenously administered RNA-lipoplexes (RNA-LPX). The LPX protects RNA from extracellular ribonucleases and mediates its efficient uptake and expression of the transgene in APCs. Most importantly, they have shown that RNA-LPX encoding tumor antigens induce strong antigen-specific effector and memory T cell responses in 3 melanoma patients [119]. The same company has also demonstrated in a phase I trial that its IVAC® MUTANOME, an individualized RNA vaccine based on patient-specific mutations, can induce anti-tumor activity in high-risk patients with late-stage melanoma [93].
Strategies to optimize future immunotherapies
As evidenced over the years, vaccines, oncoviral and immunomodulating modalities used to treat cancer patients have displayed limited clinical effectiveness when used as single agents. Aforementioned pre-clinical and clinical data indicate that combining two or more immunotherapeutic strategies is the logical evolution of the field. However, in order to fully maximize the potential of such approaches, we need carefully perform optimization tests to determine the optimal sequence of drug deliveries. Additionally, novel strategies are needed to abrogate immune suppression in the tumor microenvironment that can hamper the efficacy of anti-tumor vaccines.
Timing of future vaccine delivery strategies
A pre-clinical study by Messenheimer has indicated that the timing of anti-PD-1 delivery in combination immunotherapy may be crucial in the induction of effective anti-tumor immunity. Using agonistic anti-OX40 in combination with anti-PD-1, they have shown that concurrent addition of the two agents had a detrimental therapeutic effect when compared to anti-OX40 alone. Sequential delivery of anti-OX40 followed by anti-PD-1 (but not in the opposite order) had resulted in significantly improved therapeutic efficacy [120]. This study highlights the potential significance of the order in which combination immunotherapies are delivered.
Patient selection in vaccine trials
Currently, only a minority of patients benefit from active and passive immunotherapies. However, patients that do respond frequently display durable benefit. Consequently, selection of patients that would most likely benefit from a specific immunotherapy based on specific predictive biomarkers of clinical benefit is a priority. It would allow for timely, personalized and cost-effective way of treating individuals. Future clinical trials need to implement robust and rational immunomonitoring strategies to evaluate immune cell phenotypes and function in circulation as well as tumors, serum levels of various circulating protein and DNA factors (i.e. liquid biopsies) and tumor profile. Consequently, a multidisciplinary approach that integrates phenotypic and functional analysis of immune cells (e.g. multi-color flow cytometry; CyTOF; multi-color ELISpot assays), immunohistology (e.g. quantitative multi-color immunofluorescence), transcriptome analysis (e.g. NanoString, RNA-seq), cell metabolism analysis, liquid biopsy assessment (circulating tumor DNA, circulating proteins) and tumor mutational analysis (whole-exome sequencing) needs to be implemented in order to identify biomarkers predictive of optimal therapeutic responsiveness. There are currently many important signals (total mutation burden, CD8+ T cell infiltrate, PD-L1 expression, IFN-. expression signature) but no clear predictive biomarkers to act upon.
Abrogation of MDSC-mediated immunosuppression
Accumulation of MDSC, immature myeloid cells, in peripheral blood and within tumor lesions correlates with poor prognosis and therapy resistance [121-123]. Additionally, MDSC have a multitude of immunosuppressive functions that hamper the development and maintenance of anti-tumor immunity [123]. In vivo studies have shown that depletion of MDSCs improves the therapeutic effectiveness of vaccination in a murine lung cancer model [124]. Multiple anti-MDSC treatments have been developed target different aspects of MDSC biology (accumulation, function, recruitment or viability) [123,125]. One such modulator is INB03 (INmune Bio, Inc.), a dominant-negative TNF biologic that selectively neutralizes the soluble form of TNF (solTNF) without affecting the transmembrane variant (tmTNF). INB03 has been shown to decrease the frequency of splenic PMN- and M-MDSCs and macrophages, and inhibit MDSC-mediated immunosuppression [126]. Other suppressors of MDSC include Tadalafil [127], all-trans retinoic acid [128] and TRAIL-R2 antibody [129].
Conclusions
Cancer vaccination has a long history in melanoma, with success in driving immune responses, although objective clinical responses have only ever been detected in a minority of patients. This active area of research continues to investigate optimal vaccination platforms, and more recently, neoantigen-based vaccination and oncolytic virus combinations.
References
- 1.Nordlund JJ, Kirkwood JM, Forget BM, Milton G, Albert DM, Lerner AB. Vitiligo in patients with metastatic melanoma: a good prognostic sign. Journal of the American Academy of Dermatology 1983; 9 (5):689–696. [DOI] [PubMed] [Google Scholar]
- 2.Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. Am J Med Sci 1893; 105:487–511. [PubMed] [Google Scholar]
- 3.Homes EC, Morton DL, Eilber FR, Golub SE, Sulit HL. Immunotherapy in malignant melanomaa. Natl Cancer Inst Monogr 1976; 44:85–86. [PubMed] [Google Scholar]
- 4.Laucius JF, Bodurtha AJ, Mastrangelo JM, Bellet RE. A Phase II study of autologous irradiated tumor cells plus BCG in patients with metastatic malignant melanoma. Cancer 1977; 40 (5):2091–2093. [DOI] [PubMed] [Google Scholar]
- 5.Wallack MK, Bash J, Bartolucci A. Improvement in disease-free survival of melanoma patients in conjunction with serologic response in a phase Ia/Ib Southeastern Cancer Study Group trial of vaccinia melanoma oncolysate. Am Surg 1989; 55 (4):243–247. [PubMed] [Google Scholar]
- 6.Lipton A, Harvey HA, Lawrence B, Gottlieb R, Kukrika M, Dixon R, et al. Corynebacterium parvum versus BCG adjuvant immunotherapy in human malignant melanoma. Cancer 1983; 51 (1):57–60. [DOI] [PubMed] [Google Scholar]
- 7.Morton DL, Foshag LJ, Hoon DS, Nizze JA, Famatiga E, Wanek LA, et al. Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma vaccine. Ann Surg 1992; 216 (4):463–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Livingston PO, Adluri S, Helling F, Yao TJ, Kensil CR, Newman MJ, et al. Phase 1 trial of immunological adjuvant QS-21 with a GM2 ganglioside-keyhole limpet haemocyanin conjugate vaccine in patients with malignant melanoma. Vaccine 1994; 12 (14):1275–1280. [DOI] [PubMed] [Google Scholar]
- 9.Stingl G, Brocker EB, Mertelsmann R, Wolff K, Schreiber S, Kampgen E, et al. Phase I study to the immunotherapy of metastatic malignant melanoma by a cancer vaccine consisting of autologous cancer cells transfected with the human IL-2 gene. Human gene therapy 1996; 7 (4):551–563. [DOI] [PubMed] [Google Scholar]
- 10.Chang AE, Sondak VK, Bishop DK, Nickoloff BJ, Mulligan RC, Mule JJ. Adoptive immunotherapy of cancer with activated lymph node cells primed in vivo with autologous tumor cells transduced with the GM-CSF gene. Human gene therapy 1996; 7 (6):773–792. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proceedings of the National Academy of Sciences of the United States of America 1994; 91 (14):6458–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Traversari C, van der Bruggen P, Luescher IF, Lurquin C, Chomez P, Van Pel A, et al. A nonapeptide encoded by human gene MAGE-1 is recognized on HLA-A1 by cytolytic T lymphocytes directed against tumor antigen MZ2-E. The Journal of experimental medicine 1992; 176 (5):1453–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Eynde BD, Hainaut P, Hérin M, Knuth A, Lemoine C, Weynants P, et al. Presence on a human melanoma of multiple antigens recognized by autologous CTL. International Journal of Cancer 1989; 44 (4):634–640. [DOI] [PubMed] [Google Scholar]
- 14.Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proceedings of the National Academy of Sciences of the United States of America 1994; 91 (9):3515–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coulie PG, Brichard V, Van Pel A, Wolfel T, Schneider J, Traversari C, et al. A new gene coding for a differentiation antigen recognized by autologous cytolytic T lymphocytes on HLA-A2 melanomas. The Journal of experimental medicine 1994; 180 (1):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vujanovic L, Butterfield LH. Melanoma cancer vaccines and anti-tumor T cell responses. Journal of cellular biochemistry 2007; 102 (2):301–310. [DOI] [PubMed] [Google Scholar]
- 17.Barrow C, Browning J, MacGregor D, Davis ID, Sturrock S, Jungbluth AA, Cebon, j. Tumor antigne expression in melanoma varies according to antigen and stage. Clinical Cancer Research, Feb. 2006 [DOI] [PubMed] [Google Scholar]
- 18.Topalian SL, Rivoltini L, Mancini M, Markus NR, Robbins PF, Kawakami Y, et al. Human CD4+ T cells specifically recognize a shared melanoma-associated antigen encoded by the tyrosinase gene. Proceedings of the National Academy of Sciences of the United States of America 1994; 91 (20):9461–9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finn OJ, Jerome KR, Henderson RA, Pecher G, Domenech N, Magarian-Blander J, et al. MUC-1 epithelial tumor mucin-based immunity and cancer vaccines. Immunological reviews 1995; 145:61–89. [DOI] [PubMed] [Google Scholar]
- 20.Bakker AB, Marland G, de Boer AJ, Huijbens RJ, Danen EH, Adema GJ, et al. Generation of antimelanoma cytotoxic T lymphocytes from healthy donors after presentation of melanoma-associated antigen-derived epitopes by dendritic cells in vitro. Cancer research 1995; 55 (22):5330–5334. [PubMed] [Google Scholar]
- 21.Boon T, Cerottini JC, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. Annual review of immunology 1994; 12:337–365. [DOI] [PubMed] [Google Scholar]
- 22.Johnson DB, Puzanov I. Treatment of NRAS-mutant melanoma. Curr Treat Options Oncol, 2015; 16(4): 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boespflug A, Caramel J, Dalle S, Thomas L. Treatment of NRAS-muated advanced or metastatic melanoma: rationale, current trials and evidence to date. Ther Adv Med Oncol. 2017. July; 9(7): 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vonderheide RH, Nathanson KL. Immunotherapy at large: the road to personalized cancer vaccines. Nature medicine 2013; 19 (9):1098–1100. [DOI] [PubMed] [Google Scholar]
- 25.Finn OJ. The dawn of vaccines for cancer prevention. Nature Reviews Immunology (Review Article) 2017; 18:183. [DOI] [PubMed] [Google Scholar]
- 26.Zhao X, Bose A, Komita H, Taylor JL, Chi N, Lowe DB, et al. Vaccines targeting tumor blood vessel antigens promote CD8(+) T cell-dependent tumor eradication or dormancy in HLA-A2 transgenic mice. Journal of Immunology 2012; 188 (4):1782–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komita H, Zhao X, Taylor JL, Sparvero LJ, Amoscato AA, Alber S, et al. CD8+ T-cell responses against hemoglobin-beta prevent solid tumor growth. Cancer research 2008; 68 (19):8076–8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Disis ML, Gralow JR, Bernhard H, Hand SL, Rubin WD, Cheever MA. Peptide-based, but not whole protein, vaccines elicit immunity to HER-2/neu, oncogenic self-protein. Journal of immunology 1996; 156 (9):3151–3158. [PubMed] [Google Scholar]
- 29.Kirkwood JM, Lee S, Moschos SJ, Albertini MR, Michalak JC, Sander C, et al. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/−granulocyte-monocyte colony-stimulating factor and/or IFN-alpha2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clinical cancer research : an official journal of the American Association for Cancer Research 2009; 15 (4):1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slingluff CL Jr., Lee S, Zhao F, Chianese-Bullock KA, Olson WC, Butterfield LH, et al. A randomized phase II trial of multiepitope vaccination with melanoma peptides for cytotoxic T cells and helper T cells for patients with metastatic melanoma (E1602). Clinical cancer research : an official journal of the American Association for Cancer Research 2013; 19 (15):4228–4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Potter DM, et al. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic Acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2014; 32 (19):2050–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okada H, Butterfield LH, Hamilton RL, Hoji A, Sakaki M, Ahn BJ, et al. Induction of robust type-I CD8+ T-cell responses in WHO grade 2 low-grade glioma patients receiving peptide-based vaccines in combination with poly-ICLC. Clinical cancer research : an official journal of the American Association for Cancer Research 2015; 21 (2):286–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Butterfield LH, Ribas A, Dissette VB, Amarnani SN, Vu HT, Oseguera D, et al. Determinant spreading associated with clinical response in dendritic cell-based immunotherapy for malignant melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2003; 9 (3):998–1008. [PubMed] [Google Scholar]
- 34.Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE, et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J Clin Oncol 2011; 29 (3):330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A, et al. Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer research 2001; 61 (17):6451–6458. [PubMed] [Google Scholar]
- 36.Aranda F, Vacchelli E, Eggermont A, Galon J, Sautes-Fridman C, Tartour E, et al. Trial Watch: Peptide vaccines in cancer therapy. Oncoimmunology 2013; 2 (12):e26621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pratama A, Srivastava M, Williams NJ, Papa I, Lee SK, Dinh XT, et al. MicroRNA-146a regulates ICOS-ICOSL signalling to limit accumulation of T follicular helper cells and germinal centres. Nat Commun 2015; 6:6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welters MJ, Kenter GG, de Vos van Steenwijk PJ, Lowik MJ, Berends-van der Meer DM, Essahsah F, et al. Success or failure of vaccination for HPV16-positive vulvar lesions correlates with kinetics and phenotype of induced T-cell responses. Proceedings of the National Academy of Sciences of the United States of America 2010; 107 (26):11895–11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kenter GG, Welters MJ, Valentijn AR, Lowik MJ, Berends-van der Meer DM, Vloon AP, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. The New England journal of medicine 2009; 361 (19):1838–1847. [DOI] [PubMed] [Google Scholar]
- 40.Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW, et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. European journal of immunology 2013; 43 (10):2554–2565. [DOI] [PubMed] [Google Scholar]
- 41.Vujanovic L, Shi J, Kirkwood JM, Storkus WJ, Butterfield LH. Molecular mimicry of MAGE-A6 and Mycoplasma penetrans HF-2 epitopes in the induction of antitumor CD8(+) T-cell responses. Oncoimmunology 2014; 3 (8):e954501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Butterfield LH, Palucka AK, Britten CM, Dhodapkar MV, Hakansson L, Janetzki S, et al. Recommendations from the iSBTc-SITC/FDA/NCI Workshop on Immunotherapy Biomarkers. Clinical cancer research : an official journal of the American Association for Cancer Research 2011; 17 (10):3064–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slingluff CL Jr., Petroni GR, Chianese-Bullock KA, Smolkin ME, Ross MI, Haas NB, et al. Randomized multicenter trial of the effects of melanoma-associated helper peptides and cyclophosphamide on the immunogenicity of a multipeptide melanoma vaccine. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2011; 29 (21):2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walter S, Weinschenk T, Stenzl A, Zdrojowy R, Pluzanska A, Szczylik C, et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nature Medicine 2012; 18 (8):1254–1261. [DOI] [PubMed] [Google Scholar]
- 45.Slingluff CL, Petroni GR, Olson W, Czarkowski A, Grosh WW, Smolkin M, et al. Helper T-Cell Responses and Clinical Activity of a Melanoma Vaccine With Multiple Peptides From MAGE and Melanocytic Differentiation Antigens. Journal of Clinical Oncology 2008; 26 (30):4973–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu Y, Kim H, Blackwell CM, Slingluff CL. Long-Term Outcomes of Helper Peptide Vaccination for Metastatic Melanoma. Annals of surgery 2015; 262 (3):456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivoltini L, Squarcina P, Loftus DJ, Castelli C, Tarsini P, Mazzocchi A, et al. A Superagonist Variant of Peptide MART1/Melan A<sub>27–35</sub> Elicits Anti-Melanoma CD8<sup>+</sup> T Cells with Enhanced Functional Characteristics: Implication for More Effective Immunotherapy. Cancer research 1999; 59 (2):301. [PubMed] [Google Scholar]
- 48.Zirlik KM, Zahrieh D, Neuberg D, Gribben JG. Cytotoxic T cells generated against heteroclitic peptides kill primary tumor cells independent of the binding affinity of the native tumor antigen peptide. Blood 2006; 108 (12):3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Capasso C, Magarkar A, Cervera-Carrascon V, Fusciello M, Feola S, Muller M, et al. A novel in silico framework to improve MHC-I epitopes and break the tolerance to melanoma. Oncoimmunology 2017; 6 (9):e1319028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Palucka K, Ueno H, Fay J, Banchereau J. Dendritic cells and immunity against cancer. Journal of internal medicine 2011; 269 (1):64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12 (4):265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013; 39 (1):38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Butterfield LH. Dendritic cells in cancer immunotherapy clinical trials: are we making progress? Frontiers in immunology 2013; 4:454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butterfield LH. Dendritic cell vaccines: release assays and potency assays. Telegraft 2015; 22 (2):1–3. [Google Scholar]
- 55.Butterfield LH. Cancer vaccines. BMJ 2015; 350:h988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kaka AS, Foster AE, Weiss HL, Rooney CM, Leen AM. Using dendritic cell maturation and IL-12 producing capacity as markers of function: a cautionary tale. J Immunother 2008; 31 (4):359–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vujanovic L, Szymkowski DE, Alber S, Watkins SC, Vujanovic NL, Butterfield LH. Virally infected and matured human dendritic cells activate natural killer cells via cooperative activity of plasma membrane-bound TNF and IL-15. Blood 2010; 116 (4):575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kalinski P, Millard RB, Geskin L, Giermasz A, Nakamura Y, Storkus WJ, et al. Polarized DC1-Based Therapeutic Cancer Vaccines. Journal of Immunotherapy 2005; 28 (6):656. [Google Scholar]
- 59.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nature medicine 1996; 2 (1):52–58. [DOI] [PubMed] [Google Scholar]
- 60.Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nature medicine 1998; 4 (3):328–332. [DOI] [PubMed] [Google Scholar]
- 61.Schreibelt G, Bol KF, Westdorp H, Wimmers F, Aarntzen EHJG, Duiveman-de Boer T, et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clinical Cancer Research ( 10.1158/1078-0432.CCR-15-2205) 2016; 22 (9):2155. [DOI] [PubMed] [Google Scholar]
- 62.Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunological reviews 2011; 239 (1):27–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature medicine 2004; 10 (9):909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwartzentruber DJ, Lawson DH, Richards JM, Conry RM, Miller DM, Treisman J, et al. gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. The New England journal of medicine 2011; 364 (22):2119–2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gross S, Erdmann M, Haendle I, Voland S, Berger T, Schultz E, et al. Twelve-year survival and immune correlates in dendritic cell-vaccinated melanoma patients. JCI Insight 2017; 2 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. The New England journal of medicine 2010; 363 (5):411–422. [DOI] [PubMed] [Google Scholar]
- 67.Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proceedings of the National Academy of Sciences of the United States of America 1993; 90 (8):3539–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McBride WH, Thacker JD, Comora S, Economou JS, Kelley D, Hogge D, et al. Genetic modification of a murine fibrosarcoma to produce interleukin 7 stimulates host cell infiltration and tumor immunity. Cancer research 1992; 52 (14):3931–3937. [PubMed] [Google Scholar]
- 69.Soiffer R, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger JC, et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proceedings of the National Academy of Sciences of the United States of America 1998; 95 (22):13141–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nemunaitis J, Sterman D, Jablons D, Smith JW 2nd, Fox B, Maples P, et al. Granulocyte-macrophage colony-stimulating factor gene-modified autologous tumor vaccines in non-small-cell lung cancer. Journal of the National Cancer Institute 2004; 96 (4):326–331. [DOI] [PubMed] [Google Scholar]
- 71.Tani K, Azuma M, Nakazaki Y, Oyaizu N, Hase H, Ohata J, et al. Phase I study of autologous tumor vaccines transduced with the GM-CSF gene in four patients with stage IV renal cell cancer in Japan: clinical and immunological findings. Molecular therapy : the journal of the American Society of Gene Therapy 2004; 10 (4):799–816. [DOI] [PubMed] [Google Scholar]
- 72.Luiten RM, Kueter EW, Mooi W, Gallee MP, Rankin EM, Gerritsen WR, et al. Immunogenicity, including vitiligo, and feasibility of vaccination with autologous GM-CSF-transduced tumor cells in metastatic melanoma patients. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2005; 23 (35):8978–8991. [DOI] [PubMed] [Google Scholar]
- 73.Geskin LJ, Damiano JJ, Patrone CC, Butterfield LH, Kirkwood JM, Falo LD. Three antigen-loading methods in dendritic cell vaccines for metastatic melanoma. Melanoma Res 2018; 28 (3):211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chakraborty NG, Sporn JR, Tortora AF, Kurtzman SH, Yamase H, Ergin MT, et al. Immunization with a tumor-cell-lysate-loaded autologous-antigen-presenting-cell-based vaccine in melanoma. Cancer immunology, immunotherapy : CII 1998; 47 (1):58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Geiger J, Hutchinson R, Hohenkirk L, McKenna E, Chang A, Mule J. Treatment of solid tumours in children with tumour-lysate-pulsed dendritic cells. Lancet 2000; 356 (9236):1163–1165. [DOI] [PubMed] [Google Scholar]
- 76.Barth RJ Jr., Fisher DA, Wallace PK, Channon JY, Noelle RJ, Gui J, et al. A randomized trial of ex vivo CD40L activation of a dendritic cell vaccine in colorectal cancer patients: tumor-specific immune responses are associated with improved survival. Clinical cancer research : an official journal of the American Association for Cancer Research 2010; 16 (22):5548–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim TS, Chopra A, IS OS, Cohen EP. Enhanced immunity to breast cancer in mice immunized with fibroblasts transfected with a complementary DNA expression library from breast cancer cells: Enrichment of the vaccine for immunotherapeutic cells. Journal of immunotherapy 2006; 29 (3):261–273. [DOI] [PubMed] [Google Scholar]
- 78.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nature Reviews Drug Discovery (Review Article) 2015; 14:642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kaufman HL, Bines SD. OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future oncology 2010; 6 (6):941–949. [DOI] [PubMed] [Google Scholar]
- 80.Ross MI, Andtbacka RH, Puzanov I, Milhem MM, Collichio FA, Delman KA, et al. Patterns of durable response with intralesional talimogene laherparepvec (T-VEC): Results from a phase III trial in patients with stage IIIb-IV melanoma. Journal of Clinical Oncology 2014; 32 (15; suppl):9026–9026. [Google Scholar]
- 81.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017; 170 (6):1109–1119 e1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fecek RJ, Storkus WJ. Combination strategies to enhance the potency of monocyte-derived dendritic cell-based cancer vaccines. Immunotherapy 2016; 8 (10):1205–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zarour HM, DeLeo A, Finn OJ, Storkus WJ. Categories of Tumor Antigens In: Kufe DW, Pollock RE, Weichselbaum RR, eds. Holland-Frei Cancer Medicine 6th edition Hamilton (ON): BC Decker, 2003. [Google Scholar]
- 84.Gubin MM, Artyomov MN, Mardis ER, Schreiber RD. Tumor neoantigens: building a framework for personalized cancer immunotherapy. J Clin Invest 2015; 125 (9):3413–3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol 2016; 28 (1):22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cantwell-Dorris ER, O’Leary JJ, Sheils OM. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Molecular Cancer Therapeutics 2011; 10 (3):385–394. [DOI] [PubMed] [Google Scholar]
- 87.Avery-Kiejda KA, Bowden NA, Croft AJ, Scurr LL, Kairupan CF, Ashton KA, et al. P53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer 2011; 11:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Box NF, Vukmer TO, Terzian T. Targeting p53 in melanoma. Pigment Cell Melanoma Res 2014; 27 (1):8–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015; 348 (6236):803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017; 547 (7662):217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rubinsteyn A, Kodysh J, Hodes I, Mondet S, Aksoy BA, Finnigan JP, et al. Computational Pipeline for the PGV-001 Neoantigen Vaccine Trial. Front Immunol 2017; 8:1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017; 547 (7662):222–226. [DOI] [PubMed] [Google Scholar]
- 93.Begley S Personalized Cancer Vaccines Vanquish Melanoma in Small Study Scientific American New York, New York STAT News, 2017. [Google Scholar]
- 94.Gutierrez C, Horin L. Cancer Vaccines: How scientists are turning cancer against itself. Science in the News, Harvard Medical School 2017. [Google Scholar]
- 95.Tyzzer EE. Tumor Immunity. Cancer Research 1916; 1 (2):125–156. [Google Scholar]
- 96.Gross L Intradermal Immunization of C3H Mice Against a Sarcoma That Originated in an Animal of the Same Line. Cancer Research 1943. 3:326–333. [Google Scholar]
- 97.Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res 2012; 72 (5):1081–1091. [DOI] [PubMed] [Google Scholar]
- 98.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015; 520 (7549):692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, et al. Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med 2014; 6 (254):254ra128. [DOI] [PubMed] [Google Scholar]
- 100.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014; 371 (23):2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015; 350 (6257):207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shields BD, Mahmoud F, Taylor EM, Byrum SD, Sengupta D, Koss B, et al. Indicators of responsiveness to immune checkpoint inhibitors. Sci Rep 2017; 7 (1):807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 2013; 369 (2):134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015; 373 (13):1270–1271. [DOI] [PubMed] [Google Scholar]
- 105.Obermajer N Guidelines for the next-generation cancer vaccines. Immuno-Oncology Research Trends. 2018. [Google Scholar]
- 106.Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 2016; 536 (7614):91–95. [DOI] [PubMed] [Google Scholar]
- 107.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992; 358 (6382):155–157. [DOI] [PubMed] [Google Scholar]
- 108.Ribas A, Glaspy JA, Lee Y, Dissette VB, Seja E, Vu HT, et al. Role of dendritic cell phenotype, determinant spreading, and negative costimulatory blockade in dendritic cell-based melanoma immunotherapy. Journal of immunotherapy 2004; 27 (5):354–367. [DOI] [PubMed] [Google Scholar]
- 109.Ribas A, Timmerman JM, Butterfield LH, Economou JS. Determinant spreading and tumor responses after peptide-based cancer immunotherapy. Trends in immunology 2003; 24 (2):58–61. [DOI] [PubMed] [Google Scholar]
- 110.Disis ML, Gooley TA, Rinn K, Davis D, Piepkorn M, Cheever MA, et al. Generation of T-cell immunity to the HER-2/neu protein after active immunization with HER-2/neu peptide-based vaccines. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2002; 20 (11):2624–2632. [DOI] [PubMed] [Google Scholar]
- 111.Disis ML. Immunologic biomarkers as correlates of clinical response to cancer immunotherapy. Cancer immunology, immunotherapy : CII 2011; 60 (3):433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wierecky J, Muller MR, Wirths S, Halder-Oehler E, Dorfel D, Schmidt SM, et al. Immunologic and clinical responses after vaccinations with peptide-pulsed dendritic cells in metastatic renal cancer patients. Cancer research 2006; 66 (11):5910–5918. [DOI] [PubMed] [Google Scholar]
- 113.Hu Y, Petroni GR, Olson WC, Czarkowski A, Smolkin ME, Grosh WW, et al. Immunologic hierarchy, class II MHC promiscuity, and epitope spreading of a melanoma helper peptide vaccine. Cancer immunology, immunotherapy : CII 2014; 63 (8):779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Krauze MT, Tarhini A, Gogas H, Kirkwood JM. Prognostic significance of autoimmunity during treatment of melanoma with interferon. Seminars in immunopathology 2011; 33 (4):385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gogas H, Ioannovich J, Dafni U, Stavropoulou-Giokas C, Frangia K, Tsoutsos D, et al. Prognostic significance of autoimmunity during treatment of melanoma with interferon. The New England journal of medicine 2006; 354 (7):709–718. [DOI] [PubMed] [Google Scholar]
- 116.Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS, et al. Eradication of spontaneous malignancy by local immunotherapy. Science Translational Medicine ( 10.1126/scitranslmed.aan4488) 2018; 10 (426). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sahin TT, Kasuya H, Nomura N, Shikano T, Yamamura K, Gewen T, et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Therapy (Original Article) 2011; 19:229. [DOI] [PubMed] [Google Scholar]
- 118.Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines — a new era in vaccinology. Nature Reviews Drug Discovery (Review Article) 2018; 17:261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016; 534:396. [DOI] [PubMed] [Google Scholar]
- 120.Messenheimer DJ, Jensen SM, Afentoulis ME, Wegmann KW, Feng Z, Friedman DJ, et al. Timing of PD-1 Blockade Is Critical to Effective Combination Immunotherapy with Anti-OX40. Clinical Cancer Research ( 10.1158/1078-0432.CCR-16-2677) 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Diaz-Montero CM, Finke J, Montero AJ. Myeloid derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Seminars in oncology 2014; 41 (2):174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gebhardt C, Sevko A, Jiang H, Lichtenberger R, Reith M, Tarnanidis K, et al. Myeloid Cells and Related Chronic Inflammatory Factors as Novel Predictive Markers in Melanoma Treatment with Ipilimumab. Clinical Cancer Research 2015; 21 (24):5453–5459. [DOI] [PubMed] [Google Scholar]
- 123.Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature Communications (Review Article) 2016; 7:12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Srivastava MK, Dubinett S, Sharma S. Targeting MDSCs enhance therapeutic vaccination responses against lung cancer. Oncoimmunology 2012; 1 (9):1650–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wesolowski R, Markowitz J, Carson WE. Myeloid derived suppressor cells – a new therapeutic target in the treatment of cancer. Journal for Immunotherapy of Cancer 2013; 1:10–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sobo-Vujanovic A, Vujanovic L, DeLeo AB, Concha-Benavente F, Ferris RL, Lin Y, et al. Inhibition of Soluble Tumor Necrosis Factor Prevents Chemically Induced Carcinogenesis in Mice. Cancer Immunology Research 2016; 4 (5):441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Weed DT, Vella JL, Reis IM, De la Fuente AC, Gomez C, Sargi Z, et al. Tadalafil reduces myeloid-derived suppressor cells and regulatory T cells and promotes tumor immunity in patients with head and neck squamous cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2015; 21 (1):39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Iclozan C, Antonia S, Chiappori A, Chen DT, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer immunology, immunotherapy : CII 2013; 62 (5):909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, et al. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clinical cancer research : an official journal of the American Association for Cancer Research 2017; 23 (12):2942–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]