

SUMMARY
Repeated drug use has long-lasting effects on plasticity throughout the brain’s reward and memory systems. Environmental cues that are associated with drugs of abuse can elicit craving and relapse, but the neural circuits responsible for driving drug-cue-related behaviors have not been well delineated, creating a hurdle for the development of effective relapse prevention therapies. In this study, we used a cocaine+cue self-administration paradigm followed by cue re-exposure to establish that the strength of the drug cue association corresponds to the strength of synapses between the medial geniculate nucleus (MGN) of the thalamus and the lateral amygdala (LA). Furthermore, we demonstrate, via optogenetically induced LTD of MGN-LA synapses, that reversing cocaine-induced potentiation of this pathway is sufficient to inhibit cue-induced relapse-like behavior.
Graphical Abstract
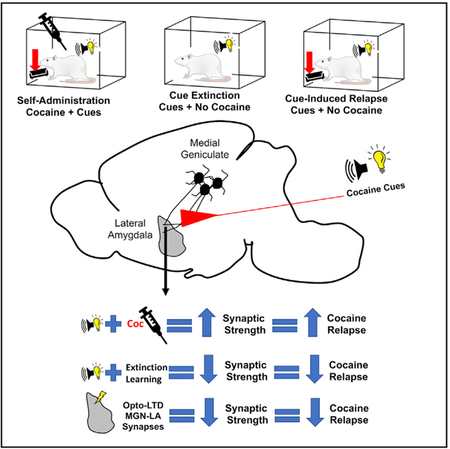
In Brief
Rich et al. report that cocaine-cue learning induces potentiation of thalamoamygdala synapses, and reversal of this plasticity, either by cue extinction or circuit-specific LTD, reduces cue-elicited cocaine seeking.
INTRODUCTION
Chronic use of cocaine leads to the formation of long-term memories of the environmental stimuli associated with the drug use experience. Over time, exposure to just the stimuli, or cues, is sufficient to induce physiological and psychological responses that drive continued use and relapse (Torregrossa and Taylor, 2013). Thus, a potential treatment strategy is to reduce the strength of drug cue memories to prevent craving and relapse. This may be possible by inducing extinction of the memory, a process whereby repeated cue exposure without the drug reduces the predictive value of the cue (Rich and Torregrossa, 2018). However, a major limitation of enacting memory-based treatments is that the neural correlates underlying drug cue memories have not been established.
In animal cocaine self-administration (SA) models, cocaine-cue memories form when an instrumental action (e.g., lever pressing) results in cocaine delivery (unconditioned stimulus [US]) paired with an audiovisual conditioned stimulus (CS). After repeated CS-US pairings, CS presentations by themselves can increase drug-seeking actions. Cue-driven drug seeking likely develops through cellular processes such as long-term potentiation (LTP) in neurons that are activated by the drug cue experience (Cruz et al., 2014). In contrast, extinction may reverse this plasticity and/or result in new learning, but this has not been determined for drug cue memories. Rather, prior animal studies have focused on the mechanisms regulating the drug-seeking action, not of the cue memory that drives relapse (Peters et al., 2008). However, most clinical efforts to extinguish drug memories have focused on the cues, leaving a large gap in the pre-clinical literature in identifying the locus and molecular mechanisms underlying the formation and extinction of drug cue memories.
On the other hand, fear-conditioned memories have been investigated extensively, where manipulation of the cue is the norm because of the purely Pavlovian nature of fear conditioning. Formation and extinction of fear memories requires plasticity at thalamic and cortical inputs to the lateral amygdala (LA) (Hong et al., 2009; Kim et al., 2007a). Thalamic projections from the medial geniculate nucleus (MGN) that synapse in the LA have been shown to be of particular importance for the establishment of auditory fear-associated memories (Ferrara et al., 2017; Kwon et al., 2014), and optogenetic induction of long-term depression (LTD) at these synapses can extinguish fear-related memories (Nabavi et al., 2014). Given that the amygdala also encodes memories with positive affective value (Carelli et al., 2003; Hsiang et al., 2014; Shabel and Janak, 2009), drug cue-associative memories, particularly those that involve an auditory component, may also be encoded at MGN-LA terminals.
Here we present evidence that cocaine-cue memory formation induces synaptic potentiation at MGN thalamo-amygdala (T-LA) but not cortico-amygdala (C-LA) synapses and that extinction of the cue memory, but not the lever pressing action, reverses this plasticity. Furthermore, optogenetic LTD of MGN-LA synapses produces physiological and behavioral changes indicative of cue extinction. Together, these results identify a specific neural correlate and cellular mechanism responsible for the acquisition and extinction of drug cue memory and present potential therapeutic approaches to prevent relapse.
RESULTS
Thalamo-amygdala Synaptic Modifications Regulate Cocaine-Cue Memories
To determine how the strength of cocaine-cue associations affects drug-seeking behavior, rats were trained on a fixed ratio 1 (FR1) schedule of reinforcement, where a single active lever press produced an infusion of cocaine (1 mg/kg/infusion) or saline, paired with an audiovisual CS (Figure 1A). Cocaine- but not saline-trained animals demonstrated reliable acquisition of SA (Figures 1B and 1C). After at least 10 days of 1-h SA sessions, rats underwent instrumental extinction (IE) for 6–10 days, where lever pressing produced no consequences (Figure 1A). 24 h after the last IE session, rats had ‘‘cue re-exposure sessions’’ during which they received passive presentations of the CS (0, 3, 60, or 120 times) without levers available and in the absence of cocaine reinforcement (Figure 1A). 24 h later, the capacity of the CS to promote cocaine-seeking behavior was determined during a cue-induced reinstatement session. Compared with non-re-exposed controls (0 CS presentations), both 60 and 120 CS presentations significantly attenuated reinstatement in a progressive manner (Figure 1D), confirming previous studies showing that relapse-promoting, cocaine-cue associations that form during cocaine SA can be extinguished by sufficient unreinforced re-exposures to the cue (Torregrossa et al., 2013).
Figure 1. Cue-Induced Reinstatement Is Attenuated by Cue Extinction.
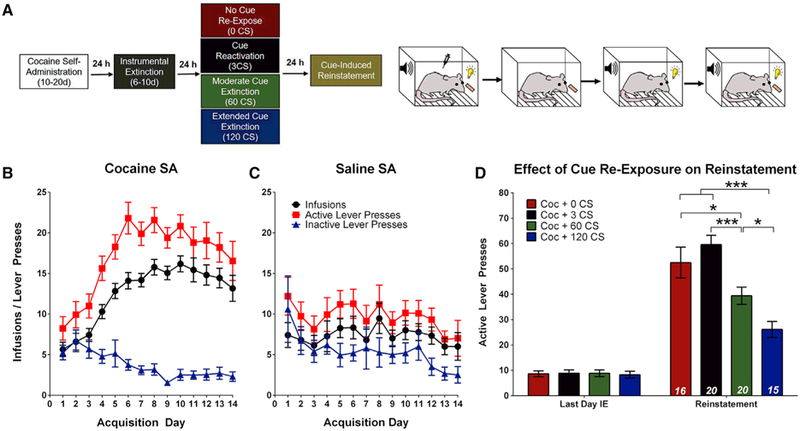
(A) Left: experimental timeline for cue-induced reinstatement experiments. Right: cartoon demonstrating the contingencies of the four experimental phases: SA, IE, cue re-exposure, and reinstatement.
(B) Cocaine SA (n = 71) animals exhibit an increasing number of infusions and active lever responses across acquisition and a steady decline in inactive lever responses.
(C) Saline SA (n = 15) animals do not exhibit an increase in infusions earned as responses on both the active and inactive lever decrease across acquisition.
(D) All cocaine SA animals made significantly more active lever presses during reinstatement compared with the last day of IE. Reinstatement is not significantly affected by brief cue re-exposure (3 CSs) but is reduced by cue extinction (60 or 120 CS re-exposure). Two-way ANOVA, main effect of group (F(3,67) = 10.38, p < 0.001) and a day x group interaction (F(3,67) = 12.01, p < 0.001); post hoc analysis: *p < 0.05, ***p < 0.001. Error bars, mean ± SEM; n in italics = number of rats; CS, conditioned stimulus; LA, lateral amygdala.
In separate groups, we tested whether cocaine-cue associations were regulated by specific LA synaptic modifications by performing ex vivo recordings in rats trained to self-administer cocaine or saline, followed by cue re-exposure either 24 h after the last SA session or after IE sessions (described below). The timing of recordings corresponded to the timing of when cue-induced reinstatement tests were conducted in the prior experiment (Figures 1A and 2A). Previous studies of auditory fear conditioning found that the strength of T-LA and C-LA synapses corresponded to fear memory strength (Hong et al., 2009; Kim et al., 2007a), so we investigated whether similar modifications underlie cocaine-cue associations. To test this, electrically evoked excitatory postsynaptic currents (EPSCs) were recorded from LA principal neurons in acute slices by stimulating either internal capsule (IC; putative thalamic afferents) or external capsule (EC; putative cortical afferents) fibers (Doron and Ledoux, 2000; Hong et al., 2009; Kim et al., 2007a; Figures 2B and 3A). We tested a series of stimulation intensities to generate an input-output relationship whereby EPSC amplitude is increased with larger stimulation intensity. These relationships have commonly been used to assess synaptic changes following extinction of conditioned fear memories (Kim et al., 2007a, 2007b; Hong et al., 2009). Relative to saline-trained controls, cocaine-trained non-CS-re-exposed rats showed an upward shift in the input-output relationship with significantly increased EPSC amplitudes at large stimulation intensities (Figures 2C and 2D). These data suggest that the formation of cocaine-cue associations potentiates T-LA synapses.
Figure 2. Cocaine SA and Cocaine-Cue Re-exposure Bidirectionally Modulate T-LA Synapses.
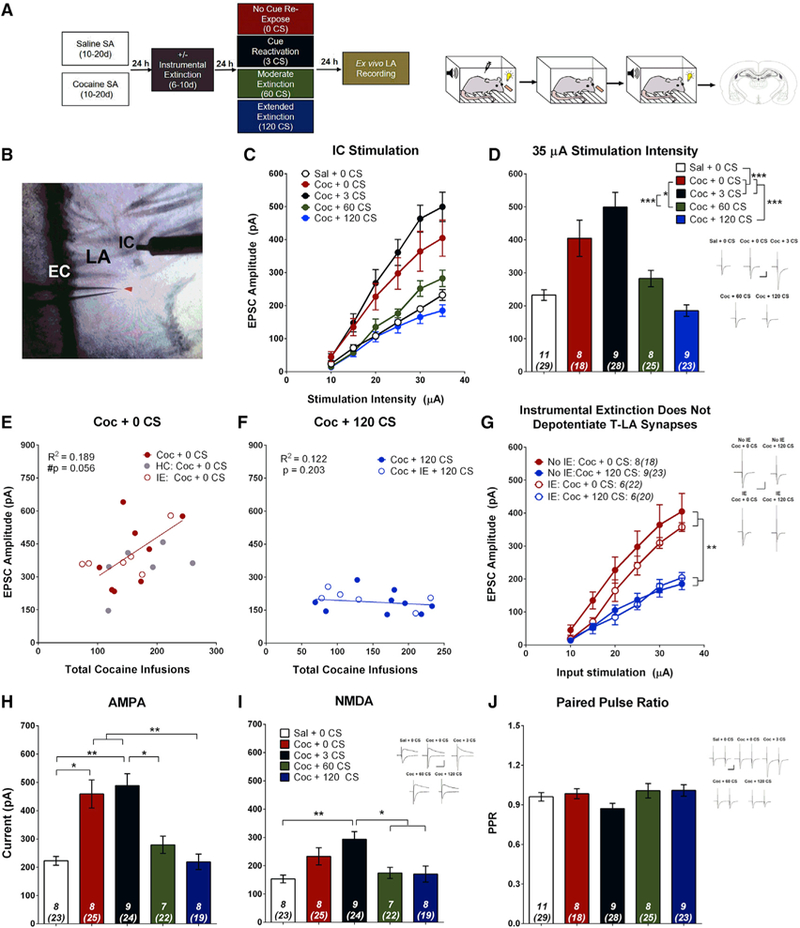
(A) Left: experimental timeline for electrophysiological recording-electrical stimulation experiments. Right: cartoon demonstrating the contingencies of the four experimental phases: SA, IE, cue re-exposure, and ex vivo electrophysiology.
(B) Image of an LA coronal section. EPSCs were evoked from LA principal neurons by stimulating the internal capsule (IC) (putative T-LA synapses). EC, external capsule.
(C) Input-output relationship for each group plotting average EPSC amplitude versus stimulation intensity. Cocaine SA increases EPSC amplitude relative to saline SA. Brief cue re-exposure (3 CSs) does not further alter EPSC amplitude, but moderate (60 CSs) and extended (120 CSs) cue extinction reverses cocaine-cue-induced potentiation. Two-way ANOVA, main effect of group (F(4,40) = 13.54, p < 0.001) and a stimulation (stim.) intensity x group interaction (F(20,200) = 8.96, p < 0.001).
(D) Bar graph showing average EPSC amplitude at 35 μA stimulation intensity. Post hoc analysis from (C): *p < 0.05, ***p < 0.001.
(E) EPSC amplitude (35 μA stimulation intensity) tends to be positively correlated with the number of cocaine infusions received during acquisition for cocaine-SA animals that did not receive cue re-exposure. The correlation includes all animals that were never re-exposed to the CS, including home cage (HC) and IE controls. r(18) = 0.434, #p = 0.056, n = 20.
(F) EPSC amplitude was not correlated with the number of cocaine infusions for animals that self-administered cocaine but received extensive cue extinction. The correlation includes all animals that received 120 CS re-exposure, including IE controls; r(5) = −0.089, p = 0.850, n = 7.
(G) IE does not alter T-LA synaptic strength. The EPSC input-output relationship was significantly higher for rats that received no cue re-exposure (0 CSs) compared with rats that received extended extinction (120 CSs), independent of IE. Two-way ANOVA, main effect of group (F(3,24) = 8.38, p < 0.001) and a stimulation intensity x group interaction (F(15,120) = 4.87, p < 0.001); post hoc analysis: **p < 0.01, ***p < 0.001.
(H) Cocaine-cue memory manipulations significantly affect AMPAR current (one-way ANOVA, F(4,34) = 12.70, p < 0.001); post hoc analysis: *p < 0.05, **p < 0.01.
(I) Cocaine-cue memory manipulations significantly affect NMDAR current (one-way ANOVA, F(4,34) = 5.77, p = 0.001); post hoc analysis: *p < 0.05, **p < 0.01.
(J) Cocaine-cue memory manipulations do not alter PPR (one-way ANOVA, F(4,40) = 1.81, p = 0.140). Insets, sample average EPSC traces evoked at reverse potential (Erev) −70 mV (AMPA, PPR) and Erev +40 mV (NMDA); scale bars, 50 ms, 200 pA; error bars, mean ± SEM, n in italics = number of rats (number of neurons). See also Figures S1–S4.
We next assessed how CS re-exposure may affect T-LA synaptic strength. As shown in Figures 2C and 2D, increasing the number of CS re-exposures progressively reduced T-LA transmission efficacy. Compared with 0 CS animals, EPSC amplitude was slightly but non-significantly increased in animals that underwent brief CS re-exposure (3 CSs), suggesting that cue memory reactivation may tend to strengthen, or at least maintain, the drug cue association through reconsolidation processes and that 3 CS presentations are insufficient to produce any evidence of extinction learning. However, further increasing the number of cue presentations (60 and 120 CSs) attenuated EPSC amplitude. Rats that underwent 120 CS re-exposures exhibited average EPSC amplitudes similar to those of saline controls, indicating that sufficient cue re-exposure depotentiates T-LA synapses and reverses the synaptic changes induced by cocaine+cue SA. To rule out the possibility that the observed synaptic potentiation was simply due to re-exposure to the SA context, a separate control group of rats underwent cocaine SA but then remained in their home cages rather than being re-exposed to the SA context. Although context-re-exposed rats show slightly higher EPSC amplitudes than home cage controls, these differences were not significant (Figure S2). Thus, our observation of synaptic strengthening after cocaine SA is a persistent effect and was not solely due to re-exposure to the cocaine context. However, context re-exposure may lead to a tendency to further strengthen T-LA synapses.
We next asked whether T-LA synaptic strength was modulated by the strength of the cocaine-cue association (Figures 2E and 2F). We found a trend of a positive correlation between the total number of cocaine infusions received during SA and the average EPSC amplitude in rats with no cue re-exposure. Conversely, in rats that underwent 120 CS extinction, there was no correlation between these factors. These data suggest that increased cocaine-cue pairing tends to be associated with more strongly potentiated T-LA synapses. However, sufficient cue re-exposure in the absence of drug reinforcement weakens T-LA synapses independent of prior drug cue experience.
As described above, previous studies have found that extinction of a drug-seeking action is associated with synaptic changes throughout the brain’s reward circuitry (Kalivas et al., 2005; Park et al., 2002). To determine whether T-LA synapses would be differentially regulated by inclusion of the IE phase of the experiment, a separate group of rats underwent 6–10 days of IE after SA. During IE, animals could press both the active and inactive levers but received no cocaine infusion or CS presentation. During the first day of IE, rats pressed the active lever frequently (Figure S3B), likely in anticipation of a cocaine infusion. However, rats quickly adapted and extinguished the lever press response. Interestingly, IE alone was insufficient to depotentiate T-LA synapses (Figure 2G). Cocaine-trained rats that underwent IE followed by 0 CS re-exposure had EPSC amplitudes similar to rats that did not undergo IE. Similarly, rats that underwent IE plus 120 CS re-exposure exhibited depotentiated T-LA synapses just as the group that did not receive IE. Together, these data demonstrate that T-LA synapses are regulated explicitly by the strength of drug cue associations and not by memories of the drug-taking action.
To identify potential mechanisms underlying changes in T-LA synaptic strength, we first examined whether alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (AMPAR) and N-methyl-d-aspartate (NMDA) receptor (NMDAR) currents changed as a result of cocaine SA and cue re-exposure. We found that synaptic changes were driven predominantly by AMPARs rather than NMDARs (Figures 2H and 2I). The AMPAR current was enhanced by cocaine SA and remained elevated following brief cue re-exposure; however, it was decreased by cue extinction. Changes in NMDARs may also contribute to the potential strengthening effects of memory reconsolidation because 3 CS-exposed rats had significantly higher AMPAR and NMDAR currents relative to saline-trained and 120 CS rats (Figures 2H and 2I). However, NMDAR currents were not significantly altered under other CS conditions. These results suggest a postsynaptic increase in AMPAR number and/or functionality following cocaine SA, which was reversed by CS re-exposure during extinction training. In addition, we compared AMPAR EPSC paired-pulse ratios (PPRs) and found no significant differences between groups (Figure 2H), suggesting that presynaptic glutamate release was not altered. Together, these data suggest that, during cocaine-cue memory formation, T-LA synapses are potentiated because of increased AMPAR transmission via postsynaptic mechanisms, which promotes drug-seeking. Sufficient cue re-exposure in the absence of cocaine reverses these changes and decreases the ability of the CS to promote drug-seeking.
Finally, we sought to determine whether C-LA synapses also encode the strength of cocaine-cue associations. To test this, we stimulated EC afferents while recording from LA principal neurons (Figure 3A). Contrary to T-LA synapses, the EPSC input-output relationship was not affected at C-LA synapses by cocaine SA or CS re-exposure (Figures 3B and 3C). Furthermore, EPSC amplitude at C-LA synapses did not correlate with the number of CS-US pairings received during training (Figures 3D and 3E). These results suggest that cortical afferents to the LA are not modulated by cocaine-cue learning. However, IE training did depotentiate AMPAR EPSCs at C-LA synapses independent of cue extinction (Figure 3F). EPSC amplitude at C-LA synapses was significantly decreased after IE both in rats that received no cue re-exposure (cocaine [Coc] + 0 CSs) and those that received extended cue re-exposure (Coc + 120 CSs). Therefore, cortical input to the amygdala appears to be regulated not by drug-cue associations but by extinction of the drug-taking action.
Figure 3. C-LA Synapses Are Altered by IE but Not by Cocaine-Cue Memory Manipulations.
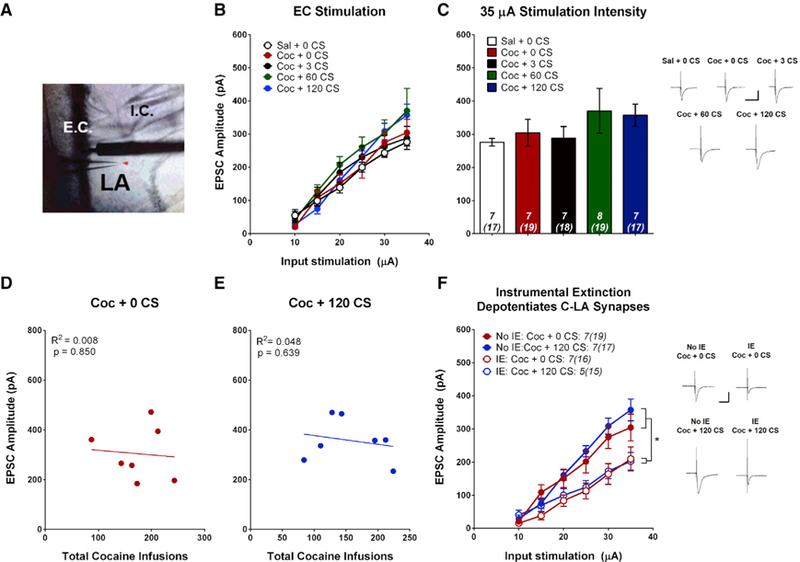
(A) Image of LA coronal section. EPSCs were evoked from LA principal neurons by stimulating the EC (putative C-LA synapses).
(B) Input-output relationship for each group, plotting average EPSC amplitude versus stimulation intensity. Neither cocaine SA nor cue re-exposure significantly alter EPSC amplitude at C-LA synapses (two-way ANOVA, F(4,31) = 0.854, p = 0.503).
(C) Bar graph showing average EPSC amplitude at 35 μA stimulation intensity.
(D) EPSC amplitude (35 μA stimulation intensity) was not correlated with the number of cocaine infusions during acquisition for cocaine-SA animals that did not receive cue re-exposure (r(5) = −0.0891, p = 0.850, n = 7).
(E) EPSC amplitude (35 μA stimulation intensity) was not correlated with the number of cocaine infusions earned during acquisition for cocaine-SA animals that received extensive cue extinction (r(5) = −0.219, p = 0.639, n = 7).
(F) C-LA synapses are depotentiated by IE. The EPSC input-output relationship was significantly lower for all rats that received IE, independent of cue re-exposure. Two-way ANOVA, main effect of group (F(3,22) = 5.11, p = 0.008) and a stimulation intensity x group interaction (F(15,110) = 3.69, p < 0.001); post hoc analysis: **p < 0.05.
Insets, sample average EPSC traces evoked at Erev −70 mV; scale bars, 50 ms, 200 pA; error bars, mean ± SEM; numbers in italics, number of rats (number of neurons).
MGN-LA Synapses Are Altered by Cocaine-Cue Associations
Projections from multiple thalamic nuclei innervate the amygdala (LeDoux et al., 1990; Nabavi et al., 2014), and studies of auditory fear conditioning suggest that connections between the MGN and LA are particularly important for mediating auditory cue-specific memories. Given that the auditory tone component of the cocaine-paired audiovisual cue is likely to have a high degree of salience in rats, we tested whether projections from the MGN to the LA are strengthened by cocaine-cue associations. We expressed a variant of channelrhodopsin in MGN neurons (AAV5.hSyn.oChIEF.tdTomato) (Lin et al., 2009; Nabavi et al., 2014), and approximately 2 weeks after viral infusions, rats underwent a similar procedure as described above (Figure 4A), where they were trained to SA cocaine or saline, followed by either 0 or 120 CS re-exposures. 24 h after cue re-exposure sessions, amygdala slices were prepared, and light-evoked (473-nm) EPSCs were recorded from LA principal neurons (Figures 4B and 4C). Optogenetic stimulation of MGN terminals within the LA revealed a shifted input-output relation to higher EPSC amplitude in cocaine-trained non-CS re-exposed animals relative to saline-trained controls, whereas 120 CS re-exposure resulted in a reversal of this potentiation (Figure 4C). Again, the changes in synaptic strength were primarily mediated by post-synaptic changes in AMPAR currents. Compared with cocaine-trained non-CS re-exposed animals, those that underwent extended cue re-exposure had a significantly reduced AMPAR current, but there were no group differences for either NMDAR current or PPR (Figures 4D–4F). These results suggest that drug-cue associations are mediated by dynamic changes in AMPAR signaling at MGN-LA synapses.
Figure 4. MGN-LA Synapses Are Bidirectionally Modulated by Cocaine SA and Cocaine-Cue Re-exposure in a Postsynaptic Manner.
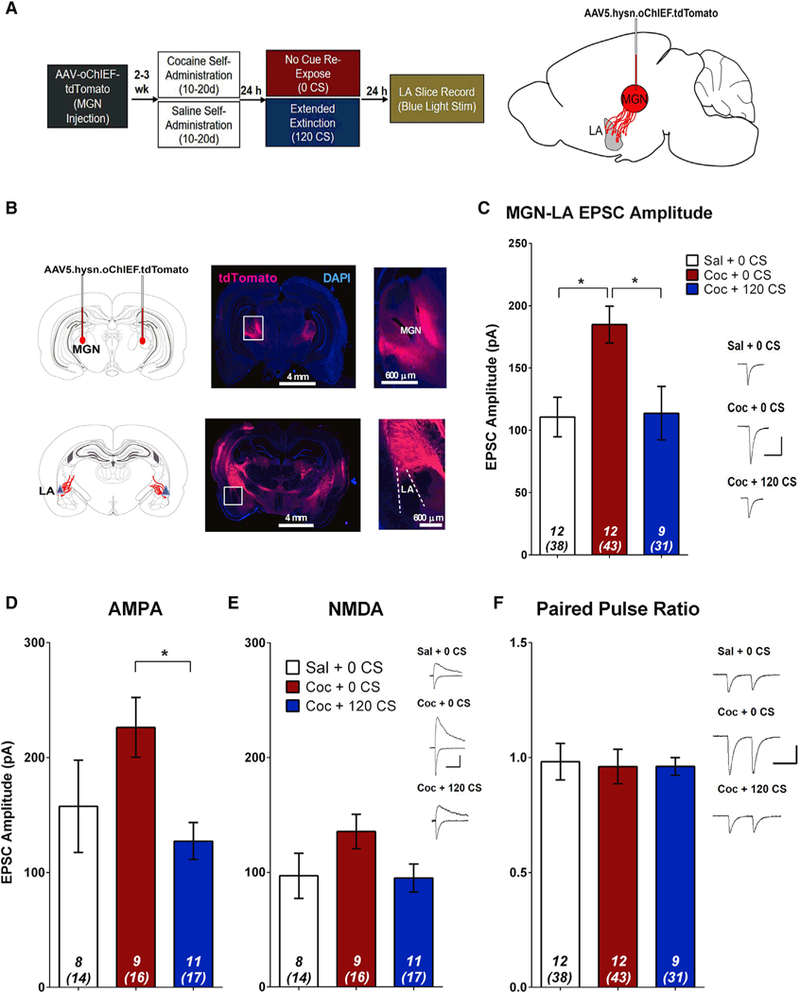
(A) Experimental design.
(B) Top: diagram and images demonstrating AAV-oChIEF targeting to the MGN. Bottom: labeling of MGN axons shows projections to the LA.
(C) EPSCs were optically evoked from LA principal neurons by stimulating AAV-infected MGN axon terminals. Cocaine-trained animals that were not re-exposed to cues had a significantly higher EPSC amplitude relative to saline-trained controls and cocaine-trained 120 CS re-exposed animals. One-way ANOVA, (F(2,25) = 6.87, p = 0.004); post hoc analysis: *p < 0.05, one-way ANOVA).
(D) Cocaine-cue memory manipulations significantly affect AMPAR current (one-way ANOVA, F(2,25) = 3.72, p = 0.039); post hoc analysis: *p < 0.05.
(E) Cocaine-cue memory manipulations do not affect NMDAR current (one-way ANOVA, F(2,25) = 2.22, p = 0.129).
(F) Cocaine-cue memory manipulations do not affect PPR (one-way ANOVA, F(2,30) = 0.03, p = 0.970). Insets, sample average EPSC traces evoked at Erev −70 mV (AMPA, PPR) and Erev +40 mV (NMDA); scale bars, 50 ms, 100 pA; error bars, mean ± SEM, numbers in italics, number of rats (number of neurons). See also Figure S4.
In Vivo Optogenetic Induction of LTD at MGN-LA Synapses Attenuates Relapse-like Behavior
We next determined whether we could mimic extinction of drug-cue memories by inducing circuit-specific LTD at MGN-LA synapses. We first demonstrated the capacity to optically induce LTD at MGN-LA synapses. Rats were injected with adeno-associated virus and a channelrhodopsin variant (AAV-oChiEF) into the MGN and were trained to SA cocaine or saline, followed by either 0 or 120 CS re-exposure. 24 h later, ex vivo electrophysiological recordings were performed. In current clamp configuration, LA-projecting MGN afferents were stimulated with 473-nm light to evoke excitatory postsynaptic potentials (EPSPs). Following LTD induction (1 Hz, 15 min), sustained suppression of EPSP slope and amplitude was reliably observed in cocaine-trained 0 CS re-exposed rats (Figures 5A, 5B, and S5A). However, LTD was not observed in saline-trained rats (Figures 5A and 5B) or in cocaine-trained 120 CS re-exposed rats (Figures 5A and 5B), presumably because LTD is occluded at non-potentiated synapses. These results suggest not only that LTD can be induced in cocaine-trained rats but that an LTD-like process is induced at MGN-LA synapses by cue memory extinction.
Figure 5. Ex Vivo Optical Low-Frequency Stimulation Induces LTD Only at Potentiated MGN-LA Synapses.
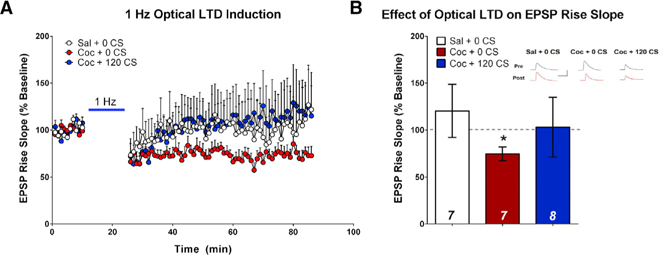
(A) Scatterplots demonstrating the effect of ex vivo optical LTD induction on EPSP rise slope at MGN-LA terminals. 15 min of 1-Hz optical stimulation induced a sustained reduction in EPSP rise slope only in cocaine-trained non-CS-re-exposed animals.
(B) Quantification of (A), demonstrating the effect of 1-Hz stimulation on EPSP rise slope as a percent change from baseline. LTD induction significantly decreased the EPSP rise slope relative to baseline only in cocaine-trained non-CS re-exposed animals (paired t test, t(6) = 3.34, *p = 0.016). Inset, sample average EPSP traces across groups during baseline (Pre, black) and 1 h after LTD induction (Post, red); scale bars, 100 ms, 10 mV; error bars, mean SEM; n in bars = number of neurons. See also Figure S5.
Finally, we tested whether in vivo optogenetic LTD induction could reduce cue-induced cocaine-seeking. Rats were injected with AAV-oChIEF or AAV-tdTomato targeting the MGN, and optic fibers were implanted, targeting the dorsal portion of the LA (Figures 6A and 6B). Following SA and IE, rather than undergoing cue re-exposure, rats received either in vivo low-frequency optical LTD (1 Hz, 15 min 473-nm light stimulation) of MGN-LA terminals or did not receive laser stimulation (sham controls). 24 h later, either ex vivo optical recordings or cue-induced reinstatement was performed. Rats that were exposed to optical LTD had significantly reduced MGN-LA EPSC amplitude relative to sham controls (Figure 6C), confirming induction of LTD by our stimulation protocol. As further evidence that this in vivo optical stimulation paradigm induced LTD, we found that we could not produce LTD in neurons from rats that received the in vivo optical stimulation but could observe LTD in neurons from rats that underwent sham stimulation (Figure S5B). Thus, in vivo LTD occluded further LTD induction in slice. We next demonstrated that in vivo optical LTD of MGN-LA terminals reduced cue-elicited drug-seeking because LTD-exposed rats made significantly fewer lever presses during reinstatement compared with both a group of rats expressing a control virus that received laser stimulation as well as sham controls (Figure 6D). Rats subsequently underwent an additional reinstatement test 7 days later, and LTD-exposed rats maintained a low level of responding that was lower than that of sham controls (Figure 6E).These data show that induction of LTD at MGN-LA synapses is sufficient to reduce drug-seeking in a manner similar to cue extinction and that this reduction can persist across multiple reinstatement tests. Together, these results indicate that drug-cue memories may be malleable to circuit-specific optogenetic manipulations.
Figure 6. In Vivo Optical LTD of the MGN-LA Circuit Inhibits Cue-Induced Reinstatement.
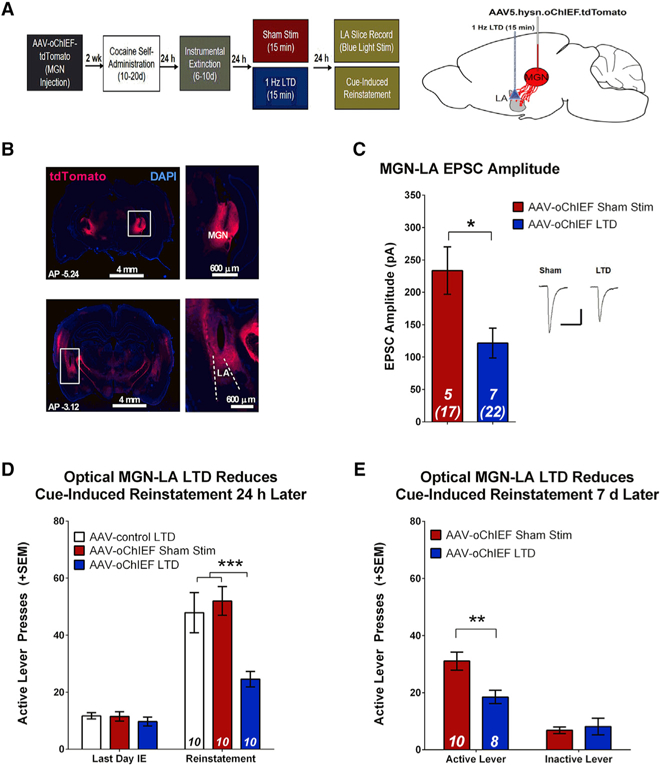
(A) Experimental design. oChIEF-expressing AAV5 was injected into the MGN, and optical fibers were implanted at the dorsal tip of the LA.
(B) Diagram and images demonstrating the position of virus injection in the MGN (top) and placement of optic fibers in the LA (bottom).
(C) In vivo dual hemisphere LTD of MGN-LA synapses attenuates EPSC amplitude relative to sham controls (unpaired t test, t(10) = 2.73, *p = 0.021). Inset, sample average EPSC traces evoked at Erev −70 mV; scale bars, 50 ms, 200 pA.
(D) In vivo dual hemisphere LTD of MGN-LA synapses attenuates reinstatement. There were no differences in active lever pressing between groups during the last day of IE. There is a significant reduction in active lever presses during reinstatement in rats that previously underwent optical LTD relative to animals that received a control virus and sham controls. Two-way ANOVA, main effect of group (F(2,27) = 7.04, p = 0.004) and a day x group interaction (F(2,29) = 8.08, p = 0.002); post hoc analysis: ***p < 0.001; error bars, mean x SEM; numbers in bars, number of rats.
(E) In vivo dual hemisphere LTD of MGN-LA synapses affects spontaneous recovery. 7 days after initial cue-induced reinstatement, rats underwent a second reinstatement test, revealing a significant reduction in active lever pressing in animals that previously underwent MGN-LA LTD relative to sham controls. Two-way ANOVA, main effect of group (F(1,32) = 5.04, p = 0.032), significant interaction (F(1,32) = 7.69, p = 0.009); post hoc analysis, **p < 0.01.
Error bars, mean ± SEM; numbers in italics, number of rats (number of neurons). See also Figure S5.
DISCUSSION
We have previously shown that re-exposure to cocaine-associated discrete cues in the absence of cocaine reinforcement can attenuate cue-elicited drug-seeking (Rich et al., 2016). In this study, we extend our findings and show that sensory input from the thalamus, but not the cortex, regulates cue-driven drug-seeking behaviors. Specifically, T-LA but not C-LA synapses are potentiated by pairing cocaine with an audiovisual cue. Importantly, this association is long-lasting because rats still showed T-LA synaptic potentiation after 7 days of IE. Conversely, re-exposure to the cue in the absence of cocaine progressively depotentiated T-LA synapses, inhibiting relapse-like behavior. Furthermore, in vivo optogenetic induction of LTD in the MGN-LA pathway of cocaine-trained rats was sufficient to reduce reinstatement. Although optical LTD of this pathway has previously been demonstrated to inhibit a fear-associated memory (Nabavi et al., 2014), this study provides direct evidence that MGN-LA circuit-specific plasticity underlies drug-cue memories.
In our study, brief cue re-exposure (3 CSs) did not alter the potentiation of T-LA synapses or cue-induced reinstatement. Brief cue re-exposure likely reactivates the drug-cue memory, initiating reconsolidation (Arguello et al., 2014; Rich et al., 2016). Reconsolidation is thought to strengthen or at least maintain memory, and our experiments showed a nonsignificant increase in T-LA EPSC amplitude relative to cocaine-trained non-CS re-exposed rats 24 h after reactivation, when memory is thought to be restabilized. The lack of statistical significance does not rule out a memory-strengthening effect. For example, the destabilization and restabilization of memory that occur after reactivation are likely to involve postsynaptic signaling, which may explain the significant increase in NMDAR current.
The importance of synaptic plasticity within the amygdala has been well documented for the regulation of other types of associative learning. Auditory fear conditioning potentiates both the T-LA and C-LA pathways, and cue extinction reverses these changes and reduces the expression of fear (Hong et al., 2009; Kim et al., 2007a). The importance of the amygdala during reward-based learning has also been demonstrated (Carelli et al., 2003; Fuchs et al., 2006; Hsiang et al., 2014; Robbins et al., 2008). For example, a small percentage of neurons within the LA are important for the formation of a cocaine place preference memory (Hsiang et al., 2014). Moreover, in vivo electrophysiology recordings have shown that a percentage of amygdala neurons fire in response to presentation of cocaine-associated cues (Carelli et al., 2003). The specific afferent inputs involved in the encoding of drug-associated memories have not been well-examined, although sucrose reward learning can strengthen T-LA and C-LA synapses. C-LA synapses, however, were only strengthened when dopamine was elevated by dopamine transporter (DAT) blockade (Tye et al., 2008, 2010). In our study, despite the DAT-blocking effects of cocaine, we did not see C-LA potentiation, although procedural differences, including the absence of a sucrose reward, may explain this seeming discrepancy.
Here we show that T-LA synapses are strengthened by the formation of a cocaine-cue association but that these alterations are reversible in a cue ‘‘dose’’-dependent manner, with 120 CS re-exposure fully reversing cocaine-cue-associated potentiation. Extinction of a fear memory also appears to be dose-dependent, with a single extinction session promoting fear inhibition through enhanced inhibitory tone in LA neurons, enhanced CS-evoked activity in the medial prefrontal cortex (mPFC), and enhanced synaptic efficacy at amygdala intercalated neurons (An et al., 2017; Amano et al., 2010; Chhatwal et al., 2005; Milad and Quirk, 2002). Multiple extinction sessions, however, produce depotentiation of T-LA synapses and seeming erasure of fear memory. Thus, our results are consistent with these fear studies, although we do find that a large number of CS presentations (120) are necessary for consistent reductions in drug-seeking.
The lack of group effects on C-LA synapses may speak to the importance of bottom-up signaling during reward-motivated behaviors that is less dependent on cognitive influence. Previous studies have demonstrated the influence of top-down circuits in extinction; however, these studies involved extinction of the drug-taking action (Augur et al., 2016; Stefanik et al., 2016). Inter- estingly, we did observe depotentiation of C-LA synapses in response to IE, suggesting a role of this circuit in IE. Our findings also do not rule out any cortical influence on drug-cue memory extinction because the C-LA pathway mostly relays input from the auditory and visual cortex (Te2 and Te3) and perirhinal cortex (McDonald, 1998), whereas other cortical areas project via separate pathways.
Our study identifies postsynaptic glutamatergic signaling as a mechanism responsible for regulating drug-cue memory. Future studies should continue to expand upon the mechanisms and circuits involved. Calcium signaling and intracellular kinases and phosphatases are important for cue-associated memories (Merlo et al., 2014; Rich et al., 2016b), and these downstream signaling mechanisms are likely involved in synaptic modifications following drug-cue re-exposure; for example, the internalization of AMPARs, which is necessary for the extinction of acquired fear (Dalton et al., 2008). Additionally, mGluR1 and mGluR2 receptors in the amygdala are critical for depotentiation and the extinction of fear-associated memories (Hong et al., 2009; Kim et al., 2007b). Although our study focuses on auditory thalamic inputs to the LA, the mediodorsal and paraventricular nuclei of the thalamus also project to the mPFC, nucleus accumbens, and amygdala (Do-Monte et al., 2017; Vertes et al., 2015). These circuits have been implicated in fear conditioning (Penzo et al., 2015), incubation of drug-seeking (Li et al., 2015; Lu et al., 2005), and the regulation of reward-seeking when an anticipated reward is omitted (Do-Monte et al., 2017) and, therefore, could also influence drug-cue memory. Given that our experiments involve use of a multimodal (i.e., audiovisual) cue, it is interesting that manipulating the MGN-LA pathway is so effective at reversing cocaine-cue-evoked plasticity. One explanation for this may be the relatively greater salience of an auditory cue to a visual cue in a rat strain with poor vision. The lateral geniculate nucleus (LGN) is the primary thalamic region involved in visual processing, but the behavioral effect of LTD in LGN-LA projections is unknown. However, it is possible that combined manipulation of LGN and MGN pathways could more completely eliminate cue-motivated lever pressing during reinstatement. Finally, because drugs of abuse act on the brain’s reward system, the role of dopaminergic inputs from the VTA in modulating LA afferent input should also be examined. Activation of DA receptors has been shown previously to enhance Te3-evoked responses in the LA (Rosenkranz and Grace, 2001). Similarly, elevated dopamine during cue-sucrose learning was sufficient to enhance C-LA plasticity when measured 30 min after training (Tye et al., 2010), so dopamine likely also influences plasticity during reconsolidation and extinction of a drug-cue memory.
Finally, one limitation of cue extinction as a therapy is that drug-seeking often spontaneously returns after a period of abstinence (Peters et al., 2008; Rescorla, 2004). Interestingly, in our study, extended cue extinction was sufficient to reduce relapse-like behavior, even in animals that had extensive drug-cue experience, suggesting that long-term cue re-exposure in the absence of drug reinforcement may be effective at preventing relapse even in chronic drug abusers. Optical induction of LTD in the MGN-LA pathway, which is occluded by prior cue extinction, results in similar decreases in reinstatement that lasts for at least a week, suggesting that cue extinction occurs via a persistent depotentiation of T-LA synapses. Together, these results support the idea that circuit-specific neuroadaptations can support the long-term inhibition or erasure of a drug-cue memory and offer an important consideration for the development of novel treatments. However, future studies should determine whether LTD-based stimulation therapy can be made selective for maladaptive drug memories or whether adaptive reward-associated memories would also be affected.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | ||
|---|---|---|---|---|
| Bacterial and Virus Strains | ||||
| AAV5.hSyn.oChIEF.tdTomato | Lin et al., 2009; Nabavi et al., 2014 | Duke Viral Vector Core (via Roger Tsien): #268 | ||
| AAV5.hSyn.tdTomato (Control) | Lin et al., 2009; Nabavi et al., 2014 | Duke Viral Vector Core Control | ||
|
Chemicals, Peptides, and Recombinant Proteins | ||||
| Cocaine HCl | NIDA Drug Supply Program | 9041–001 | ||
| 0.9% Saline | Fisher Scientific | NC0291799 | ||
| Ketamine HCl | Henry Schein | 055853 | ||
| Xylazine | Butler Schein | 033198 | ||
| Rimadyl | Henry Schein | 024751 | ||
| Lactated Ringer’s | Henry Schein | 009846 | ||
| Betadine | Butler Schein | 038250 | ||
| Ethanol | University of Pittsburgh Chemistry Stockroom | 200C5000 | ||
| Gentamicin | Henry Schein | 006913 | ||
| Heparin | Henry Schein | 055737 | ||
| N-methyl-D-glucamine | Sigma-Aldrich | M2004 | ||
| Sodium chloride | Sigma-Aldrich | S7653 | ||
| Potassium chloride | Sigma-Aldrich | P9333 | ||
| Sodium phosphate | Sigma-Aldrich | S9638 | ||
| Sodium bicarbonate | Sigma-Aldrich | S5761 | ||
| HEPES | Sigma-Aldrich | H3375 | ||
| D-Glucose | Sigma-Aldrich | G8270 | ||
| Sodium L-Ascorbate | Sigma-Aldrich | A7631 | ||
| Thiourea | Sigma-Aldrich | T8656 | ||
| Sodium pyruvate | Sigma-Aldrich | P2256 | ||
| Magnesium sulfate | Sigma-Aldrich | 203726 | ||
| Calcium chloride | Fisher Scientific | C1016 | ||
| Hydrochloric Acid | Fisher Scientific | 0219405490 | ||
| Cesium methanesulfonate | Fisher Scientific | C1426 | ||
| Cesium chloride | Fisher Scientific | 289329 | ||
| EGTA | Fisher Scientific | E3889 | ||
| TEA-Chloride | Fisher Scientific | T2265 | ||
| ATP Magnesium Salt | Fisher Scientific | A9187 | ||
| GTP Sodium Salt | Fisher Scientific | G8877 | ||
| QX-314-Cl | Alomone Labs | Q-150 | ||
| Sodium phosphocreatine | Fisher Scientific | P7936 | ||
| L-glutathione | Fisher Scientific | G4251 | ||
| Potassium methanesulfonate | Fisher Scientific | 83000 | ||
| Cesium hydroxide | Fisher Scientific | 516988 | ||
| Potassium hydroxide | Fisher Scientific | P5958 | ||
| Isoflurane | Henry Schein | 029405 | ||
| DMSO | Fisher Scientific | BP231–1 | ||
| Picrotoxin | Fisher Scientific | AC131210010 | ||
| Sodium Pentobarbital | Henry Schein | 024352 | ||
| Paraformaldehyde | Sigma-Aldrich | P6148 | ||
| Fluoroshield with DAPI | Sigma-Aldrich | F6057 | ||
| Lidocaine | Butler Schein | 014583 | ||
|
Experimental Models: Organisms/Strains | ||||
| R. norvegicus: Sprague Dawley | Envigo | https://www.envigo.com | ||
| Order Code #002 | ||||
|
Software and Algorithms | ||||
| MedPC IV | MedAssociates | http://www.medassociates.com | ||
| Clampex | Molecular Devices | https://www.moleculardevices.com/ | ||
| Clampfit 10.3 | Molecular Devices | https://www.moleculardevices.com/ | ||
| GraphPad Prism 6 | GraphPad | https://www.graphpad.com/ | ||
| Virtual Slide Fluorescence Software (VS-ASW FL) | Olympus | http://www.olympus-lifescience.com/en/ | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Mary Torregrossa (torregrossam@upmc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Naive, adult male Sprague-Dawley rats (Envigo/Harlan), weighing 275–325 g on arrival, were used in all studies (total n for all experiments = 213). All rats were housed in a temperature- and humidity-controlled room, in auto-ventilated racks with an automated watering system. Animals were housed in pairs, given ad libitum access to food and water, and maintained on a 12 h light-dark cycle. Prior to surgical procedures, rats were given at least 5 d to acclimate to the facility. Rats were food-deprived 24 h prior to the start of behavioral experiments and maintained at ~90% of their free-feeding body weight (~20 g of chow per day) for the duration of testing. All behavioral experiments were run during the light-cycle. Animals were allocated to groups following cocaine self-administration (SA) and, when applicable, instrumental extinction (IE), based on a matching procedure that ensured no significant differences between acquisition and IE behavior (See Figures S1 and S3). All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh’s Institutional Animal Care and Use Committee.
Viral Vector Construct
Adeno-associated virus serotype 5 (AAV5) vectors were constructed to deliver oChIEF, a variant of the blue-light sensitive opsin channelrhodopsin (ChR2). oCHIEF is a mammalian codon version of ChIEF, with stronger expression in mammalian cells and an additional N-terminal amino acid residue that can respond to both low and high frequency stimulations (Nabavi et al., 2014; Lin et al., 2009). oChIEF was flanked downstream by the fluorescent marker tdTomato and expression of oChIEF was driven by the neuron-specific synapsin (hSyn) promoter (Lin et al., 2009). In some experiments, a control tdTomato expressing AAV5 was used. The oChIEF construct was donated by Dr. Roger Tsien and processed for packaging and purification by the Duke Viral Vector Core.
METHOD DETAILS
Drugs
Cocaine hydrochloride (generously provided by the Drug Supply Program of the National Institute on Drug Abuse, Research Triangle Park, NC) was dissolved in sterile 0.9% saline (2 mg/ml) and filter-sterilized for SA.
Rodent Intravenous Catheterization
Rats were fully anesthetized with ketamine hydrochloride (87.5–100 mg/kg, i.m.) and xylazine hydrochloride (5 mg/kg, i.m.) and then received an analgesic (Rimadyl, 5 mg/kg, s.c.) and 5 mL of Lactated Ringer’s (s.c.) prior to surgery. Betadine and 70% ethanol were applied to all incision sites. All rats received a chronic indwelling intravenous (i.v.) catheter as described previously (Rich et al., 2016). Catheters were implanted into the right jugular vein, then fed subcutaneously to the midscapular region, where they exited through a round incision. After surgery, rats recovered on a heating pad. Rats were then individually housed and given at least 7 d to recover before behavioral training. Rimadyl (5 mg/kg; s.c.) was administered for the first two days after surgery and catheters were kept patent by daily infusions of sterile saline containing gentamicin (5 mg/ml) and heparin (30 USP/ml).
Virus Delivery and Optic Fiber Implantation
For experiments involving viral infection of MGN neurons, rats were placed in a stereotaxic frame immediately following catheter surgery. They were given a small injection (~0.2–0.3 ml) of lidocaine (Henry Schein) to the scalp as a local anesthetic. A 26-gauge stainless steel injection cannula connected to a Hamilton syringe was used to bilaterally inject 1 μL of concentrated AAV solution into the medial portion of MGN (in mm from bregma, anterior and posterior (AP): −5.4; medial and lateral (ML): ± 3.0; dorsal and ventral (DV): −6.6) through a pump (Harvard Apparatus) at a flow rate of 0.1 mL/min. Cannula were left in place for 5 min after infusions were complete before being slowly withdrawn. For experiments involving in vivo optogenetic control of MGN-LA terminals, two 200-μm optic fibers (0.5 NA, Thor Labs) were implanted (See Sparta et al., 2011) at the dorsal portion of the lateral amygdala (in mm from bregma, AP: −3.0; ML: ± 5.1; DV: −7.9 mm). Fibers were lowered at a rate of 2 mm/min, then secured to the skul lwith screws, Loctite instant adhesive (Henkel Corp) and OrthoJet dental cement (Lang Dental).
Rodent Cocaine or Saline Self-Administration
Rats were trained to SA cocaine in standard operant conditioning chambers (MedAssociates), as described previously (Rich et al., 2016). Rats administered saline (0.9%) or cocaine (2 mg/ml) during daily sessions for 1 h, on a fixed ratio 1 (FR1) schedule of reinforcement with a 10 s timeout. The designated active lever (counterbalanced across left and right levers) produced a cocaine or saline infusion paired with a 10 s compound light and tone cue. Pump durations were adjusted daily according to body weight in order to deliver the correct dose of drug (1.0 mg/kg of body weight per infusion). Responses on the other, inactive, lever were recorded, but had no programmed consequences. Rats underwent training for at least 10 d and until they administered at least 8 infusions per day over 3 consecutive days. Rats that did not meet acquisition criteria by 20 d were excluded from the study. The program was controlled by and data were collected using MedPC IV (MedAssociates).
Instrumental Lever Extinction
After successful acquisition of SA, rats underwent IE for 6–10 d. During these daily 1 h sessions, responses on both the active and inactive levers were recorded but had no programmed consequences. IE continued until extinction criteria had been met (an average of < 25 lever presses on the last two days of extinction). Throughout IE, rats received no cocaine or cocaine-associated cue reinforcement, thus reducing responses to a stable, low rate. This reduces the motivational value of other cues in the SA context, so that subsequent reinstatement testing or physiological assessment specifically isolates the memory for the discrete cue associated with cocaine infusion.
Pavlovian Cue Re-Exposure
Cue re-exposure occurred as described previously (Rich et al., 2016). Briefly, rats were returned to the SA context 24 h after the final day of SA (or IE). During this 1 hr session, levers were unavailable, so responses could not be made. Rats that had undergone cocaine SA received noncontingent presentations of the previously drug-paired cues: either 0, 3, 60, or 120. A separate group of cocaine-trained rats were left in their home cages undisturbed as a control for re-exposure to the training context. Saline-trained animals were returned to the training context and did not undergo cue re-exposure (0 cue presentations). During re-exposure sessions, the cocaine-associated cue was presented for 10 s, with each presentation separated by 30 s. The timing of cue delivery was such that each group of animals received their final cue presentation at the end of the session.
Cue-Induced Reinstatement
24 h after cue re-exposure, cue-induced reinstatement was assessed during a 1 h session that took place in the original SA context. A lever press on the active lever produced a 10 s presentation of the cocaine-associated cue on an FR1 schedule, but no drug reinforcement. Lever presses on the inactive lever were recorded but had no programmed consequences. In the optical LTD experiment, to measure spontaneous recovery of drug-seeking, rats underwent a second cue-induced reinstatement test 7 d later.
Ex vivo Slice Preparation
Slices were prepared as described previously (Huang et al., 2008), with slight modifications, and using methods designed to improve neuronal health in adult rodents (Ting et al., 2014). Briefly, 24 h after cue re-exposure sessions, rats were deeply anesthetized with isoflurane. Rats were then briefly perfused with ice-cold cutting solution containing (in mM): 92 N-methyl-d-glucamine (NMDG), 2.5 KCl, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 10 MgSO4, and 0.5 CaCl2, saturated with carbogen (95% O2/5% CO2), pH adjusted to 7.4 with HCl. Rats were then decapitated and brains removed. Acute coronal slices of the amygdala (250 μm thick) were obtained (normally 4–6 slices were obtained from each rat) using a VT1200S vibratome (Leica, Weltzar, Germany) in 4 °C cutting solution. Slices were placed in a holding chamber filled with the same cutting solution, and incubated at 37°C for 10–15 min before being transferred to a beaker of HEPES-based holding solution containing (in mM): 86 NaCl, 2.5 KCl, 1.2 NaH2PO4, 35 NaHCO3, 20 HEPES, 25 glucose, 5 sodium ascorbate, 2 thiourea, 3 sodium pyruvate, 1 MgCl2, and 2 CaCl2, saturated with carbogen. Slices were allowed to recover for > 30 min at room temperature before experimentation.
Ex vivo Electrophysiological Recordings
Slices were transferred to an Olympus BX51WI upright microscope equipped with Dodt Gradient Contrast infrared optics. The LA was identified using a 4X objective and this region was then magnified for identification of neurons with a 40X water immersion lens. Whole-cell recordings were obtained from individual principal neurons in the LA using glass pipettes (3–5 MΩ). Principal neurons were identified by morphology, and in voltage clamp, typically showing low levels of spontaneous activity. For a subset of cells, principal neurons could be confirmed in current clamp by injecting current pulses to elicit action potentials, as described elsewhere (Kim et al., 2007a; See Figure S4A). Voltage-clamp experiments used pipettes filled with a cesium-based internal solution [in mM, cesium methanesulfonate 108, CsCl 15, CsEGTA 0.4, TEA-Cl 5, HEPES 20, Mg-ATP 2.5, Na-GTP 0.25, QX-314-Cl 1, sodium phosphocreatine 7.5, and L-glutathione 1, and pH to 7.3 with CsOH] and current-clamp experiments used pipettes filled with a potassium-based internal solution [in mM, potassium methanesulfonate 108, KCl 20, K-EGTA 0.4, HEPES 10, Mg-ATP 2.5, Na-GTP 0.25, sodium phosphocreatine 7.5, L-glutathione 1, MgCl2 2, and pH to 7.3 with KOH]. During recordings, slices were superfused with aCSF that was heated to 31–33 °C by passing the solution through a feedback-controlled in-line heater (Warner, CT) before entering the chamber. External perfusion consisted of a modified artificial cerebrospinal fluid (ACSF), containing, in mM NaCl 119, KCl 2.5, NaHCO3 26, NaH2PO4 1.2, glucose 12.5, HEPES 5, MgSO4 1, CaCl2 2, saturated with 95% O2/5% CO2. Neurons were voltage-clamped at −70 mV. For experiments involving electrical stimulation, a concentric bipolar stimulating electrode (FHC, Bowdoin, ME) was placed over axon fibers emerging from the internal capsule (putative thalamic afferents) or external capsule (putative sensory cortical afferents). Projections to the LA were stimulated using 0.1 ms pulses at predetermined series of intensities (10–35 μA) using an isolated constant current stimulator (A-M instruments; Digitimer Ltd, Hertfordshire, England), and the evoked excitatory postsynaptic currents (EPSCs) were recorded. Picrotoxin (100 μM; dissolved in DMSO) was included to inhibit GABAA receptor-mediated currents in all experiments. For paired pulse delivery, each pulse was separated by a 50 ms interpulse interval. AMPAR currents were elicited at reversal potential (ERev)-70 mV holding potential and mixed AMPAR+NMDAR currents were elicited at a ERev+40 mV holding potential. NMDAR amplitude was operationally defined as the amplitude of the ERev+40 mV current 35 ms after the peak of the AMPAR current (ERev-70 mV); at this time point, AMPAR-mediated currents have subsided (Huang et al., 2008, 2009). To elicit action potential firing, in current-clamp mode, depolarizing current pulses of −100 to +200 pA (20 pA steps, 1 s duration) were delivered.
For experiments involving optical stimulation, AAV-infected MGN projections were identified using fluorescence and then stimulated using a blue light (473-nm) DPSS laser (IkeCool), driven by TTL signals generated using the Clampex software (Molecular Devices) and a pulse generator (A-M Systems). Collimated laser light was coupled to a fluorescent port of the Olympus BX51WI microscope, and focused onto the slice through the objective. Optical stimulations of 1 ms duration were used for paired-pulse or AMPA/NMDA ratio measurements. Neurons receiving inputs from AAV-infected MGN neurons exhibited reliable EPSCs in response to stimulations (See Figure S4B). Likewise, under current-clamp conditions, AAV-infected MGN neurons generated action potentials in response to various frequencies of 473-nm-light stimulations (See Figure S4C). Ex vivo LTD experiments were performed in cur- rent-clamp mode, with the bridge balanced routinely. Optically-evoked EPSPs were recorded at 0.1 Hz for 10 minutes prior to LTD induction [900 2-ms pulses of 473-nm light, at 1 Hz (15 min induction protocol)]. Following LTD induction, EPSPs were continuously recorded at 0.1 Hz for the next 60 minutes. For all experiments, series resistance was 10–25 MΩ, uncompensated, and monitored continuously during recording. Cells with a change in series resistance beyond 20% were not accepted for data analysis. Synaptic currents were recorded with a MultiClamp 700B amplifier (Molecular Devices), filtered at 3 kHz, amplified 5 times, and then digitized at 20 kHz.
In vivo Optogenetic Procedures
Rats were transferred to a clean standard housing cage. Bilateral optic fiber implants were connected to an optic fiber patch cord, which was connected to a 473-nm blue laser diode (IkeCool) via a rotary joint (Prismatix). The light intensity through the patch cord, which was measured by a light sensor (S130A; Thor Labs), was adjusted to ~5–7 mW. Rats were allowed to explore the environment for 3 min prior to LTD induction. LTD was induced using the paradigm described above (900 2-ms pulses of 473-nm light delivered at 1 Hz). After induction rats remained in the cage for 3 min, before being placed back in their home cages. Control rats either expressed the AAV5-tdTomato control virus and received the same stimulation procedure, or expressed oChIEF and had a sham optic fiber patch cord attached to the head-mounted optic fiber for the same duration as the LTD induction. 24 hours after in vivo optogenetic stimulations, rats were assessed for drug-seeking in a standard cue-induced reinstatement session (See above).
Staining, Fluorescence, and Imaging
Animals were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.). Rats were perfused through the aorta with 1X PBS for 5 min followed by 4% paraformaldehyde in 1X PBS, pH 7.4 for 10 min. The brains were extracted, postfixed in 4% paraformaldehyde for 24 h, and transferred to 30% sucrose solution. Brains were sectioned at 50 μm using a cryostat (Leica). Slices containing the LA or MGN were mounted onto glass slides, and coverslipped with Fluoroshield with DAPI (for nuclear identification) mounting media (Sigma-Aldrich). Slices were imaged using an Olympus BX61VS epifluorescent slide-scanning microscope to verify AAV-oChIEF-tdTomato expression in the MGN and its projections to the LA (See Figure S6). Additionally, position of the optic fiber over the LA was verified. Rats lacking expression of AAV in MGN or LA and those in which the optic fiber was not correctly positioned were removed from the study.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed using GraphPadPrism for Windows and results are expressed as mean ± SEM. Rats were distributed into groups based on a matching procedure that ensured that each group had no statistical differences in their cocaine infusions acquired over days, or differences in instrumental extinction behavior. For behavioral experiments, reinstatement tests were analyzed by two-way ANOVA with repeated-measures, with the within-subjects factor being responding on the last day of instrumental extinction versus reinstatement responding and the between-subjects factor being cue re-exposure condition. For electrophysiological experiments, data were coded such that experimenters were not aware of treatment groups when performing data analysis, and then decoded for final results. It was not always possible for experimenters to be blind to condition during electrophysiology recordings, but a standard protocol was used for all recording sessions. Data were analyzed offline using ClampFit 10.3. For experiments in which the end points were from individual cells, such as EPSCs, we used the averaged value of a parameter from all cells recorded from an animal to represent the parameter of this animal. For electrical stimulation experiments EPSC amplitude was calculated at each stimulation intensity and compared between groups using two-way ANOVA with repeated-measures, with the within-subjects factor being stimulation intensity and the between-subjects factor being cue re-exposure condition. For correlation analyses, Pearson’s correlation coefficients were calculated, with number of infusions as the independent variable and EPSC amplitude as the dependent variable. Paired pulse ratio (PPR) was calculated as the ratio of the peak current of the second EPSC to the first EPSC. AMPAR current was calculated as the peak current at Erev −70mV and NMDAR current was calculated as the peak current at Erev+40 mV, 35 ms after peak AMPAR current. For AMPA, NMDA, and PPR, comparisons were made using one-way ANOVA. All data points were an average of 10 trials. For optical stimulation experiments, EPSC amplitude, PPR, and AMPA:NMDA ratios were calculated as described above and compared using one-way ANOVA or unpaired t test. For LTD experiments, peak EPSP amplitude and EPSP rise slope were calculated for every trial and six consecutive trials were averaged together for each data point. For comparisons of pre- and post-LTD comparisons, data points across the last 7 minutes of baseline were compared to the last 7 minutes of post-LTD recordings with a paired t test. Each experiment was replicated in at least 5–6 rats (1–5 cells were recorded from each rat) for electrophysiological analysis and at least 6 rats for behavioral tests. For all analyses, significant effects were further analyzed by Tukey’s or Bonferroni’s post hoc tests, with significance set at p < 0.05. All data were determined to be normally distributed using the Shapiro-Wilk test, and Bartlett’s test was used to determine that there were no significant differences in the estimated variance between groups. Statistical parameters for each analysis can be found in the corresponding figure legends.
Supplementary Material
Highlights.
Cocaine-cue memory formation strengthens thalamoamygdala, not cortico-amygdala synapses
Cocaine-cue extinction reduces relapse and depotentiates thalamo-amygdala synapses
Medial geniculate thalamic (MGN) projections to amygdala (LA) regulate cocaine memory
Optogenetic LTD at MGN-LA synapses is sufficient to block cocaine-cue-induced relapse
ACKNOWLEDGMENTS
We would like to acknowledge T. Cahanap, A. Miller, and E. Compagnoni for assistance with behavioral experiments and Y. Wang, R. Guo, and Z. Liu for technical advice. The research was supported by USPHS grants K01DA031745 (to M.M.T.), R01DA042029 (to M.M.T.), R01DA035805 (to Y.H.H.), F31DA039646 (to M.T.R.), and T32031111 (to M.T.R.) and the Pennsylvania Department of Health.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Amano T, Unal CT, and Paré D (2010). Synaptic correlates of fear extinction in the amygdala. Nat. Neurosci 13, 489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An B, Kim J, Park K, Lee S, Song S, and Choi S (2017). Amount of fear extinction changes its underlying mechanisms. eLife 6, e25224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello AA, Hodges MA, Wells AM, Lara H 3rd, Xie X, and Fuchs RA (2014). Involvement of amygdalar protein kinase A, but not calcium/calmodulin-dependent protein kinase II, in the reconsolidation of cocaine-related contextual memories in rats. Psychopharmacology (Berl.) 231, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augur IF, Wyckoff AR, Aston-Jones G, Kalivas PW, and Peters J (2016). Chemogenetic Activation of an Extinction Neural Circuit Reduces Cue-Induced Reinstatement of Cocaine Seeking. J. Neurosci 36, 10174–10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carelli RM, Williams JG, and Hollander JA (2003). Basolateral amygdala neurons encode cocaine self-administration and cocaine-associated cues. J. Neurosci 23, 8204–8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhatwal JP, Myers KM, Ressler KJ, and Davis M (2005). Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J. Neurosci 25, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz FC, Babin KR, Leao RM, Goldart EM, Bossert JM, Shaham Y, and Hope BT (2014). Role of nucleus accumbens shell neuronal ensembles in context-induced reinstatement of cocaine-seeking. J. Neurosci 34, 7437–7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton GL, Wang YT, Floresco SB, and Phillips AG (2008). Disruption of AMPA receptor endocytosis impairs the extinction, but not acquisition of learned fear. Neuropsychopharmacology 33, 2416–2426. [DOI] [PubMed] [Google Scholar]
- Do-Monte FH, Minier-Toribio A, Quiñones-Laracuente K, Medina-Colón EM, and Quirk GJ (2017). Thalamic Regulation of Sucrose Seeking during Unexpected Reward Omission. Neuron 94, 388–400.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron NN, and Ledoux JE (2000). Cells in the posterior thalamus project to both amygdala and temporal cortex: a quantitative retrograde double-labeling study in the rat. J. Comp. Neurol 425, 257–274. [PubMed] [Google Scholar]
- Ferrara NC, Cullen PK, Pullins SP, Rotondo EK, and Helmstetter FJ (2017). Input from the medial geniculate nucleus modulates amygdala encoding of fear memory discrimination. Learn. Mem 24, 414–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs RA, Feltenstein MW, and See RE (2006). The role of the basolateral amygdala in stimulus-reward memory and extinction memory consolidation and in subsequent conditioned cued reinstatement of cocaine seeking. Eur. J. Neurosci 23, 2809–2813. [DOI] [PubMed] [Google Scholar]
- Hong I, Song B, Lee S, Kim J, Kim J, and Choi S (2009). Extinction of cued fear memory involves a distinct form of depotentiation at cortical input synapses onto the lateral amygdala. Eur. J. Neurosci 30, 2089–2099. [DOI] [PubMed] [Google Scholar]
- Hsiang H-L, Epp JR, van den Oever MC, Yan C, Rashid AJ, Insel N, Ye L, Niibori Y, Deisseroth K, Frankland PW, and Josselyn SA (2014). Manipulating a ‘‘cocaine engram’’ in mice. J. Neurosci 34, 14115–14127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Brown TE, Han MH, Saal DB, Neve RL, Zukin RS, Sorg BA, Nestler EJ, Malenka RC, and Dong Y (2008). CREB modulates the functional output of nucleus accumbens neurons: a critical role of N-methyl-D-aspartate glutamate receptor (NMDAR) receptors. J. Biol. Chem 283, 2751–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, Marie H, Liu W, Yan Z, Sorg BA, et al. (2009). In vivo cocaine experience generates silent synapses. Neuron 63, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Volkow N, and Seamans J (2005). Unmanageable motivation in addiction: a pathology in prefrontal-accumbens glutamate transmission. Neuron 45, 647–650. [DOI] [PubMed] [Google Scholar]
- Kim J, Lee S, Park K, Hong I, Song B, Son G, Park H, Kim WR, Park E, Choe HK, et al. (2007a). Amygdala depotentiation and fear extinction. Proc. Natl. Acad. Sci. USA 104, 20955–20960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Lee S, Park H, Song B, Hong I, Geum D, Shin K, and Choi S (2007b). Blockade of amygdala metabotropic glutamate receptor subtype 1 impairs fear extinction. Biochem. Biophys. Res. Commun 355, 188–193. [DOI] [PubMed] [Google Scholar]
- Kwon J-T, Nakajima R, Kim H-S, Jeong Y, Augustine GJ, and Han J-H (2014). Optogenetic activation of presynaptic inputs in lateral amygdala forms associative fear memory. Learn. Mem 21, 627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE, Farb C, and Ruggiero DA (1990). Topographic organization of neurons in the acoustic thalamus that project to the amygdala. J. Neurosci 10, 1043–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zeric T, Kambhampati S, Bossert JM, and Shaham Y (2015). The central amygdala nucleus is critical for incubation of methamphetamine craving. Neuropsychopharmacology 40, 1297–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Lin MZ, Steinbach P, and Tsien RY (2009). Characterization of engineered channelrhodopsin variants with improved properties and kinetics. Biophys. J 96, 1803–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Hope BT, Dempsey J, Liu SY, Bossert JM, and Shaham Y (2005). Central amygdala ERK signaling pathway is critical to incubation of cocaine craving. Nat. Neurosci 8, 212–219. [DOI] [PubMed] [Google Scholar]
- McDonald AJ (1998). Cortical pathways to the mammalian amygdala. Prog. Neurobiol 55, 257–332. [DOI] [PubMed] [Google Scholar]
- Merlo E, Milton AL, Gooze´ e ZY, Theobald DE, and Everitt BJ (2014). Reconsolidation and extinction are dissociable and mutually exclusive processes: behavioral and molecular evidence. J. Neurosci 34, 2422–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milad MRR, and Quirk GJJ (2002). Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420, 70–74. [DOI] [PubMed] [Google Scholar]
- Nabavi S, Fox R, Proulx CD, Lin JY, Tsien RY, and Malinow R (2014). Engineering a memory with LTD and LTP. Nature 511, 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, and Pierce RC (2002). Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J. Neurosci 22, 2916–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzo MA, Robert V, Tucciarone J, De Bundel D, Wang M, Van Aelst L, Darvas M, Parada LF, Palmiter RD, He M, et al. (2015). The paraventricular thalamus controls a central amygdala fear circuit. Nature 519, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, LaLumiere RT, and Kalivas PW (2008). Infralimbic prefrontal cortex is responsible for inhibiting cocaine seeking in extinguished rats. J. Neurosci 28, 6046–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA (2004). Spontaneous recovery varies inversely with the training-extinction interval. Learn. Behav 32, 401–408. [DOI] [PubMed] [Google Scholar]
- Rich MT, and Torregrossa MM (2018). Molecular and synaptic mechanisms regulating drug-associated memories: Towards a bidirectional treatment strategy. Brain Res. Bull 141, 58–71. [DOI] [PubMed] [Google Scholar]
- Rich MT, Abbott TB, Chung L, Gulcicek EE, Stone KL, Colangelo CM, Lam TT, Nairn AC, Taylor JR, and Torregrossa MM (2016). Phos- phoproteomic Analysis Reveals a Novel Mechanism of CaMKIIa Regulation Inversely Induced by Cocaine Memory Extinction versus Reconsolidation. J. Neurosci 36, 7613–7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Ersche KD, and Everitt BJ (2008). Drug addiction and the memory systems of the brain. Ann. N Y Acad. Sci 1141, 1–21. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, and Grace AA (2001). Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J. Neurosci 21, 4090–4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabel SJ, and Janak PH (2009). Substantial similarity in amygdala neuronal activity during conditioned appetitive and aversive emotional arousal. Proc. Natl. Acad. Sci. USA 106, 15031–15036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparta DR, Stamatakis AM, Phillips JL, Hovelsø N, van Zessen R, and Stuber GD (2011). Construction of implantable optical fibers for long-term optogenetic manipulation of neural circuits. Nat. Protoc 7, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanik MT, Kupchik YM, and Kalivas PW (2016). Optogenetic inhibition of cortical afferents in the nucleus accumbens simultaneously prevents cue-induced transient synaptic potentiation and cocaine-seeking behavior. Brain Struct. Funct 221, 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, and Feng G (2014). Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods Mol. Biol 1183, 221–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrossa MM, and Taylor JR (2013). Learning to forget: manipulating extinction and reconsolidation processes to treat addiction. Psychopharmacology (Berl.) 226, 659–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torregrossa MM, Gordon J, and Taylor JR (2013). Double dissociation between the anterior cingulate cortex and nucleus accumbens core in encoding the context versus the content of pavlovian cocaine cue extinction. J. Neurosci 33, 8370–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Stuber GD, de Ridder B, Bonci A, and Janak PH (2008). Rapid strengthening of thalamo-amygdala synapses mediates cue-reward learning. Nature 453, 1253–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Tye LD, Cone JJ, Hekkelman EF, Janak PH, and Bonci A (2010). Methylphenidate facilitates learning-induced amygdala plasticity. Nat. Neurosci 13, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vertes RP, Linley SB, and Hoover WB (2015). Limbic circuitry of the midline thalamus. Neurosci. Biobehav. Rev 54, 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.