

Abstract
Eukaryotic translation initiation factor 4E (eIF4E) is a key player in the initiation of cap-dependent translation through recognition of the m7GpppX cap at the 5’ terminus of coding mRNAs. As eIF4E overexpression has been observed in a number of human diseases, most notably cancer, targeting this oncogenic translation initiation factor has emerged as a promising strategy for the development of novel anti-cancer therapeutics. Toward this end, in the present study, we have rationally designed a series of Bn7GxP-based PROTACs for the targeted degradation of eIF4E. Herein we describe our synthetic efforts, in addition to biochemical and cellular characterization of these compounds.
Keywords: eIF4E, PROTACs, m7G cap, cancer, proteasomal degradation
Graphical Abstract
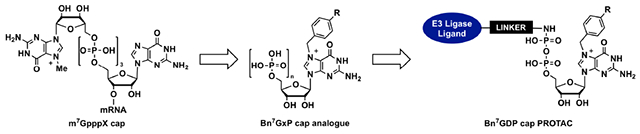
1. INTRODUCTION
Dysregulation of cap-dependent translation is a hallmark of many cancers [1,2,3]. Initiation of cap-dependent translation is dependent upon recognition of the m7GpppX cap (Figure 1A) at the 5’-terminus of a mRNA by eukaryotic translation initiation factor 4E (eIF4E) [4]. Binding of eIF4E to the mRNA cap results in recruitment of the translation machinery, which then initiates protein synthesis at the transcript’s start codon. eIF4E plays an essential role in controlling cellular growth and proliferation [5], and overexpression of this translation initiation factor is observed in many malignant cell lines and primary tumors, including breast [6], head and neck [7], and non-Hodgkin’s lymphomas [8]. In previous literature reports, it has been shown that inhibition of eIF4E function slows tumor growth and induces apoptosis [9]. Thus, development of selective eIF4E inhibitors or degradation-inducing molecules could be a promising approach for the treatment of many cancers, in addition to providing valuable chemical tools for studying cap-dependent translation.
Figure 1.
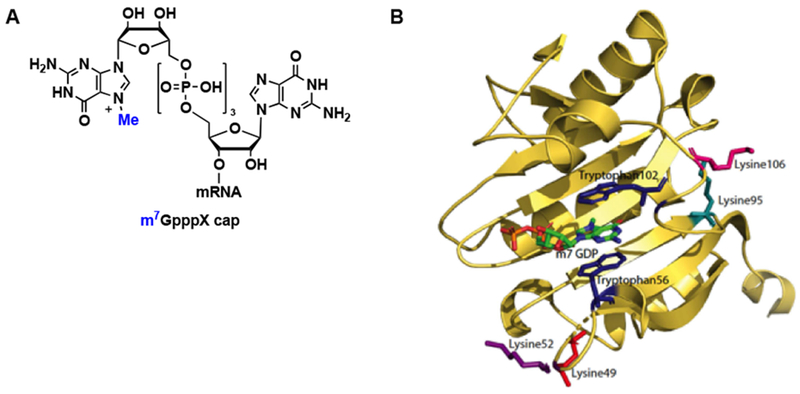
eIF4E biological activity and structure. (A) Structure of the m7GpppX cap. (B) Crystal structure of eIF4E bound to the m7GpppX cap (PDB: 1EJ1). The key tryptophan residues for cap binding are highlighted, in addition to lysines close to the cap-binding pocket.
Molecular recognition of the m7GpppX cap by eIF4E involves cation-π interactions, in which Trp56 and Trp102 of eIF4E sandwich the positively-charged methylated guanine nucleobase of the cap (Figure 1B) [10]. This interaction is of utmost significance, as the binding affinity of m7GTP has been shown to be ~100-fold tighter than that of GTP itself (Kds of 0.15 and 14 μM, respectively) [10]. Based on the demonstrated importance of eIF4E in cellular and disease biology [3,11], many structure-activity relationship (SAR) campaigns have been carried out to rationally design m7G-based competitive inhibitors of eIF4E binding to capped mRNA (Figure 2) [12,13,14,15] These studies have revealed that analogues containing benzyl or substituted benzyl groups at the 7-position (~10-fold enhancement in binding affinity) and at least one phosphate group are optimal inhibitors of eIF4E-cap binding [16,17,18]. In addition, efforts have been made to improve the cellular permeability of cap analogues through various pro-drug strategies (4Ei-1,2,3 in Figure 2) [19,20,21].
Figure 2.
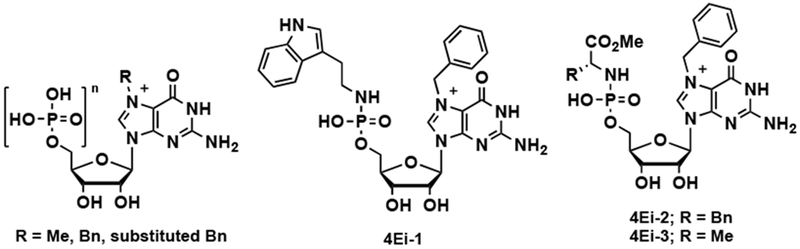
Structures of previously reported m7G cap analogues [21].
Recently, PROteolysis-TArgeting Chimeras (PROTACs) [22,23] have emerged as a powerful alternative to traditional small molecule-based drug discovery approaches in that these bifunctional molecules can trigger intracellular protein degradation by marking proteins for proteosomal digestion [24,25,26,27,28]. In short, a small molecule ligand for the protein-of-interest is conjugated to an E3 ubiquitin ligase ligand, such as lenalidomide or VHL peptide, generating a novel two warhead probe [29] to enable the directed ubiquitination, and subsequent degradation, of the target protein [30]. To date, this strategy has been successfully extended to BET family [25,31,32,33], Sirtuin 2 [34], CDK9 [35], Skp1-cullin-F-box [36], BCR-ABL [37] and ERRα proteins [38]. Thus, based on the previous successes of m7G cap analogues as inhibitors of eIF4E, we became interested in developing PROTACs based upon this scaffold for the targeted proteasomal degradation of eIF4E [14,39,40]. To this end, 4Ei-1 has been previously shown to induce eIF4E proteasomal degradation in a dose-dependent manner; however, high concentrations (100-500 μM) of the compound were required to see an effect [41]. By using a PROTAC strategy, our goal was to achieve this biological effect with lower concentrations due to the catalytic nature of these conjugates, thereby, providing in-roads toward the development of these molecules for therapeutic applications. Herein we disclose our synthetic efforts toward cap-competitive eIF4E-targeted PROTACs and their preliminary in vitro and cellular characterization.
2. RESULTS AND DISCUSSION
2.1. Assessing eIF4E as a Candidate for PROTAC Development
Little is known about the endogenous turnover of the eIF4E protein [42,43,44]. To date, only the C-terminus of Hsc-70 interacting protein (CHIP) and cellular inhibitor of apoptosis protein-1 (cIAP1) E3 ligases have been implicated in eIF4E ubiquitination and degradation under conditions of cellular stress [43,44]. eIF4E contains 16 total lysine residues, 13 of which have been shown to be solvent accessible via X-ray crystallography (Lys21, 36, 49, 52, 54, 65, 106, 108, 119, 159, 162, 192, 212) [45] for possible ubiquitination. Notably, several of these residues are within 15 Å of the cap-binding pocket (Figure 1B) [45], which is an essential requirement for the use of PROTAC technology [46]. Thus, we were encouraged that eIF4E degradation could be induced with appropriate Bn7GxP-based PROTAC design.
2.2. Design and Synthesis of GMP-based PROTACs
As an initial PROTAC design strategy, we generated a small library of guanosine monophosphate (GMP) derivatives conjugated to lenalidomide (1), a ligand for the well-known E3 ligase, cereblon [47,48]. GMP was chosen as the Bn7GxP scaffold based on its successful use in previously reported inhibitors of eIF4E (Figure 2) [21,39,40,49]. The synthetic route is shown in Scheme 1 and began with HATU-mediated coupling of 1 with Boc-6-aminohexanoic acid to form Boc-protected amide 2 in 89% yield, followed by Boc deprotection in quantitative yield. The unmasked free amine 3 was then subjected to coupling with GMP disodium salt to provide GMP-lenalidomide conjugate 4 in 31% yield [18,50]. The low reaction yield for this step was due to solvent incompatibility between the hydrophobic lenalidomide derivative 3 and hydrophilic GMP. Alkylation at the 7-position with benzyl bromide or 4-fluorobenzyl bromide [18,51] yielded Bn7GMP PROTACs 5a and 5b in 4% and 29% yields, respectively. 4-Fluorobenzyl substitution was also examined, as this modification was previously shown to further enhance binding affinity to eIF4E [10]. Again, yields were low due to poor solubility.
Scheme 1.
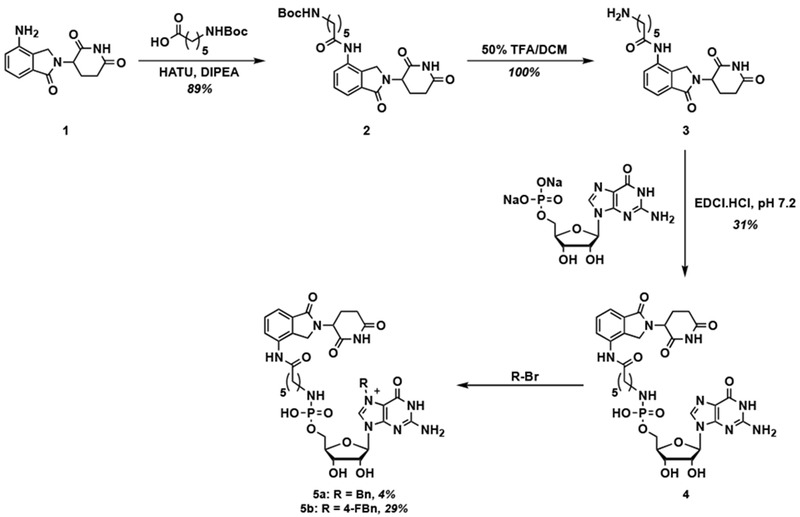
Synthesis of Bn7GMP-based lenalidomide conjugates.
2.3. Design and Synthesis of Bioisoteres of GMP-based PROTACs
Aryl sulfamates have previously been reported as bioisosteres of phosphate groups [51]. Thus, we were also interested in the synthesis of such non-hydrolyzable Bn7GMP-based PROTACs. The synthesis, which is shown in Scheme 2, began with TBS protection of guanosine to yield 6 in 70% yield. Sulfamoylation at the 5’ position was then carried out using sulfamoyl chloride [51] generated in situ from formic acid and chlorosulfonyl isocyanate [51] to yield 7 in 95% yield. Coupling with N-hydroxysuccinic (NHS) ester 8 in the presence of DBU, followed by deprotection of the tert-butyl protecting group resulted in 10. Interestingly, conjugation of the free acid of this compound with lenalidomide (1) under HATU- or PyBrOP-mediated coupling conditions did not yield the desired product. Since we speculated that this may be due to the weak basicity of 1, we utilized the lenalidomide derivative 3, which resulted in the formation of 11 in 40% yield. Subsequent deprotection of the TBS groups with TBAF and alkylation at the 7-position produced the target PROTACs 13a and 13b in 45% and 55% yields, respectively. Yields for the alkylation of sulfamate derivative 12 were better than that of the corresponding GMP due to its enhanced solubility in DMSO.
Scheme 2.
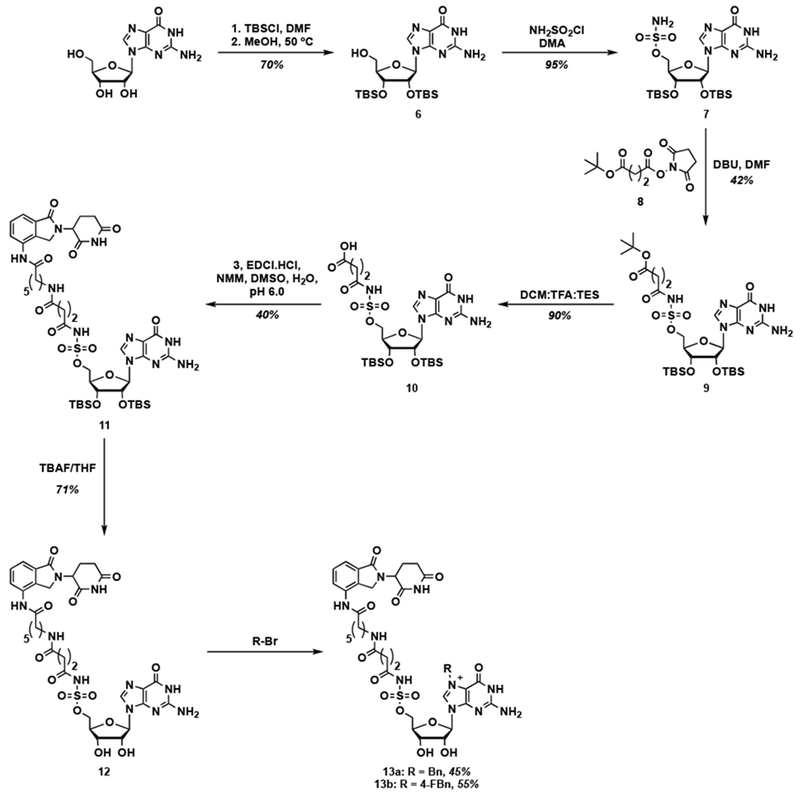
Synthesis of sulfamate-containing Bn7GMP-based lenalidomide conjugates.
2.4. Biochemical Characterization of GMP-based PROTACs
To test the ability of the Bn7GMP-based PROTACs to bind to eIF4E and compete with the m7GpppX cap, we developed an in vitro cap competition assay (Figure 3A). In this assay, cell lysate is first incubated with m7GxP (GMP or GDP) agarose resin [52] to enable the affinity purification of eIF4E. The preparation of this resin is shown in Figure 3B and uses aniline-catalyzed reductive amination chemistry for chemical conjugation. Resin-immobilized eIF4E is then incubated with compounds, such as m7GxP or a PROTAC. If the compound is active, eIF4E will be competed off the resin; however, if there is no inhibitory activity, eIF4E will be retained and detected by Western blot analysis.
Figure 3.
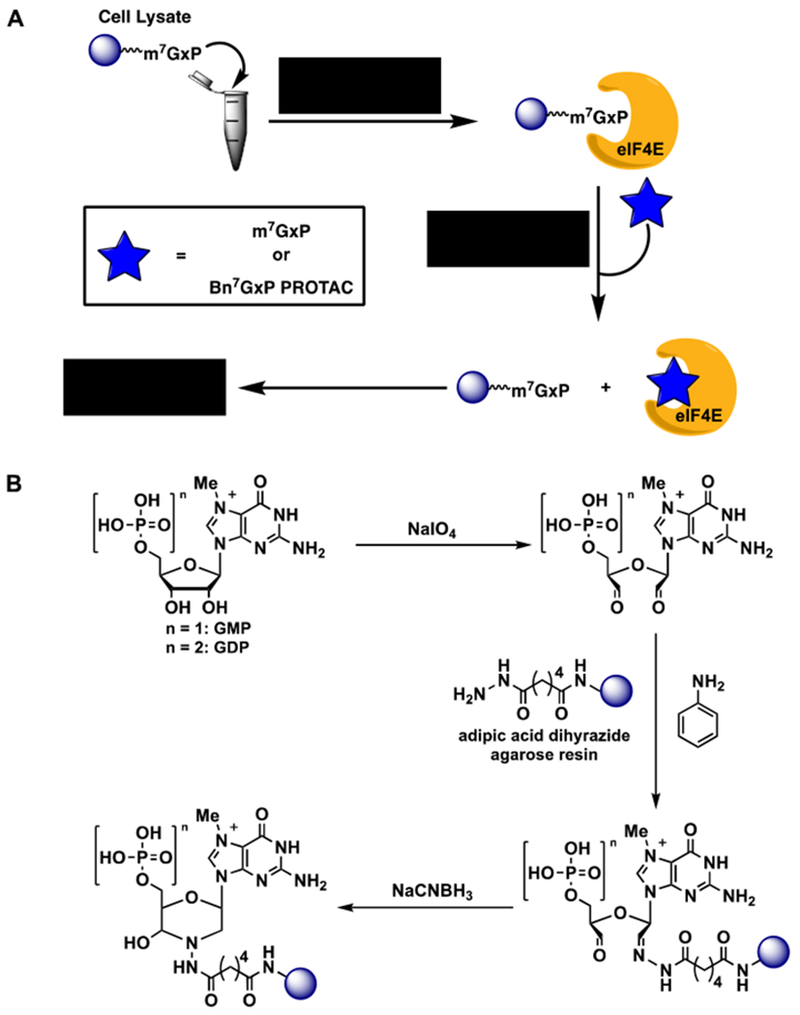
Cap-competition assay. (A) Assay overview. (B) Synthesis of m7GxP agarose resin.
Each of the synthesized Bn7GMP-based PROTACs (5a, 5b, 13a, 13b), in addition to the GMP-lenalidomide conjugates 4 and 12, were tested in the m7GMP cap competition assay using m7GMP as a positive control. As shown in Figure 4, upon treatment of eIF4E immobilized from HEK293 cell lysate, negligible inhibition was observed with either the Bn7GMP- or sulfamate-based PROTACs in comparison to the control. No inhibition with the GMP derivatives was observed as expected. We hypothesize that this is due to the decreased or absent negative charge on the phosphoramide and sulfamate moieties, respectively, as a similar effect has been observed with other GxP-binding proteins [53].
Figure 4.

Western blot analysis of eIF4E following the m7GMP cap competition assay. Immobilized eIF4E was treated with 500 μM of m7GMP or a GMP- or Bn7GMP-based PROTAC. DMSO was used as a negative control.
2.5. Design and Synthesis of GDP-based PROTACs
As m7GDP has been shown to exhibit 50-fold greater inhibitory activity against eIF4E-cap binding as compared to m7GMP [17], we next synthesized Bn7GDP-based PROTACs with hopes that they would exhibit activity in the cap competition assay. In this case, we chose to incorporate both lenalidomide and VHL ligand conjugates as part of our PROTAC design, in addition to varying linker lengths, which has been shown to play a crucial role in the success of this approach [26],[54].
For the lenalidomide conjugates, we followed a similar synthetic route to those used for the GMP compounds (Scheme 3). The synthesis began with HATU-mediated coupling of lenalidomide (1) with alkyl and PEG linkers to form Boc-protected amides 14–16 in 18-89% yields. In case of the PEG7 analogue 16, we suspect that the observed low yield was due to its aggregation propensity. Following deprotection, the unmasked free amines 17–19 were subjected to EDCI-mediated coupling with the sodium salt of GDP to yield conjugates 20–22 in 32-35% yields [18,50]. Alkylation was performed with benzyl or 4-fluorobenzyl bromide [18,51], and resulted in Bn7GDP-based PROTACs 23–25 in 9-20% yields. Low yields were observed due to both poor solubility and minor hydrolysis of GDP.
Scheme 3.
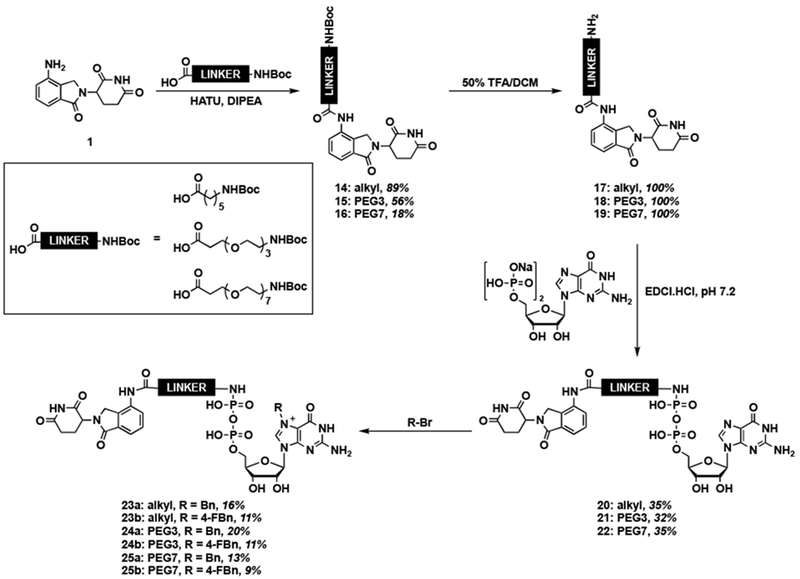
Synthesis of Bn7GDP-based lenalidomide conjugates.
The VHL ligand (26) was conjugated to GDP through a similar synthetic route (Scheme 4). In this case, however, only the alkyl linker was used. HATU-mediated coupling of 26 with Boc-6-aminohexanoic acid to yield Boc-protected amide 27 in 75% yield. After deprotection, amine 28 was conjugated to GDP via known EDC coupling [21], which produced GDP-VHL conjugate 29 in 32% yield. Subsequent alkylation [18,51] then generated the target PROTACs 30a and 30b in 11% and 41% yields, respectively. Similar challenges with solubility and stability were encountered.
Scheme 4.
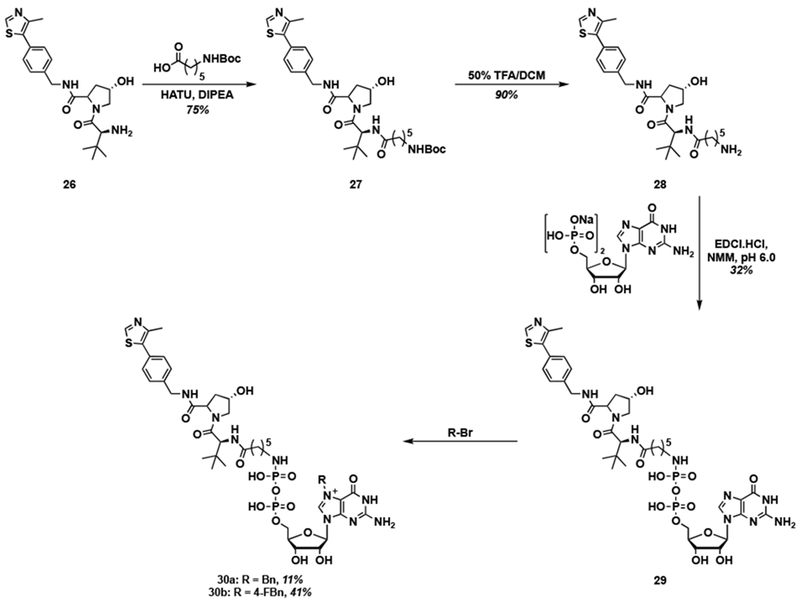
Synthesis of Bn7GDP-based VHL conjugates.
2.6. Biochemical Characterization of GDP-based PROTACs:
Each of the synthesized GDP- (20–22 and 29) and Bn7GDP-based PROTACs (23–25 and 30) was tested for eIF4E inhibitory activity in the m7GDP cap competition assay (Figure 3A), in this case, using m7GDP as a positive control. To our delight, each of the Bn7GDP conjugates was found to be active (Figure 5A), while the GDP derivatives showed no activity as expected. Within the alkyl linker series (Figure 5A, top), no observable inhibitory activity differences were observed between the lenalidomide and VHL conjugates. Interestingly, however, the PEG3 lenalidomide (21, 24a-b) PROTAC showed enhanced activity in comparison the PEG7 analogues (22, 25a-b) (Figure 5A, bottom). Inhibition of eIF4E-cap binding was also found to be dose-dependent for each of the PROTACs, and representative data for 23a is shown in Figure 5B.
Figure 5.
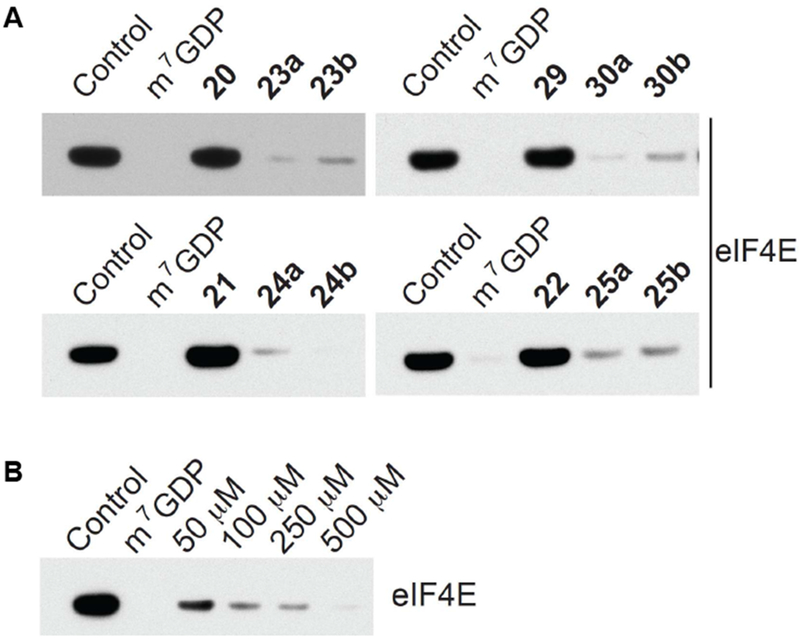
Western blot analysis of eIF4E following the m7GDP cap competition assay in HEK293 cell lysate. (A) Immobilized eIF4E was treated with 500 μM of m7GDP or a GDP- or Bn7GDP-based PROTAC. DMSO was used as a negative control. (B) Dose-dependent inhibition by 23a.
2.7. Cellular Characterization of GDP-based PROTACs:
Encouraged by the promising biochemical data obtained with the Bn7GDP-based PROTACs, we proceeded to cellular assays to determine if these compounds were able to induce the proteasomal degradation of eIF4E. MDA-MB-231 breast cancer cells were treated with varying concentrations (25–200 μM) of PROTACs for 24–72 h. Unfortunately, decreased eIF4E protein levels were not detected with any of the conjugates (data not shown). Because we suspected that low cellular permeability may be a problem, we next tested the PROTACs in K562 cells, which have been found to more readily uptake purine nucleotides [55]. Across a similar set of conditions, again we saw no intracellular degradation of eIF4E (representative data in Figure 6). Thus, we hypothesize that the permeability of these compounds is still low, as has been observed with other cap analogues[14,18,21] or that ternary complex formation between eIF4E and the E3 ligase is not possible with the designed molecules. Future efforts should be focused on examining additional linkers, as this has been shown to play a crucial role in successful PROTAC design, in addition to the exploration of additional E3 ligase ligands, such as those for cIAP and MDM2 [26, 54].
Figure 6.
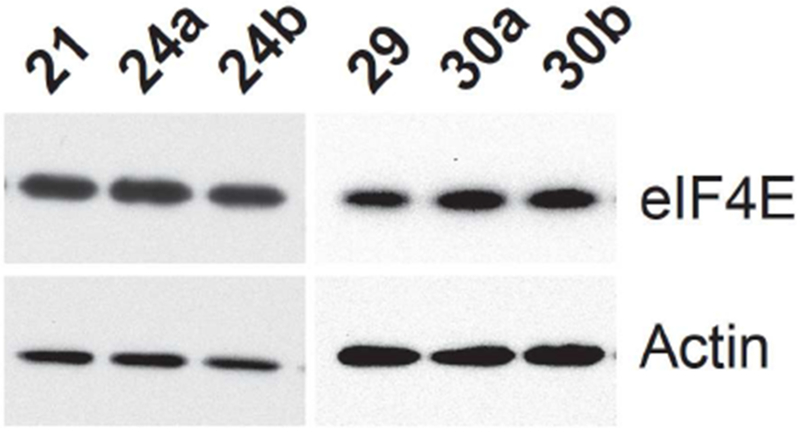
Western blot of total eIF4E following the treatment of K562 cells with Bn7GDP-based PROTACs. Actin was used as a loading control.
3. CONCLUSION
In conclusion, we have synthesized a library of Bn7GxP-based PROTACs for the targeted degradation of eIF4E, the m7GpppX cap-binding translation initiation factor. The targeting of aberrant eIF4E activity with m7G cap analogues is rich with history [2,11,13], yet continues to be plagued with issues of cellular permeability due the necessity for nucleotide-based scaffolds [18,39,40]. Unfortunately, our efforts described herein were similarly unsuccessful in translating to cellular assays. While pro-drugs have been developed (Figure 2), the requirement for high micromolar concentrations in cells dampens enthusiasm for clinical development [19,20, 41]. Thus, future efforts in targeting eIF4E should focus on the discovery of non-nucleotide small molecule competitive antagonists of cap binding or inhibiting its protein-protein interactions, which are also crucial for its activity in initiating cap-dependent translation [1,3].
4. EXPERIMENTAL
4.1. General Materials and Methods:
Lenalidomide (1) was purchased from AstaTech and used as received. VHL ligand (26) was purchased from Abosyn and used as received. GMP and GDP were purchased from ChemImpex and used as received. All solvents and reagents were used without further purification. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials, unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25-mm SiliCycle silica gel plates (60F-254) using UV-light (254 nm). Flash chromatography was performed using SiliaFlash P60 silica gel. Analytical RP-HPLC was performed using an Agilent 1260 Infinity HPLC equipped with a ZORBAX Eclipse XDB-C18 column (4.6 × 150 mm; 5 μm) at a flow rate of 1 mL/min, with detection at 214 and 254 nm. Semi-preparative HPLC was carried out on Agilent 1260 Infinity HPLC equipped with a PrepHT XDB-C18 column (21.2 × 150 mm; 5 μm) at a flow rate of 18 mL/min using 95% water as a mobile phase and detection at 254 nm and 214 nm. NMR spectra were performed on a 300 MHz Bruker instrument and calibrated using a solvent peak as an internal reference. Spectra were processed using MestReNova software. Mass spectrometry (HRMS) was performed using an Agilent 6520 Accurate-Mass Q-TOF LC/MS spectrometer using ESI ionization with less than 5-ppm error for all HRMS analyses.
4.2. General procedure A:
Boc-protected acids (1.1 mmol, 1.1 equiv) were added to a stirring solution of lenalidomide/VHL ligand (1.0 equiv), HATU (1.2 equiv) and DIPEA (2.0 equiv) in dry DMF (0.22 M) at 25 °C. The reaction mixture was allowed to stir at 25 °C for 12 h. Upon completion of the reaction as determined via TLC, the mixture was acidified with dilute HCl (5%, 5 mL) and extracted with EtOAc (3×). The organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure in vacuo. The crude reaction mixture was purified by silica gel chromatography using EtOAc in hexanes (30–100% EtOAc) to yield the desired products as a white solid in 18–56% yields.
4.3. General procedure B:
To an ice-cold stirring solution of Boc-protected derivatives (1 equiv) in DCM (5 mL) was added trifluoroacetic acid (TFA) (5 mL). The reaction mixture was warmed to 25 °C and stirred for an additional 2 h. Upon completion of the reaction, the solvent was evaporated under reduced pressure in vacuo. The crude product was dried further under high vacuum for 4 h and was used directly for the next step.
4.4. General procedure C:
To a stirring solution of GMP or GDP sodium salt (1.0 equiv) in water at pH 7.2 (0.06 M) was added N-methyl morpholine (10 equiv), EDCI. HCl (5 equiv), and free amine derivatives (1.0 equiv) in DMSO (0.06 M). The reaction mixture was stirred overnight at 25 °C. Upon completion, the mixture was diluted with water and purified by semi-preparative HPLC (2–20% acetonitrile in water). Product-containing fractions were pooled and lyophilized to dryness to yield the desired products as white solids.
4.5. General procedure D:
To a stirring solution of guanosine analogues (1.0 equiv) in dry DMSO (0.006 M) was added alkyl bromide (10 equiv). The reaction mixture was stirred overnight at 25 °C. Upon completion, the mixture was diluted with water and extracted with diethyl ether (3×). The combined water layers were lyophilized and purified by semi-preparative HPLC (2–40% acetonitrile in water). Product containing fractions were pooled and lyophilized to dryness to yield the Bn7GxP analogues as white solids.
4.6. Compound characterization:
tert-Butyl(6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxohexyl) carbamate (2).
General procedure A, 89% yield. Rf 0.40 (EtOAc). 1H NMR (500 MHz, DMSO-d6) δ 11.01 (s, 1H), 9.78 (s, 1H), 7.81 (d, J = 7.2 Hz, 1H), 7.47 (d, J = 11.7 Hz, 2H), 6.75 (d, J = 5.4 Hz, 1H), 5.13 (d, J = 18.4 Hz, 1H), 4.43–4.27 (m, 2H), 2.89 (d, J = 24.4 Hz, 4H), 2.35 (s, 3H), 2.01 (dd, J = 9.7, 4.5 Hz, 1H), 1.58 (d, J = 14.8 Hz, 2H), 1.42–1.36 (m, 2H), 1.34 (s, 9H), 1.27 (p, J = 7.5, 6.9 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 173.28, 171.78, 171.49, 168.29, 162.74, 156.03, 134.26, 134.12, 133.09, 129.02, 125.67, 119.39, 77.75, 52.00, 46.97, 40.44, 40.22, 36.22, 31.66, 29.76, 28.71, 26.45, 25.30, 23.09. HRMS (ESI-TOF) m/z calcd for C24H32N4O6 [M + H]+ 473.2400, found 473.2421.
6-Amino-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)hexanamide (3).
General procedure B, 100% yield. Rf 0.2 (EtOAc). HRMS (ESI-TOF) m/z calcd for C19H24N4O4 [M + H]+ 373.1876, found 373.1882.
((2S,3R,4S,5S)-5-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methylhydrogen(6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxohexyl)phosphoramidate (4).
General procedure C, 31% yield. 1H NMR (500 MHz, D2O) δ 7.94 (t, J = 3.5 Hz, 1H), 7.57–7.52 (m, 1H), 7.46–7.34 (m, 2H), 5.73–5.63 (m, 1H), 5.06–4.96 (m, 1H), 4.57–4.49 (m, 1H), 4.35–4.29 (m, 1H), 4.25–4.13 (m, 3H), 3.93–3.82 (m, 2H), 3.01 (d, J = 50.7 Hz, 5H), 2.87–2.75 (m, 2H), 2.61 (d, J = 3.2 Hz, 2H), 2.44–2.34 (m, 1H), 2.26–2.16 (m, 3H), 1.75 (d, J = 6.9 Hz, 2H), 1.41 (d, J = 5.4 Hz, 2H), 1.30 (d, J = 3.2 Hz, 2H), 1.19–1.00 (m, 3H), 0.93 (d, J = 17.5 Hz, 2H). 31P NMR (202 MHz, D2O) δ 9.18 (d, J = 6.1 Hz). HRMS (ESI-TOF) m/z calcd for C29H35N9O11P [M − H]− 716.2194, found 716.2234.
2-Amino-7-benzyl-9-((2S,3S,4R,5S)-5-(((((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxohexyl)amino)(hydroxy)phosphoryl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-7-ium (5a).
General procedure D, 4% yield. HRMS (ESI-TOF) m/z calcd for C36H41N9O11P [M − H]− 806.2658, found 806.2683.
2-Amino-9-((2S,3S,4R,5S)-5-(((((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxohexyl)amino)(hydroxy)phosphoryl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-7-(4-fluorobenzyl)-6-oxo-6,9-dihydro-1H-purin-7-ium (5b).
General procedure D, 29% yield. HRMS (ESI-TOF) m/z calcd for C36H40FN9O11P [M − H]− 824.2563, found 824.2593.
2-Amino-9-((2R,3R,4R,5R)-3,4-bis((tert-butyldimethylsilyl)oxy)-5-(hydroxymethyl)tetra hydrofuran-2-yl)-1,9-dihydro-6H-purin-6-one (6).
To a stirring solution of guanosine (10 g, 35.3 mmol, 1.0 equiv) and imidazole (19 g, 283 mmol, 8 equiv) in dry DMF (70 mL) was added TBSCl (21.2 g, 142 mmol, 4 equiv) under nitrogen atmosphere. The resulting solution was stirred overnight at 25 °C. The reaction mixture was diluted with water (200 mL) and extracted with DCM (3 × 50 mL). The combined DCM layers were washed with sat. aq. NH4Cl (1 × 25 mL), water (1 ×50 mL), dried over sodium sulfate and concentrated under vacuum. The resultant white solid was suspended in 80% acetic acid (300 mL) and the reaction mixture was heated at 60 °C for 12 h. Upon completion, the solvent was evaporated under vacuum and the crude product was purified by silica gel column chromatography using DCM/methanol (0–20% methanol) to yield 6 as a white solid (12.7 g, 70% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.62 (s, 1H), 7.98 (s, 1H), 6.44 (s, 3H), 5.73 (d, J = 7.3 Hz, 1H), 5.29–5.19 (m, 2H), 4.69 (dd, J = 7.3, 4.5 Hz, 1H), 4.27–4.18 (m, 1H), 3.92 (ddd, J = 5.0, 3.5, 1.3 Hz, 1H), 3.67 (dt, J = 11.9, 5.2 Hz, 1H), 3.56 (ddd, J = 11.9, 5.9, 3.5 Hz, 1H), 0.91 (s, 10H), 0.73 (s, 9H), 0.11 (d, J = 8.3 Hz, 6H), −0.09 (s, 3H), −0.31 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 157.10, 154.09, 151.87, 136.14, 117.18, 87.17, 86.16, 75.43, 73.57, 61.71, 40.48, 40.31, 40.14, 39.97, 39.81, 39.64, 39.47, 26.26, 26.20, 25.96, 18.26, 18.02, −2.74, −4.21, −4.30, −5.09. HRMS (ESI-TOF) m/z calcd for C22H42N5O5Si2 [M + H]+ 512.2724, found 512.2724.
Sulfamoyl chloride [51].
To an ice-cold stirring solution of chlorosulfonyl isocyanate (600 μL, 6.85 mmole, 1 equiv.), formic acid (285.5 μL, 6.85 mmole, 1 equiv.) was added drop-wise. After 10 min, the reaction mixture was warmed to 25 °C and stirred for 1 h. It was used directly for the synthesis of 7.
((2R,3R,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-bis((tert-butyldimethylsilyl)oxy)tetrahydrofuran-2-yl)methyl sulfamate (7).
To an ice-cold stirring solution of 6 (1.5 g, 3.0 mmol, 1.0 equiv) in dry DMF (10 mL) was added sulfamoyl chloride (345 mg, 3.3 mmol, 1.1 equiv) under nitrogen atmosphere. The resulting solution was warmed to 25 °C and stirred for 1 h. Triethyl amine (3 mL) was then added and the reaction was stirred for 10 min. The reaction mixture was cooled to 0 °C, quenched with methanol (5 mL) and concentrated under vacuum. The reaction mixture was dissolved in ethyl acetate (50 mL), washed with brine and water (200 mL), dried over sodium sulfate and concentrated under vacuum. The crude reaction mixture was purified by silica gel column chromatography using DCM/methanol (0–20% methanol with 0.1% TEA) to yield 7 as a white solid (1.64 g, 95% yield). 1H NMR (500 MHz, DMSO-d6) δ 10.66 (d, J = 6.0 Hz, 1H), 7.89 (s, 1H), 7.63 (s, 2H), 6.43 (s, 2H), 5.74 (d, J = 7.4 Hz, 1H), 4.76 (dd, J = 7.3, 4.4 Hz, 1H), 4.29 (dd, J = 11.3, 5.2 Hz, 1H), 4.26–4.19 (m, 2H), 4.13 (t, J = 5.2 Hz, 1H), 0.90 (s, 9H), 0.71 (s, 9H), 0.11 (d, J = 10.5 Hz, 6H), −0.08 (s, 3H), −0.30 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 157.14, 154.13, 151.98, 136.09, 117.25, 86.26, 83.39, 74.49, 73.12, 68.50, 40.46, 40.30, 40.13, 39.96, 39.80, 39.63, 39.46, 26.16, 25.93, 18.19, 17.97, −4.28, −4.30, −4.40, −5.09. HRMS (ESI-TOF) m/z calcd for C22H43N6O7SSi2 [M + H]+ 591.2452 found 591.2460.
tert-butyl-4-(((((2R,3R,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-bis((tertbutyldimethylsilyl)oxy)tetrahydrofuran-2-yl)methoxy)sulfonyl)amino)-4-oxobutanoate (9).
To a stirring solution of 4-(tert-butoxy)-4-oxobutanoic acid (693 mg, 3.0 mmol, 1 equiv) in DCM (5.0 mL), DIPEA (1.04 mL, 6.0 mmol, 2 equiv), and N-hydroxysuccinimide (379.5 mg, 3.3 mmol, 1.1 equiv) was added HATU (1.25 g, 3.3 mmol, 1.1 equiv). The resulting solution was stirred under argon at 25 °C for 4 h. Upon completion of reaction, sat. brine (20 mL) was added and the aqueous layer was extracted with DCM. The combined DCM layers were dried over anhydrous sodium sulfate and concentrated under vacuum. The flask was left under high vacuum for 2 h and crude 8 was used without further purification. 8 (1.7 g, 3.0 mmol, 1.5 equiv) and compound 7 (1.8 g, 2.0 mmol, 1.0 equiv) was re-dissolved in dry DMF (5 mL) and was cooled at 0 °C. DBU (492 μL, 3.3 mmol, 1.1 equiv) was added and the reaction mixture was stirred at 25 °C overnight. Next, the reaction mixture was dried under high vacuum and purified by silica gel column chromatography using DCM/methanol (0–20% methanol) to yield 9 as a white solid (939 mg, 42% yield). 1H NMR (500 MHz, DMSO-d6) δ 7.92 (d, J = 9.5 Hz, 1H), 7.76 (s, 1H), 6.49 (s, 2H), 5.75 (d, J = 7.4 Hz, 1H), 4.77 (dd, J = 7.2, 4.7 Hz, 1H), 4.54–4.42 (m, 2H), 4.40–4.32 (m, 1H), 4.21 (dd, J = 21.1, 4.7 Hz, 2H), 4.13 (s, 1H), 2.49–2.40 (m, 3H), 1.38 (d, J = 5.8 Hz, 9H), 0.91 (s, 9H), 0.72 (s, 9H), 0.11 (s, 7H), −0.07 (s, 3H), −0.30 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 171.87, 171.56, 157.19, 154.13, 152.00, 136.10, 117.17, 92.79, 86.18, 83.29, 82.85, 80.28, 77.40, 74.47, 73.20, 72.14, 70.95, 53.56, 40.48, 40.31, 40.15, 39.98, 39.81, 39.65, 39.48, 31.39, 29.78, 28.22, 28.14, 26.21, 26.15, 25.92, 25.67, 18.30, 18.19, 18.11, 17.95, −4.00, −4.23, −4.32, −4.41, −4.45, −4.51, −5.17. HRMS (ESI-TOF) m/z calcd for C30H54N6O10SSi2 [M + H]+ 747.3239 found 747.3232.
4-(((((2R,3R,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-bis((tert-butyldimethylsilyl)oxy)tetrahydrofuran-2-yl)methoxy)sulfonyl)amino)-4-oxobutanoic acid (10).
Compound 9 (185 mg, 0.26 mmol) was dissolved in DCM/TFA/Et3SiH (2:1:0.1, 2 mL) and stirred for 2 h. The solution was concentrated to dryness and left on high vacuum for 2 h. It was used directly for next reaction (90% yield). HRMS (ESI-TOF) m/z calcd for C26H47N6O10SSi2 [M + H]+ 691.2613 found 691.2610.
((2R,3R,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-bis((tert-butyldimethylsilyl)oxy)tetrahydrofuran-2-yl)methyl(4-((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxohexyl)amino)-4-oxobutanoyl)sulfamate (11).
To a stirring solution of 10 (100 mg, 0.26 mmol, 1 equiv), 3 (185 mg, 0.26 mmol, 1 equiv) and NMM (289 μL, 2.6 mmol, 10 equiv) in DMSO/water at pH 7.0 (6 mL, 1:1) was added EDC.HCl (248.3 mg, 1.3 mmol, 5 equiv). The resultant solution was stirred overnight at 25 °C. The reaction mixture was diluted with water and purified by semi-preparative HPLC (20–80% acetonitrile in water) to yield 11 as a white solid (112 mg, 40%). 1H NMR (500 MHz, DMSO-d6) δ 10.99 (s, 1H), 10.67 (s, 1H), 9.73 (s, 1H), 7.89 (s, 1H), 7.85 (q, J = 5.6 Hz, 1H), 7.81–7.78 (m, 1H), 7.51–7.41 (m, 2H), 6.43 (s, 2H), 5.73 (d, J = 7.5 Hz, 1H), 5.12 (dd, J = 13.3, 5.1 Hz, 1H), 4.73 (dd, J = 7.4, 4.5 Hz, 1H), 4.50 (dd, J = 10.9, 5.3 Hz, 1H), 4.40 (dd, J = 10.7, 4.7 Hz, 2H), 4.37–4.27 (m, 2H), 4.21 (d, J = 4.7 Hz, 1H), 4.11 (t, J = 5.2 Hz, 1H), 3.03 (d, J = 6.6 Hz, 2H), 2.91 (dd, J = 13.5, 5.3 Hz, 2H), 2.58 (d, J = 16.8 Hz, 1H), 2.50–2.46 (m, 3H), 2.33 (q, J = 7.1, 6.6 Hz, 5H), 2.03–1.96 (m, 1H), 1.57 (p, J = 7.5 Hz, 2H), 1.43–1.35 (m, 2H), 1.32–1.24 (m, 2H), 0.88 (s, 9H), 0.69 (s, 9H), 0.09 (d, J = 9.9 Hz, 6H), −0.09 (s, 3H), −0.32 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 173.28, 171.72, 171.56, 171.49, 170.84, 168.27, 157.14, 154.15, 152.00, 135.99, 134.25, 134.08, 133.10, 129.02, 125.64, 119.39, 117.18, 86.12, 83.12, 74.46, 73.11, 71.35, 51.98, 46.93, 40.87, 40.46, 40.29, 40.13, 39.96, 39.79, 39.62, 39.46, 38.99, 36.20, 31.66, 31.36, 29.64, 29.36, 26.59, 26.20, 26.14, 25.90, 25.28, 23.09, 18.16, 17.94, −4.32, −4.35, −4.46, −5.19. HRMS (ESI-TOF) m/z calcd for C45H68N10O13SSi2 [M + H]+ 1045.4305 found 1045.4295.
((2R,3S,4R,5R)-5-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl(4-((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxo hexyl)amino)-4-oxobutanoyl)sulfamate (12).
To an ice-cold stirring solution of 11 (60 mg, 0.06 mmol, 1 equiv) in THF (3.0 mL) was added 1M TBAF (171 μL, 0.2 mmol, 3 equiv). The resultant solution was warmed to 25 °C and stirring was continued for 5 min. The reaction was diluted with methanol and concentrated under vacuum. The crude residue was purified by silica gel chromatography (20% methanol in DCM) followed by further purification via semi-preparative HPLC (20–80% acetonitrile in water) to yield 12 as white solid (35 mg, 71%). 1H NMR (500 MHz, DMSO-d6) δ 11.00 (s, 1H), 9.82 (s, 2H), 8.36 (s, 2H), 7.90 (s, 1H), 7.86–7.75 (m, 3H), 7.46 (d, J = 6.8 Hz, 2H), 6.82 (s, 2H), 5.68 (d, J = 6.5 Hz, 1H), 5.14–5.06 (m, 1H), 4.50–4.29 (m, 5H), 4.12–3.90 (m, 5H), 3.01 (d, J = 6.5 Hz, 3H), 2.93–2.83 (m, 2H), 2.64–2.56 (m, 1H), 2.48 (s, 2H), 2.36 (dt, J = 22.2, 5.9 Hz, 4H), 2.04–1.96 (m, 1H), 1.55 (s, 9H), 1.52 (d, J = 7.7 Hz, 8H), 1.44–1.35 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 177.14, 173.28, 172.49, 171.90, 171.46, 168.29, 165.79, 157.42, 154.49, 151.97, 135.87, 134.35, 134.11, 133.09, 128.96, 125.60, 119.24, 116.92, 86.69, 83.14, 73.87, 71.40, 67.52, 57.99, 52.06, 47.07, 38.79, 36.22, 35.32, 32.64, 31.66, 29.36, 26.53, 25.32, 23.05. HRMS (ESI-TOF) m/z calcd for C33H41N10O13S [M + H]+ 817.2575 found 817.2558.
2-Amino-7-benzyl-9-((2R,3R,4S,5R)-5-(((N-(4-((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoiso indolin-4-yl)amino)-6-oxohexyl)amino)-4-oxobutanoyl)sulfamoyl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-7-ium (13a).
General procedure D, 45%. 1H NMR (500 MHz, DMSO-d6) δ 10.99 (s, 1H), 10.12 (d, J = 66.0 Hz, 1H), 9.73 (s, 1H), 7.85–7.21 (m, 9H), 5.85 (d, J = 25.5 Hz, 1H), 5.61 (t, J = 13.1 Hz, 3H), 5.47 (s, 1H), 5.11 (d, J = 12.5 Hz, 2H), 4.64–3.75 (m, 11H), 3.08–2.83 (m, 2H), 2.59 (d, J = 16.3 Hz, 1H), 2.41–2.19 (m, 6H), 2.04–1.89 (m, 1H), 1.63–1.19 (m, 7H). HRMS (ESI-TOF) m/z calcd for C40H45N10O13S [M − H]− 905.2883 found 905.2874.
2-Amino-9-((2R,3R,4S,5R)-5-(((N-(4-((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxohexyl)amino)-4-oxobutanoyl)sulfamoyl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-7-(4-fluorobenzyl)-6-oxo-6,9-dihydro-1H-purin-7-ium (13b).
General procedure D, 55%. 1H NMR (500 MHz, DMSO-d6) δ 11.01 (s, 1H), 10.18 (s, 1H), 9.75 (s, 2H), 8.16 (s, 1H), 7.84–7.80 (m, 1H), 7.78–7.69 (m, 4H), 7.50 (d, J = 6.4 Hz, 2H), 7.17 (t, J = 8.8 Hz, 3H), 5.83 (s, 1H), 5.71–5.52 (m, 3H), 5.13 (d, J = 18.4 Hz, 2H), 4.43–4.30 (m, 4H), 4.21 (s, 1H), 4.12 (d, J = 17.4 Hz, 2H), 4.02 (d, J = 10.9 Hz, 1H), 3.19–3.12 (m, 2H), 2.94 (d, J = 35.4 Hz, 3H), 2.61 (d, J = 18.0 Hz, 2H), 2.41–2.20 (m, 9H), 2.10–1.98 (m, 2H). 19F NMR (470 MHz, DMSO-d6) δ −148.28, −148.33. HRMS (ESI-TOF) m/z calcd for C40H46FN10O13S [M + H]+ 925.2945 found 925.2951.
tert-Butyl(2-(2-(2-(3-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-3-oxopropoxy)ethoxy)ethoxy)ethyl)carbamate (15).
General procedure A, 56% yield. 1H NMR (500 MHz, DMSO-d6) δ 11.01 (s, 1H), 9.82 (s, 1H), 7.81 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 10.3 Hz, 2H), 6.72 (t, J = 5.3 Hz, 1H), 5.15–5.08 (m, 1H), 4.44–4.27 (m, 2H), 3.70 (t, J = 6.3 Hz, 2H), 3.53–3.46 (m, 6H), 3.46–3.42 (m, 2H), 3.34 (s, 6H), 3.03 (q, J = 5.9 Hz, 2H), 2.59 (d, J = 6.3 Hz, 2H), 1.35 (s, 9H). 13C NMR (126 MHz, DMSO-d6) δ 173.28, 171.49, 169.78, 168.25, 156.01, 134.09, 133.11, 129.09, 125.63, 119.53, 78.03, 70.15, 70.08, 69.93, 69.59, 67.04, 51.97, 46.88, 40.46, 40.30, 40.30, 40.13, 39.96, 39.80, 39.63, 39.46, 37.04, 31.65, 28.67, 23.12. HRMS (ESI-TOF) m/z calcd for C27H39N4O9 [M + H]+ 563.2717, found 563.2708.
tert-Butyl(24-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-24-oxo-3,6,9,12,15,18,21-heptaoxatetracosyl)carbamate (16).
General procedure A, 18% yield. 1H NMR (500 MHz, D2O) δ 7.61 (t, J = 8.5 Hz, 1H), 7.50 (dt, J = 15.2, 7.4 Hz, 2H), 5.05 (dd, J = 13.3, 5.3 Hz, 1H), 4.44–4.28 (m, 2H), 3.75 (t, J = 5.8 Hz, 2H), 3.65–3.35 (m, 26H), 3.10 (t, J = 5.3 Hz, 2H), 2.90–2.70 (m, 3H), 2.62 (t, J = 5.8 Hz, 2H), 2.48–2.32 (m, 2H), 2.24–2.09 (m, 2H), 1.27 (s, 9H). 13C NMR (126 MHz, D2O) δ 175.70, 172.89, 172.22, 170.72, 158.07, 136.24, 131.84, 131.64, 129.52, 128.42, 121.59, 69.51, 69.46, 69.32, 66.57, 52.39, 47.58, 39.57, 36.11, 30.72, 27.60, 22.13. HRMS (ESI-TOF) m/z calcd for C35H54N4O13 [M + K]+ 777.3354, found 777.3316.
3-(2-(2-(2Aaminoethoxy)ethoxy)ethoxy)-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propenamide (18).
General procedure B, 100%. HRMS (ESI-TOF) m/z calcd for C22H31N4O7 [M + H]+ 463.2193, found 463.2177.
1-Amino-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-3,6,9,12,15,18,21-heptaoxa tetracosan-24-amide (19).
General procedure B, 100%. HRMS (ESI-TOF) m/z calcd for C30H46N4O11 [M + H]+ 639.3241, found 639.3239.
6-(((((((2S,3R,4S,5S)-5-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)oxy)(l1-oxidaneyl)phosphoryl)amino)-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)hexanamide (20).
General procedure C, 35% yield. 1H NMR (500 MHz, DMSO-d6) δ 11.00 (s, 1H), 10.85 (s, 1H), 10.01 (s, 1H), 7.95 (s, 2H), 7.82 (d, J = 6.5 Hz, 1H), 7.50–7.41 (m, 2H), 5.69 (d, J = 5.8 Hz, 1H), 5.10 (dd, J = 18.8, 6.4 Hz, 1H), 4.54–4.47 (m, 1H), 4.43 (s, 1H), 4.41–4.38 (m, 1H), 4.35 (s, 1H), 4.31 (s, 1H), 4.19 (s, 1H), 4.01 (d, J = 16.1 Hz, 3H), 3.02 (s, 2H), 3.00–2.92 (m, 5H), 2.41–2.29 (m, 3H), 2.03–1.94 (m, 1H), 1.60–1.53 (m, 3H), 1.43 (s, 1H), 1.36–1.24 (m, 3H). 31P NMR (202 MHz, DMSO-d6) δ −2.48 (d, J = 20.7 Hz), −10.98 (d, J = 20.8 Hz). HRMS (ESI-TOF) m/z calcd for C29H36N9O14P2 [M − H]− 796.1857, found 796.1867.
3-(2-(2-(2-(((((((2S,3R,4S,5S)-5-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(l1-oxidaneyl)phosphoryl)oxy)(hydroxy) phosphoryl)amino)ethoxy)ethoxy)ethoxy)-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propenamide (21).
General procedure C, 32% yield. 1H NMR (500 MHz, D2O) δ 9.03 (d, J = 10.8 Hz, 1H), 7.67–7.50 (m, 3H), 5.97 (d, J = 31.4 Hz, 2H), 5.12 (d, J = 13.3 Hz, 2H), 4.61 (d, J = 10.6 Hz, 1H), 4.45 (d, J = 17.1 Hz, 2H), 4.37 (d, J = 17.4 Hz, 3H), 4.23 (d, J = 10.8 Hz, 1H), 3.84 (t, J = 5.7 Hz, 2H), 3.73–3.57 (m, 10H), 3.15 (t, J = 6.4 Hz, 4H), 3.12–3.06 (m, 5H), 2.72 (t, J = 5.7 Hz, 2H), 1.89–1.77 (m, 4H). 13C NMR (126 MHz, D2O) δ 175.67, 172.24, 170.62, 162.49, 160.43, 155.22, 154.75, 135.97, 131.75, 129.39, 128.14, 121.36, 119.64, 117.32, 115.00, 89.82, 83.67, 74.67, 74.50, 69.36, 66.52, 66.22, 55.24, 52.36, 47.49, 42.65, 38.95, 36.34, 34.88, 30.69, 24.88, 22.07; 31P NMR (202 MHz, D2O) δ −0.15, −0.19, −11.11, −11.21, −11.77, −23.59, −23.65. HRMS (ESI-TOF) m/z calcd for C32H42N9O17P2 [M + NH4]+ 886.2174, found 886.2175.
1-(((((((2S,3R,4S,5S)-5-(2-Amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-dihydroxytetra hydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)oxy)(l1-oxidaneyl)phosphoryl)amino)-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-3,6,9,12,15,18,21-heptaoxatetracosan-24-amide (22).
General procedure C, 35% yield. 1H NMR (500 MHz, DMSO-d6) δ 11.01 (s, 1H), 10.56 (s, 2H), 10.06 (s, 1H), 9.89 (s, 1H), 7.95–7.86 (m, 2H), 7.83 (d, J = 7.2 Hz, 1H), 7.76 (s, 1H), 7.45 (dd, J = 18.2, 5.7 Hz, 2H), 7.21 (s, 1H), 6.61 (d, J = 45.8 Hz, 4H), 5.96 (s, 3H), 5.65 (d, J = 5.4 Hz, 1H), 5.12 (dd, J = 13.3, 5.0 Hz, 1H), 4.78–4.69 (m, 1H), 4.65–4.56 (m, 2H), 4.39 (ddd, J = 45.1, 25.5, 10.9 Hz, 4H), 4.24 (s, 1H), 4.02–3.89 (m, 3H), 3.73–3.54 (m, 5H), 3.13 (s, 2H), 3.03–2.93 (m, 3H), 2.91 (s, 1H), 2.87 (s, 2H), 2.71 (s, 1H), 2.62 (d, J = 5.0 Hz, 3H), 2.52 (s, 2H), 2.32 (d, J = 25.4 Hz, 5H), 2.06 (d, J = 6.2 Hz, 2H), 1.86 (s, 2H), 1.59–1.50 (m, 2H), 1.09 (d, J = 42.2 Hz, 3H). 31P NMR (202 MHz, DMSO-d6) δ −1.69, −10.05. HRMS (ESI-TOF) m/z calcd for C40H58N9O21P2 [M − H]− 1062.3222, found 1062.3217.
2-Amino-7-benzyl-9-((2S,3S,4R,5S)-5-(((((((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-6-oxohexyl)amino)(hydroxy)phosphoryl)oxy)(hydroxy)phosphoryl)oxy) methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-7-ium (23a).
General procedure D, 16% yield. HRMS (ESI-TOF) m/z calcd for C36H42N9O14P2 [M − H]− 886.2321, found 886.2361.
2-Amino-9-((2S,3S,4R,5S)-5-(((((((6-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl) amino)-6-oxohexyl)amino)(hydroxy)phosphoryl)oxy)(hydroxy)phosphoryl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-7-(4-fluorobenzyl)-6-oxo-6,9-dihydro-1H-purin-7-ium (23b).
General procedure D, 11% yield. HRMS (ESI-TOF) m/z calcd for C36H41FN9O14P2 [M − H]− 904.2227, found 904.2212.
2-Amino-7-benzyl-9-((2S,3S,4R,5S)-5-(((((((2-(2-(2-(3-((2-(2,6-dioxopiperidin-3-yl)-1-oxo isoindolin-4-yl)amino)-3-oxopropoxy)ethoxy)ethoxy)ethyl)amino)(hydroxy)phosphoryl) oxy)(hydroxy)phosphoryl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-7-ium (24a).
General procedure D, 20% yield. HRMS (ESI-TOF) m/z calcd for C39H48N9O17P2 [M − H]− 976.2638, found 976.2677.
2-Amino-9-((2S,3S,4R,5S)-5-(((((((2-(2-(2-(3-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)amino)-3-oxopropoxy)ethoxy)ethoxy)ethyl)amino)(hydroxy)phosphoryl)oxy)(hydroxy) phosphoryl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-7-(4-fluorobenzyl)-6-oxo-6,9-dihydro-1H-purin-7-ium (24b).
General procedure D, 11% yield). HRMS (ESI-TOF) m/z calcd for C39H47FN9O17P2 [M − H]− 994.2544, found 994.2543.
2-Amino-7-benzyl-9-((2R,3R,4S,5R)-5-(((((((24-((2-(2,6-dioxopiperidin-3-yl)-1-oxo isoindolin-4-yl)amino)-24-oxo-3,6,9,12,15,18,21-heptaoxatetracosyl)amino)(hydroxy) phosphoryl)oxy)(hydroxy)phosphoryl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-7-ium (25a).
General procedure D, 13% yield. HRMS (ESI-TOF) m/z calcd for C47H64N9O21P2 [M − H]− 1152.3686, found 1152.3695.
2-Amino-9-((2R,3R,4S,5R)-5-(((((((24-((2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl) amino)-24-oxo-3,6,9,12,15,18,21-heptaoxatetracosyl)amino)(hydroxy)phosphoryl)oxy) (hydroxy)phosphoryl)oxy)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-7-(4-fluorobenzyl)-6-oxo-6,9-dihydro-1H-purin-7-ium (25b).
General procedure D, 9% yield. HRMS (ESI-TOF) m/z calcd for C47H63FN9O21P2 [M − H]− 1170.3592, found 1170.3599.
tert-Butyl(6-(((2S)-1-((4R)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)carbamate (27).
General procedure A, 75% yield. 1H NMR (500 MHz, Chloroform-d) δ 8.60 (s, 1H), 7.88 (d, J = 36.1 Hz, 1H), 7.17 (s, 1H), 6.57–6.43 (m, 1H), 4.69–4.58 (m, 2H), 4.52–4.39 (m, 3H), 4.24 (dd, J = 15.1, 5.2 Hz, 2H), 4.03 (d, J = 11.1 Hz, 1H), 3.62–3.48 (m, 1H), 3.01–2.91 (m, 2H), 2.86 (s, 1H), 2.79 (s, 1H), 2.42 (s, 3H), 2.39–2.20 (m, 2H), 2.10 (dd, J = 13.6, 7.3 Hz, 3H), 1.56–1.42 (m, 2H), 1.31 (s, 9H), 1.17 (d, J = 11.7 Hz, 3H), 0.85 (s, 9H). 13C NMR (126 MHz, Chloroform-d) δ 174.67, 174.11, 171.92, 170.91, 162.69, 156.06, 150.43, 148.27, 138.45, 138.12, 131.70, 130.79, 129.45, 128.02, 79.16, 77.27, 77.01, 76.76, 70.00, 58.89, 57.52, 56.97, 43.17, 40.33, 36.10, 34.94, 31.49, 29.57, 28.40, 26.39, 26.10, 25.16, 20.81, 15.91. HRMS (ESI-TOF) m/z calcd for C33H49N5O6S [M + H]+ 644.3482, found 644.3476.
(4R)-1-((S)-2-(6-Aminohexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl) pyrrolidine-2-carboxamide (28).
General procedure B, 90%. 1H NMR (500 MHz, DMSO-d6) δ 8.97 (s, 1H), 8.61 (s, 1H), 8.44 (s, 1H), 7.89 (d, J = 9.2 Hz, 1H), 7.45–7.32 (m, 5H), 4.53 (d, J = 9.3 Hz, 1H), 4.48–4.38 (m, 3H), 4.34 (s, 2H), 4.21 (dd, J = 15.8, 5.2 Hz, 2H), 3.65 (q, J = 12.2, 11.5 Hz, 3H), 2.72 (t, J = 7.4 Hz, 3H), 2.25 (dd, J = 14.1, 7.3 Hz, 2H), 2.12 (dt, J = 14.2, 7.1 Hz, 2H), 2.07–1.99 (m, 1H), 1.97–1.81 (m, 2H), 1.49 (dd, J = 17.0, 7.7 Hz, 6H), 1.27 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 172.41, 170.13, 166.85, 151.88, 148.14, 139.95, 131.61, 130.07, 129.30, 129.07, 128.52, 127.85, 69.31, 59.15, 56.81, 56.78, 42.10, 40.43, 40.35, 40.26, 40.09, 39.93, 39.76, 39.59, 39.43, 39.18, 38.42, 35.66, 35.27, 35.11, 27.60, 27.59, 26.83, 26.03, 25.90, 25.52, 25.39, 16.38. HRMS (ESI-TOF) m/z calcd for C28H41N5O4 [M + H]+ 544.2958, found 544.2939.
(4S)-1-((2S)-2-(6-(((((((2S,3R,4S,5S)-5-(2-amino-6-oxo-1,6-dihydro-9H-purin-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(hydroxy)phosphoryl)oxy)(λ1-oxidaneyl) phosphoryl)amino)hexanamido)-3,3-dimethylbutanoyl)-4-hydroxy-N-(4-(4-methylthiazol-5-yl)benzyl)pyrrolidine-2-carboxamide (29).
General procedure C, 32% yield. 1H NMR (500 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.97 (d, J = 4.3 Hz, 2H), 8.59–8.53 (m, 1H), 7.87 (d, J = 9.2 Hz, 2H), 7.48–7.29 (m, 7H), 6.61 (d, J = 52.9 Hz, 2H), 5.68 (d, J = 4.8 Hz, 1H), 4.58–4.15 (m, 9H), 3.69–3.57 (m, 3H), 3.49–3.23 (m, 8H), 2.43 (d, J = 3.0 Hz, 4H), 2.32–2.17 (m, 2H), 2.17–2.07 (m, 1H), 2.05 (s, 1H), 1.95–1.83 (m, 2H), 1.59–1.41 (m, 4H), 1.32–1.20 (m, 2H). 13C NMR (126 MHz, DMSO-d66) δ 172.38, 170.13, 169.28, 152.02, 151.89, 148.33, 148.15, 139.95, 138.97, 131.60, 131.42, 130.63, 130.07, 129.40, 129.07, 128.25, 127.86, 69.87, 69.31, 59.15, 58.69, 56.79, 54.11, 42.51, 42.09, 40.46, 40.29, 40.12, 39.96, 39.79, 39.62, 39.46, 38.42, 35.68, 35.10, 27.24, 26.84, 26.00, 25.36, 16.40. HRMS (ESI-TOF) m/z calcd for C38H53N10O14P2S [M+H]+ 969.3095, found 969.3085.
2-Amino-7-benzyl-9-((2S,3S,4R,5S)-3,4-dihydroxy-5-(((hydroxy((hydroxy((6-(((2S)-1-((4S)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)amino)phosphoryl)oxy)phosphoryl)oxy)methyl)tetrahydrofuran-2-yl)-6-oxo-6,9-dihydro-1H-purin-7-ium (30a).
General procedure D, 11% yield. HRMS (ESI-TOF) m/z calcd for C45H59N10O14P2S [M − H]− 1057.3403, found 1057.3411
2-Amino-9-((2S,3S,4R,5S)-3,4-dihydroxy-5-(((hydroxy((hydroxy((6-(((2S)-1-((4S)-4-hydroxy-2-((4-(4-methylthiazol-5-yl)benzyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-6-oxohexyl)amino)phosphoryl)oxy)phosphoryl)oxy)methyl)tetrahydrofuran-2-yl)-7-(4-fluorobenzyl)-6-oxo-6,9-dihydro-1H-purin-7-ium (30b).
General procedure D, 41% yield. HRMS (ESI-TOF) m/z calcd for C45H59FN10O14P2S [M − H]− 1075.3308, found 1075.3315.
((2R,3S,4R,5R)-5-(2-Amino-7-methyl-6-oxo-1,6-dihydro-9H-purin-7-ium-9-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl phosphate (m7GMP).
General procedure D, 75% yield. 1H NMR (500 MHz, D2O) δ 9.12 (s, 1H), 6.06 (d, J = 3.4 Hz, 1H), 4.67–4.63 (m, 1H), 4.45 (t, J = 5.2 Hz, 1H), 4.41–4.37 (m, 1H), 4.28–4.21 (m, 1H), 4.14–4.11 (m, 1H), 4.10 (s, 3H). 13C NMR (126 MHz, D2O) δ 155.49, 154.99, 149.47, 136.43, 108.39, 89.67, 83.99, 74.67, 69.14, 63.28, 35.87. 31P NMR (202 MHz, D2O) δ 0.12. HRMS (ESI-TOF) m/z calcd for C11H15N5O8P [M − H]− 376.0664, found 376.0669.
4.7. Preparation of m7GxP Agarose Resin
The synthesis was adapted from that of Edery et al. [52]. m7GMP (synthesized; see below) or m7GDP (Sigma) sodium salts was dissolved in water (10 mM final), and a solution of sodium periodate (1.0 equiv) in sodium acetate buffer (0.1 M, pH 6; 51 mM final) was added. The resulting mixture was agitated at 25 °C for 30 min protected from light. Adipic acid dihydrazide agarose (1 mL packed; Sigma) was washed with water (1× 20 mL) followed by sodium acetate buffer (1× 20mL), and then resuspended in sodium acetate buffer (2 mL). To this slurry was added aniline (20 equiv) and the oxidized m7GxP solution. The resin mixture was then shaken at 25 °C for 45 min before adding sodium cyanoborohydride (16 equiv). The mixture was agitated overnight at 4 °C. The resin was then washed with NaCl (1M; 5× 5 mL), equilibrated in buffer A (5 mL; 50 mM HEPES, pH 7, 200 mM KCl), and stored at 4 °C.
4.8. Cell Culture
HEK293 cells were grown in DMEM supplemented with 10% FBS and 1 mM Glutamine. MDA-MB-231 cells were grown in RPMI-1640 supplemented with 10% FBS and 1 mM Glutamine. K562 cells were grown in Iscove’s Modified Dulbecco’s Medium supplemented with 10% FBS and 1 mM Glutamine. Cells were grown at 37 °C with 5% CO2 in a humidified incubator. All cell lines were authenticated by STR profiling and regularly tested for mycoplasma contamination
4.9. m7G Cap Competition Assay and Western Blot
The cap competition assay was carried out as previously described (Yanagiya et al., 2012). Briefly, HEK293 cells were lysed in cap pull-down buffer (50 mM HEPES-KOH pH 7.5, 150 mM KCl, 1 mM EDTA, 2 mM DTT and 0.1% Tween 20) containing protease inhibitors and freeze-thawed thrice. Cell lysate was centrifuged at 20,000 × g for 25 min. Total protein in the supernatant was measured using the BCA reagent (Thermo Scientific). 350 μg of total protein from the cell lysate was then incubated for 2 h at 4 °C with m7GMP or m7GDP agarose resin. The resin was washed 3× with cap pull-down buffer followed by 1× Tris Buffered Saline (TBS) and 1× Buffer A (50 mM HEPES-KOH pH 7.5, 150 mM KCl). The resin was then incubated with compounds (in Buffer A) overnight. The resin was again washed 1× with cap pull down buffer followed by 1× TBS and 1× water. Proteins were then eluted from the resin by boiling in 2× LDS sample buffer (10 min at 70 °C) resolved on a 4–12% Bis-Tris gel, and transferred to PVDF membrane in Towbin’s Buffer. The membrane was blocked in 5% milk for 1 h at 25 °C, and then incubated with a primary antibody (overnight at 4 °C) and secondary antibody (1 h at 25 °C). Proteins were visualized by autoradiography. Anti-eIF4E antibody was purchased from Cell Signaling Technology (cat# 9742). Figures were formatted in Adobe Illustrator.
Supplementary Material
HIGHLIGHTS:
New Bn7GMP- and Bn7GDP-based proteolysis targeting chimeras (PROTACs) were synthesized for the targeted degradation of eIF4E
New m7GMP- and m7GDP-based cap competition assays were developed
Discovery of Bn7GDP-based PROTACs with in vitro inhibitory activity of eIF4E-cap binding
ACKNOWLEDGMENTS:
We would like to thank Sydney Rosenblum for giving creative inputs while preparing the manuscript. This work was supported by the NIH (R01 CA202018 to A.L.G).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING INTERESTS:
The authors declare no competing interests.
SUPPORTING INFORMATION:
Spectra for all synthesized compounds.
REFERENCES:
- [1].Sonenberg N, Hinnebusch AG, Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets, Cell. 136 (2009) 731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Mamane Y, Petroulakis E, Rong L, Yoshida K, Ler LW, Sonenberg N, eIF4E – from translation to transformation, Oncogene. 23 (2004) 3172–3179. doi: 10.1038/sj.onc.1207549. [DOI] [PubMed] [Google Scholar]
- [3].Jia Y, Polunovsky V, Bitterman PB, Wagner CR, Cap-Dependent Translation Initiation Factor eIF4E: An Emerging Anticancer Drug Target, Medicinal Research Reviews. 32 (2012) 786–814. doi: 10.1002/med.21260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Sonenberg N, Gingras A-C, The mRNA 5′ cap-binding protein eIF4E and control of cell growth, Current Opinion in Cell Biology. 10 (1998) 268–275. doi: 10.1016/S0955-0674(98)80150-6. [DOI] [PubMed] [Google Scholar]
- [5].Avdulov S, Li S, Van Michalek, Burrichter D, Peterson M, Perlman DM, Manivel JC, Sonenberg N, Yee D, Bitterman PB, Polunovsky VA, Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells, Cancer Cell. 5 (2004) 553–563. doi: 10.1016/j.ccr.2004.05.024. [DOI] [PubMed] [Google Scholar]
- [6].Kerekatte V, Smiley K, Hu B, Smith A, Gelder F, Benedetti AD, The protooncogene/translation factor eIF4E: A survey of its expression in breast carcinomas, International Journal of Cancer. 64 (1995) 27–31. doi: 10.1002/ijc.2910640107. [DOI] [PubMed] [Google Scholar]
- [7].Nathan C-AO, Liu L, Li BD, Abreo FW, Nandy I, De Benedetti A, Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer, Oncogene. 15 (1997) 579–584. doi: 10.1038/sj.onc.1201216. [DOI] [PubMed] [Google Scholar]
- [8].Wang S, Rosenwald IB, Hutzler MJ, Pihan GA, Savas L, Chen J-J, Woda BA, Expression of the Eukaryotic Translation Initiation Factors 4E and 2α in Non-Hodgkin’s Lymphomas, The American Journal of Pathology. 155 (1999) 247–255. doi: 10.1016/S0002-9440(10)65118-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, Schwier P, Capen A, Goode RL, Dowless MS, Chen Y, Zhang H, Sissons S, Cox K, McNulty AM, Parsons SH, Wang T, Sams L, Geeganage S, Douglass LE, Neubauer BL, Dean NM, Blanchard K, Shou J, Stancato LF, Carter JH, Marcusson EG, Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity, J Clin Invest. 117 (2007) 2638–2648. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Brown CJ, McNae I, Fischer PM, Walkinshaw MD, Crystallographic and Mass Spectrometric Characterisation of eIF4E with N7-alkylated Cap Derivatives, Journal of Molecular Biology. 372 (2007) 7–15. doi: 10.1016/j.jmb.2007.06.033. [DOI] [PubMed] [Google Scholar]
- [11].Fischer PM, Cap in hand: Targeting eIF4E, Cell Cycle. 8 (2009) 2535–2541.doi: 10.4161/cc.8.16.9301. [DOI] [PubMed] [Google Scholar]
- [12].Liu W, Zhao R, McFarland C, Kieft J, Niedzwiecka A, Jankowska-Anyszka M, Stepinski J, Darzynkiewicz E, Jones DNM, Davis RE, Structural Insights into Parasite eIF4E Binding Specificity for m7G and m2,2,7G mRNA Caps, J. Biol. Chem 284 (2009) 31336–31349. doi: 10.1074/jbc.M109.049858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Merrick WC, eIF4F: A Retrospective, J. Biol. Chem 290 (2015) 24091–24099. doi: 10.1074/jbc.R115.675280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Soukarieh F, Nowicki MW, Bastide A, Pöyry T, Jones C, Dudek K, Patwardhan G, Meullenet F, Oldham NJ, Walkinshaw MD, Willis AE, Fischer PM, Design of nucleotide-mimetic and non-nucleotide inhibitors of the translation initiation factor eIF4E: Synthesis, structural and functional characterisation, European Journal of Medicinal Chemistry. 124 (2016) 200–217. doi: 10.1016/j.ejmech.2016.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ziemniak M, Strenkowska M, Kowalska J, Jemielity J, Potential therapeutic applications of RNA cap analogs, Future Medicinal Chemistry. 5 (2013) 1141–1172. doi: 10.4155/fmc.13.96. [DOI] [PubMed] [Google Scholar]
- [16].Guimarães CRW, Kopecky DJ, Mihalic J, Shen S, Jeffries S, Thibault ST, Chen X, Walker N, Cardozo M, Thermodynamic Analysis of mRNA Cap Binding by the Human Initiation Factor eIF4E via Free Energy Perturbations, J. Am. Chem. Soc 131 (2009) 18139–18146. doi: 10.1021/ja9064359. [DOI] [PubMed] [Google Scholar]
- [17].Cai A, Jankowska-Anyszka M, Centers A, Chlebicka L, Stepinski J, Stolarski R, Darzynkiewicz E, Rhoads RE, Quantitative Assessment of mRNA Cap Analogues as Inhibitors of in Vitro Translation, Biochemistry. 38 (1999) 8538–8547. doi: 10.1021/bi9830213. [DOI] [PubMed] [Google Scholar]
- [18].Chen X, Kopecky DJ, Mihalic J, Jeffries S, Min X, Heath J, Deignan J, Lai S, Fu Z, Guimaraes C, Shen S, Li S, Johnstone S, Thibault S, Xu H, Cardozo M, Shen W, Walker N, Kayser F, Wang Z, Structure-Guided Design, Synthesis, and Evaluation of Guanine-Derived Inhibitors of the eIF4E mRNA–Cap Interaction, J. Med. Chem 55 (2012) 3837–3851. doi: 10.1021/jm300037x. [DOI] [PubMed] [Google Scholar]
- [19].Mehellou Y, Rattan HS, Balzarini J, The ProTide Prodrug Technology: From the Concept to the Clinic, J. Med. Chem 61 (2018) 2211–2226. doi: 10.1021/acs.jmedchem.7b00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF, Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs, Chem. Rev 114 (2014) 9154–9218.doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ghosh B, Benyumov AO, Ghosh P, Jia Y, Avdulov S, Dahlberg PS, Peterson M, Smith K, Polunovsky VA, Bitterman PB, Wagner CR, Nontoxic Chemical Interdiction of the Epithelial-to-Mesenchymal Transition by Targeting Cap-Dependent Translation, ACS Chem. Biol 4 (2009) 367–377. doi: 10.1021/cb9000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ohoka N, Shibata N, H. T and Naito M, Protein Knockdown Technology: Application of Ubiquitin Ligase to Cancer Therapy, Current Cancer Drug Targets. (2016). http://www.eurekaselect.com/136884/article (accessed November 20, 2018). [DOI] [PubMed] [Google Scholar]
- [23].Ottis P, Crews CM, Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy, ACS Chem. Biol 12 (2017) 892–898. doi: 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- [24].Lu J, Qian Y, Altieri M, Dong H, Wang J, Raina K, Hines J, Winkler JD, Crew AP, Coleman K, Crews CM, Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4, Chemistry & Biology. 22 (2015) 755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Winter GE, Buckley DL, Paulk J, Roberts JM, Souza A, Dhe-Paganon S, Bradner JE, Phthalimide conjugation as a strategy for in vivo target protein degradation, Science. 348 (2015) 1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lai AC, Crews CM, Induced protein degradation: an emerging drug discovery paradigm, Nature Reviews Drug Discovery. 16 (2017) 101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].An S, Fu L, Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs, EBioMedicine. 36 (2018) 553–562. doi: 10.1016/j.ebiom.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bondeson DP, Smith BE, Burslem GM, Buhimschi AD, Hines J, Jaime-Figueroa S, Wang J, Hamman BD, Ishchenko A, Crews CM, Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead, Cell Chemical Biology. 25 (2018) 78–87. e5. doi: 10.1016/j.chembiol.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Corson TW, Aberle N, Crews CM, Design and Applications of Bifunctional Small Molecules: Why Two Heads Are Better Than One, ACS Chem. Biol 3 (2008) 677–692. doi: 10.1021/cb8001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Galdeano C, Gadd MS, Soares P, Scaffidi S, Van Molle I, Birced I, Hewitt S, Dias DM, Ciulli A, Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities, J. Med. Chem 57 (2014) 8657–8663. doi: 10.1021/jm5011258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Zhou B, Hu J, Xu F, Chen Z, Bai L, Fernandez-Salas E, Lin M, Liu L, Yang C-Y, Zhao Y, McEachern D, Przybranowski S, Wen B, Sun D, Wang S, Discovery of a Small-Molecule Degrader of Bromodomain and Extra-Terminal (BET) Proteins with Picomolar Cellular Potencies and Capable of Achieving Tumor Regression, J. Med. Chem 61 (2018) 462–481. doi: 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zengerle M, Chan K-H, Ciulli A, Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4, ACS Chem. Biol 10 (2015) 1770–1777. doi: 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Raina K, Lu J, Qian Y, Altieri M, Gordon D, Rossi AMK, Wang J, Chen X, Dong H, Siu K, Winkler JD, Crew AP, Crews CM, Coleman KG, PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer, PNAS. 113 (2016) 7124–7129. doi: 10.1073/pnas.1521738113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Schiedel M, Herp D, Hammelmann S, Swyter S, Lehotzky A, Robaa D, Oláh J, Ovádi J, Sippl W, Jung M, Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals), J. Med. Chem 61 (2018) 482–491. doi: 10.1021/acs.jmedchem.6b01872. [DOI] [PubMed] [Google Scholar]
- [35].Robb CM, Contreras JI, Kour S, Taylor MA, Abid M, Sonawane YA, Zahid M, Murry DJ, Natarajan A, Rana S, Chemically induced degradation of CDK9 by a proteolysis targeting chimera (PROTAC), Chem. Commun 53 (2017) 7577–7580. doi: 10.1039/C7CC03879H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ, Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation, PNAS. 98 (2001) 8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lai AC, Toure M, Hellerschmied D, Salami J, Jaime-Figueroa S, Ko E, Hines J, Crews CM, Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL, Angewandte Chemie International Edition. 55 (2016) 807–810. doi: 10.1002/anie.201507634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bondeson DP, Mares A, Smith IED, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, Zinn N, Grandi P, Shimamura S, Bergamini G, Faelth-Savitski M, Bantscheff M, Cox C, Gordon DA, Willard RR, Flanagan JJ, Casillas LN, Votta BJ, den Besten W, Famm K, Kruidenier L, Carter PS, Harling JD, Churcher I, Crews CM, Catalytic in vivo protein knockdown by small-molecule PROTACs, Nature Chemical Biology. 11 (2015) 611–617. doi: 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Jia Y, Chiu T-L, Amin EA, Polunovsky V, Bitterman PB, Wagner CR, Design, synthesis and evaluation of analogs of initiation factor 4E (eIF4E) cap-binding antagonist Bn7-GMP, European Journal of Medicinal Chemistry. 45 (2010) 1304–1313. doi: 10.1016/j.ejmech.2009.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ghosh P, Park C, Peterson MS, Bitterman PB, Polunovsky VA, Wagner CR, Synthesis and evaluation of potential inhibitors of eIF4E cap binding to 7-methyl GTP, Bioorganic & Medicinal Chemistry Letters. 15 (2005) 2177–2180. doi: 10.1016/j.bmcl.2005.01.080. [DOI] [PubMed] [Google Scholar]
- [41].Li S, Jia Y, Jacobson B, McCauley J, Kratzke R, Bitterman PB, Wagner CR, Treatment of Breast and Lung Cancer Cells with a N-7 Benzyl Guanosine Monophosphate Tryptamine Phosphoramidate Pronucleotide (4Ei-1) Results in Chemosensitization to Gemcitabine and Induced eIF4E Proteasomal Degradation, Molecular Pharmaceutics. 10 (2013) 523–531. doi: 10.1021/mp300699d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Othumpangat S, Kashon M, Joseph P, Eukaryotic Translation Initiation Factor 4E Is a Cellular Target for Toxicity and Death Due to Exposure to Cadmium Chloride, J. Biol. Chem 280 (2005) 25162–25169. doi: 10.1074/jbc.M414303200. [DOI] [PubMed] [Google Scholar]
- [43].Murata T, Shimotohno K, Ubiquitination and Proteasome-dependent Degradation of Human Eukaryotic Translation Initiation Factor 4E, J. Biol. Chem 281 (2006) 20788–20800. doi: 10.1074/jbc.M600563200. [DOI] [PubMed] [Google Scholar]
- [44].Seo TW, Lee JS, Choi YN, Jeong DH, Lee SK, Yoo SJ, A novel function of cIAP1 as a mediator of CHIP-driven eIF4E regulation, Scientific Reports. 7 (2017) 9816. doi: 10.1038/s41598-017-10358-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Marcotrigiano J, Gingras A-C, Sonenberg N, Burley SK, Cocrystal Structure of the Messenger RNA 5′ Cap-Binding Protein (eIF4E) Bound to 7-methyl-GDP, Cell. 89 (1997) 951–961. doi: 10.1016/S0092-8674(00)80280-9. [DOI] [PubMed] [Google Scholar]
- [46].Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, Crews CM, Small-Molecule Inhibitors of the Interaction between the E3 Ligase VHL and HIF1α, Angewandte Chemie International Edition. 51 (2012) 11463–11467. doi: 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ito T, Handa H, Cereblon and its downstream substrates as molecular targets of immunomodulatory drugs, International Journal of Hematology. 104 (2016) 293–299. doi: 10.1007/s12185-016-2073-4. [DOI] [PubMed] [Google Scholar]
- [48].Mori T, Ito T, Liu S, Ando H, Sakamoto S, Yamaguchi Y, Tokunaga E, Shibata N, Handa H, Hakoshima T, Structural basis of thalidomide enantiomer binding to cereblon, Scientific Reports. 8 (2018) 1294. doi: 10.1038/s41598-018-19202-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Natarajan A, Moerke N, Fan Y-H, Chen H, Christ WJ, Wagner G, Halperin JA,Synthesis of fluorescein labeled 7-methylguanosinemonophosphate, Bioorganic & Medicinal Chemistry Letters. 14 (2004) 2657–2660. doi: 10.1016/j.bmcl.2004.02.090. [DOI] [PubMed] [Google Scholar]
- [50].Chou T-F, Baraniak J, Kaczmarek R, Zhou X, Cheng J, Ghosh B, Wagner CR, Phosphoramidate Pronucleotides: A Comparison of the Phosphoramidase Substrate Specificity of Human and Escherichia coli Histidine Triad Nucleotide Binding Proteins, Mol. Pharmaceutics. 4 (2007) 208–217. doi: 10.1021/mp060070y. [DOI] [PubMed] [Google Scholar]
- [51].Shah R, Strom A, Zhou A, Maize KM, Finzel BC, Wagner CR, Design, Synthesis, and Characterization of Sulfamide and Sulfamate Nucleotidomimetic Inhibitors of hHint1, ACS Med. Chem. Lett 7 (2016) 780–784. doi: 10.1021/acsmedchemlett.6b00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Edery I, Altmann M, Sonenberg N, High-level synthesis in Escherichia coli of functional cap-binding eukaryotic initiation factor eIF-4E and affinity purification using a simplified cap-analog resin, Gene. 74 (1988) 517–525. doi: 10.1016/0378-1119(88)90184-9. [DOI] [PubMed] [Google Scholar]
- [53].Xiong Y, Lu J, Hunter J, Li L, Scott D, Choi HG, Lim SM, Manandhar A, Gondi S, Sim T, Westover KD, Gray NS, Covalent Guanosine Mimetic Inhibitors of G12C KRAS, ACS Med Chem Lett. 8 (2017) 61–66. doi: 10.1021/acsmedchemlett.6b00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Churcher I, Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones?, J. Med. Chem 61 (2018) 444–452. doi: 10.1021/acs.jmedchem.7b01272. [DOI] [PubMed] [Google Scholar]
- [55].Boonstra J, Verkleij AJ, Regulation of enzyme activity in vivo is determined by its cellular localization, Advances in Enzyme Regulation. 44 (2004) 61–73. doi: 10.1016/j.advenzreg.2003.11.008. [DOI] [PubMed] [Google Scholar]
- [56].Yanagiya A, Suyama E, Adachi H, Svitkin YV, Aza-Blanc P, Imataka H, Mikami S, Martineau Y, Ronai ZA, Sonenberg N, Translational Homeostasis via the mRNA Cap-Binding Protein, eIF4E, Molecular Cell. 46 (2012) 847–858. doi: 10.1016/j.molcel.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.