

Supplemental Digital Content is available in the text.
Keywords: apoptosis, cell proliferation, endothelial cells, mutation, umbilical vein
Abstract
Objective—
Venous malformations (VMs) arise from developmental defects of the vasculature and are characterized by massively enlarged and tortuous venous channels. VMs grow commensurately leading to deformity, obstruction of vital structures, bleeding, and pain. Most VMs are associated with the activating mutation L914F in the endothelial cell (EC) tyrosine kinase receptor TIE2. Therapeutic options for VM are limited and ineffective while therapy with the mammalian target of rapamycin inhibitor rapamycin shows moderate efficacy. Here, we investigated novel therapeutic targets promoting VM regression.
Approach and Results—
We performed an unbiased screen of Food and Drug Administration–approved drugs in human umbilical vein ECs expressing the TIE2-L914F mutation (HUVEC-TIE2-L914F). Three ABL (Abelson) kinase inhibitors prevented cell proliferation of HUVEC-TIE2-L914F. Moreover, c-ABL, common target of these inhibitors, was highly phosphorylated in HUVEC-TIE2-L914F and VM patient–derived ECs with activating TIE2 mutations. Knockdown of c-ABL/ARG in HUVEC-TIE2-L914F reduced cell proliferation and vascularity of murine VM. Combination treatment with the ABL kinase inhibitor ponatinib and rapamycin caused VM regression in a xenograft model based on injection of HUVEC-TIE2-L914F. A reduced dose of this drug combination was effective in this VM murine model with minimal side effects. The drug combination was antiproliferative, enhanced cell apoptosis and vascular channel regression both in vivo and in a 3-dimensional fibrin gel assay.
Conclusions—
This is the first report of a combination therapy with ponatinib and rapamycin promoting regression of VM. Mechanistically, the drug combination enhanced AKT inhibition compared with single drug treatment and reduced PLCγ (phospholipase C) and ERK (extracellular signal-regulated kinase) activity.
Highlights.
c-ABL (Abelson) is constitutively active downstream of mutated TIE2-L914F and patient-derived venous malformation-endothelial cell.
ABL kinase inhibitors prevented proliferation of TIE2-L914F human umbilical vein endothelial cell.
-
Combination treatment with ABL kinase inhibitor ponatinib and mammalian target of rapamycin inhibitor rapamycin-induced lesion regression in xenograft models based on human umbilical vein endothelial cell–TIE2-L914F by:
Reducing cell proliferation.
Increasing cell apoptosis.
Mechanistically, by concomitantly enhancing AKT (protein kinase B) inhibition and reducing PLCγ (phospholipase C)-ERK (extracellular signal-regulated kinase) signaling.
Venous malformations (VMs) are slow-flow vascular anomalies with an estimated incidence of ≈1/10 000 of the population.1–3 VMs are compressible bluish lesions that can grow commensurately with the developing child causing disfiguration and morbidity.1,4 Pathologically, VM lesions are characterized by ectatic endothelial cell (EC)-lined channels covered by rare and irregularly distributed smooth muscle cells.5 Expansion of VM can cause extensive disfigurement, organ dysfunction, and other chronic manifestations, including bleeding, oozing, and pain.6,7 Elevated D-dimer levels (>0.5 μg/mL), spontaneous thrombosis, and localized intravascular coagulopathy are reported in ≈50% of VM patients.8,9 To reduce the thrombosis events and the pain caused by phleboliths, tailored compression garments, and low molecular weight heparin are often used. Sclerotherapy, alone or in combination with surgery, is the standard first-line therapy. However, these procedures are highly invasive and need to be performed repeatedly because of lesion recurrence.10
According to the International Society for the Study of Vascular Anomalies, VMs are subdivided into sporadic VM, inherited cutaneomucosal VM, blue rubber bleb nevus syndrome, and glomuvenous malformations.11 Up to 60% of sporadic VM are associated with activating mutations in the EC tyrosine kinase receptor TIE2-encoding gene (TEK).12,13 Most TIE2 mutations occur in the intracellular, tyrosine kinase, or kinase insert domain, with the most common substitution being p.L914F. Expression of TIE2 mutations in human umbilical vein ECs (HUVECs) causes ligand-independent phosphorylation of TIE2, although to varying degrees.12,13 Activation of TIE2 increases the phosphorylation of downstream pathways including PI3K (phosphoinositide-3-kinase)/AKT (protein kinase B) to promote EC proliferation and survival.12,14 We previously showed that the TIE2-activating mutation p.L914F is sufficient to induce HUVEC to form VM lesions in nude mice.15 These murine VMs are bluish lesions that expand over time and have similar histological characteristics to patients’ VM. Recent studies reported somatic activating mutations in the catalytic subunit of class I phosphoinositide 3-kinase (PIK3CA) in association with ≈25% of VM cases.12,14,16,17 PIK3CA mutations have been reported in several types of cancer,18 overgrowth syndromes,19–21 and lymphatic malformations.22–24
The mammalian target of rapamycin (mTOR) integrates signals from the PI3K/AKT pathway to regulate multiple cellular processes, including cell growth and proliferation.25 Enhanced mTOR signaling can increase expression of VEGF (vascular endothelial growth factor) thus promoting pathological angiogenesis. The mTOR inhibitor rapamycin suppresses TIE2-L914F–induced AKT phosphorylation and inhibits murine VM lesion expansion although it fails to promote regression.15 Clinical trials of rapamycin in patients with difficult-to-treat VM and complicated vascular anomalies reported improved clinical symptoms and tolerated toxicity.6,7 Hence, rapamycin has become a new therapeutic option for VM patients who are refractory to standard care, although lesion regression does not occur.
The ABL (Abelson) family of nonreceptor tyrosine kinases (c-ABL and ARG) has critical roles in regulating cytoskeletal reorganization, cell proliferation, and survival.26 Enhanced ABL expression occurs as a consequence of chromosomal translocation of BCR (breakpoint cluster region protein)-ABL1 fusion proteins, which promotes constitutive ABL kinase activity and drives human leukemia. Imatinib is the first ABL kinase inhibitor approved for the treatment of chronic myelogenous leukemia and Philadelphia-positive leukemia.27 ATP-competitive ABL kinase inhibitors ponatinib, nilotinib, and bosutinib improved efficacy and overcame resistance to imatinib in patients. These inhibitors are also under investigation to treat diverse pathologies characterized by hyperactive ABL kinases.26
Here, to identify new and improved therapies for VM, we performed an unbiased screening of Food and Drug Administration–approved drugs and found that mTOR and ABL kinase inhibitors were the most potent compounds decreasing HUVEC-TIE2-L914F cell proliferation. Hence, we hypothesized that ABL kinase inhibitors combined with rapamycin would be more efficacious in causing regression of VM lesions. We found that c-ABL is constitutively phosphorylated downstream of TIE2-L914F and in patient-derived ECs expressing activating TIE2 mutations. We determined that c-ABL is required for HUVEC-TIE2-L914F cell proliferation and VM formation by using the c-ABL inhibitor ponatinib and genetic knockdown of c-ABL/ARG. Finally, our studies determined that ponatinib combined with rapamycin is a novel targeted therapy to induce VM regression.
Materials and Methods
Reagents
The Food and Drug Administration–approved drug library was provided by the National Cancer Institute Development Therapeutics Program (https://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm). Ponatinib and rapamycin were purchased from LC Laboratories. Wortmannin was purchased from Selleckchem. An equal volume of dimethyl sulfoxide (DMSO) was used as control/vehicle.
Cell Culture
HUVEC and retrovirally transfected HUVEC expressing full-length TIE2-wild type (WT) or TIE2-L914F were previously described.15 Cells were expanded in culture on 1% (w/v) gelatin/PBS-coated plates and EC growth medium (EGM-2; Lonza)/10% fetal bovine serum (HyClone). ECs from VM patients numbers 1 and 2 (VM-EC) were collected from freshly resected VM lesions as previously described.14 Briefly, VM tissue was digested, and ECs were purified using CD31 immunomagnetic beads (Dako). VM-ECs were grown on fibronectin (0.5 μg/cm2; Millipore) coated plates in EGM-2/20% fetal bovine serum medium.
Tissue Samples
Patient tissue samples were obtained from participants after informed consent from the collection and repository of tissue samples and data from patients with tumors and vascular anomalies (Institutional Review Board no. 2008-2001 and Institutional Review Board no. 2016-3878 per institutional policies) at Cincinnati Children’s Hospital Medical Center, Cancer and Blood Disease Institute and with approval of the Committee on Clinical Investigation. Collected data and identifying names were stored in a secure database maintained by the Cancer and Blood Disease Institute. This was further deidentified by creating a patient ID for use in this study.
c-ABL/ARG Double Knockdown
The sequences in the shRNA vectors targeting c-ABL and ARG are as follows. c-ABL (1): CCGGGCTGAAATCCACCAAGCCTTTCTCGAGAAAGGCTTGGTGGATTTCAGCTTTTTG, c-ABL (2): CCGGGCAGTCATGAAAGAGATCAAACTCGAGTTTGATCTCTT TCATGA CTGCTTTTTG, ARG (1): CCGGGTTCCATGACTCCAGCATTTCCTCGAGGAAATGCTG GAGTCATGGAACTTTTTTG, ARG (2): CCGGCCTATGGAATGTCACCATATCCTCGAG GATATGGTGACATTCCATAGGTTTTTTG. Lentivirus was packaged in 293T cells. Stable c-ABL and ARG shRNA double knockdown (sh c-ABL/ARG) and shRNA scramble knockdown cell lines (sh Scramble) were established by infecting HUVEC-TIE2-L914F with 2 different sets of c-ABL and ARG targeting virus (target1: c-ABL (1)+ARG (1) and target2: c-ABL (2)+ARG (2)) or scramble sequence virus, then selected in medium containing puromycin (1.5 μg/mL). The knockdown efficiencies were evaluated by real-time polymerase chain reaction and immunoblotting.
Cell Proliferation Assay and Combination Index Calculation
Cell proliferation was measured by sulforhodamine B assays,28 and the optical density (OD) value was read at 540 nm using SpectraMax i3x Multi-Mode Detection Platform (Molecular Devices). The percent (%) inhibitory rate of compounds was calculated as (OD540 control−OD540 compound)/OD540 control×100 while the % inhibitory rate of combination rapamycin was calculated as (OD540 rapamycin−OD540 combination)/OD540 rapamycin×100. Combination index (CI) values were calculated as previously described.29
Immunoblotting
Cells were washed with PBS then lysed using radioimmunoprecipitation assay buffer with protease inhibitor and phosphatase inhibitor cocktail (Roche). Cell lysates were analyzed by immunoblotting with antibodies against the following: phospho-c-ABL (Y245), c-ABL, phospho-TIE2, phospho-AKT (Ser473 and Thr308), AKT, phospho-PDK1, PDK1, phospho-ERK1/2 (extracellular signal-regulated kinase), ERK1/2, phospho-PLCγ (phospholipase C; Tyr783) and PLCγ (Cell Signaling), ARG (Novus Biologicals), TIE2 (Abcam), and Tubulin or β-Actin (Sigma). Bands were quantified with ImageJ software. For antibody concentration, please see the Major Resources Table in the online-only Data Supplement.
Murine Model of VM
A total of 2.5×106 HUVEC-TIE2-L914F were suspended in 200 μL of Matrigel (Corning) and injected subcutaneously on both flanks of 6 to 7 weeks old male athymic nu/nu mice (Envigo; n=8 mice with 2 injections/mouse) as previously described.15 All the procedures were approved by the Animal Care and Use Committee of the Cincinnati Children’s Hospital. Nude mice are only acting as a host for the grafted cells, using male mice that do not have the hormone cycling is the best strategy to study vasculogenesis. Animals received oral gavage daily of either 200 μL vehicle (citric buffer pH 2.5; 25 mmol/L—ethanol 30% [v/v] solution), ponatinib (30 mg/kg), rapamycin (2 mg/kg), ponatinib and rapamycin combination, and reduced-dose combination (RD combo; 20 mg/kg ponatinib+1 mg/kg rapamycin). For lesion expansion studies, daily oral administration by gavage feeding started from day 1. For regression studies, treatment started when average lesion size in each group exceeded 110 or 130 mm2. The size of the lesions was measured using a caliper. Lesions were dissected, weighed, and fixed in 10% formalin. Sections were stained with Hematoxylin and Eosin. Images were acquired with EVOS cell imaging system (Invitrogen). Five images were taken randomly per section, and vascular area (%) was quantified by a blinded operator with ImageJ software, as previously reported.15
For c-ABL/ARG double knockdown studies 2.5×106 sh Scramble or sh c-ABL/ARG HUVEC-TIE2-L914F were injected in mice as described above.
Immunohistochemistry
Paraffin sections of murine VM lesions were stained with UEA I (Ulex europaeus Agglutinin I; Vector Laboratories), cleaved caspase-3 (Cell Signaling), and anti-human Ki-67 (Abcam). This was followed by peroxidase-conjugated secondary antibody and staining with diaminobenzidine peroxidase (horseradish peroxidase) substrate (Vector Laboratories). Slides were counterstained using hematoxylin (Vector Laboratories).
Blood and Chemical Profiling
Peripheral blood was processed at the Cincinnati Children’s Hospital Medical Center Veterinary Laboratory. Analysis included complete blood counts, liver, and kidney function tests. D-dimer levels were measured in plasma, in duplicate, using an Asserachrom D-dimer kit (Stago) per manufacturer’s instructions.
Topical Sirolimus (Rapamycin) Treatment of Murine VM Lesions
Sirolimus and vehicle (without sirolimus) creams were custom compounded in a liposomal cream base (Lipoderm) and provided by Chemistry Rx pharmacy. The vehicle or 1% w/w sirolimus cream was gently applied topically with a cotton swab on the VM lesions twice/day for 2 weeks.
Apoptosis Analysis
HUVEC-TIE2-L914F (3×105/well) were seeded in 6-well plates and incubated with compounds or vehicle for 72 hours. Cells were collected according to the protocol for fluorescein isothiocyanate (FITC) Annexin V Apoptosis Detection Kit (BioLegend). Briefly, cells were resuspended in cold (4°C) binding buffer and incubated for 15 minutes at room temperature following addition of 5 mL of Annexin V-FITC and 5 mL of 7-AAD solutions. Flow cytometry analysis was performed using FACSCalibur2 and analyzed with CellQuest Pro software.
Cell Cycle Analysis
HUVEC-TIE2-L914F (4×105/well) were seeded in 6-well plates and treated with tested compounds for 48 hours. Cells were collected and fixed in 70% ice-cold ethanol. Before the analysis, cells were treated with Propidium Iodide Flow Cytometry Kit (Abcam) according to the manufacturer’s protocol. Cell cycle distribution and sub-G1 DNA content were measured with FACSCalibur2 and analyzed with ModFit-LT3.0 software (Verity Software House).
Cell Migration
Chemotaxis experiments were performed using a Boyden chamber technique (24-well chemotaxis chamber) and 8 μm pore size polycarbonate membranes (Costar). HUVEC-TIE2-L914F (105) were seeded in 100 μL EGM-2 with DMSO, ponatinib, rapamycin, or combination into the upper surface of the membrane insert. After incubation at 37°C for 6 hours, the upper surface of the filter was gently scraped to remove cells that had not migrated. Cells were fixed with 10% formalin and stained with 1% crystal violet. Experiments were performed in duplicate, and the number of cells present in 5 fields per well was counted at ×10 magnification in a blinded manner using an EVOS microscope (Invitrogen).
Three-Dimensional Fibrin Gel Assay
Three-dimensional (3D) fibrin gel assays were performed as previously described.30 Briefly, Cytodex 3 microcarrier beads (Sigma) were seeded with HUVEC-GFP (green fluorescent protein) or HUVEC-TIE2-L914F-GFP cells (2500 beads with 1×106 cells) and mixed in fibrinogen (Sigma) solution at a concentration of ≈500 beads/mL. Then 0.625 U/mL of thrombin (Sigma), and 0.5 mL beads/fibrinogen suspension were added per well in 24-well plate. After clotting in the 37°C incubator, 1 mL of EGM-2/10% fetal bovine serum medium and 20 000 fibroblasts per well were added on the top. EGM-2/10% fetal bovine serum medium with DMSO, ponatinib, rapamycin, or combination was added on the wells and replaced every other day. Images were acquired with EVOS cell imaging system (Invitrogen).
Human Phospho-Kinase Array
Cells were lysed according to Proteome Profiler Human Phospho-Kinase Array Kit protocol (R&D systems). Membranes were subjected to ChemiDoc MP Imaging System (Biorad), and bands were quantified by Image Lab software.
Sanger DNA Sequencing
DNA was extracted from cultured patient VM-EC using QIAamp DNA Mini Kit (Qiagen) according to manufacturer’s protocol and quantified on a Nanodrop 2000c Spectrophotometer (ThermoFisher). Approximately 200 ng of DNA was used for polymerase chain reaction using GoTaq Polymerase Master Mix (Promega) as per manufacturer’s instructions. Forward (5′ TGGTGTTGCTAGATGTGTTT) and reverse (5′ TTTTGGCTCAAGTAGTCCAT) primers were used to amplify and assess mutations in exon 17 coding sequence (Integrated DNA Technologies). Product amplification was confirmed by gel electrophoresis on a 2% agarose gel and further purified by gel excision using QIAquick Gel Extraction Kit (Qiagen) following manufacturer’s protocol. Purified polymerase chain reaction product was sequenced at Cincinnati Children’s Hospital Medical Center DNA Sequencing and Genotyping Core. Patient number 1 VM-EC expressed a TIE2 p.L914F mutation and patient number 2 VM-EC a TIE2 p.R915C mutation.
Immunofluorescent Staining
Immunofluorescence staining was performed using UEA (Vector Laboratories) and anti-CD31 (Dako) on paraffin sections and VM-EC monolayers, respectively. FITC/Texas Red-conjugated secondary antibodies (Vector Laboratories) were used. Samples were mounted using Prolong Gold with DAPI (Thermofisher). Images were acquired using Nikon C2 confocal microscope.
Statistical Analysis
Data for in vivo and in vitro experiments are expressed as mean±SD or SEM and analyzed by Student t test or parametric 1-way ANOVA after normal distribution and equal variance were assessed. When these assumptions failed, the nonparametric Wilcoxon rank-sum test or 1-way ANOVA with post hoc tests was used. Pairwise comparisons from the same ANOVA model were controlled for multiple comparisons. In lesion regression experiments, for data with correlated observations, longitudinal and multiple lesions on each mouse, a linear mixed effects model was used. To fit normality assumptions, the log10 of size increase and the log10 of weight were used (where applicable, log10 data analysis is shown in the online-only Data Supplement). All calculations were performed using GraphPad Prism or SAS version 9.3. Differences were considered significant at P value <0.05.
Data
The data that support the findings of this study are available from the corresponding author on reasonable request.
Results
ABL Kinase Inhibitors Suppress HUVEC-TIE2-L914F Cell Proliferation
Retrovirally transfected TIE2-L914F HUVEC (HUVEC-TIE2-L914F) were proven to be a powerful tool for the preclinical testing of rapamycin for the treatment of murine and human VM.15 HUVEC-TIE2-L914F proliferate faster than HUVEC-TIE2-WT and patient VM tissue show Ki67 positive cells lining the enlarged vascular channels (Figure I in the online-only Data Supplement). To identify drugs that induce VM regression, we performed an unbiased screening of Food and Drug Administration–approved oncology drugs and tested their ability to suppress the cell proliferation of HUVEC-TIE2-L914F. We identified 11 targeted, nonchemotherapeutic candidate drugs that were more selective (P<0.05) inhibitors of HUVEC-TIE2-L914F over HUVEC-TIE2-WT and nontransfected HUVEC (Table I in the online-only Data Supplement). Three mTOR inhibitors (sirolimus, temsirolimus, and everolimus) showed dose-independent (30 nmol/L—10 μmol/L) inhibition curves of HUVEC-TIE2-L914F, and the half-maximal inhibitory concentration (IC50) values were <30 nmol/L (Table II in the online-only Data Supplement), consistent with previous studies.29,31 Intriguingly, IC50 values of 3 ABL kinase inhibitors ponatinib, bosutinib, and nilotinib were 0.42±0.03, 3.54±1.14, and 3.61±0.03 μmol/L, respectively, whereas their IC50 values were higher in HUVEC-TIE2-WT and HUVEC nontransfected (Table II in the online-only Data Supplement). These results indicated that ABL kinase inhibitors are candidate drugs for the treatment of murine VM and that ABL kinase may play an important role in VM expansion.
c-ABL Is Constitutively Phosphorylated in HUVEC-TIE2-L914F
We assessed whether ABL kinase was activated in HUVEC-TIE2-L914F. Phosphorylation of c-ABL was higher in HUVEC-TIE2-L914F compared with HUVEC-TIE2-WT and HUVEC nontransfected (Figure 1A). The 3 ABL kinase inhibitors are ATP-competitive inhibitors that can also target multiple tyrosine kinases.26 Ponatinib showed higher efficacy than bosutinib and nilotinib in suppressing c-ABL phosphorylation (Figure II in the online-only Data Supplement) and inhibiting HUVEC-TIE2-L914F cell proliferation (Table II in the online-only Data Supplement). We tested ponatinib on HUVEC-TIE2-L914F and it reduced phosphorylation of c-ABL, suppressed TIE2 and its downstream effector AKT (Figure 1B). These results show that the ABL kinase inhibitor ponatinib inhibits TIE2-L914F-induced activation of c-ABL and PI3K/AKT signaling.
Figure 1.
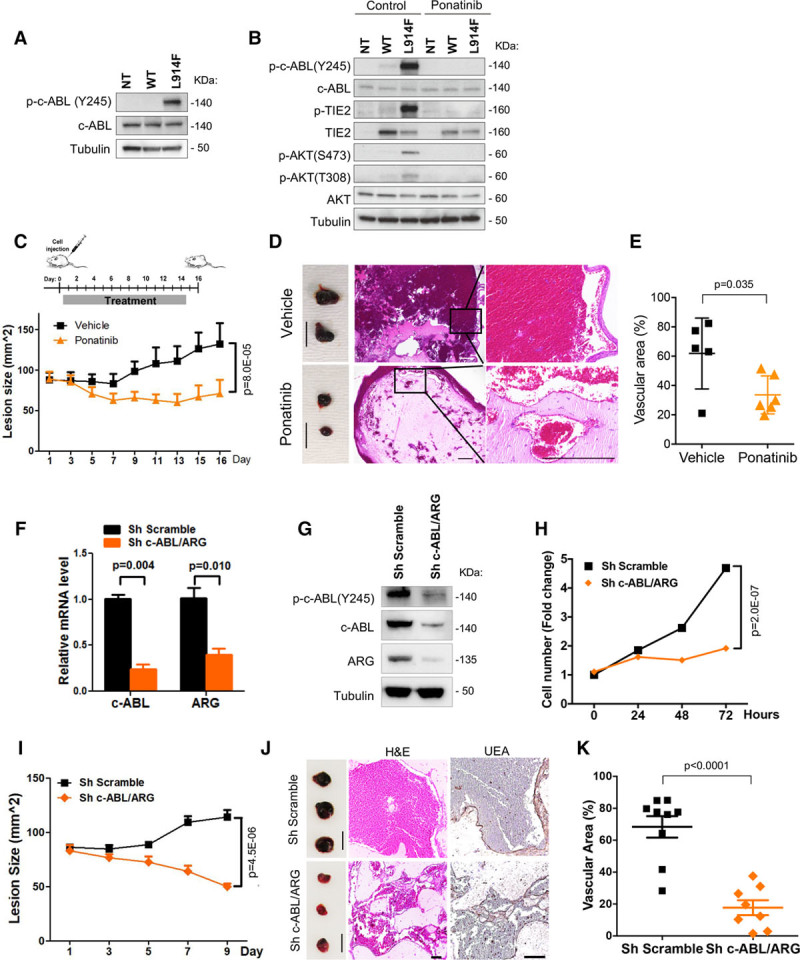
Ponatinib inhibits venous malformation (VM) formation and expansion. A, Immunoblot analysis of nontransfected human umbilical vein endothelial cell (HUVEC-NT), HUVEC transfected with wild-type TIE2 (WT) or with mutant TIE2 (L914F) probed with indicated antibodies. B, Immunoblot analysis of HUVEC-NT, -TIE2-WT, and -TIE2-L914F probed with indicated antibodies. Cells were treated with dimethyl sulfoxide (control) or 500 nmol/L ponatinib for 1 h. C, Treatment scheme: HUVEC-TIE2-L914F injected mice were treated daily with vehicle or ponatinib (30 mg/kg per day, oral gavage) for 14 d. Lesion size measured by caliper every 2 d; Data expressed as mean±SD, linear mixed-effect model (n=5–6 mice with 2 lesions/group). D, Representative images of lesions; scale bar: 1 cm (left). Representative images of Hematoxylin and Eosin (H&E) stained lesion sections; scale bar: 500 μm (right). E, Quantification of vascular area (%). Data expressed as average value for 2 lesions on each mouse, mean shown by horizontal bars, Student t test (n=5–6 mice). F, Relative mRNA levels of c-ABL and ARG in sh scramble and sh c-ABL/ARG (target 1) HUVEC-TIE2-L914F measured 48 h after shRNA induction. G, Immunoblot of HUVEC-TIE2-L914F sh Scramble and sh c-ABL/ARG (target 1) at 48 h. H, Proliferation rates of HUVEC-TIE2-L914F sh Scramble and sh c-ABL/ARG (target 1). Data expressed as mean±SEM, linear mixed-effect model. I, HUVEC-TIE2-L914F sh Scramble and sh c-ABL/ARG (target 1) cells were injected into nude mice and lesion size measured by caliper every 2 days; Data expressed as mean±SEM, linear mixed-effect model (n=8–9 mice with 2 lesions/group). J, Representative images of lesions (left) and sections stained for H&E and UEA (Ulex europaeus agglutinin I; right). Scale bar: 1 cm (lesions) and 100 μm (H&E and UEA staining). K, Quantification of vascular area. Data expressed as average value for 2 lesions on each mouse, mean shown by horizontal bars, Student t test (n=8–9 mice).
Ponatinib Inhibits VM Formation In Vivo
We further assessed ponatinib efficacy in a VM murine model.15 In previous studies, ponatinib treatment at 30 mg/kg was effective in preventing BCR-ABLT315I–induced tumor growth in murine xenografts.27 In our VM murine model, ponatinib (30 mg/kg) treatment prevented VM lesion growth (P=8.0×10−05; Figure 1C). Hematoxylin and Eosin staining of lesion explants showed that ponatinib treatment prevented enlargement of vessels (P=0.035; Figure 1D and 1E). These data demonstrate that ponatinib inhibits VM lesion growth and vascular expansion in HUVEC-TIE2-L914F injected mice.
ABL Kinase Knockdown Diminishes Cell Proliferation and VM Lesion Vascularity
To determine the role of ABL kinase in TIE2 mutation-dependent VM formation, shRNA-mediated silencing was used to downregulate both ABL kinases c-ABL (ABL1) and ARG (ABL2) in HUVEC-TIE2-L914F (Figure 1F). Decreased expression of both c-ABL and ARG resulted in reduced c-ABL phosphorylation (Figure 1G) and cell proliferation as compared with the scrambled control shRNA (Figure 1H). HUVEC-TIE2-L914F in which c-ABL/ARG was knocked down were injected into mice. Lesions did not grow in size (Figure 1I) and were noticeably less vascularized at day 9 (Figure 1J). Both Hematoxylin and Eosin staining and immunostaining with the human-specific lectin UEA showed that the sh c-ABL/ARG HUVEC-TIE2-L914F-derived vessels and vascular area were smaller (P=0.0001) compared with the sh Scramble control (Figure 1J and 1K). Furthermore, 2 different sets of shRNA templates (targets 1 and 2) inhibited vascular/tube area formation in a 3D fibrin gel assay30 (Figure III in the online-only Data Supplement). These results suggest that ABL kinase activation is required for TIE2-L914F–derived VM lesions to form vessels and expand.
Combination of Ponatinib With Rapamycin Induces VM Lesion Regression
As the mTOR inhibitor rapamycin caused modest VM lesion regression in clinical trials,6,7,15 we investigated whether combined treatment with rapamycin and ABL kinase inhibitor ponatinib was more potent than single drug treatment alone. To assess in vitro synergism between the 2 drugs, we calculated the CI (combination index) value. In our study, rapamycin caused a dose-independent inhibition of proliferation (Table II in the online-only Data Supplement) that is an ineligible prerequisite for the Chou-Talalay method, the gold standard for CI evaluation. Based on published studies, we defined drug synergism for a more stringent CI value ≤0.8 (and not CI ≤1 as proposed by the Chou-Talalay method).29,32 The proliferation inhibition curve for drug combination (ponatinib with rapamycin) minus rapamycin alone (deducting effect of rapamycin from the combination) showed a significant shift of IC50 to a lower value in HUVEC-TIE2-L914F when compared with ponatinib alone (Figure IV in the online-only Data Supplement). The mean CI value was 0.67±0.26 (Table III in the online-only Data Supplement), indicative of a synergistic antiproliferative effect on the HUVEC-TIE2-L914F.
Therefore, we first tested the effects of ponatinib combined with rapamycin in the formation and expansion of murine VM lesions generated by injection of HUVEC-TIE2-L914F.15 Combination treatment started 1 day after cell injection and suppressed VM lesion expansion compared with vehicle and ponatinib monotherapy while caused a mild weight loss in the mice (Figure V in the online-only Data Supplement). To identify the minimal effective dose of the combination, we then tested 2 reduced-dose regimens (RD1 and RD2). RD1 combo (to which we will further refer to as RD combo: 20 mg/kg ponatinib+1 mg/kg rapamycin) showed efficacy similar to full-dose combination and did not cause weight loss in treated animals (Figures VI and VII in the online-only Data Supplement).
Next, to assess whether combination treatment causes a significant regression of preformed VM lesions, drug treatment was initiated on day 9, when the average VM lesion size of each group reached about 110 mm2 (schematic in Figure 2A). Full-dose combination and RD combo treatment caused around 88% to 81% of lesions to regress (14/16 and 13/16, respectively), while only 31% to 13% of lesions (5/16 and 2/16) shrunk in the ponatinib and rapamycin monotherapy groups, respectively (Figure 2B; Figure VIII in the online-only Data Supplement). In accordance with these results, VM lesion weight in full-dose combination and RD combo treated mice was lower than in monotherapy and vehicle groups (Figure 2C). Analysis of lesion histology showed that full-dose combination and RD combo treatments induced vascular regression (Figure 2D and 2E). Both combination treatments showed a trend to reduced Ki-67-expressing proliferative cells (Figure 2F and 2G) and increased the number of apoptotic cells (cleaved caspase-3-positive; Figure 2F and 2H) compared with monotherapy. These results suggest (1) drug combination is more effective than monotherapy and (2) reduced-dose drug combination achieved a similar effect on inducing lesion regression as the full-dose combination but without affecting mouse body weight.
Figure 2.
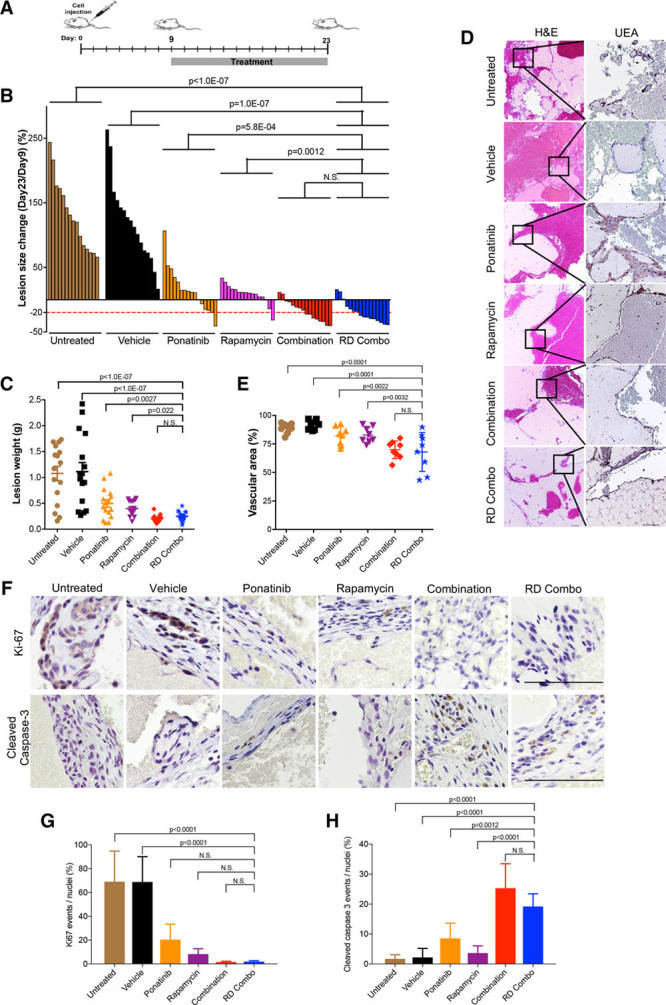
Ponatinib combined with rapamycin induces regression of murine venous malformation (VM). A, Treatment scheme: Human umbilical vein endothelial cell (HUVEC)-TIE2-L914F were injected into mouse at day 0 and lesions expanded until their size reached 110 mm2. Treatment was started (day 9) by daily oral gavage with vehicle, ponatinib (30 mg/kg), rapamycin (2 mg/kg), combination (ponatinib 30 mg/kg + rapamycin 2 mg/kg) and reduced-dose combination (RD combo, ponatinib 20 mg/kg + rapamycin 1 mg/kg) for 14 d. B, Waterfall plot of % lesion size change (day 23/day 9). Data expressed as single value for each lesion, linear mixed-effect model (n=8 mice with 2 lesions/group). C, Lesion weight: Data expressed as single value for each lesion, mean shown by horizontal bars, linear mixed-effect model (n=8 mice with 2 lesions/group). D, Representative Hematoxylin and Eosin (H&E) and UEA (Ulex europaeus agglutinin I) stained lesion sections. Scale bar: 100 μm. E, Quantification of vascular area. Data expressed as average value for 2 lesions on each mouse, mean shown by horizontal bars, 1-way ANOVA for multiple comparisons (n=8 mice with 2 lesions/group). F, Representative images of lesion sections stained with cleaved caspase-3 and Ki-67. Scale bar: 500 μm. G, Quantification of Ki-67 events and (H) quantification of cleaved caspase-3 events. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (n=5).
Reduced-Dose Drug Combination Results in VM Lesion Regression With Minimal Side Effects
We assessed the efficacy and safety of long-term drug combination treatment in mice. Treatment started on day 12 when average lesion size in all groups reached 130 mm2 and continued for 4 weeks. Full-dose combination and RD combo induced marked lesion regression, and no lesion rebound during treatment (Figure 3A and 3B; Figure IX in the online-only Data Supplement). After 4 weeks of treatment, mouse body weight in the RD combo group was similar to the vehicle-treated animals, and no deaths occurred (Figure 3C). Reduced hemoglobin and red blood cell levels were previously reported in mice with VM lesions.15 Drug combination treatments normalized both hemoglobin levels and erythrocyte number (Figure 3D). In the full-dose combination and RD combo treated mice, serum levels of alanine aminotransferase, aspartate aminotransferase, and blood urea nitrogen were in the normal range for liver and kidney function, as in unchallenged mice (no lesion; Figure 3E). Elevated blood cholesterol (Figure 3E) was consistent with hypercholesterolemia reported in the rapamycin clinical trial for lymphangioleiomyomatosis patients.33 D-dimer levels were elevated in mice with VM. In response to drug combination treatment, D-dimer levels were comparable to mice without lesions (Figure 3F). These results indicate that long-term treatment with drug combination caused lesion regression without major side effects and normalized the hemoglobin and D-dimer levels.
Figure 3.
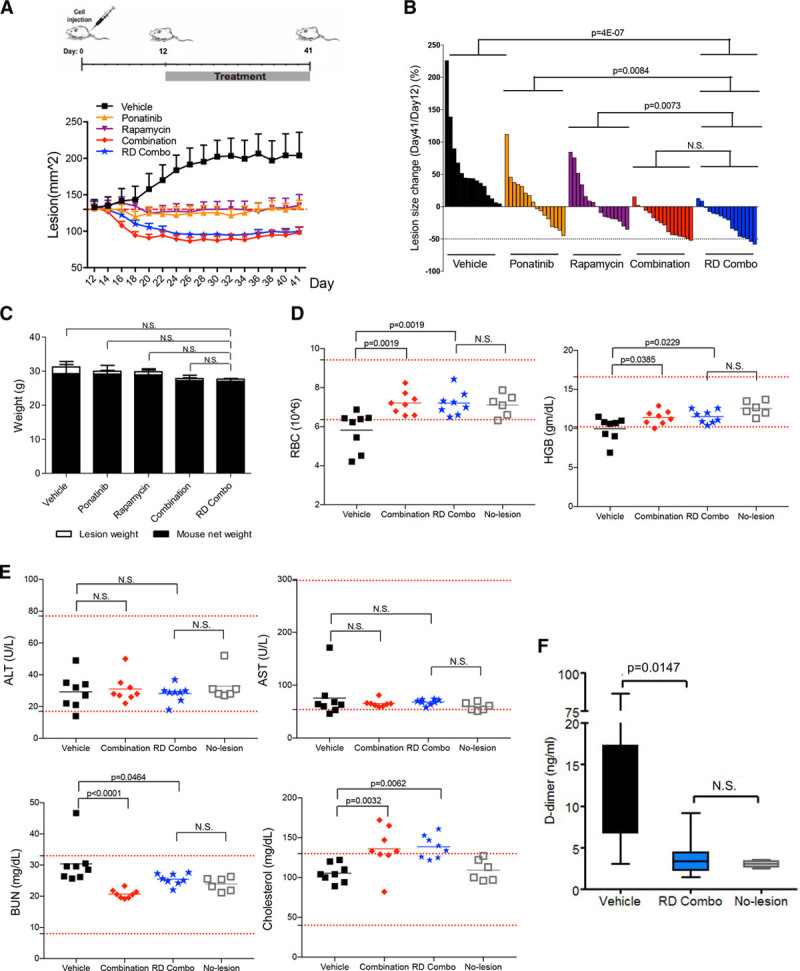
Long-term combination treatment with ponatinib and rapamycin (4 wk) shows minimal side effects in mice bearing venous malformation (VM). A, Treatment scheme: Human umbilical vein endothelial cell (HUVEC)-TIE2-L914F were injected into mouse at day 0 and lesions expanded until their size reached 130 mm2. Treatment was started (day 12) by daily oral gavage with vehicle, ponatinib (30 mg/kg), rapamycin (2 mg/kg), combination (ponatinib 30 mg/kg + rapamycin 2 mg/kg), and reduced-dose combination (RD combo, ponatinib 20 mg/kg + rapamycin 1 mg/kg) for 4 wk. Lesion size of each group measured every other day. Data expressed as mean±SEM. B, Waterfall plot of % lesion size change (day 41/day 12). Data expressed as single value for each lesion, linear mixed-effect model (n=8 mice with 2 lesions/group). C, Mouse body weight at day 41. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (n=6–8 mice/group). D and E, Analysis of red blood cells (RBC), hemoglobin (HBG), alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN) in the peripheral blood of vehicle, combination, RD combo, and no lesion mouse groups. Red dashed lines indicated reference value and gray square-holes show values in mice without VM lesions (unchallenged, no cell injection). Data expressed as single value for each mouse, mean shown by horizontal bars, 1-way ANOVA for multiple comparisons (n=6–8 mice/group). F, D-dimer levels were tested in vehicle, RD combo, and no lesion group. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (n=6–8 mice/group).
Topical Rapamycin Prevents Lesion Rebound After Drug Combination Withdrawal
Topical rapamycin was reported to prevent microcystic lymphatic malformation growth in patients, without notable side effects.34,35 Here, we investigated if we could stop the drug combination therapy and maintain its effects (reduced lesion size) with a topical rapamycin treatment. We first injected HUVEC-TIE2-L914F into mice and then treated these animals with the drug combination ponatinib+rapamycin (RD combo and oral treatment) from day 14 to 28. On day 28, oral combination treatment was suspended, and we applied topical rapamycin 2 times/day (schematic in Figure 4A) for 2 weeks. VM lesions regressed during the oral combination treatment (day 14–28) but recurred when treatment was discontinued (day 29–42; Figure 4B). However, VM lesions treated with topical rapamycin did not increase in size and remained similar or became smaller compared with day 28, the end of oral combination treatment (Figure 4B through 4D; Figure X in the online-only Data Supplement). Hence, topical rapamycin prevented murine VM lesion rebound after discontinuing drug combination therapy.
Figure 4.
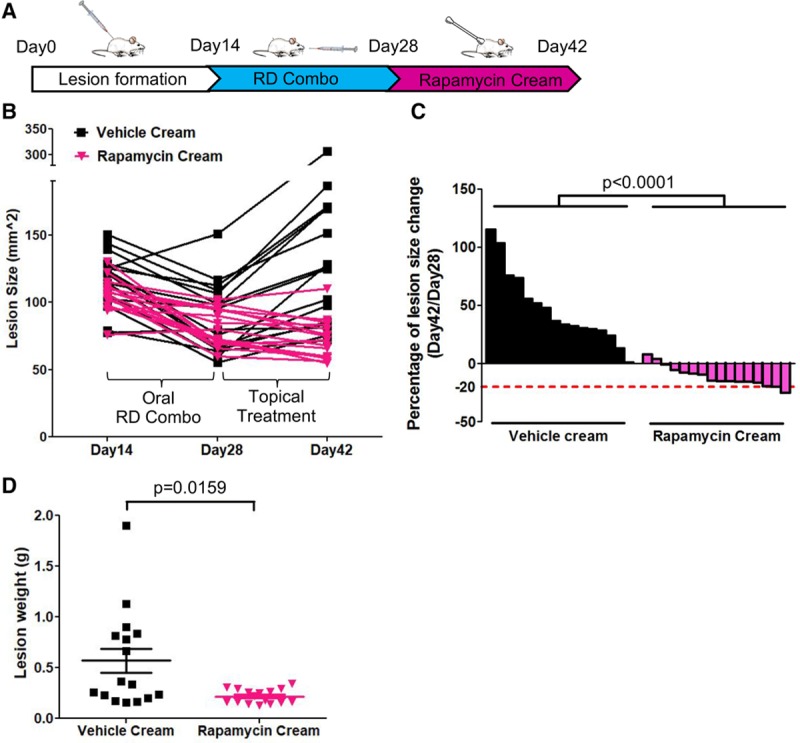
Topical rapamycin treatment prevents lesion rebound. A, Treatment scheme: Human umbilical vein endothelial cell (HUVEC)-TIE2-L914F were injected into mice to form venous malformation (VM) lesions for 14 d, then treated with oral reduced-dose combination (RD combo) until day 28. Combination treatment was discontinued, and topical vehicle cream or rapamycin cream (1%) was applied on the surface of the lesions twice/day for 14 d. Lesion images were taken at day 42. B, Lesion size from day 14–28 and to day 42. Each line represents 1 lesion. C, Waterfall plot of lesion size change (day 42/day 28). Data expressed as single value for each lesion, linear mixed-effect model (n=8 mice with 2 lesions/group). D, Lesion weight at day 42. Data expressed as single value for each lesion, mean shown by horizontal bars, linear mixed-effect model (n=8 mice with 2 lesions/group).
Drug Combination Enhances Cell Apoptosis and Suppresses Cell Migration in HUVEC-TIE2-L914F
To study the cellular functions affected by the drug combination, we analyzed cell apoptosis, cell cycle, and migration. Although previous studies showed rapamycin treatment alone promotes cell death in both human umbilical artery ECs and normal HUVEC,36,37 in our studies it did not induce apoptosis in HUVEC-TIE2-L914F (Figure 5A and 5B; Figure XI in the online-only Data Supplement). In contrast, ponatinib treatment resulted in apoptosis of HUVEC-TIE2-L914F (25.43±4.67%) and, when combined with rapamycin, this effect was significantly greater than with ponatinib alone (53.47±5.02%; P=0.0018).
Figure 5.
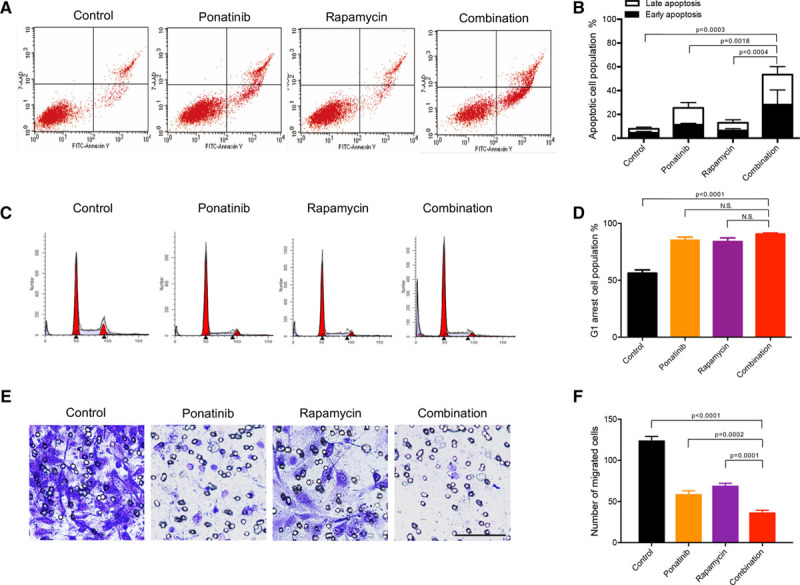
Ponatinib combined with rapamycin enhances apoptosis and inhibits cell migration in human umbilical vein endothelial cell (HUVEC)-TIE2-L914F. A, Flow cytometric analysis of apoptosis. HUVEC-TIE2-L914F cells were treated with control (dimethyl sulfoxide [DMSO]), 100 nmol/L ponatinib, 10 nmol/L rapamycin, or combination for 72 h. B, Quantification of % apoptotic cell populations. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (representative of 2 independent experiments, n=4). C, Representative images of cell cycle analysis. HUVEC-TIE2-L914F cells were treated with control (DMSO), 100 nmol/L ponatinib, 10 nmol/L rapamycin, or combination for 48 h. D, Quantification of % G0-G1 cell cycle cell population. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (representative of 2 independent experiments, n=4). E, Representative images of cells migrated through the 8 μm pore filter (Transwell migration assay). HUVEC-TIE2-L914F cells were treated with control (DMSO), 100 nmol/L ponatinib, 10 nmol/L rapamycin, or combination for 6 h and then stained with crystal violet. Scale bar: 100 μm. F, Quantification of migrated cells. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (n=3 independent experiments).
We next investigated the effects of drug combination on cell cycle arrest. Rapamycin was previously reported to block HUVEC cell cycle in the G1 phase.37 Consistent with these data, rapamycin treatment induced G0/G1 phase arrest (83.99±4.58%) compared with vehicle (DMSO; 56.29±4.06%; Figure 5C and 5D). Similarly, ponatinib treatment induced HUVEC-TIE2-L914F to accumulate in G0/G1 phase (85.41±3.57%). Drug combination did not show any additional effect when compared with single drug treatments (90.02±1.53%).
Mutant TIE2 HUVEC display increased cell motility and migration compared with HUVEC-TIE2-WT.13 Drug combination reduced HUVEC-TIE2-L914F migration in a 3D Boyden chamber (Figure 5E and 5F) and was more effective than single drug treatment. These data indicate that, compared with single drug treatment, the combination of ponatinib with rapamycin enhanced cell apoptosis and decreased cell migration.
Drug Combination Promotes Regression of HUVEC-TIE2-L914F–Derived Vascular Channels
3D systems such as the fibrin gel assay can recapitulate the fundamental steps of the angiogenesis process in vitro.30 In this assay, when compared with normal HUVEC that formed vascular tubes, HUVEC-TIE2-L914F formed large and ectatic saccular channels similar to patient VM lesions (Figure 6A). To test the effect of drug treatment on 3D fibrin gel channel formation, rapamycin, ponatinib, or combination were added on day 1 (Figure 6B). At the end of treatment (day 11), in the vehicle (DMSO) group vascular/tube area increased to reach ≈75% of the total area. In both mono and combination treatments, tube formation was almost completely suppressed (tube/channel area was <1%; Figure 6C; Figure XIIA in the online-only Data Supplement). There was no statistical difference between mono and combination treatments.
Figure 6.
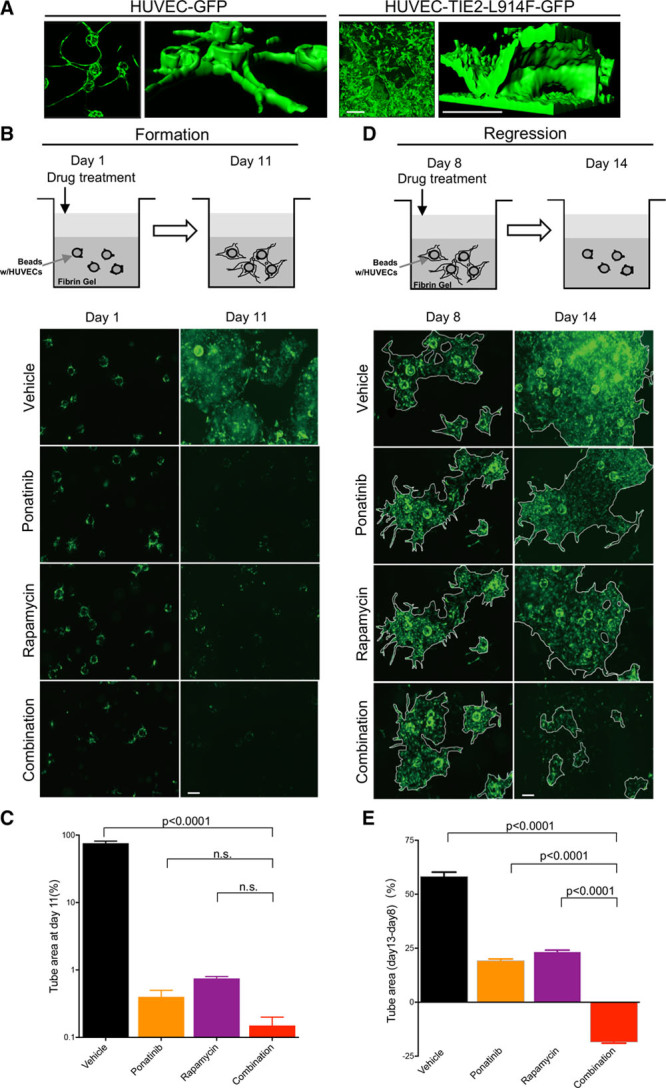
Ponatinib combined with rapamycin induces vascular channel regression in a 3-dimensional (3D) fibrin gel assay. A, Representative 2-dimensional (2D) images (left) and 3D structural models (right) of tubular networks formed by human umbilical vein endothelial cell (HUVEC)-GFP or HUVEC-TIE2-L914F-GFP. Scale bar: 200 μm. B, Treatment scheme of vascular network formation and representative 2D images taken on the same field at day 1 and 11. HUVEC-TIE2-L914F-GFP cells were treated with vehicle (dimethyl sulfoxide [DMSO]), 100 nmol/L ponatinib, 10 nmol/L rapamycin, or combination starting at day 1, when beads coated with cells were embedded in the fibrin gel. Scale bar: 200 μm. C, Quantification of tube/channel area. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (n=3 independent experiments). D, Treatment scheme of vascular network regression and representative 2D images taken on the same field at day 8 and 14. HUVEC TIE2-L914F-GFP cells were treated with vehicle (DMSO), 100 nmol/L ponatinib, 10 nmol/L rapamycin, or combination starting day 8 when tubular networks were already established. Scale bar: 200 μm. E, Quantification of vascular tube area change from day 8–14. Data expressed as mean±SD, 1-way ANOVA for multiple comparisons (representative of 2 independent experiments, n=3 wells each with 2–3 vascular channels analyzed).
Next, we evaluated the effect of the drug combination on the regression of existing VM ectatic channels. Compounds were added on day 8 when the HUVEC-TIE2-L914F–derived vascular network was already established (Figure 6D). The vascular/tube area expanded from day 8 to 14 by 58.31±1.94% in the vehicle group and 19.33±0.7% and 23.48±0.62% in ponatinib and rapamycin single treatment groups, respectively (Figure 6E; Figure XIIB in the online-only Data Supplement). Regression of vascular/tube area was recorded only in response to combination treatment, by 18.57±0.4%. These data suggest that the combination of ponatinib with rapamycin was responsible for the regression of vascularity seen in the in vivo experiments.
Combination of Ponatinib and Rapamycin Enhances AKT Suppression and Reduces PLCγ-ERK1/2 Activity in HUVEC-TIE2-L914F
To identify signaling pathways implicated in the combination treatment response, we assessed the phosphorylation of 45 tyrosine kinase sites in HUVEC-TIE2-WT and HUVEC-TIE2-L914F. As shown in Figure 7A, phosphorylation of PLCγ, AKT (Ser473), and ERK1/2 was elevated in HUVEC-TIE2-L914F relative to HUVEC-TIE2-WT. Upregulation of these signaling pathways in the HUVEC-TIE2-L914F was also confirmed by immunoblotting (Figure 7B).
Figure 7.
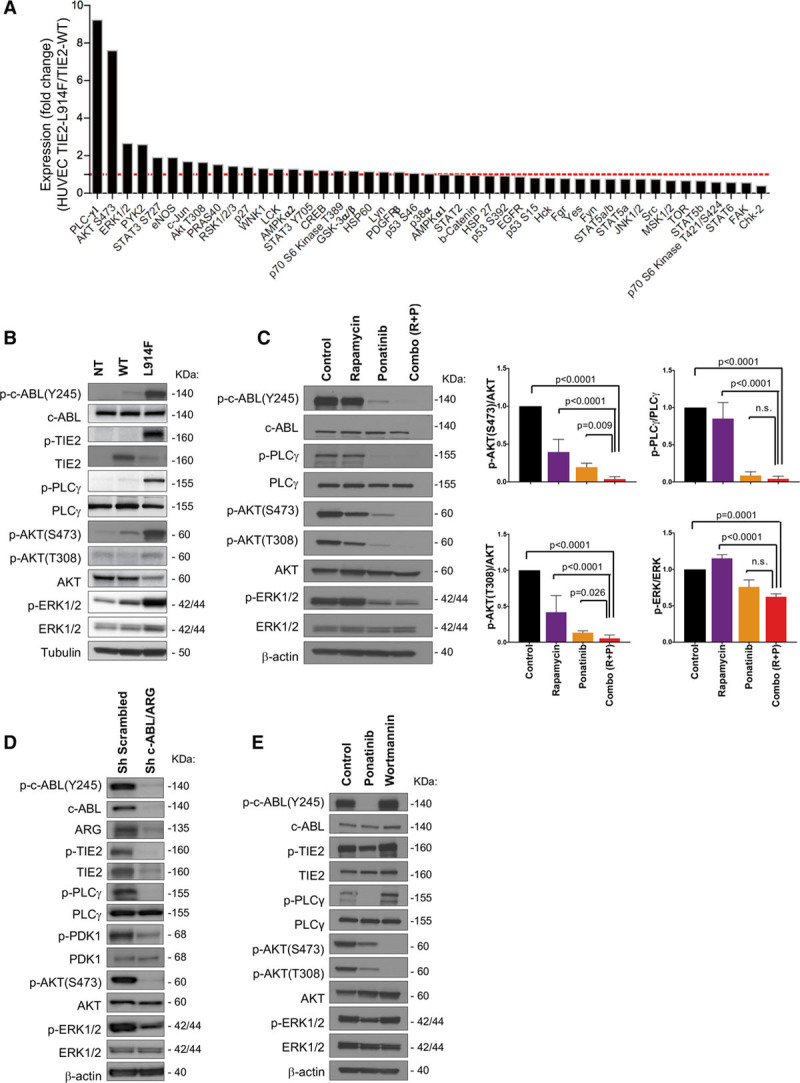
Ponatinib combined with rapamycin enhances AKT (protein kinase B) suppression and ERK1/2 (extracellular signal-regulated kinase) activity. A, Phosphorylation changes of 45 kinases in human umbilical vein endothelial cell (HUVEC)-TIE2-L914F (L914F) relative to HUVEC-TIE2-wild type (WT). B, Immunoblot analysis of HUVEC nontransfected (NT), HUVEC-TIE2-WT, and HUVEC-TIE2-L914F (L914F) with indicated antibodies. C, Immunoblot analysis of HUVEC-TIE2-L914F with listed antibodies. Cells were treated with DMSO (Control), 100 nmol/L ponatinib, 10 nmol/L rapamycin, or combination for 48 h. Immunoblot bands were quantified, data expressed as mean±SD, 1-way ANOVA for multiple comparisons (n=3–4 independent experiments). D, Immunoblot analysis of HUVEC-TIE2-L914F treated with shRNA for c-ABL/ARG or with scrambled sequence (sh Scrambled). E, Western blotting analysis of HUVEC-TIE2-L914F with indicated antibodies. Cells were treated with DMSO (Control), 100 nmol/L ponatinib or 10 µmol/L Wortmannin for 1 h.
We analyzed phosphorylation of these kinases after treatment with rapamycin, ponatinib or combination in HUVEC-TIE2-L914F. Rapamycin treatment diminished the phosphorylation of AKT (Ser473) but did not affect the activity of PLCγ that was reduced with ponatinib. While rapamycin or ponatinib alone inhibited AKT, the drug combination significantly enhanced this suppression (P<0.0001 and P=0.009, respectively; Figure 7C). In addition, the drug combination reduced ERK1/2 phosphorylation that tends to increase with rapamycin treatment alone. Thus, combining rapamycin with ponatinib not only enhanced inhibition of AKT (Ser473) but also suppressed PLCγ and ERK1/2 activity. shRNA-mediated suppression of c-ABL/ARG downregulated the pathways targeted by ponatinib: c-ABL and its downstream effectors PDK1 (pyruvate dehydrogenase kinase 1)-AKT and PLCγ-ERK1/2 activity (Figure 7D).
Next, to determine if c-ABL is activated directly downstream of TIE2 or indirectly through PI3K, we treated HUVEC-TIE2-L914F with the PI3K inhibitor Wortmannin. Wortmannin ablated phospho-AKT but had no effect on c-ABL activation and its downstream effectors PLCγ and ERK1/2 that were instead effectively targeted by ponatinib (Figure 7E). These results suggest a novel mechanism by which TIE2-L914F signals and drives VM lesion formation through concomitant activation of the PI3K/AKT and c-ABL/PLCγ/MAPK pathways.
Combination of Ponatinib and Rapamycin Induces VM Lesion Regression in a Patient-Derived Cell Xenograft Model
To establish that the signaling pathways affected by the drug combination were upregulated in patient’s VMs, ECs were isolated from freshly resected patient lesion tissue (VM-EC). VM-EC exhibited EC morphology (cobblestone phenotype) and expressed the EC marker CD31 at the cell membrane (Figure 8A). VM-EC from patient number 1 and number 2 expressed activating TIE2 mutations resulting in constitutive activation of c-ABL and downstream effectors AKT, PDK1, PLCγ, and ERK1/2 (Figure 8B; Figure XIII in the online-only Data Supplement). ANGPT1 (Angiopoietin 1) treatment (15 minutes) enhanced phospho-c-ABL in VM-EC, as well as in normal HUVEC, suggesting that c-ABL activation is a direct effector of TIE2 activation.
Figure 8.
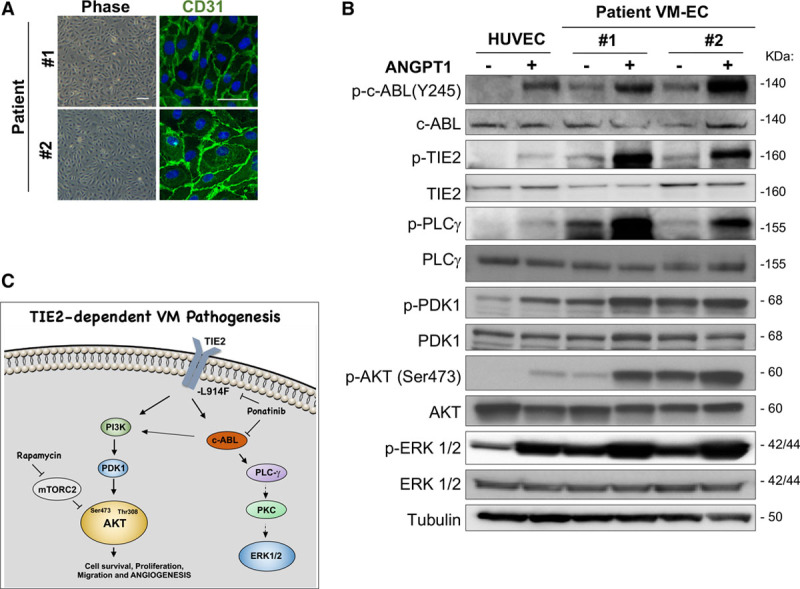
Ponatinib combined with rapamycin induces venous malformation (VM) lesion regression in a patient-derived cell xenograft model. A, Representative phase-contrast images of VM-patient-derived endothelial cell (VM-EC) cell morphology and stained with endothelial cells marker CD31 (green) and nuclei with DAPI (blue). Scale bar: phase 100 μm, IF 50 μm. B, Immunoblotting analysis of human umbilical vein endothelial cell (HUVEC), patient no. 1 and no. 2 VM-EC with indicated antibodies. Cells were treated with or without 500 nmol/L ANGPT1 (angiopoietin 1) for 15 min. C, Scheme of signaling pathways downstream of TIE2-L914F and mechanisms of action of the drug combination rapamycin+ponatinib. Rapamycin inhibited AKT (protein kinase B) activation in EC by prolonged treatment. Here, we show that ponatinib can target c-ABL but also TIE2, resulting in inhibition of AKT most likely by c-ABL effect on PI3K (phosphoinositide-3-kinase)/PDK1. Ponatinib also affected PLCγ (phospholipase C) and ERK1/2 (extracellular signal-regulated kinase), both were also decreased upon c-ABL/ARG knockdown.
To assess the efficacy of the drug combination on a patient-derived cell xenograft model, we injected VM-EC into immune-compromised mice, as previously reported.14 When lesions were evident and bluish in color, treatment was started. The average vascular density in the RD combo was reduced compared with vehicle and monotherapy groups (Figure XIV in the online-only Data Supplement). These results are a proof-of-concept suggesting the efficacy of ponatinib combined with rapamycin in inducing regression of TIE2-mutated VM lesions.
In conclusion, we summarize here our proposed model of TIE2-dependent VM pathogenesis and drug combination efficacy (Figure 8C; Figure XV in the online-only Data Supplement). Prolonged treatment with rapamycin inhibited AKT activation in EC by targeting mTORC2, as reported in previous studies.15,38,39 Here, we show that ponatinib can target c-ABL and TIE2, resulting in inhibition of AKT most likely by c-ABL effect on PI3K subunit p85, as reported in Gotoh et al40 and Skorski et al,41 and as suggested by the reduced PDK1 activity in c-ABL/ARG knockdown HUVEC-TIE2-L914F. Ponatinib also affected PLCγ and ERK1/2, both were also decreased on c-ABL/ARG knockdown. PLCγ was previously shown to be regulated by the oncogenic form of c-ABL.40,42 Here, we hypothesize that PLCγ activates ERK1/2 as a previous link between the 2 signaling molecules has previously been established in the context of VEGF signaling.43,44
Discussion
Treatment of VM patients remains a major challenge with no targeted therapies currently available to induce lesions to regress. In our study, combination treatment with ponatinib and rapamycin induced murine VM regression with minimal side effects. Drug combination withdrawal induced a VM lesion rebound that was prevented by topical rapamycin. Furthermore, we identified a novel mechanism by which mutated TIE2-L914F signals in HUVEC-TIE2-L914F and in patient-derived EC, through constitutive c-ABL phosphorylation. Targeting c-ABL with ponatinib or c-ABL/ARG knockdown significantly suppressed HUVEC-TIE2-L914F proliferation. Combination treatment of ponatinib with rapamycin had a greater effect on HUVEC-TIE2-L914F proliferation, apoptosis, and migration, resulting in vascular regression in VM. The principal mechanism of combined drug treatment involved enhanced suppression of AKT (Ser473 and Thr308) and inhibition of PLCγ and ERK1/2 activity.
ABL kinases are involved in the regulation of cell proliferation, cytoskeletal dynamics, membrane, and organelle trafficking.26 In ECs, ABL kinase regulates EC survival and vascular stability.45 In the mouse, specific Abl/Arg double-knockout induced late-stage embryonic and perinatal lethality with hepatic necrosis, localized loss of vasculature, and hemorrhage. Other reports indicate that ABL kinase activity is required to maintain EC permeability46,47 and normal vascular development.47,48 Our studies showed that knockdown of c-ABL/ARG or treatment with multikinase inhibitors with high affinity for c-ABL (ponatinib, nilotinib, and bosutinib) was efficacious in decreasing proliferation of HUVEC-TIE2-L914F. In our drug screen, ponatinib showed higher inhibition rate compared with MEK or PI3K inhibitors, we hypothesize this is because of its inhibition of both phospho-AKT and phospho-PLCγ/ERK signaling pathways. Additionally, c-ABL/ARG knockdown in HUVEC-TIE2-L914F resulted in smaller, less vascularized VM lesions, and suppressed TIE2-L914F upregulated pathways, including PDK1-AKT and PLCγ and ERK1/2, the same pathways targeted by ponatinib in our model. In addition, we confirmed that c-ABL is a major downstream effector of TIE2 as ANGPT1 treatment enhances c-ABL activation, in both mutant EC and normal HUVEC. While our study focused on the most frequent mutation found in VM patients, TIE2-L914F, whether other somatic or inherited TIE2 mutations induce ABL activation merits further investigation.
The mTOR inhibitor rapamycin (sirolimus) inhibited lesion expansion in murine VM, as well as in clinical trials.6,7,15 In our in vitro drug screening, 2 rapamycin analogs, temsirolimus (Torisel) and everolimus (Afinitor), showed similar efficacy as rapamycin in preventing the proliferation of HUVEC-TIE2-L914F. Recently, somatic activating mutations in PIK3CA, that encodes the p110α catalytic subunit of PI3K, were identified in VM patient samples.12,14,16,17 EC expressing these PIK3CA mutations have increased levels of AKT phosphorylation but not ERK1/2 activation.12,14 Furthermore, Pik3caH1047R knockin murine models exhibited venous slow-flow lesions, similar to patient VM.16,17 Treatment with 4 mg/kg of rapamycin decreased lesion size by 25%, supporting the use of rapamycin for VM caused by PIK3CA mutations. This rapamycin concentration is higher than the one used here (1–2 mg/kg) and in clinical studies (0.8 mg/m2).6,7,33 In these studies, Pik3caH1047R-driven murine VM expansion was also suppressed by the PI3Kα inhibitor BYL719, that increased the number of cleaved caspase-3 positive cells, whereas everolimus prevented lesion growth but did not induce cell apoptosis. Taken together, these data indicate that the PI3K-AKT-mTOR signaling pathway is a promising therapeutic target for treatment of VM.16 It remains unclear if VM patient response to rapamycin treatment differs in TIE2-mutated VM compared with PIK3CA-mutated VM.
Recently, we reported that in TIE2-mutated EC there is constitutive activation of both PI3K/AKT and MAPK/ERK signaling.14 In the present study, combination treatment of rapamycin with ponatinib in HUVEC-TIE2-L914F enhanced inhibition of AKT (Ser473 and Thr308) and suppressed PLCγ and ERK1/2, suggesting this is the mechanism leading to regression of the VM lesions. This implies that concomitant suppression of AKT and ERK1/2 generated by the combined drug therapy is more effective than monotherapy, especially in VM driven by the endothelial TIE2 p.L914F mutation. We also speculate that ERK1/2 signaling may not be necessary for VM formation/expansion as hyperactive PI3KCA mutations can induce VM formation. Further investigations could reveal the importance of the PLCγ/ERK1/2 signaling for the VM genesis.
Our data shows the patient-derived VM-EC upregulated the activated pathways targeted by the drug combination. Furthermore, our proof-of-concept results obtained with the VM-EC xenograft model supports the therapeutic benefits of this drug combination. Moreover, future studies with the use of patient-derived xenograft models are needed to assess the efficacy of the drug combination for VM patients that express other mutations in TIE2 or PIK3CA. Patient cells with mutations other than TIE2-L914F should be analyzed in vitro to screen for drugs or compounds effective in targeting the specific mutated protein/s, thus enabling the development of personalized treatment options.
In our studies, ponatinib was the most potent ABL kinase inhibitor when used alone and in combination with rapamycin. A recent clinical trial comparing the effectiveness of 45 mg ponatinib versus imatinib for chronic myelogenous leukemia showed earlier molecular responses to ponatinib but an increased risk for vascular occlusive events.49 Lower doses of ponatinib are still under clinical investigation for efficacy and safety (URL: http://www.clinicaltrials.gov. Unique identifiers: NCT02467270 and NCT02627677). Other trials comparing imatinib with dasatinib, nilotinib, ponatinib, or bosutinib determined that the risk of vascular occlusive events was increased with all ABL kinase inhibitors, except imatinib and bosutinib.50 In our xenograft studies, there were limited side effects and no animal deaths in response to ponatinib or combination treatment. Additionally, a recent study determined the efficacy of ponatinib treatment in models of cerebral cavernous malformation51 based on endothelial loss of cerebral cavernous malformations 1 or 2. These exciting findings suggest ponatinib should be investigated as a candidate for the treatment of different types of vascular anomalies. Furthermore, in our study, we report an additional effective drug combination strategy: 2 weeks of oral combination therapy to induce lesions to regress followed by topical rapamycin administration to prevent lesion rebound. Two recent studies reported that topical rapamycin action is mediated by suppression of AKT activation.38,52 This therapeutical strategy could further reduce side effects and lesion rebound growth in patients that require long-term treatment. Furthermore, given that most VM form slow-flow, localized cutaneous lesions, combined treatment with rapamycin, and ponatinib could further be optimized by direct lesional injection or topical application to reduce systemic toxicity.
In conclusion, to the best of our knowledge, this is the first study reporting the efficacy and mechanism of combined therapy with ponatinib and rapamycin for the treatment of VM. This targeted combined therapy induced regression of VM in xenograft models with minimal adverse effects. These findings represent a highly promising and novel therapeutical approach to induce regression of difficult-to-treat VM induced by activating TIE2 mutations.
Acknowledgments
We thank Dr Lauri Eklund (University of Oulu, Finland) and Dr Miikka Vikkula (Université Catholique de Louvain, Brussels, Belgium) for generating and providing retrovirally transfected human umbilical vein endothelial cells expressing full-length TIE2-wild type (WT) or TIE2-L914F. We thank Omar Khalfaoui for performing blinded vascular area measurements and Nora Lakes for reviewing this article. We acknowledge Drs Adrienne Hammill (Cincinnati Children’s Hospital Medical Center) and Denise Adams (Boston Children’s Hospital) for critical discussions. We are grateful to the National Cancer Institute Development Therapeutics Program for providing Food and Drug Administration–approved drug library. We thank Research Flow Cytometry Core, Confocal Imaging Core, and DNA Sequencing Core at Cincinnati Children’s Hospital Medical Center for providing state-of-the-art instrumentation, services, training, and education. E. Boscolo and X. Li conceived the project. E. Boscolo and T.D. Le Cras supervised the research. X. Li, Y. Cai, and J. Goines performed the in vitro and in vivo experiments. Y. Cai optimized the working conditions for the migration and fibrin gel bead assays. J. Goines performed in vivo experiments, analysis, staining, and sanger sequencing of the venous malformation-endothelial cell (VM-EC). P. Pastura performed immunoblotting for VM-EC. L. Brichta helped with experiments with topical rapamycin. A. Lane performed the statistical analysis. J. Goines, X. Li, Y. Cai, A. L, and E. Boscolo analyzed the data. X. Li and Y. Cai prepared the figures. X. Li and E. Boscolo wrote the article. T.D. Le Cras, Y. Cai, J. Goines, L. Brichta, and A. Lane reviewed the article.
Sources of Funding
This research reported in this article was supported by the National Heart, Lung, and Blood Institute, under Award Number R01 HL117952 (E. Boscolo), part of the National Institutes of Health. This project was also supported by the Charles H. Hood Foundation (E. Boscolo), by the National Center for Advancing Translational Sciences of the National Institutes of Health, under Award Number 5UL1TR001425-03 (E. Boscolo) and a grant from the Lymphatic Malformation Institute (T.D. Le Cras). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- 3D
- 3-dimensional
- ABL
- Abelson
- ANGPT1
- angiopoietin 1
- CI
- combination index
- DMSO
- dimethyl sulfoxide
- EC
- endothelial cell
- EGM
- EC growth medium
- FITC
- fluorescein isothiocyanate
- HUVEC
- human umbilical vein endothelial cell
- mTOR
- mammalian target of rapamycin
- PI3K
- phosphoinositide 3-kinase
- PIK3CA
- class I phosphoinositide 3-kinase
- RD combo
- reduced-dose combination
- TEK
- TIE2-encoding gene
- UEA
- Ulex europaeus Agglutinin I
- VEGF
- vascular endothelial growth factor
- VM
- venous malformation
- WT
- wild type
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/ATVBAHA.118.312315.
References
- 1.Behravesh S, Yakes W, Gupta N, Naidu S, Chong BW, Khademhosseini A, Oklu R. Venous malformations: clinical diagnosis and treatment. Cardiovasc Diagn Ther. 2016;6:557–569. doi: 10.21037/cdt.2016.11.10. doi: 10.21037/cdt.2016.11.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boon LM, Mulliken JB, Vikkula M, Watkins H, Seidman J, Olsen BR, Warman ML. Assignment of a locus for dominantly inherited venous malformations to chromosome 9p. Hum Mol Genet. 1994;3:1583–1587. doi: 10.1093/hmg/3.9.1583. [DOI] [PubMed] [Google Scholar]
- 3.Soblet J, Limaye N, Uebelhoer M, Boon LM, Vikkula M. Variable somatic TIE2 mutations in half of sporadic venous malformations. Mol Syndromol. 2013;4:179–183. doi: 10.1159/000348327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dompmartin A, Vikkula M, Boon LM. Venous malformation: update on aetiopathogenesis, diagnosis and management. Phlebology. 2010;25:224–235. doi: 10.1258/phleb.2009.009041. doi: 10.1258/phleb.2009.009041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vikkula M, Boon LM, Carraway KL, III, Calvert JT, Diamonti AJ, Goumnerov B, Pasyk KA, Marchuk DA, Warman ML, Cantley LC, Mulliken JB, Olsen BR. Vascular dysmorphogenesis caused by an activating mutation in the receptor tyrosine kinase TIE2. Cell. 1996;87:1181–1190. doi: 10.1016/s0092-8674(00)81814-0. [DOI] [PubMed] [Google Scholar]
- 6.Adams DM, Trenor CC, III, Hammill AM, et al. Efficacy and safety of sirolimus in the treatment of complicated vascular anomalies. Pediatrics. 2016;137:e20153257. doi: 10.1542/peds.2015-3257. doi: 10.1542/peds.2015-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammill AM, Wentzel M, Gupta A, Nelson S, Lucky A, Elluru R, Dasgupta R, Azizkhan RG, Adams DM. Sirolimus for the treatment of complicated vascular anomalies in children. Pediatr Blood Cancer. 2011;57:1018–1024. doi: 10.1002/pbc.23124. doi: 10.1002/pbc.23124. [DOI] [PubMed] [Google Scholar]
- 8.Dompmartin A, Acher A, Thibon P, Tourbach S, Hermans C, Deneys V, Pocock B, Lequerrec A, Labbé D, Barrellier MT, Vanwijck R, Vikkula M, Boon LM. Association of localized intravascular coagulopathy with venous malformations. Arch Dermatol. 2008;144:873–877. doi: 10.1001/archderm.144.7.873. doi: 10.1001/archderm.144.7.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dompmartin A, Ballieux F, Thibon P, Lequerrec A, Hermans C, Clapuyt P, Barrellier MT, Hammer F, Labbé D, Vikkula M, Boon LM. Elevated D-dimer level in the differential diagnosis of venous malformations. Arch Dermatol. 2009;145:1239–1244. doi: 10.1001/archdermatol.2009.296. doi: 10.1001/archdermatol.2009.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horbach SE, Lokhorst MM, Saeed P, de Goüyon Matignon de Pontouraude CM, Rothová A, van der Horst CM. Sclerotherapy for low-flow vascular malformations of the head and neck: a systematic review of sclerosing agents. J Plast Reconstr Aesthet Surg. 2016;69:295–304. doi: 10.1016/j.bjps.2015.10.045. doi: 10.1016/j.bjps.2015.10.045. [DOI] [PubMed] [Google Scholar]
- 11.Wassef M, Blei F, Adams D, Alomari A, Baselga E, Berenstein A, Burrows P, Frieden IJ, Garzon MC, Lopez-Gutierrez JC, Lord DJ, Mitchel S, Powell J, Prendiville J, Vikkula M ISSVA Board and Scientific Committee. Vascular anomalies classification: recommendations from the International Society for the Study of Vascular Anomalies. Pediatrics. 2015;136:e203–e214. doi: 10.1542/peds.2014-3673. doi: 10.1542/peds.2014-3673. [DOI] [PubMed] [Google Scholar]
- 12.Limaye N, Kangas J, Mendola A, Godfraind C, Schlögel MJ, Helaers R, Eklund L, Boon LM, Vikkula M. Somatic activating PIK3CA mutations cause venous malformation. Am J Hum Genet. 2015;97:914–921. doi: 10.1016/j.ajhg.2015.11.011. doi: 10.1016/j.ajhg.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nätynki M, Kangas J, Miinalainen I, Sormunen R, Pietilä R, Soblet J, Boon LM, Vikkula M, Limaye N, Eklund L. Common and specific effects of TIE2 mutations causing venous malformations. Hum Mol Genet. 2015;24:6374–6389. doi: 10.1093/hmg/ddv349. doi: 10.1093/hmg/ddv349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goines J, Li X, Cai Y, Mobberley-Schuman P, Metcalf M, Fishman SJ, Adams DM, Hammill AM, Boscolo E. A xenograft model for venous malformation. Angiogenesis. 2018;21:725–735. doi: 10.1007/s10456-018-9624-7. doi: 10.1007/s10456-018-9624-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boscolo E, Limaye N, Huang L, et al. Rapamycin improves TIE2-mutated venous malformation in murine model and human subjects. J Clin Invest. 2015;125:3491–3504. doi: 10.1172/JCI76004. doi: 10.1172/JCI76004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castel P, Carmona FJ, Grego-Bessa J, Berger MF, Viale A, Anderson KV, Bague S, Scaltriti M, Antonescu CR, Baselga E, Baselga J. Somatic PIK3CA mutations as a driver of sporadic venous malformations. Sci Transl Med. 2016;8:332ra42. doi: 10.1126/scitranslmed.aaf1164. doi: 10.1126/scitranslmed.aaf1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Castillo SD, Tzouanacou E, Zaw-Thin M, et al. Somatic activating mutations in Pik3ca cause sporadic venous malformations in mice and humans. Sci Transl Med. 2016;8:332ra43. doi: 10.1126/scitranslmed.aad9982. doi: 10.1126/scitranslmed.aad9982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson JK, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 19.Kurek KC, Luks VL, Ayturk UM, Alomari AI, Fishman SJ, Spencer SA, Mulliken JB, Bowen ME, Yamamoto GL, Kozakewich HP, Warman ML. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–1115. doi: 10.1016/j.ajhg.2012.05.006. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maclellan RA, Luks VL, Vivero MP, Mulliken JB, Zurakowski D, Padwa BL, Warman ML, Greene AK, Kurek KC. PIK3CA activating mutations in facial infiltrating lipomatosis. Plast Reconstr Surg. 2014;133:12e–19e. doi: 10.1097/01.prs.0000436822.26709.7c. [DOI] [PubMed] [Google Scholar]
- 21.Rivière JB, Mirzaa GM, O’Roak BJ, et al. Finding of Rare Disease Genes (FORGE) Canada Consortium. De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat Genet. 2012;44:934–940. doi: 10.1038/ng.2331. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boscolo E, Coma S, Luks VL, Greene AK, Klagsbrun M, Warman ML, Bischoff J. AKT hyper-phosphorylation associated with PI3K mutations in lymphatic endothelial cells from a patient with lymphatic malformation. Angiogenesis. 2015;18:151–162. doi: 10.1007/s10456-014-9453-2. doi: 10.1007/s10456-014-9453-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luks VL, Kamitaki N, Vivero MP, et al. Lymphatic and other vascular malformative/overgrowth disorders are caused by somatic mutations in PIK3CA. J Pediatr. 2015;166:1048.e1–1054.e1. doi: 10.1016/j.jpeds.2014.12.069. doi: 10.1016/j.jpeds.2014.12.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osborn AJ, Dickie P, Neilson DE, Glaser K, Lynch KA, Gupta A, Dickie BH. Activating PIK3CA alleles and lymphangiogenic phenotype of lymphatic endothelial cells isolated from lymphatic malformations. Hum Mol Genet. 2015;24:926–938. doi: 10.1093/hmg/ddu505. doi: 10.1093/hmg/ddu505. [DOI] [PubMed] [Google Scholar]
- 25.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 26.Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci. 2016;129:9–16. doi: 10.1242/jcs.175521. doi: 10.1242/jcs.175521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orellana EA, Kasinski AL. Sulforhodamine B (SRB) assay in cell culture to investigate cell proliferation. Bio Protoc. 2016;6:e1984. doi: 10.21769/BioProtoc.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Tong LJ, Ding J, Meng LH. Systematic combination screening reveals synergism between rapamycin and sunitinib against human lung cancer. Cancer Lett. 2014;342:159–166. doi: 10.1016/j.canlet.2013.08.046. doi: 10.1016/j.canlet.2013.08.046. [DOI] [PubMed] [Google Scholar]
- 30.Nakatsu MN, Hughes CC. An optimized three-dimensional in vitro model for the analysis of angiogenesis. Methods Enzymol. 2008;443:65–82. doi: 10.1016/S0076-6879(08)02004-1. doi: 10.1016/S0076-6879(08)02004-1. [DOI] [PubMed] [Google Scholar]
- 31.Houghton PJ, Morton CL, Gorlick R, Lock RB, Carol H, Reynolds CP, Kang MH, Maris JM, Keir ST, Kolb EA, Wu J, Wozniak AW, Billups CA, Rubinstein L, Smith MA. Stage 2 combination testing of rapamycin with cytotoxic agents by the pediatric preclinical testing program. Mol Cancer Ther. 2010;9:101–112. doi: 10.1158/1535-7163.MCT-09-0952. doi: 10.1158/1535-7163.MCT-09-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 33.McCormack FX, Inoue Y, Moss J, et al. National Institutes of Health Rare Lung Diseases Consortium; MILES Trial Group. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364:1595–1606. doi: 10.1056/NEJMoa1100391. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garcia-Montero P, Del Boz J, Sanchez-Martinez M, Escudero Santos IM, Baselga E. Microcystic lymphatic malformation successfully treated with topical rapamycin. Pediatrics. 2017;139:e20162105. doi: 10.1542/peds.2016-2105. [DOI] [PubMed] [Google Scholar]
- 35.Ivars M, Redondo P. Efficacy of topical sirolimus (Rapamycin) for the treatment of microcystic lymphatic malformations. JAMA Dermatol. 2017;153:103–105. doi: 10.1001/jamadermatol.2016.3697. doi: 10.1001/jamadermatol.2016.3697. [DOI] [PubMed] [Google Scholar]
- 36.Barilli A, Visigalli R, Sala R, Gazzola GC, Parolari A, Tremoli E, Bonomini S, Simon A, Closs EI, Dall’Asta V, Bussolati O. In human endothelial cells rapamycin causes mTORC2 inhibition and impairs cell viability and function. Cardiovasc Res. 2008;78:563–571. doi: 10.1093/cvr/cvn024. doi: 10.1093/cvr/cvn024. [DOI] [PubMed] [Google Scholar]
- 37.Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol. 2004;24:200–216. doi: 10.1128/MCB.24.1.200-216.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Du W, Gerald D, Perruzzi CA, Rodriguez-Waitkus P, Enayati L, Krishnan B, Edmonds J, Hochman ML, Lev DC, Phung TL. Vascular tumors have increased p70 S6-kinase activation and are inhibited by topical rapamycin. Lab Invest. 2013;93:1115–1127. doi: 10.1038/labinvest.2013.98. doi: 10.1038/labinvest.2013.98. [DOI] [PubMed] [Google Scholar]
- 39.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 40.Gotoh A, Miyazawa K, Ohyashiki K, Toyama K. Potential molecules implicated in downstream signaling pathways of p185BCR-ABL in Ph+ ALL involve GTPase-activating protein, phospholipase C-gamma 1, and phosphatidylinositol 3’-kinase. Leukemia. 1994;8:115–120. [PubMed] [Google Scholar]
- 41.Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood. 1995;86:726–736. [PubMed] [Google Scholar]
- 42.Markova B, Albers C, Breitenbuecher F, Melo JV, Brümmendorf TH, Heidel F, Lipka D, Duyster J, Huber C, Fischer T. Novel pathway in Bcr-Abl signal transduction involves Akt-independent, PLC-gamma1-driven activation of mTOR/p70S6-kinase pathway. Oncogene. 2010;29:739–751. doi: 10.1038/onc.2009.374. doi: 10.1038/onc.2009.374. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T, Shibuya M. The 230 kDa mature form of KDR/Flk-1 (VEGF receptor-2) activates the PLC-gamma pathway and partially induces mitotic signals in NIH3T3 fibroblasts. Oncogene. 1997;14:2079–2089. doi: 10.1038/sj.onc.1201047. doi: 10.1038/sj.onc.1201047. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi T, Ueno H, Shibuya M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999;18:2221–2230. doi: 10.1038/sj.onc.1202527. doi: 10.1038/sj.onc.1202527. [DOI] [PubMed] [Google Scholar]
- 45.Chislock EM, Ring C, Pendergast AM. Abl kinases are required for vascular function, Tie2 expression, and angiopoietin-1–mediated survival. Proc Natl Acad Sci USA. 2013;110:12432–12437. doi: 10.1073/pnas.1304188110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fantin A, Lampropoulou A, Senatore V, Brash JT, Prahst C, Lange CA, Liyanage SE, Raimondi C, Bainbridge JW, Augustin HG, Ruhrberg C. VEGF165-induced vascular permeability requires NRP1 for ABL-mediated SRC family kinase activation. J Exp Med. 2017;214:1049–1064. doi: 10.1084/jem.20160311. doi: 10.1084/jem.20160311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raimondi C, Fantin A, Lampropoulou A, Denti L, Chikh A, Ruhrberg C. Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. J Exp Med. 2014;211:1167–1183. doi: 10.1084/jem.20132330. doi: 10.1084/jem.20132330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Charng WL, Chen CA, et al. Germline mutations in ABL1 cause an autosomal dominant syndrome characterized by congenital heart defects and skeletal malformations. Nat Genet. 2017;49:613–617. doi: 10.1038/ng.3815. doi: 10.1038/ng.3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lipton JH, Chuah C, Guerci-Bresler A, et al. EPIC investigators. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: an international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17:612–621. doi: 10.1016/S1470-2045(16)00080-2. doi: 10.1016/S1470-2045(16)00080-2. [DOI] [PubMed] [Google Scholar]
- 50.Haguet H, Douxfils J, Mullier F, Chatelain C, Graux C, Dogné JM. Risk of arterial and venous occlusive events in chronic myeloid leukemia patients treated with new generation BCR-ABL tyrosine kinase inhibitors: a systematic review and meta-analysis. Expert Opin Drug Saf. 2017;16:5–12. doi: 10.1080/14740338.2017.1261824. doi: 10.1080/14740338.2017.1261824. [DOI] [PubMed] [Google Scholar]
- 51.Choi JP, Wang R, Yang X, Wang X, Wang L, Ting KK, Foley M, Cogger V, Yang Z, Liu F, Han Z, Liu R, Baell J, Zheng X. Ponatinib (AP24534) inhibits MEKK3-KLF signaling and prevents formation and progression of cerebral cavernous malformations. Sci Adv. 2018;4:eaau0731. doi: 10.1126/sciadv.aau0731. doi: 10.1126/sciadv.aau0731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tan W, Jia W, Sun V, Mihm MC, Jr, Nelson JS. Topical rapamycin suppresses the angiogenesis pathways induced by pulsed dye laser: molecular mechanisms of inhibition of regeneration and revascularization of photocoagulated cutaneous blood vessels. Lasers Surg Med. 2012;44:796–804. doi: 10.1002/lsm.22101. doi: 10.1002/lsm.22101. [DOI] [PMC free article] [PubMed] [Google Scholar]