

Abstract
Cyclometallated ruthenium complexes typically exhibit red‐shifted absorption bands and lower photolability compared to their polypyridyl analogues. They also have lower symmetry, which sometimes makes their synthesis challenging. In this work, the coordination of four N,S bidentate ligands, 3‐(methylthio)propylamine (mtpa), 2‐(methylthio)ethylamine (mtea), 2‐(methylthio)ethyl‐2‐pyridine (mtep), and 2‐(methylthio)methylpyridine (mtmp), to the cyclometallated precursor [Ru(bpy)(phpy)(CH3CN)2]+ (bpy=2,2′‐bipyridine, Hphpy=2‐phenylpyridine) has been investigated, furnishing the corresponding heteroleptic complexes [Ru(bpy)(phpy)(N,S)]PF6 ([2]PF6–[5]PF6, respectively). The stereoselectivity of the synthesis strongly depended on the size of the ring formed by the Ru‐coordinated N,S ligand, with [2]PF6 and [4]PF6 being formed stereoselectively, but [3]PF6 and [5]PF6 being obtained as mixtures of inseparable isomers. The exact stereochemistry of the air‐stable complex [4]PF6 was established by a combination of DFT, 2D NMR, and single‐crystal X‐ray crystallographic studies. Finally, [4]PF6 was found to be photosubstitutionally active under irradiation with green light in acetonitrile, which makes it the first cyclometallated ruthenium complex capable of undergoing selective photosubstitution of a bidentate ligand.
Keywords: chelates, coordination isomers, cyclometallation, photoactivated chemotherapy, photosubstitution, ruthenium
Introduction
Cyclometallated complexes are metal complexes containing a metallacycle in which at least one of the donor atoms in the first coordination sphere is either an sp2 or an sp3 carbon atom. Upon replacing a neutral pyridine ligand of a polypyridyl metal complex with its cyclometallated monoanionic phenylene analogue, the symmetry of the resulting molecule is usually reduced, leading to more coordination isomers than for the starting polypyridyl complex. The separation of such isomers is typically challenging, and it has been claimed that developing efficient synthetic strategies towards geometrically complex coordination compounds is a prerequisite for many applications, notably in biology.1, 2, 3, 4, 5, 6 In the last two decades, cycloruthenated complexes have been extensively studied, in particular in biological contexts.7, 8, 9 They usually show higher cytotoxicity towards cancer cells compared to their non‐cyclometallated analogues.9, 10, 11 This is attributed to their higher lipophilicity, higher cellular uptake, and lower RuIII/II redox potential, which causes cytotoxic interactions with proteins such as those of oxido‐reductase enzymes.12 These advantages, together with their ability to generate reactive oxygen species (ROS) upon light irradiation, make them good candidates for photodynamic therapy (PDT), an anticancer therapy in which the compound needs to be excited to its metal‐to‐ligand charge‐transfer (MLCT) state by visible light irradiation.13, 14 Furthermore, destabilization of the t2g orbitals of the ruthenium(II) centre due to the π‐donor character of the metal‐bound carbon atom, as in the monoanionic chelate 2‐pyridylphenylene (phpy−), shifts the 1MLCT absorption band of a cyclometallated polypyridyl complex to lower energies compared to its non‐cyclometallated analogue.8 This property is particularly attractive in the phototherapy field, whereby photoactive complexes should absorb light in the phototherapeutic window (600–1000 nm), as such light penetrates deeper into biological tissues.
To date, however, the utility of cycloruthenated complexes has remained limited in photoactivated chemotherapy (PACT), an oxygen‐independent anticancer therapy that relies on prodrug activation by photosubstitution.15 In PACT, the binding of a toxic metal complex to biological molecules is typically prevented (or “caged”) by coordination of a thermally inert ligand, and then “photouncaging” is obtained by light irradiation of the compound‐containing tissue, which triggers photosubstitution of the protecting ligand. Cycloruthenated complexes usually show low photosubstitution quantum yields, in particular for the photosubstitution of bidentate ligands, which makes these complexes difficult to use for PACT.16, 17, 18 Destabilization of the eg orbitals in cyclometallated ruthenium(II) complexes due to the excellent σ‐donor character of the metal‐bound carbon atom increases the gap between the π* orbital of the polypyridyl ligands and the eg orbitals of the complex, which disfavours thermal population of the triplet metal‐to‐ligand (3MC) excited state from the photochemically generated metal‐to‐ligand charge‐transfer (3MLCT) state.19, 20 In non‐cyclometallated complexes, a common strategy for enhancing the photoreactivity is to lower the energy of the 3MC state by increasing the distortion of the coordination octahedron through the use of, for example, hindering polypyridyl ligands.21, 22 However, this strategy proved to be inapplicable in the case of [Ru(biq)2(phpy)]PF6 (biq=2,2′‐biquinoline), as this complex is not photoreactive in either CH3CN or water.23 To date, the only cyclometallated ruthenium compound that has been reported to be capable of photosubstitution is [Ru(phen)(phpy)(CH3CN)2]PF6 ([1 a]PF6, phen=1,10‐phenanthroline), which photoreleases a monodentate ligand.9, 24
In this work, we have sought to prepare cycloruthenated complexes capable of undergoing photosubstitution of a bidentate ligand. Indeed, for PACT, bidentate ligands offer better caging in the dark than monodentate ligands.25, 26 For polypyridyl complexes such as [Ru(bpy)2(mtmp)]2+ (bpy=2,2′‐bipyridine) or [Ru(Ph2phen)2(mtmp)]2+ (Ph2phen=4,7‐diphenyl‐1,10‐phenanthroline), thioether‐containing bidentate chelates such as 2‐(methylthio)methylpyridine (mtmp) have recently been shown to be selectively photosubstituted by two water molecules upon irradiation with blue light in water.27 Here, we first investigated whether mtmp and three analogues thereof, 3‐(methylthio)propylamine (mtpa), 2‐(methylthio)ethylamine (mtea), and 2‐(methylthio)ethyl‐2‐pyridine (mtep), could be stereoselectively coordinated to cage the cyclometallated precursor [Ru(bpy)(phpy)(CH3CN)2]PF6 ([1 a]PF6) and thereby prepare well‐characterized heteroleptic complexes [Ru(bpy)(phpy)(N,S)]PF6 ([2]PF6–[5]PF6) (Scheme 1). In a second step, we investigated the stability of the resulting heteroleptic complexes, both in the dark and under light irradiation, and found that only one of these four complexes could be prepared stereoselectively and undergo photosubstitution of the caging N,S bidentate ligand.
Scheme 1.
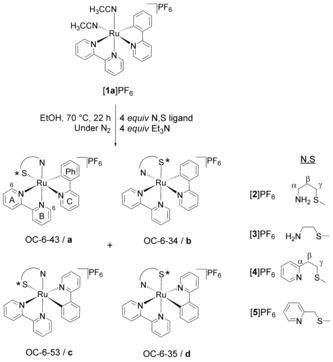
Synthesis of [2]PF6, [3]PF6, [4]PF6, and [5]PF6 from [1 a]PF6 and the structures of the four possible coordination isomers obtained. Note that, in order to obtain isomers c and d, the polypyridyl ligands must rearrange so the σ‐bound C atom becomes trans to the entering N,S ligand. For clarity, only the Λ isomers are shown, but all samples were obtained as racemic Δ/Λ mixtures.
Results and Discussion
Synthesis
The four cycloruthenated complexes [2]PF6–[5]PF6 were prepared as shown in Scheme 1, following the synthetic route established by the Pfeffer group.28, 29 The dimer [(η6‐C6H6)RuCl(μ‐Cl)]2 was heated in CH3CN at 45 °C together with NaOH, KPF6, and Hphpy to yield the cycloruthenated complex [Ru(phpy)(CH3CN)4]PF6. After purification by column chromatography on alumina using CH2Cl2 as eluent, the complex was further reacted with 0.8 equiv of bpy in CH2Cl2 at room temperature for 20 h to afford cis‐[Ru(bpy)(phpy)(CH3CN)2]PF6 ([1 a]PF6), with the carbon donor atom trans to bpy. Achieving the controlled coordination of only one equivalent of bpy is not straightforward, as [Ru(bpy)2(phpy)]PF6 is readily formed in this reaction. To avoid formation of this product, only 0.8 equiv of bpy was added to the reaction mixture. As shown by Ryabov et al., only the isomer having the σ‐bound C atom trans to bpy ([1 a]PF6) is thereby obtained.30 Coordination of the respective N,S bidentate ligand (mtpa, mtea, mtep, or mtmp) was performed by heating the precursor [1 a]PF6 at 70 °C in EtOH in the presence of about 4 equiv. each of the N,S ligand and Et3N (to ensure coordination of the amine group of the N,S ligand) for 22 h under N2. The syntheses of [2]PF6 and [3]PF6 proved to be very air‐sensitive, with a trace of air leading to formation of a dark‐green ruthenium(III)‐containing solid. No such oxidation occurred when preparing complexes [4]PF6 and [5]PF6, which indicated the influence of the nature of the nitrogen ligand of the N,S chelate (amine vs. pyridine) on the redox properties of the cyclometallated complexes. After recrystallization by vapour diffusion of diethyl ether into the crude mixture, [3]PF6, [4]PF6, and [5]PF6 were obtained in yields between 44 % and 58 %, but [2]PF6 was often contaminated with unknown amounts of RuIII impurities, as demonstrated by an absorption band of variable intensity in the 700 nm region (Figure 4; Figures S4a and S5a). In all cases, mass spectrometry clearly showed the molecular peaks corresponding to [2]+–[5]+ (see the Experimental Section).
Figure 4.
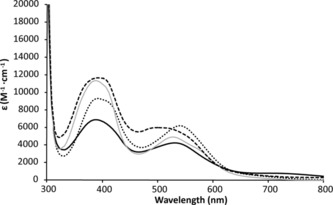
Electronic absorption spectra of compounds [2]PF6 (black continuous, not oxidized), [3]PF6 (dots), [4]PF6 (grey continuous), and [5]PF6 (dashes) in CH3CN.
Stereoselectivity of the coordination
Octahedral complexes bearing three different bidentate ligands, two of which are dissymmetric, have many isomers. The carbon donor atom can be either trans or cis to the nitrogen donor atoms of the bpy ligand, and in each of these cases the nitrogen atom of the N,S ligand can be trans to either the bpy or the phpy− ligand, generating up to four coordination isomers, each of which exists as an enantiomeric pair Λ/Δ (Scheme 1). Following the IUPAC configuration index convention, these four coordination isomers are designated as (OC‐6‐43)‐[Ru(bpy)(phpy)(N,S)]PF6, (OC‐6‐34)‐[Ru(bpy)(phpy)(N,S)]PF6, (OC‐6‐53)‐[Ru(bpy)(phpy)(N,S)]PF6, and (OC‐6‐35)‐[Ru(bpy)(phpy)(N,S)]PF6, but for easier reading we refer to them herein as a, b, c, and d, respectively (Scheme 1). Besides the different configurations of the coordination sphere created by the dissymmetric ligands, the coordinating sulfur atom is a prochiral centre, which, after coordination to ruthenium, can adopt either an R or an S configuration (Scheme 2). Thus, for each of the four Λ coordination isomers, a pair of diastereoisomers Λ‐R and Λ‐S may exist, giving a total of eight possible Λ‐isomers for [3]+ and [5]+. Finally, for the complexes [2]+ and [4]+, the N,S chelate creates a six‐membered ring that can switch between two chair conformations, in which the methyl group of the thioether is in either an equatorial (eq) or an axial (ax) position (Scheme 2).31 These configurations are not identical, and hence there are as many as 16 possible Λ‐isomers for these two complexes.
Scheme 2.
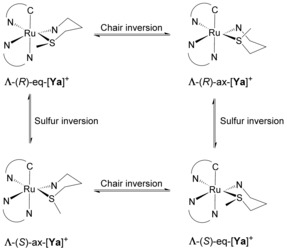
Stereoisomers of [Ya]+ (Y=2 or 4) as a result of the inversion of either the chirality of the sulfur atom (R or S) or the conformation of the six‐membered chair. For clarity, only the Λ isomers are shown, but all samples were obtained as racemic Δ/Λ mixtures.
Detailed 1H NMR analysis was undertaken to assess whether coordination of the dissymmetric N,S chelate showed any stereoselectivity. Quite incredibly, according to the 1H NMR spectra in [D6]acetone, [2]PF6 and [4]PF6 were obtained as single Λ/Δ enantiomeric pairs of isomers. Characteristic doublets corresponding to the proton in the 6‐position of bpy were found at δ=9.82 and 9.44 ppm for [2]PF6 and [4]PF6, respectively, as shown in Figure 1. Single sets of peaks in the aromatic region corresponding to 16 and 20 H, respectively, were found. On the contrary, as shown in Figure 1, the 1H NMR spectrum of [3]PF6 in [D6]acetone showed two doublets at δ=9.02 and 9.11 ppm with an integration ratio of 1:0.8, indicative of two diastereoisomers. For [5]PF6, three doublets were observed in the 1H NMR spectrum at δ=9.16, 9.39, and 9.49 ppm in a ratio of 0.3:1:0.2 (Figure 1), indicating the formation of at least three isomers. Thus, N,S chelates having the appropriate number of carbon atoms (3) between the N and S atoms allowed the stereoselective preparation of the tris‐heteroleptic cyclometallated complexes [2]PF6 and [4]PF6, whereas a shorter linker led to inseparable mixtures of isomers of [3]PF6 and [5]PF6.
Figure 1.
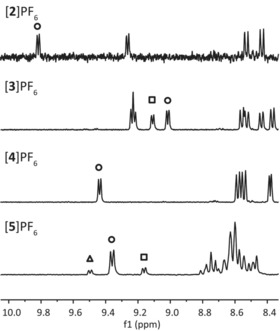
1H NMR spectra of solutions of [2]PF6, [3]PF6, [4]PF6, and [5]PF6 in [D6]acetone. Peaks corresponding to the H6 proton on the bpy ligand (see Scheme 1) of the major isomer are marked with a circle, those for the minor isomers (if any) with a square and a triangle. For complexes [3]PF6 and [5]PF6, mixtures of isomers were obtained, probably due to the higher flexibility of five‐membered metallacycles compared to six‐membered rings. For complexes [2]PF6 and [4]PF6, only one of the 16 possible isomers was obtained (see Schemes 1 and 2). The stereoselectivty of the reaction is explained in the text.
Structural characterization
Characterization of the configurations of [2]PF6 and [4]PF6 was challenging. A more detailed explanation of the deductive process can be found in the Supporting Information. First, density functional theory (DFT) minimization of all isomers of both complexes was performed in water using COSMO to simulate solvent effects (see the Experimental Section). In all calculations, only the Λ enantiomers with the six‐membered chelate ring in a chair conformation were considered, but the sulfur atom was placed in either the R or S configuration. To reduce the number of structures, only those isomers with the methyl group in the equatorial position were calculated.31 The optimized structures and their energies in water are given in Figures S1 and S2 and Table S1, respectively. For [2]+, the isomer Λ‐(S)‐eq‐[2 d]+, in which the σ‐bound C atom is trans to the amine group of mtpa, was found to be the most stable, with the other isomers at energies in a narrow range from +2.6 to +10.0 kJ mol−1 (Table S1). Likewise, for complex [4]+, isomer Λ‐(S)‐eq‐[4 d]+ was found to be the most stable in water. However, the other S isomers were found at higher energies ranging from +8.8 to +10.6 kJ mol−1, and the R isomers at energies ranging from +10.9 to +21.2 kJ mol−1 (Table S1). In this case, the energy differences between the most and least stable isomers are significantly larger than for [2]+, which highlights the different geometric requirements of the sp2 carbon and nitrogen atoms in [4]+ with respect to those of the sp3 atoms in [2]+. Notably, the six‐membered ring involving the mtep ligand was found to be in a pseudo‐chair conformation in the minimized structures, due to the different orbital hybridization of the N and C atoms of the pyridine ring. For example, in Λ‐(S)‐eq‐[4 d]+, the angle Cβ‐Cα‐N is 120.17°, whereas in Λ‐(S)‐eq‐[2 d]+ it is 113.54°.
A potential reason for the increased stabilization of isomer d of [4]+ is that the electron‐rich carbon ligand is trans to the π‐accepting pyridine ligand of mtep, whereas in [2 d]+ the trans primary amine cannot accept the excess electron density. Overall, all of the isomers of both complexes with the sulfur atom in R configuration and the methyl group in an equatorial position show very short distances between this methyl group and the closest proton at the 6‐position of bpy or phpy− (ca. 2.1 Å, Table S1 and Figure 2), whereas in the S configuration the corresponding distance is much longer (ca. 3.5 Å, Table S1). In the latter configuration, the methyl group is positioned above the centre of either the bpy or the phpy− ligand (referred to as ancillary ligands), lowering steric repulsion and thus explaining the general preference for an S configuration of the sulfur atom (Figure 2). Although the structures having the methyl group in the axial position were not minimized, a similar trend is expected. As we have shown in earlier publications,31 inversion of the pseudo‐chair does not change the configuration of the sulfur atom, but it changes the position of the methyl group from equatorial to axial and vice versa. This inversion does not affect the position of the methyl group with respect to the ancillary ligands and the corresponding steric effects.
Figure 2.
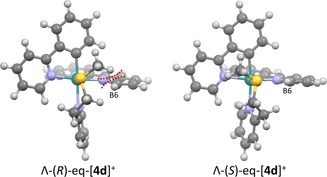
DFT structures of stereoisomers Λ‐(R)‐eq‐[4 d]+ and Λ‐(S)‐eq‐[4 d]+. The isomer with the sulfur in R configuration (left) shows a very short distance between the methyl group and proton B6 on bipyridine, whereas in the isomer with the sulfur in S configuration the methyl group is much further from B6 and resides above the centre of the phpy− ligand, which lowers steric repulsion. This difference in steric interactions is observed in all computed R/S pairs.
To corroborate these DFT analyses experimentally, comprehensive 1H NMR studies in [D6]acetone were performed to assign the stereochemistry of the complexes in solution. These studies, which are explained in detail in the Supporting Information (Figure S3), indicated that, among all possible isomers, the one that best fits the reported off‐diagonal signals is Λ‐(S)‐eq‐[4 c]+ (with the sulfur atom trans to the σ‐bound C), with distances between A6 and Hγ and between C6 and Hβ of 3.101 and 2.253 Å, respectively. Isomer Λ‐(S)‐ax‐[4 d]+, with an axial S‐methyl group, would also fit the reported off‐diagonal signals. Single crystals suitable for X‐ray structure determination were obtained for complexes [4]PF6 by slow vapour diffusion of diethyl ether into a solution of the respective complex in acetone. The crystal structure is a racemate of a single isomer of [Ru(bpy)(phpy)(mtep‐κN,κS)]PF6, and is found in the centrosymmetric space group Pbca, containing both configurations Λ‐(S) and Δ‐(R), with the pyridine moiety of the N,S ligand trans to the σ‐bound C donor atom and the methyl group in a pseudo‐axial position. Thus, the obtained structure corresponds to the isomer Λ‐(S)‐ax‐[4 d]PF6, confirming the geometry predicted by NOESY studies in solution. The structure, shown in Figure 3, features a longer Ru−S bond (2.3331(8) Å, Table 1) compared to the Ru−N bonds of the ancillary ligands (between 2.049(3) and 2.085(3) Å), as expected from the higher ionic radius of sulfur compared to nitrogen. The Ru−N bond (2.239(3) Å) trans to the Ru−C bond (2.027(3) Å) is also significantly longer than the other Ru−N bonds, which is consistent with the expected trans influence of the electron‐rich carbon donor atom.
Figure 3.
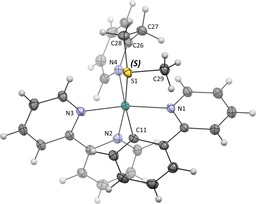
Displacement ellipsoid plot (50 % probability level) of the Λ enantiomer of the cationic complex in the crystal structure of the pair Λ‐(S)/Δ‐(R)‐ax‐[4 d]PF6 at 110(2) K. The hexafluorophosphate counteranion has been omitted for clarity.
Table 1.
Selected bond lengths (Å) and dihedral angle (°) for Λ‐(S)/Δ‐(R)‐ax‐[4 d]PF6.
| Λ‐(S)‐ax‐[4 d]PF6 | |||
|---|---|---|---|
| Ru1−S1 | 2.3311(8) | Ru1−N3 | 2.049(3) |
| Ru1−N1 | 2.085(3) | Ru1−N4 | 2.239(3) |
| Ru1−N2 | 2.060(3) | Ru1−C11 | 2.027(3) |
| S1‐C28‐C26‐N4 | 26.4(2) | ||
In the literature, it is generally accepted that in complexes of the type [Ru(bpy)(phpy)(N,N)]+, synthesized from [Ru(bpy)(phpy)(CH3CN)2]+, any third bidentate N,N ligand would coordinate to ruthenium by simply substituting the CH3CN molecules without isomerization, that is, cis to the carbon donor atom of phpy−.32 However, Pfeffer et al. recently showed that this structural assignment might not be correct.29 CH3CN is a very good ligand for ruthenium(II), and in order to turn it into a good leaving group the complex must first isomerize (either thermally or photochemically) so that one CH3CN ligand becomes trans to the carbon ligand, which is a very reactive position due to the trans effect of the C donor atom.29 This mechanism also seems to occur for the coordination of N,S ligands, as the obtained isomer of [4]+ has the N atom of the last incoming ligand trans to the carbon atom of phpy−, as proven by NOESY studies and X‐ray diffraction analysis.
Electronic spectroscopy and electrochemistry
The UV/Vis absorption spectra of compounds [2]PF6–[5]PF6 in CH3CN are provided in Figure 4, and their absorption maxima (λ max) and molar extinction coefficients (ϵ) are listed in Table 2. It should be noted that for complexes [3]PF6 and [5]PF6, mixtures of two or three isomers were used. A common feature of all of the absorption spectra is the presence of two main bands in the MLCT region: one with λ max at around 390 nm and a broader band between 450 and 650 nm with a lower molar absorption coefficient, with a tail reaching the 700 nm region. According to Bomben et al.,8 the first band corresponds to a 1MLCT transition involving the coordinated carbon atom of the phpy− ligand, whereas the broad band at lower energy corresponds to a Ru→bpy transition. This broader MLCT band compared to those of the non‐cyclometallated analogues is a result of the lower symmetry of the cyclometallated compound.8 The lower‐energy MLCT band has a λ max of 530 and 540 nm for primary amine‐based complexes [2]PF6 and [3]PF6, respectively, whereas pyridine‐based complexes [4]PF6 and [5]PF6 show a blue‐shifted band with λ max at 526 and 501 nm, respectively. Furthermore, the latter two compounds show bands with a shoulder, which could be ascribed to an overlapping Ru→py transition. The higher energy of the MLCT band, that is, the larger gap between the HOMO and the LUMO for [4]PF6 and [5]PF6 compared to those for [2]PF6 and [3]PF6, is another indication of the stabilization of the HOMO in the pyridine‐based complexes. For complex [2]PF6, a band with λ max at 725 nm was also visible, which may have been due to its oxidation. Degradation of [2]PF6 may also explain the much lower molar absorption coefficient of its Ru→phpy− band (6900 m −1 cm−1) compared to those of the other three complexes (around 10 000 m −1 cm−1).
Table 2.
Wavelengths of the MLCT transitions (λ abs/nm) and molar absorptivities (ϵ/m −1 cm−1) of [2]PF6, [3]PF6, [4]PF6, and [5]PF6 in CH3CN, and their redox potentials measured by cyclic voltammetry.[a]
| Complex | λ abs [nm] (ϵ [m −1 cm−1]) | E 1/2 (RuIII/II) [V][a] | ΔE p [V][a] |
|---|---|---|---|
| [2]PF6 | 530 (4300), 389 (6900) | −0.03 | 0.060 |
| [3]PF6 [b] | 540 (6200), 392 (9300) | 0.00 | 0.071 |
| [4]PF6 | 526 (4900), 388 (11400) | +0.16 | 0.00 |
| [5]PF6 [c] | 501 (6000), 395 (11700) | +0.16 | 0.090 |
[a] Measurement conditions: 1 mm complexes in 0.1 m Bu4NPF6/CH3CN, scanning rate 100 mV s−1. The potentials are referenced to Fc+/0. [b] A mixture of two isomers in a ratio of 1:0.8 was used. [c] A mixture of three isomers in a ratio of 0.3:1:0.2 was used.
The electrochemical properties of complexes [2]PF6–[5]PF6 in CH3CN were investigated by cyclic voltammetry in order to gain more insight into their redox stabilities (Figure S4). For complexes [2]PF6 and [3]PF6, the reversible oxidation waves corresponding to the RuIII/RuII couple were observed at potentials E1/2 of −0.03 and 0.00 V vs. Fc+/0, respectively, whereas complexes [4]PF6 and [5]PF6 showed reversible peaks at a significantly higher potential E1/2 of +0.16 V vs. Fc+/0 (Table 2), highlighting the π‐acceptor property of the pyridine‐based N,S chelating ligand, which stabilizes the HOMOs of complexes [4]PF6 and [5]PF6 compared to those of [2]PF6 and [3]PF6. In practice, oxidation of the former compounds is more difficult, which makes them more stable in air, whereas compounds [2]PF6 and [3]PF6 are easily oxidized during synthesis.
Thermal and photochemical stabilities
Thermal stability is an important feature of photoactivatable prodrugs, and the stability in the dark of all four complexes was studied in CH3CN by UV/Vis spectrophotometry.33 Under air, solutions of complexes [3]PF6, [4]PF6, and [5]PF6 in CH3CN did not show any significant changes in their UV/Vis spectra over 10 h, except for a general increase in absorbance due to evaporation of the solvent (Figure S5a–d). Thus, in CH3CN in the dark, neither oxidation nor thermal substitution of the N,S ligand by solvent molecules occurred. For [2]PF6, however, samples showed different degrees of oxidation to RuIII, as demonstrated by an absorption band around 700 nm, and low signal‐to‐noise ratios in all NMR spectra.
The photoreactivities of the complexes were studied in CH3CN by means of UV/Vis spectrophotometry, mass spectrometry, and NMR spectroscopy. When a solution of [3]PF6 in CH3CN was irradiated with light from a green (521 nm) LED at a photon flux of around 6×10−8 mol s−1 under N2, the UV/Vis spectrum did not show any change of the absorption bands. Only a general increase of absorbance was observed due to slow evaporation of the solvent (Figure S6b). Thus, [3]PF6 was not photoreactive in CH3CN, as reported by Turro et al. for [Ru(biq)2(phpy)]+. Irradiation of solutions of [2]PF6 or [5]PF6 showed very slow changes in their UV/Vis spectra over time, with blue shifts of the MLCT bands; isosbestic points were observed at 421 and 552 nm for [2]PF6 (Figure S6a) and at 474 and 546 nm for [5]PF6 (Figure S6c). Although the photoreactions did not reach a steady state, highlighting the very low photosubstitution quantum yields for these two complexes, mass spectra recorded after several hours of irradiation showed a peak at m/z 494.1, corresponding to [Ru(bpy)(phpy)(CH3CN)2]+ (calcd m/z 494.1). For both complexes, photosubstitution of the N,S bidentate ligand occurred, albeit very slowly (Scheme 3, see the Supporting Information).
Scheme 3.
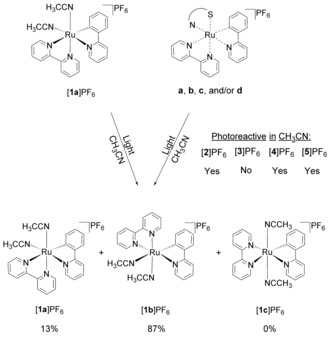
Summary of the products obtained upon irradiation of solutions of complexes [1 a]PF6, [2]PF6, [3]PF6, [4]PF6, and [5]PF6 in CH3CN, performed either with a 521 nm LED with a photon flux of ca. 7×10−8 mol s−1 (0.1 mm) and monitored by UV/Vis spectrophotometry, or with an Xe lamp (2 mm) and monitored by 1H NMR. According to 1H NMR spectra, a mixture of isomers [1 a]+ and [1 b]+ in yields of 13 and 87 %, respectively, was always obtained, with no trace of the trans isomer [1 c]+. For clarity, only the Λ isomers are shown, but all samples were obtained as racemic Δ/Λ mixtures. See the Supporting Information for a discussion on which isomer is obtained.
In contrast, [4]PF6 proved to be much more photolabile. Under the same conditions as above, this complex showed hypsochromic shifts of the absorption maxima of both MLCT bands from 526 and 388 nm to 516 and 376 nm, respectively, reaching a steady state after 6 h (Figure 5). Mass spectrometry at this point showed the peak of the bis‐acetonitrile photoproduct at m/z 494.1, indicative of complete photosubstitution of mtep by two solvent molecules (Scheme 3, see the Supporting Information). Since complex [4]PF6 was the only air‐stable complex of the series that was obtained as a pure isomer, the quantum yield for the photosubstitution of mtep (Φ PR) by acetonitrile could be determined using Glotaran34 global fitting (see the Supporting Information). Although the value obtained, Φ PR=0.00035 (Figure S7), is lower than that of [Ru(bpy)2(mtmp)]Cl2 in water (0.0030),27 [4]+ is the first cyclometallated ruthenium compound capable of quantitatively undergoing photosubstitution of a bidentate ligand. In this complex, as in [2]+, the N,S ligand generates an unfavourable six‐membered ring that probably lowers the ligand field splitting and thus the energy of the 3MC levels, which permits photosubstitution. In contrast, N,S complexes with five‐membered rings, such as [3]+ and [5]+, show very low photoconversion rates. We suggest that these low photosubstitution rates are due to higher ligand field splitting energies, and possibly also faster rechelation (also referred to as recaptation) of the five‐membered ring after initial cleavage of a first Ru−N or Ru−S bond, as has been proposed for [Ru(bpy)3]2+ and [Ru(bpy)2(glutamate‐κN,κO)]2+.35, 36
Figure 5.
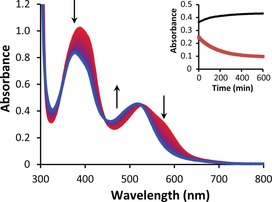
Evolution of the UV/Vis spectra of a solution of [4]PF6 in CH3CN upon irradiation with a 521 nm LED (photon flux 6.80×10−8 mol s−1) under N2. Inset: black dots represent the absorbance at 500 nm vs. time, and red squares represent the absorbance at 590 nm vs. time.
Conclusion
The development of stereoselective syntheses is an important but challenging goal in coordination and organometallic chemistry, whereby the number of isomers of octahedral complexes bearing dissymmetric bidentate chelate ligands can be very high. In this work, the ring size resulting from the coordination of a dissymmetric N,S bidentate ligand to a cyclometallated ruthenium complex was found to have a critical influence on the number of isomers obtained in syntheses of [2]+–[5]+, as well as on their photoreactivities. Under the same conditions, tris‐heteroleptic complexes bearing a six‐membered ring ([2]PF6 and [4]PF6) were stereoselectively obtained as racemic Λ/Δ mixtures, in spite of their exceptional configurational complexity. For the air‐stable compound [4]PF6, the obtained isomer could be unequivocally characterized as Λ‐(S)/Δ‐(R)‐ax‐[4 d]PF6 through a combination of DFT calculations, NMR spectrometry, and single‐crystal X‐ray crystallography. In contrast, the complexes containing five‐membered N,S chelate rings ([3]PF6 and [5]PF6) were obtained as inseparable mixtures of diastereoisomers due to the higher flexibility of the five‐membered metallacycle. Thus, the stereochemical complexity imparted by the two dissymmetric chelates can be controlled by choosing the appropriate ring size in the final synthetic step. Those with a six‐membered N,S chelate ring showed selective substitution of this ligand in CH3CN upon irradiation with green light, since rechelation is slow. Overall, this study has demonstrated that highly dissymmetric, air‐stable cyclometallated complexes that undergo well‐defined photosubstitution of a bidentate ligand with red‐shifted light, can be stereoselectively prepared by fine‐tuning the ligand ring size and controlling the order of ligand coordination.
Experimental Section
Synthesis
General: The ligands 2‐(methylthio)ethylamine (mtea) and 3‐(methylthio)propylamine (mtpa), as well as bis[(benzene)dichlororuthenium] ([η6‐(C6H6)RuCl2]2) and sodium hydroxide (NaOH), were purchased from Sigma–Aldrich. 2‐Phenylpyridine (Hphpy), 2,2′‐bipyridine (bpy), and potassium hexafluorophosphate (KPF6) were purchased from Alfa‐Aesar. All reactants and solvents were used without further purification. [Ru(phpy)(CH3CN)4]PF6, [Ru(bpy)(phpy)(CH3CN)2]PF6 ([1 a]PF6), 2‐(methylthio)methylpyridine (mtmp), and 2‐(methylthio)ethyl‐2‐pyridine (mtep) were synthesized according to literature procedures.28, 30, 37
Electrospray ionization mass spectra (ESI‐MS) were recorded on an MSQ Plus spectrometer. UV/Vis spectra were recorded on a Cary Varian spectrometer. All 1H NMR spectra were recorded on Bruker DPX‐300 or DMX‐400 spectrometers. Chemical shifts are indicated in ppm relative to the residual solvent peak.
Synthesis of complexes [2]PF6, [3]PF6, [4]PF6, and [5]PF6: General procedure: In a two‐necked flask, [1 a]PF6 (1 equiv), N,S ligand (4 equiv), and Et3N (4 equiv) were dissolved in deaerated EtOH (2–5 mL), and the mixture was heated in an oil bath at 70 °C for 22 h under N2. A Schlenk flask containing diethyl ether was then attached to the flask containing the reaction mixture in order to obtain a crystalline dark precipitate by slow vapour diffusion. The solid was collected by filtration, washed with diethyl ether, and stored at −20 °C.
[Ru(bpy)(phpy)(mtpa)]PF6 ([2]PF6): According to the general procedure, a solution of [1]PF6 (40 mg, 0.063 mmol), mtpa (28 μL, 0.25 mmol), and Et3N (40 mg, 0.29 mmol) in EtOH (5 mL) was heated at 70 °C for 22 h under N2. After 6 days of slow vapour diffusion of diethyl ether into the solution, a crystalline dark precipitate was obtained (23 mg, 52 %). 1H NMR (300 MHz, [D6]acetone): δ=9.81 (dt, J=5.8, 1.1 Hz, 1 H), 9.26 (ddd, J=5.7, 1.6, 0.8 Hz, 1 H), 8.52 (dt, J=8.2, 1.2 Hz, 1 H), 8.43 (dt, J=8.1, 1.1 Hz, 1 H), 8.16 (dt, J=8.1, 1.1 Hz, 1 H), 8.07–8.02 (m, 1 H), 7.97 (ddd, J=8.2, 7.3, 1.6 Hz, 1 H), 7.81 (ddd, J=8.2, 7.5, 1.5 Hz, 1 H), 7.74 (ddd, J=8.8, 7.5, 1.4 Hz, 2 H), 7.67 (dd, J=5.7, 0.8 Hz, 1 H), 7.46 (ddd, J=7.2, 5.7, 1.4 Hz, 1 H), 7.24 (ddd, J=7.3, 5.7, 1.4 Hz, 1 H), 6.73–6.66 (m, 1 H), 6.59 (td, J=7.3, 1.4 Hz, 1 H), 6.40 (dd, J=7.4, 1.2 Hz, 1 H), 1.15 ppm (s, 3 H); high‐resolution ESI‐MS: m/z (calcd): 517.09851 (517.09944, [2]+); UV/Vis (CH3CN): λ (ϵ)=530 (4300), 389 nm (6900 m −1 cm−1).
[Ru(bpy)(phpy)(mtea)]PF6 ([3]PF6): According to the general procedure, a solution of [1]PF6 (15 mg, 0.024 mmol), mtea (8.5 μL, 0.095 mmol), and Et3N (7.0 μL, 0.095 mmol) in EtOH (2 mL) was heated at 70 °C for 22 h under N2. After 5 days of slow vapour diffusion of diethyl ether into the solution, a crystalline dark precipitate was obtained (9.0 mg, 58 %). Two isomers A/B in a ratio of 1:0.8 were obtained. 1H NMR (400 MHz, [D6]acetone): δ=9.26–9.19 (m, 1 HA + 1 HB), 9.11 (d, J=5.1 Hz, 1 HB), 9.02 (d, J=5.8 Hz, 1 HA), 8.56 (d, J=8.0 Hz, 1 HA), 8.53 (d, J=8.0 Hz, 1 HB), 8.44 (dt, J=6.5, 1.0 Hz, 1 HB), 8.37 (dt, J=8.0, 1.2 Hz, 1 HA), 8.16 (d, J=8.1 Hz, 1 HB), 8.14–8.07 (m, 2 HA), 8.05–8.00 (m, 1 HB), 7.97 (ddd, J=8.2, 7.3, 1.5 Hz, 1 HB), 7.91–7.85 (m, 2 HA), 7.83 (d, J=5.8 Hz, 1 HB), 7.82–7.73 (m, 2 HA + 2 HB), 7.71–7.64 (m, 1 HA + 1 HB), 7.43 (ddd, J=7.2, 5.7, 1.4 Hz, 1 HB), 7.38 (ddd, J=7.3, 5.7, 1.5 Hz, 1 HA), 7.22 (ddd, J=7.3, 5.7, 1.4 Hz, 1 HB), 7.11 (ddd, J=7.3, 5.8, 1.4 Hz, 1 HA), 6.77 (ddd, J=7.7, 7.1, 1.4 Hz, 1 HA), 6.74–6.67 (m, 1 HA + 1 HB), 6.64 (td, J=7.3, 1.4 Hz, 1 HB), 6.52–6.47 (m, 1 HA + 1 HB), 4.24 (d, J=11.4 Hz, 1 H), 4.12 (d, J=12.1 Hz, 1 H), 3.50–3.35 (m, 1 H), 3.20–3.00 (m, 4 H), 2.97–2.86 (m, 3 H), 2.76–2.65 (m, 1 H), 1.67 (s, 3 H), 1.27 ppm (s, 3 H); high‐resolution ESI‐MS: m/z (calcd): 503.08379 (503.08585, [3]+); UV/Vis (CH3CN): λ (ϵ)=540 (6200), 392 nm (9300 m −1 cm−1).
[Ru(bpy)(phpy)(mtep)]PF6 ([4]PF6): According to the general procedure, a solution of [1]PF6 (30 mg, 0.046 mmol), mtep (27 mg, 0.18 mmol), and Et3N (26 μL, 0.19 mmol) in EtOH (5 mL) was heated at 70 °C for 22 h under N2. After 4 days of slow vapour diffusion of diethyl ether into the solution, a crystalline dark precipitate was obtained (14 mg, 44 %) as a pure single isomer. 1H NMR (400 MHz, [D6]acetone): δ=9.44 (d, J=5.8 Hz, 1 H; A6), 8.58 (dt, J=7.9 Hz, 1 H; A3), 8.54 (dt, J=8.1, 1.1 Hz, 1 H; B3), 8.38 (ddd, J=5.7, 1.6, 0.8 Hz, 1 H; C6), 8.21 (dt, J=8.4, 1.2 Hz, 1 H; C3), 8.06–8.00 (m, 1 H; A4), 7.99–7.94 (m, 1 H; C4), 7.94–7.88 (m, 1 H; B4), 7.84–7.78 (m, 2 H; Py5 + Ph3), 7.68–7.63 (m, 2 H; A5 + B6), 7.56–7.51 (m, 2 H; Py3 + Py6), 7.36 (ddd, J=7.4, 5.7, 1.4 Hz, 1 H; B5), 7.30 (ddd, J=7.3, 5.7, 1.4 Hz, 1 H; C5), 7.08 (ddd, J=7.3, 5.7, 1.4 Hz, 1 H; Py4), 6.78 (ddd, J=7.7, 7.2, 1.3 Hz, 1 H; Ph4), 6.68 (td, J=7.3, 1.4 Hz, 1 H; Ph5), 6.42–6.36 (m, 1 H; Ph6), 3.46–3.41 (m, 2 H; β), 3.09–2.99 (m, 2 H; γ), 1.28 ppm (s, 3 H; CH3S‐); 13C NMR (101 MHz, [D6]acetone): δ=152.98, 152.33, 152.06, 150.15, 138.38, 137.17, 137.08, 135.80, 134.97, 128.95, 128.19, 127.52, 127.19, 124.72, 124.59, 124.43, 124.06, 123.46, 121.61, 120.16, 34.95, 32.00, 15.20 ppm; high‐resolution ESI‐MS: m/z (calcd): 565.10083 (565.09944, [4]+); elemental analysis calcd (%) for C29H27F6N4PRuS: C 49.08, H 3.84, N 7.90; found: C 48.84, H 3.99, N 7.65; UV/Vis (CH3CN): λ (ϵ): 526 (4900), 388 nm (11 300 m −1 cm−1).
[Ru(bpy)(phpy)(mtmp)]PF6 ([5]PF6): According to the general procedure, a solution of [1]PF6 (20 mg, 0.031 mmol), mtmp (17 mg, 0.12 mmol), and Et3N (20 μL, 0.14 mmol) in EtOH (5 mL) was heated at 70 °C for 22 h under N2. After 5 days of slow vapour diffusion of diethyl ether into the solution, a crystalline dark precipitate was obtained (9.7 mg, 45 %). 1H NMR of three isomers labelled as A, B, and C (300 MHz, [D6]acetone): δ=9.50 (d, J=5.6 Hz, 1 HA), 9.36 (dd, J=5.5, 1.2 Hz, 1 HB), 9.16 (d, J=5.8 Hz, 1 HB), 8.83–8.74 (m, 1 HA + 1 HC), 8.62 (dt, J=7.4, 1.8 Hz, 2 HB), 8.53–8.46 (m, 1 HA + 1 HC), 8.31 (d, J=6.9 Hz, 1 HC), 8.24–8.19 (m, 1 HA + 1 HB + 1 HC), 8.17–8.09 (m, 1 HC), 8.07–8.00 (m, 1 HB), 8.02–7.93 (m, 1 HA + 1 HB + 1 HC), 7.87–7.79 (m, 5 H), 7.74–7.67 (m, 1 HA + 1 HC), 7.61 (ddd, J=7.3, 5.8, 1.4 Hz, 1 HB), 7.49 (d, J=5.5 Hz, 1 HB), 7.39 (ddd, J=7.3, 5.7, 1.4 Hz, 1 HB), 7.29–7.25 (m, 1 HA + 1 HC), 7.22 (t, J=8.0 Hz, 1 HB), 7.08–7.02 (m, 1 HA + 1 HC), 6.99 (d, J=9.0 Hz, 1 HA), 6.91–6.84 (m, 1 HC), 6.83–6.76 (m, 1 HB), 6.68 (td, J=7.3, 1.4 Hz, 1 HB), 6.48 (dd, J=7.4, 1.3 Hz, 1 HB), 6.42 ppm (d, J=6.8 Hz, 1 HA); ESI‐MS: m/z (calcd): 551.1 (551.1, [5]+); UV/Vis (CH3CN): λ (ϵ)=501 (6000), 395 nm (11 700 m −1 cm−1).
Cyclic voltammetry
Electrochemical measurements were performed at room temperature under argon using an Autolab PGstat10 potentiostat controlled by NOVA software. A three‐electrode cell system was used, with a glassy carbon working electrode, a platinum counter electrode, and an Ag/AgCl reference electrode. All electrochemistry experiments were conducted in CH3CN solution with tetrabutylammonium hexafluorophosphate as the supporting electrolyte. Ferrocene was used as a reference after the measurement.
Photochemistry
General: For experiments involving irradiation of NMR tubes, the light of an LOT 1000 W xenon arc lamp fitted with 400 nm long‐pass and IR filters was used. For NMR experiments under N2, NMR tubes with PTFE stoppers were used. UV/Vis experiments were performed on a Cary 50 Varian spectrometer. When following photoreactions by UV/Vis spectrophotometry and mass spectrometry, an LED light source (λ ex=521 nm; full‐width at half‐maximum of 33 nm) with a photon flux of between 6.09 and 8.62×10−8 mol s−1 was used.
Experiments monitored by 1 H NMR spectroscopy: A stock solution of either [1 a]PF6 (2.61 mm), [2]PF6 (4.47 mm), or [4]PF6 (1.84 mm) in CD3CN was prepared and deaerated under N2. An aliquot (660 μL) of this solution was then transferred, under N2, to an NMR tube. The tube was irradiated at room temperature with light from an LOT 1000 W xenon lamp equipped with IR short‐pass and 400 nm long‐pass filters. In addition, a control experiment was performed without white light irradiation. The reactions were monitored by recording 1H NMR spectra at various time intervals.
Experiments monitored by UV/Vis spectrophotometry and MS: UV/Vis spectrophotometry was performed with a UV/Vis spectrophotometer equipped with a temperature controller set to 298 K and a magnetic stirrer. The irradiation experiments were performed with quartz cuvettes containing 3 mL of solution. A stock solution of the requisite complex was prepared in CH3CN, which was then diluted in the cuvette to a working solution concentration. When the experiment was carried out under N2, the sample was deaerated for 15 min by gentle bubbling of N2 and the atmosphere was kept inert during the experiment by a gentle flow of N2 on top of the cuvette. A UV/Vis spectrum was measured every 30 s for the first 10 min, every 1 min for the next 10 min, and eventually every 10 min until the end of the experiment. Data were analyzed with Microsoft Excel.
Single‐crystal X‐ray crystallography
General: All reflection intensities were measured at 110(2) K on a SuperNova diffractometer (equipped with an Atlas detector) employing CuKα radiation (λ=1.54178 Å), using the program CrysAlisPro (Version CrysAlisPro 1.171.39.29c, Rigaku OD, 2017). The same program was used to refine the cell dimensions and for data reduction. The structure was solved with the program SHELXS‐2014/7 and refined against F 2 with SHELXL‐2014/7.38 Analytical numeric absorption correction based on a multifaceted crystal model was applied using CrysAlisPro. The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). H atoms were placed in calculated positions (unless otherwise specified) using the instructions AFIX 23, AFIX 43, or AFIX 137 with isotropic displacement parameters having values 1.2 times U eq of the attached C atoms.
Crystal growth: [4]PF6 (1.0 mg) was dissolved in acetone (1 mL, 1.2 mm) and an aliquot (300 μL) of this solution was transferred to a GC vial, which was placed in a larger vial that contained diethyl ether (3 mL) as a counter solvent. The large vial was stoppered. After a few days, quality crystals suitable for X‐ray structure determination were obtained by vapour diffusion.
Crystallographic data: The structure of [4]PF6 is ordered. 0.26×0.07×0.02 mm3, orthorhombic, Pbca, a=10.95531(15), b=15.6391(3), c=32.2937(4) Å, V=5532.92(15) Å3, Z=8, μ=6.46 mm−1, T min–T max: 0.254–0.886. 28 523 reflections were measured up to a resolution of (sin θ/λ)max=0.617 Å−1. 5427 reflections were unique (R int =0.044), of which 4742 were observed [I>2σ(I)]. 380 parameters were refined. R 1/wR 2 [I>2σ(I)]: 0.0347/0.0797. R 1/wR 2 (all reflections): 0.0411/0.0829. S=1.09. Residual electron density found between −0.61 and 0.77 e Å−3. CCDC 1885015 ([4]PF6) contains the supplementary crystallographic data. These data can be obtained free of charge by The Cambridge Crystallographic Data Centre
Density functional theory calculations
Electronic structure calculations were performed using DFT, as implemented in the ADF program (SCM). The structures of all possible isomers of [2]+ and [4]+ were optimized in water using COSMO to simulate the effect of the solvent. The PBE0 functional and a triple‐ζ potential basis set (TZP) were used for all calculations.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
The European Research Council is kindly acknowledged for an ERC starting grant to S.B. The NWO is kindly acknowledged for a VIDI grant to S.B. The COST action CM1105 is acknowledged for stimulating scientific discussions. Prof. E. Bouwman is kindly acknowledged for support and scientific discussions.
J.-A. Cuello-Garibo, C. C. James, M. A. Siegler, S. L. Hopkins, S. Bonnet, Chem. Eur. J. 2019, 25, 1260.
References
- 1. Dörr M., Meggers E., Curr. Opin. Chem. Biol. 2014, 19, 76–81. [DOI] [PubMed] [Google Scholar]
- 2. Feng L., Geisselbrecht Y., Blanck S., Wilbuer A., Atilla-Gokcumen G. E., Filippakopoulos P., Kräling K., Celik M. A., Harms K., Maksimoska J., Marmorstein R., Frenking G., Knapp S., Essen L.-O., Meggers E., J. Am. Chem. Soc. 2011, 133, 5976–5986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burke C. S., Keyes T. E., RSC Adv. 2016, 6, 40869–40877. [Google Scholar]
- 4. Spiccia L., Deacon G. B., Kepert C. M., Coord. Chem. Rev. 2004, 248, 1329–1341. [Google Scholar]
- 5. Thummel R., Lefoulon F., Chirayil S., Inorg. Chem. 1987, 26, 3072–3074. [Google Scholar]
- 6. von Zelewsky A., Gremaud G., Helv. Chim. Acta 1988, 71, 1108–1115. [Google Scholar]
- 7. Wadman S. H., Kroon J. M., Bakker K., Lutz M., Spek A. L., van Klink G. P. M., van Koten G., Chem. Commun. 2007, 1907–1909. [DOI] [PubMed] [Google Scholar]
- 8. Bomben P. G., Robson K. C. D., Sedach P. A., Berlinguette C. P., Inorg. Chem. 2009, 48, 9631–9643. [DOI] [PubMed] [Google Scholar]
- 9. Leyva L., Sirlin C., Rubio L., Franco C., Le Lagadec R., Spencer J., Bischoff P., Gaiddon C., Loeffler J.-P., Pfeffer M., Eur. J. Inorg. Chem. 2007, 3055–3066. [Google Scholar]
- 10. Huang H., Zhang P., Chen H., Ji L., Chao H., Chem. Eur. J. 2015, 21, 715–725. [DOI] [PubMed] [Google Scholar]
- 11. Zeng L., Chen Y., Huang H., Wang J., Zhao D., Ji L., Chao H., Chem. Eur. J. 2015, 21, 15308–15319. [DOI] [PubMed] [Google Scholar]
- 12. Fetzer L., Boff B., Ali M., Xiangjun M., Collin J.-P., Sirlin C., Gaiddon C., Pfeffer M., Dalton Trans. 2011, 40, 8869–8878. [DOI] [PubMed] [Google Scholar]
- 13. Peña B., David A., Pavani C., Baptista M. S., Pellois J.-P., Turro C., Dunbar K. R., Organometallics 2014, 33, 1100–1103. [Google Scholar]
- 14. Sainuddin T., McCain J., Pinto M., Yin H., Gibson J., Hetu M., McFarland S. A., Inorg. Chem. 2016, 55, 83–95. [DOI] [PubMed] [Google Scholar]
- 15. Bonnet S., Dalton Trans. 2018, 47, 10330–10343. [DOI] [PubMed] [Google Scholar]
- 16. Pinnick D. V., Durham B., Inorg. Chem. 1984, 23, 1440–1445. [Google Scholar]
- 17. Salassa L., Garino C., Salassa G., Gobetto R., Nervi C., J. Am. Chem. Soc. 2008, 130, 9590–9597. [DOI] [PubMed] [Google Scholar]
- 18. Wagenknecht P. S., Ford P. C., Coord. Chem. Rev. 2011, 255, 591–616. [Google Scholar]
- 19. Kreitner C., Heinze K., Dalton Trans. 2016, 45, 13631–13647. [DOI] [PubMed] [Google Scholar]
- 20. Collin J.-P., Beley M., Sauvage J.-P., Barigelletti F., Inorg. Chim. Acta 1991, 186, 91–93. [Google Scholar]
- 21. Collin J.-P., Jouvenot D., Koizumi M., Sauvage J.-P., Inorg. Chem. 2005, 44, 4693–4698. [DOI] [PubMed] [Google Scholar]
- 22. Mobian P., Kern J.-M., Sauvage J.-P., Angew. Chem. 2004, 116, 2446–2449. [Google Scholar]
- 23. Albani B. A., Pena B., Dunbar K. R., Turro C., Photochem. Photobiol. Sci. 2014, 13, 272–280. [DOI] [PubMed] [Google Scholar]
- 24. Palmer A. M., Pena B., Sears R. B., Chen O., El Ojaimi M., Thummel R. P., Dunbar K. R., Turro C., Philos. Trans. R. Soc. London Ser. A 2013, 371, 20120135. [DOI] [PubMed] [Google Scholar]
- 25. Kohler L., Nease L., Vo P., Garofolo J., Heidary D. K., Thummel R. P., Glazer E. C., Inorg. Chem. 2017, 56, 12214–12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hidayatullah A. N., Wachter E., Heidary D. K., Parkin S., Glazer E. C., Inorg. Chem. 2014, 53, 10030–10032. [DOI] [PubMed] [Google Scholar]
- 27. Cuello-Garibo J. A., Meijer M. S., Bonnet S., Chem. Commun. 2017, 53, 6768–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fernandez S., Pfeffer M., Ritleng V., Sirlin C., Organometallics 1999, 18, 2390–2394. [Google Scholar]
- 29. Boff B., Ali M., Alexandrova L., Espinosa-Jalapa N. Á., Saavedra-Díaz R. O., Le Lagadec R., Pfeffer M., Organometallics 2013, 32, 5092–5097. [Google Scholar]
- 30. Ryabov A. D., Le Lagadec R., Estevez H., Toscano R. A., Hernandez S., Alexandrova L., Kurova V. S., Fischer A., Sirlin C., Pfeffer M., Inorg. Chem. 2005, 44, 1626–1634. [DOI] [PubMed] [Google Scholar]
- 31. Cuello-Garibo J. A., James C. C., Siegler M. A., Bonnet S., Chem. Sq. 2017, 1, 2. [Google Scholar]
- 32. Peña B., Leed N. A., Dunbar K. R., Turro C., J. Phys. Chem. C 2012, 116, 22186–22195. [Google Scholar]
- 33. Cuello-Garibo J.-A., Pérez-Gallent E., van der Boon L., Siegler M. A., Bonnet S., Inorg. Chem. 2017, 56, 4818–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Snellenburg J. J., Laptenok S. P., Seger R., Mullen K. M., van Stokkum I. H. M., J. Stat. Softw. 2012, 49, 1–22. [Google Scholar]
- 35. Arakawa R., Tachiyashiki S., Matsuo T., Anal. Chem. 1995, 67, 4133–4138. [Google Scholar]
- 36. Zayat L., Filevich O., Baraldo L. M., Etchenique R., Philos. Trans. R. Soc. London Ser. A 2013, 371, 20120330. [DOI] [PubMed] [Google Scholar]
- 37. Reisner E., Abikoff T. C., Lippard S. J., Inorg. Chem. 2007, 46, 10229–10240. [DOI] [PubMed] [Google Scholar]
- 38. Sheldrick G. M., Acta Crystallogr. Sect. A 2008, 64, 112–122. [DOI] [PubMed] [Google Scholar]