

Abstract
Tandem mass spectrometry can provide structural information on intact protein assemblies, generating mass fingerprints indicative of the stoichiometry and quaternary arrangement of the subunits. However, in such experiments, collision-induced dissociation yields restricted information due to simultaneous subunit unfolding, charge rearrangement, and subsequent ejection of a highly charged unfolded single subunit. Alternative fragmentation strategies can potentially overcome this and supply a deeper level of structural detail. Here, we implemented ultraviolet photodissociation (UVPD) on an Orbitrap mass spectrometer optimized for native MS and benchmark its performance to HCD fragmentation using various protein oligomers. We investigated dimeric β-lactoglobulin, dimeric superoxide dismutase, dimeric and tetrameric concanavalin A, and heptameric GroES and Gp31; ranging in molecular weight from 32 to 102 kDa. We find that, for the investigated systems, UVPD produces more symmetric charge partitioning than HCD. While HCD spectra show sporadic fragmentation over the full protein backbone sequence of the subunits with a bias toward fragmenting labile bonds, UVPD spectra provided higher sequence coverage. Taken together, we conclude that UVPD is a strong addition to the toolbox of fragmentation methods for top-down proteomics experiments, especially for native protein assemblies.
Introduction
Most biological processes in cells involve in time and space regulated noncovalent interactions between proteins. These interactions functionalize molecular machines, providing more complex behavior than the sum of the individual parts would allow.1,2 An estimated 80% of all expressed proteins engage in such interactions, which for the human proteome with its 20 000 genes may result in 650 000 protein–protein interactions within the cell.3,4 This rich and highly dynamic level of complexity in cellular processes can be investigated with high throughput methods like affinity-purification combined with mass spectrometry (AP-MS), producing large catalogues of interacting proteins5−7 in short timeframes.8 Although very useful to provide a snapshot of active protein interactions, these methods identify the involved proteins with varying degrees of confidence and provide very global insight into the detected interactions. A higher level of detail requires use of different structure based methods such as X-ray crystallography, electron microscopy, and NMR. However, these methods exhibit inherent restrictions as they typically require high amounts of analyte and have low throughput. Thus, alternative methods would be beneficial.
Native mass spectrometry is a complementary technique that allows for the investigation of proteins and their interaction partners in their native state.9−11 With this technique, it is possible to extract information with regard to the quaternary structure and subunit stoichiometry of the assembly,9 as well as, sequence information from individual subunits.12 While stoichiometry can often be determined by the intact mass spectrum through the use of high-resolution mass spectrometers, determination of quaternary structure requires additional techniques like MS fragmentation methods. Subjecting the protein assembly ion to increasing collisional energies leads to the sequential ejection of individual subunits, providing insight into the quaternary structure of the assembly.13 From the available data so far, it has been shown that collision based fragmentation methods almost exclusively eject a protein monomer regardless of the protein assembly size, structure or subunit organization.14−19 Moreover, the ejected monomer is visible in the mass spectrum at high charge states, indicative of protein monomer unfolding and charge relocalization prior to its ejection,20 which leads to a loss of information about the structure of the precursor protein assembly.21,22 It has been postulated that the monomer unfolding occurs due to an increase in internal energy followed by transfer of several protons to relieve coulombic repulsion, which in turn promotes further unfolding and finally monomer ejection.15,23,24 The remaining (n – 1)-meric assembly is present in the mass spectrum with the remainder of the charges, leading to the assumption that it remains in a more folded state.15 Overall, this results in charge partitioning that is asymmetric with respect to the mass of each product, as the charges are distributed roughly proportional to the surface area.23,25 This mechanism of fragmentation limits its usefulness in determining structural information as the unfolded monomer does not retain significant amounts of topological information.23 Additionally, the highly charged nature of the monomer can limit our ability to resolve structural heterogeneity which may be present within the primary sequence of the protein and, in some cases, drive the ions outside the mass and/or transmission range of the mass spectrometer due to the limited m/z window of the ion optics.26 Previous investigations have indicated that the asymmetric dissociation process depends on several factors including the charge state of the molecule, but also on the timescales involved in the fragmentation process.17 This suggests that alternative and faster means of activating protein assemblies may be beneficial, especially when they open up dissociation channels other than the ejection of a single, unfolded monomer.
An interesting alternative fragmentation method explored is surface induced dissociation (SID). Noteworthy, SID was shown to produce preferably symmetric charge partitioning for protein assemblies, which was attributed to its more prompt, high-energy fragmentation mechanism.25,27,28 Another newcomer in the field of protein assembly dissociation techniques is ultraviolet photodissociation (UVPD), which utilizes the natural chromophores present in the backbone of peptides and proteins to absorb highly energetic photons (λ = 193 nm) emitted from a laser.29−35 The technique may provide prompt fragmentation at an activation time scale close to the energy deposition in SID,36 and could thus potentially prove to be a beneficial method for probing of assembly composition and topology. In this study, we report the modification of an Orbitrap-based mass spectrometer with Extended Mass Range (EMR) capabilities to support UVPD. We introduce further optimizations to make our previously reported UVPD strategy work for these large assemblies.35,37 The new UVPD capabilities of the instrument were applied to the investigation of multimeric protein assemblies in their native state and compared to the dissociation behavior with HCD. Studied systems include dimeric β-lactoglobulin (β-Lac), dimeric Cu, Zn-superoxide dismutase (Cu,Zn-SOD), dimeric and tetrameric concanavalin A (ConA), and heptameric GroES and Gp31 assemblies. These protein assemblies range in mass from 32 to 102 kDa. Each system was subjected to a range of collision energies, which were selected in such a way that the lowest value is the onset of precursor depletion while the highest value completely depletes the precursor. For these systems, HCD fragmentation data, in agreement with literature, shows largely asymmetric charge partitioning and ejection of monomers. However, we also find that the asymmetric charge partitioning of HCD is diminished when structural constraints like disulfide bonds are present.15,38 In contrast, UVPD experiments, where photons are absorbed by the precursor ions during the fixed short 5 ns pulse of the laser, lead more to the ejection of a compact, low-charge monomer via a high-energy deposition pathway, similar to SID. Investigation of these phenomena as a function of system size, degree of stabilizing interactions, and complexity revealed that there is likely a natural limit to UVPD in its utility for symmetric partitioning based on the size of the subunits and the stability of the binding interfaces between the subunits. Additionally, we find that UVPD for all investigated systems outperforms HCD in terms of backbone sequence coverage. Overall, these data demonstrate that UVPD provides a simple, versatile method for the structural analysis of protein assemblies by native mass spectrometry, adding to the toolbox for top-down proteomics.
Results and Discussion
Implementation of UVPD on the Orbitrap-based EMR
For a detailed description of the instrument modifications please refer to the Methods section. In brief, we modified a standard Orbitrap-based Exactive Plus mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) to support the analysis of large protein assemblies under native MS conditions.39 The implementation of the UV laser is largely similar to the one described previously.35 In addition, to compensate for the pressure drop caused by the removal of the electrometer, a CaF2 Teflon-sealed viewport was constructed, which sealed the HCD cell and served as an optical aperture for collimation of the laser beam (Figure 1; inset).
Figure 1.

Implementation of UVPD on the modified Orbitrap-based Exactive Plus mass spectrometer. The laser is guided into the HCD cell through a viewport fitted to the back-end of the HCD cell. Triggering of the laser is done through read-out of the split lens. The inset shows the newly designed viewport, with the Teflon sealing ring highlighted in yellow.
The performance of UVPD on our system was benchmarked on the often used model system ubiquitin, for which our setup produces fragments covering 100% of the sequence and results in significantly better N-terminal backbone coverage when compared to HCD (Figure S1.1). With these modifications, protein fragments generated for native state proteins with precursor masses of up to at least 100 kDa can be transmitted and analyzed (Figure S1.2). Following this successful benchmark testing, we subsequently set out to investigate the performance of HCD and UVPD on native noncovalently bound protein assemblies, starting from dimers to higher oligomers, as described in the following paragraphs.
Dimeric Protein Assemblies
As an initial system to investigate the difference between HCD and UVPD for dissociation of protein assemblies, the lectin concanavalin A (ConA) was selected. This lectin naturally occurs as both a homodimer (51 kDa) and a homotetramer (102 kDa) and has previously been investigated by tandem MS using CID,40 SID,41 and electron transfer dissociation (ETD).42 The dimer was investigated first as it offers the least noncovalent intermolecular interactions between the subunits making it the simpler system. During tandem MS analysis, the 15+ charge state (z = 15+) of the homodimer was isolated and subjected to either HCD or UVPD fragmentation, at a variety of collision energies and laser energies with values of 10–140 V and 0.5–2.5 mJ pulse–1, respectively.
When subjected to low collision energy HCD (10 V), the tandem mass spectrum (Figure 2a) is dominated by the remaining precursor and shows monomeric dissociation products with z = 8+ and 7+. Interestingly, these products are consistent with symmetric charge partitioning of the homodimer, indicating that the initially accessed fragmentation pathway is the disruption of the noncovalent interactions between the two subunits. When the collision energy is increased to 100 V, the tandem mass spectrum shows the emergence of two distinct charge state envelopes (Figure 2a, bottom; red and orange dashed lines) and several covalent fragments present at m/z values of <2000 Th. The first envelope, denoted by the orange dashed line, comprises of the monomeric dissociation products at z = 8+ and 7+, which are consistent with the symmetric dissociation products observed at low collision energies. This envelope also shows the presence of the monomeric dissociation products at z = 9+ and 6+, suggesting that charge relocalization and thus asymmetric dissociation is starting to occur. This is further supported by the presence of a second charge state envelope (denoted by the red dashed line), which is centered on the monomeric dissociation product at z = 11+ and is consistent with the full onset of asymmetric dissociation and monomer unfolding. This transition from symmetric to asymmetric dissociation is clearly visible by studying the dissociation products as a function of collision energy (Figure 2b). At the lowest collision energy, the symmetric dissociation pathway dominates, as shown by the high intensity of the monomeric dissociation product at z = 8+. At a collision energy of 80 V, the asymmetric dissociation pathway is increasingly accessed as shown by the appearance of the monomeric dissociation product at z = 11+, reaching maximum intensity at 100 V collision energy, and a sharp decrease in intensity of z = 8+. At collision energies higher than 100 V, both the 11+ and 8+ charge states are reduced in relative intensity and there is an increase in sequence coverage, which indicates the onset of covalent bond cleavage. Taken together we hypothesize that the dissociation pathway for dimeric ConA with HCD displays the following energy dependent order. At low energies, the protein assembly undergoes symmetric dissociation. As the collision energy is increased, the protein assembly dissociates in a more asymmetric fashion. Further increase of the collision energy enhances the asymmetric dissociation behavior and finally leads to covalent bond cleavage.
Figure 2.

Annotated spectra representing (a) low and high energy HCD and (c) low and high energy UVPD of dimer ConA (z = 15+). Symbols corresponding to the form of ConA are described on the right side of the figure. Normalized intensities of the z = 8+ and 11+, representations of the symmetric and asymmetric dissociation pathway, respectively, are plotted as a function of (b) collisional energies for HCD and (d) laser energies for UVPD. Sequence coverage (seq. cov.) is provided on the right-hand vertical axis. The dashed lines indicate the different peak envelopes.
In contrast to HCD, the UVPD fragmentation mass spectra show only symmetric dissociation prior to covalent bond cleavage. At the lowest laser energy investigated, the formation of the monomer dissociation products at z = 8+ and 7+ are dominant (Figure 2c, top). As the laser energy is increased to 2 mJ pulse–1, these monomeric product ions remain the predominant pathway of dissociation, as shown by the stable charge state envelope over the range of energies (green dashed line), and the absence of charge states consistent with asymmetric dissociation (Figure 2c, bottom). Moreover, covalent bond cleavages are readily occurring at almost all laser energies, indicating that symmetric dissociation leads directly to backbone fragmentation with high sequence coverage, while the asymmetric dissociation pathway is not accessed at any appreciable amount. At all investigated laser energies, the symmetric dissociation products are the dominant pathway as suggested by an absence of the monomeric dissociation product at z = 11+. Although, as the energy increases, the relative intensity of the monomeric dissociation product at z = 8+ decreases and there is a small increase in sequence coverage after 1 mJ pulse–1 (Figure 2d). These data demonstrate that UVPD does not lead to appreciable monomeric unfolding upon fragmentation for this dimeric assembly.
For the ConA dimer, UVPD somewhat outperforms HCD in backbone coverage, covering 50% and 45% of the sequence, respectively. The differences in activation, however, do result in differences in the observed fragmentation patterns (Figure S2.1). The observed sporadic cleavages over the full backbone for HCD are likely caused by the preference of collision-induced dissociation techniques toward cleaving the labile bonds first, resulting in more readily detectable fragments associated with those bonds. UVPD on the other hand tends to provide most of the cleavages from the surface-exposed regions, which are known to be both termini in the case of dimeric ConA (PDB reference: dimeric concanavalin A, 1GKB). Such behavior was previously reported for ETD and electron capture dissociation (ECD),12 however these methods are hampered, in the analysis of proteins under native conditions, by their strong charge dependence. When combining single HCD and UVPD deconvoluted spectra the backbone sequence coverage reaches 66%, an increase of 32% and 46% as compared to UVPD and HCD alone, respectively (Figure S2.1). Comparison of the UVPD fragmentation patterns between the monomeric and dimeric form provides insights into the binding interface. Based on a previously reported spectroscopy study, the regions stabilized by hydrogen bonds tend to favor proton transfer, which can potentially lead to a lower degree of fragmentation.43 Here, we indeed find that UVPD of the monomer provided longer N-terminal fragments than for the dimer, highlighting that this region is potentially involved in subunit interaction. The UVPD data of the monomer additionally showed reduced C-terminal coverage, suggesting that this part is surface-exposed upon binding.
The differences in fragmentation pathways between HCD and UVPD for dimeric protein assemblies have been attributed to the unfolding of the monomeric subunit prior to ejection with HCD versus the ejection of the folded monomer with UVPD.44 As such, it is of interest to explore the differences in fragmentation pathways for an assembly that possesses structural constraints restricting its ability to unfold.45 The protein assembly β-lactoglobulin (β-Lac) is present as a homodimer (36 kDa); however, unlike ConA, β-Lac contains two intrasubunit disulfide bridges that restrict the conformational flexibility and the ability to unfold.46 The 13+ charge state of the homodimer was isolated and subjected to HCD and UVPD fragmentation at a variety of collision and laser energies with values of 25–150 V and 0.5–2.5 mJ pulse–1, respectively. At the lowest collision energy (25 V) the spectrum is dominated by monomeric dissociation products at z = 7+ and z = 6+, which are consistent with symmetric charge partitioning (Figure 3a, top). At 100 V collision energy, the production of these ion species remains the dominant pathway and the formation of backbone fragmentation products starts to occur (Figure 3a, bottom). Consistent with the onset of backbone fragmentation, the monomeric dissociation product at z = 7+ decreases in relative intensity at energies higher than 100 V (Figure S3.1, top panel). UVPD fragmentation of the β-Lac homodimer at the lowest laser energy of 0.5 mJ pulse–1 shows minor dissociation products, nevertheless, the monomeric dissociation products at z = 7+ and 6+, indicative of symmetrical charge partitioning, are already visible (Figure 3b, top panel). As the energy is increased to 2 mJ pulse–1, the amount of precursor depletion increases but the symmetrical dissociation products remain dominant (Figure 3b, bottom panel) and backbone fragments below 2000 m/z start to appear. For variable laser energy studies, the symmetric dissociation pathway remains dominant at all investigated laser energies, as shown by the minor decrease in normalized intensity of the monomeric dissociation product at z = 7+ (Figure S3.1, bottom panel).
Figure 3.
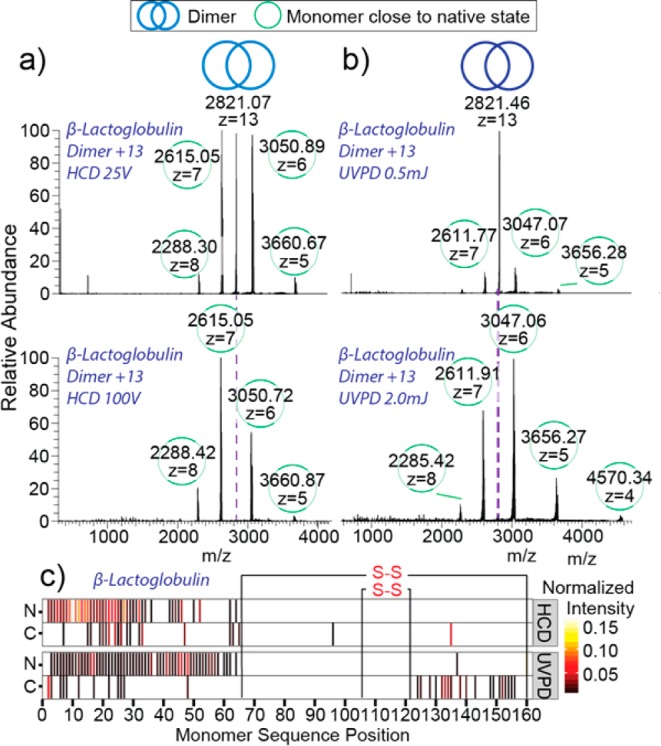
(a) HCD and (b) UVPD of dimeric β-lactoglobulin are represented by spectra recorded at low (top) and high (bottom) collisional and laser energies. Fragmentation heat maps (c) display cleavage positions produced via HCD and UVPD as well as normalized intensities of the respective fragments.
Comparison of covalent bond cleavages generated by HCD and UVPD fragmentation show that for both methods there are very few fragments within the region demarked by the disulfide bridges on the monomer and the majority of assigned fragments correspond to the unrestricted N-terminal region of the protein (Figure 3c). Within this N-terminal region, UVPD produces greater sequence coverage than that produced by HCD. Additionally, UVPD shows some fragment ions within the disulfide protected region, indicating that the disulfide bond is cleaved during laser irradiation, consistent with previously reported results by O’Brien et al.44 and subsequent covalent bond cleavages occur. This process is largely absent for HCD fragmentation. Collectively, the fragmentation data for the β-Lac homodimer generated with HCD and UVPD appear largely similar in terms of symmetric versus asymmetric dissociation, indicating that the structural rigidity supplied by the disulfide bonds limits the extent of monomeric unfolding that occurs with HCD. Similar behavior was also observed for the superoxide dismutase (Cu–Zn–SOD) dimer, which also contains a disulfide bridge offering it structural rigidity (Figure S3.2).
Tetrameric Protein Assemblies
The tetrameric assembly of ConA (102 kDa) provides enhanced stability between the subunits owing to increased numbers of noncovalent intermolecular interactions, potentially resulting in differences between dissociation with UVPD and HCD as compared to the homodimer. When tetrameric ConA at z = 21+ is analyzed with UVPD at 3 mJ pulse–1, the resulting mass spectrum shows two charge envelopes (Figure 4a). The first envelope shows monomeric dissociation products ranging from z = 6+ to 13+, while the second envelope corresponds to the complementary trimeric dissociation products ranging from z = 8+ to 12+. These data indicate that the preferred pathway of dissociation of the tetramer is the ejection of a monomeric subunit as opposed to formation of two dimers. Moreover, the range of charge states for the ejected monomer suggests that a combination of symmetric (z = 6+ and 7+) dissociation, as well as, asymmetric dissociation (z = 13+, 12+, and 11+) occurs and that structural rearrangement may potentially also occur with UVPD. In comparison, the HCD generated mass spectrum contains more highly charged monomeric dissociation products (Figure 4b). It is of interest to note that the HCD mass spectrum shows no monomeric dissociation products at charge states that correspond to the symmetric dissociation pathway. Additionally, HCD appears to produce a bimodal charge state envelope for the monomer (red and orange dashed lines). The elevated charge states in the second distribution suggest that an additional structural transition occurs leading to a likely less compact structural gas-phase conformation of the monomer.47 In contrast, UVPD results in an envelope with a smaller average charge, suggesting a more compact gas-phase conformation. The fragmentation map for tetrameric ConA shows few N-terminal fragments (Figure S4.1), potentially explained by additional stabilization by the Ca2+ and Mn2+ bound at the N-terminus.48
Figure 4.
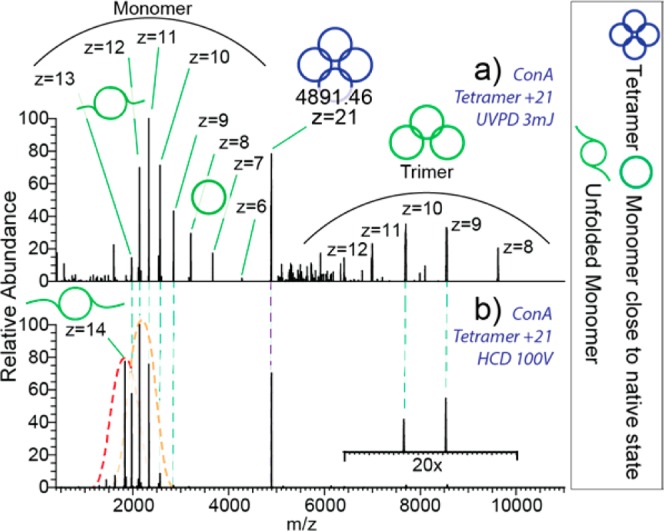
(a) UVPD spectrum represent partitioning of tetrameric ConA of z = 21+ into monomers of broad charge range and complementary trimers. (b) HCD spectrum is dominated by highly charged monomers. The dotted lines indicate the peak envelopes.
Heptameric Protein Assemblies
As a next step in our investigation we analyzed the GroES heptamer, which is a molecular co-chaperonin found for instance in E. coli. In complex with the chaperonin GroEL it acts as a macromolecular machine whose main function is to assist the correct folding of the proteins in the cell.49,50 Under physiological conditions the 10.4 kDa monomers of GroES assemble into stable ring-shaped heptamers with a molecular weight of 73 kDa.51 The z = 18+ charge state of heptameric GroES was isolated and subjected to fragmentation at a range of collision and laser energies; for HCD: 20–200 V, and for UVPD: 0.5–4 mJ pulse–1. At low energies, both photon-induced activation and collisional activation resulted in ejection of the monomer with charge states ranging from z = 4+ to 8+ displaying a remarkable bimodal charge distribution (Figure S5.1). The bimodal distribution, observed for the heptameric GroES dissociation spectra is similar to that observed for the tetrameric ConA HCD spectra, which suggests a gas-phase conformational change of the ejected monomer. At higher laser energies the photon-induced activation prompted the ejection of a low-charged monomer (z = 3+) that was never observed in the HCD spectra (Figure 5a). This represents a unique UVPD dissociation pathway that is consistent with symmetric charge partitioning upon dissociation. The subunits that were ejected following this pathway were likely to partly retain their tertiary structure, indicative of the fast deposition of a large amount of energy into the ion.25,52
Figure 5.

(a) Dissociation spectra of heptameric GroES (z = +18) activated by UVPD at 2 mJ pulse–1 (top) and with HCD at 140 V (bottom). (b) Dissociation spectra of Gp31 heptamer (z = +21) activated by UVPD at 2 mJ pulse–1 (top) and with HCD at 50 V.
Gp31 is a bacteriophage T4 structural homologue of GroES, which following infection of E. coli competes with GroES for binding to GroEL to favor the folding of the bacteriophage proteins.53 The three-dimensional structure of Gp31 closely resembles that of the GroES with slightly larger subunits and thus a higher molecular weight of the intact heptamer of 84 kDa.54 We found that the Gp31 heptamer exhibits lower stability compared to GroES both in solution and in the gas phase. Both UVPD and HCD activation of the isolated Gp31 heptamer at z = 21+ leads to ejection of a monomer displaying asymmetric charge partitioning (Figure 5b). However, UVPD resulted in a lower average charge, as the highly charged monomer dissociation products observed in the HCD spectra (z = +9, + 8) are absent in the UVPD spectra. This is indicative of a relatively more compact state of the subunits dissociated via UVPD. Interestingly, due to the weak intersubunit interactions ejection of a compact monomer from the Gp31 complex becomes energetically more favorable relative to the subunit unfolding. For this co-chaperone system both HCD and UVPD display symmetrical charge partitioning pathway, producing z = 3+ monomers from the z = 21+ heptameric precursor. This highlights the role of intersubunit interactions in the interplay between the monomer ejection and unfolding upon collision- and photon-induced activation, but also reveals that dissociation pathways of very alike systems (GroEL and Gp31 heptamers) may be distinct, and indicative of the biochemical properties of their native precursors.
At the higher energies, UVPD of Gp31 resulted in improved sequence coverage compared to HCD fragmentation. As described above, the average charge states for UVPD are lower, indicating that this method is capable of retaining a higher degree of structural stability for weakly interacting subunits, and generate covalent fragments from a more compact state of the molecule (Figure S6.1).
Comparison of Experimental Charge Partitioning with Theoretical Predictions
To describe the charge partitioning upon assembly dissociation, a number of models have been proposed that suggest that the charge state of the ejected subunit can serve as a predictor of the degree of its unfolding.24 Additionally, the ejected subunit and the remaining (n – 1)-mer divide the number of charges roughly proportional to their exposed surface areas,17 which has been shown to often be the case with SID fragmentation.25 Thus, if dissociation occurs on a time scale shorter than the time scale of gas-phase conformational rearrangement, the fraction of the precursor ion charge retained by the ejected monomer can be roughly estimated as Smon/(Smon + S(n–1)mer), where Smon and S(n–1)mer are exposed surface areas of the ejected monomer and the remaining (n – 1)-mer, respectively. Our implementation of UVPD allows energy deposition on a time scale close to that of SID,36 enabling it to more readily achieve symmetric charge partitioning. We indeed find that UVPD produces ejected monomers with charge states more consistent with symmetric charge partitioning as compared to those generated by HCD, which is especially true for the dimers (Figure 6). For assemblies with more than two subunits we however cannot expect fully symmetric charge distributions, as the smaller subunit has a larger surface area relative to its mass than the remaining (n – 1)-mer. Overall, we find that in the case of UVPD the ConA dimer displays fully symmetrical charge partitioning. For the tetramers and the heptamers the average charge of the ejected monomer is higher than expected for the symmetric partitioning, indicating that the energy deposited by the UV photons also leads to subunit unfolding, although to a smaller extent than HCD.
Figure 6.
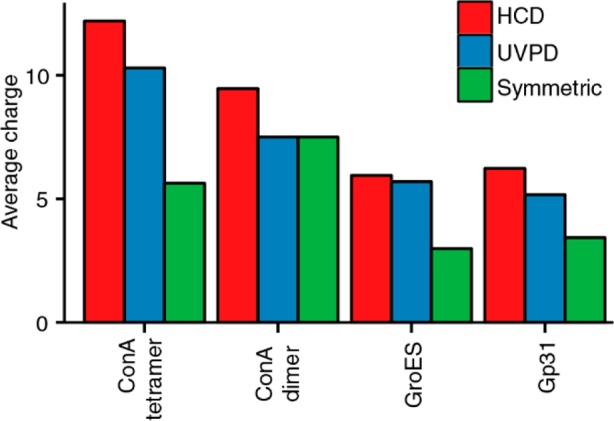
Average charges of the ejected monomers produced by HCD and UVPD, compared to the expected charge in symmetric dissociation. The symmetric values were calculated from the X-ray structure based on the exposed surface areas following deletion of individual subunits.
Conclusions
Here, we report new modifications to an Exactive Plus mass spectrometer with EMR capabilities, enabling it to perform both HCD and UVPD fragmentation on native protein assemblies with molecular weights up to at least 100 kDa. We compare the UVPD performance to the built-in HCD fragmentation capabilities on a set of oligomeric protein assemblies, ranging from dimers to heptamers, and in mass from 32 to 102 kDa. As expected, HCD leads to mostly asymmetric dissociation products, consistent with structural unfolding during the dissociation process. However, UVPD showed more symmetrical dissociation behavior, resembling, in some cases, SID-like behavior.
While UVPD did lead to a higher degree of symmetric dissociation for all systems investigated, we also show that UVPD depends on both the size of the protein complex as well as the stability of the intersubunit interface. This is reflected in that higher laser energies were required to produce more symmetric dissociation products. However, this intersubunit stability dependence of UVPD may be structurally informative for certain oligomeric protein complexes. For the same oligomeric state of GroES and Gp31, we show that the protein complex with the lower intersubunit stability, Gp31, exhibits more symmetric dissociation products than GroES with UVPD. Furthermore, the ability of UVPD to offer both symmetric dissociation products, while at the same time producing significant backbone coverage makes the technique an attractive one-stop method for simultaneous probing protein assembly structure and stability and subunit sequence. This will provide further confidence in protein identification and ligand/PTM site localization. Collectively, our results demonstrate that UVPD is poised to become a strong addition to the top-down proteomics toolbox as it produces higher subunit backbone coverage, a high percentage of symmetric dissociation products as compared to HCD, and that the partitioning between symmetric and asymmetric pathways may be reflective of the biochemical and biophysical nature of that particular protein complex.
Methods
Instrument Modifications
As previously described, an Exactive Plus mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) was optimized for transmission and detection of ions with m/z up to 50 kTh.37,39,55 A dedicated gauge was installed controlling the pressure of the collisional gas for more efficient cooling and desolvation of heavy ions. Furthermore, the operating frequencies of the front-end RF guides and the HCD cell were lowered to improve ion transmission and reduce loss of ions during activation at high energies; a preamplifier with lower high-pass filter cutoff was used to improve transmission of lower frequency image current signals originating from ions with higher m/z values. For isolation we added a standard quadrupole mass filter from a Q Exactive instrument (Thermo Fisher Scientific, Bremen, Germany) with a modified electronic board featuring a decreased resonance frequency of 284 kHz enabling an upper mass-selection limit above 20 kTh.
Introduction of the laser into the mass spectrometer was done as described before.35 A parallel coherent beam of 193 nm UV photons was generated with an ExciStar XS 500 series excimer laser (Coherent, Santa Clara, CA) filled with an ArF gas mixture. The laser produces 5 ns pulses at a maximum repetition rate of 500 Hz, with functional energies ranging from 0.5 to 5 mJ pulse–1 (∼10–60 photons per nm2). The laser beam was guided into the high vacuum region of the mass spectrometer via a periscope assembly, equipped with 45° UV mirrors (Edmund Optics, Barrington, NJ) mounted on micropositioners. The back end flange of the HCD cell was modified by replacing the equipped electrometer with a fused silica vacuum viewport (Kurt J. Lesker Company, Hustings, England). The laser beam was aligned to the longitudinal axis of the HCD cell to maximize the overlap with the trapped ion cloud and avoid irradiation of the ion optics components. Energy transmission through the viewport was measured as 3.5% at the energy range of 2–10 mJ pulse–1. Removal of the electrometer breaks the AGC functionality of the mass spectrometer, which we resolved by optimizing fixed injection time for each protein system. It also removes the vacuum seal between the HCD cell and the high-vacuum chamber. To mitigate the resulting loss of pressure, we designed a custom viewport with a CaF2 window and Teflon ring (Thorlabs, Newton, NJ) to completely seal the opening. The measured energy transmission through the viewport was 93% at the energy range of 2–10 mJ pulse–1.
To synchronize the laser pulses with the presence of the trapped ion cloud inside the HCD cell a purpose-built trigger pulse generator (TPG) was designed. By reading the pulse sequence from the split lens of the mass spectrometer the TPG determines the moment when injection of the ions in the HCD cell is completed. It then generates one (or multiple) 50 μs TTL pulse(s) that trigger(s) the laser emission. The number of pulses as well as the delay between the end of the injection and the trigger pulse can be adjusted.
Studied Proteins
All proteins were purchased from Sigma (Zwijndrecht, The Netherlands) unless otherwise stated; acetonitrile (ACN) was purchased from Biosolve (Valkenswaard, The Netherlands). Gp31 and GroES were recombinantly expressed in E. coli and purified as previously described.56,57 Native MS analysis was performed on three dimeric protein assemblies: bovine β-lactoglobulin (β-Lac), concanavalin A (ConA), and Cu, Zn-superoxide dismutase (Cu, Zn-SOD); a tetrameric protein assembly: ConA; and two heptameric protein assemblies: Gp31 and GroES. Lyophilized protein assemblies were dissolved to stock concentration of 1 mg/mL in aqueous ammonium acetate (10–300 mM) with pH ranging from 6.5 to 7.5 depending on the most stable conditions reported for each protein assembly. Proteins were desalted in centrifugal filters (Amicon Ultra, Merck, Germany) with 10 kDa molecular weight cutoff. Prior to mass spectrometric analysis stock solutions of protein assemblies were diluted in aqueous ammonium acetate solution to final monomer concentrations of 5 μM (β-Lac, Cu,Zn-SOD, GroES, and ConA) and 7 μM (Gp31).
Data Acquisition
Electrospray ionization for native MS on the modified Exactive Plus mass spectrometer was performed using in-house pulled borosilicate capillaries coated with gold using a static nanoESI source. Capillary voltage, source fragmentation voltage, front-end transfer parameters, and injection times were optimized for each analyte individually. Nitrogen pressure inside HCD cell was optimized indirectly by monitoring the ultrahigh vacuum (UHV) read-out in the Orbitrap chamber. For all tandem MS experiments the resolution of the Orbitrap mass analyzer was set to 140 000 at 400 m/z. The most abundant charge state of each protein assembly was isolated with a 1–10 Th window for subsequent analysis via UVPD or HCD. All data were collected as a single scan of 500 microscans.
Data Analysis
Native protein fragmentation spectra were deconvoluted with Xtract58 incorporated into Protein Deconvolution 4.1 (Thermo Fisher Scientific, Bremen, Germany), with the following settings: a signal-to-noise ratio (S/N) threshold of 2, a fit factor of 80%, and a remainder threshold of 25%. The resulting deconvoluted spectra were further processed with the in-house developed intact protein data analysis environment top-down lab (Brunner et al., publication in preparation). Shortly, as a first step, we determined which ions both fragmentation techniques produce for noncovalently bound assemblies by looking at frequently found mass differences to reference points for each amino acid position in the fragmentation spectrum. We calculated these reference points for N-terminal as b − H; and for C-terminal as y + H. After binning the found mass differences to the reference points in 40 ppm bins, the number of occurrences in each bin was calculated. With this unbiased method we found the predominant fragment ions for HCD: y and b (validating the approach); and for UVPD: x, y, a, a+, b, and c (Figure S7.1). After configuring the environment with these fragment ion types, dynamic mass calibration is applied to each spectrum individually based on all annotatable fragment ions for the used fragmentation technique at ±20 ppm. The median of the mass deviation of all annotated fragment ions is consequently used as correction factor. After calibration the global mass cutoff for all spectra is dynamically calculated by estimating the boundaries of the normally distributed mass deviations; the resulting narrow mass filter prevents false positives in assignment. Further statistical analysis of the resulting peptide fragment annotations was performed in R, extended by ggplot2 for data visualization.59,60 The protein exposed surface areas were calculated using POPS algorithm.61
Acknowledgments
We thank the group of Jennifer Brodbelt (University of Texas, Austin, TX) for advice in implementing the UV laser in our setup. We also thank our colleagues in the BioMS group, especially Arjan Barendregt for fruitful discussions and technical support. A.J.R.H. and A.M. acknowledge support through the European Union Horizon 2020 program FET-OPEN project MSmed, Project Number 686547. This work forms part of the Roadmap Initiative Proteins@Work (Project Number 184.032.201) financed by The Netherlands Organisation for Scientific Research (NWO).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b05147.
Benchmarking of the modified instrument; behavior of dimeric assemblies; behavior of dimeric assemblies with structural constraints; behavior of tetrameric assemblies; behavior of heptameric assemblies; sequencing multimeric assemblies; UVPD fragment ion investigation (PDF)
Author Contributions
∥ S.T. and A.D. contributed equally.
The authors declare the following competing financial interest(s): A.M. declares competing financial interests as he is an employee of Thermo Fisher Scientific, the manufacturer of the Exactive Plus instrument used in this research.
Supplementary Material
References
- Alberts B. Cell 1998, 92 (3), 291. 10.1016/S0092-8674(00)80922-8. [DOI] [PubMed] [Google Scholar]
- Robinson C. V.; Sali A.; Baumeister W. Nature 2007, 450 (7172), 973. 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- Stumpf M. P. H.; Thorne T.; de Silva E.; Stewart R.; An H. J.; Lappe M.; Wiuf C. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (19), 6959. 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan K.; Rual J.-F.; Vazquez A.; Stelzl U.; Lemmens I.; Hirozane-Kishikawa T.; Hao T.; Zenkner M.; Xin X.; Goh K.-I.; Yildirim M. A.; Simonis N.; Heinzmann K.; Gebreab F.; Sahalie J. M.; Cevik S.; Simon C.; de Smet A.-S.; Dann E.; Smolyar A.; Vinayagam A.; Yu H.; Szeto D.; Borick H.; Dricot A.; Klitgord N.; Murray R. R.; Lin C.; Lalowski M.; Timm J.; Rau K.; Boone C.; Braun P.; Cusick M. E.; Roth F. P.; Hill D. E.; Tavernier J.; Wanker E. E.; Barabási A.-L.; Vidal M. Nat. Methods 2009, 6 (1), 83. 10.1038/nmeth.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.-C.; Gstaiger M.; Raught B.; Aebersold R. Nat. Rev. Mol. Cell Biol. 2007, 8 (8), 645. 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- Mellacheruvu D.; Wright Z.; Couzens A. L.; Lambert J.-P.; St-Denis N. A.; Li T.; Miteva Y. V.; Hauri S.; Sardiu M. E.; Low T. Y.; Halim V. A.; Bagshaw R. D.; Hubner N. C.; Al-Hakim A.; Bouchard A.; Faubert D.; Fermin D.; Dunham W. H.; Goudreault M.; Lin Z.-Y.; Badillo B. G.; Pawson T.; Durocher D.; Coulombe B.; Aebersold R.; Superti-Furga G.; Colinge J.; Heck A. J. R.; Choi H.; Gstaiger M.; Mohammed S.; Cristea I. M.; Bennett K. L.; Washburn M. P.; Raught B.; Ewing R. M.; Gingras A.-C.; Nesvizhskii A. I. Nat. Methods 2013, 10 (8), 730. 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein M. Y.; Hubner N. C.; Poser I.; Cox J.; Nagaraj N.; Toyoda Y.; Gak I. A.; Weisswange I.; Mansfeld J.; Buchholz F.; Hyman A. A.; Mann M. Cell 2015, 163 (3), 712. 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Hosp F.; Scheltema R. A.; Eberl H. C.; Kulak N. A.; Keilhauer E. C.; Mayr K.; Mann M. Mol. Cell. Proteomics 2015, 14 (7), 2030. 10.1074/mcp.O115.049460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck A. J. R. Nat. Methods 2008, 5 (11), 927. 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]
- Snijder J.; Heck A. J. R. Annu. Rev. Anal. Chem. 2014, 7, 43. 10.1146/annurev-anchem-071213-020015. [DOI] [PubMed] [Google Scholar]
- Mehmood S.; Allison T. M.; Robinson C. V. Annu. Rev. Phys. Chem. 2015, 66, 453. 10.1146/annurev-physchem-040214-121732. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Cui W.; Wen J.; Blankenship R. E.; Gross M. L. J. Am. Soc. Mass Spectrom. 2010, 21 (12), 1966. 10.1016/j.jasms.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Heuvel R. H. H.; van Duijn E.; Mazon H.; Synowsky S.; Lorenzen K.; Versluis C.; Brouns S. J. J.; Langridge D.; van der Oost J.; Hoyes J.; Heck a. J. R. Anal. Chem. 2006, 78 (21), 7473. 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- Felitsyn N.; Kitova E. N.; Klassen J. S. Anal. Chem. 2001, 73 (19), 4647. 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- Jurchen J. C.; Garcia D. E.; Williams E. R. J. Am. Soc. Mass Spectrom. 2004, 15 (9), 1408. 10.1016/j.jasms.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch J. L. P.; Aquilina J. A.; Ruotolo B. T.; Sobott F.; Robinson C. V. Chem. Biol. 2006, 13 (6), 597. 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Wanasundara S. N.; Thachuk M. J. Am. Soc. Mass Spectrom. 2007, 18 (12), 2242. 10.1016/j.jasms.2007.09.022. [DOI] [PubMed] [Google Scholar]
- Light-Wahl K. J.; Springer D. L.; Winger B. E.; Edmonds C. G.; Camp D. G.; Thrall B. D.; Smith R. D. J. Am. Chem. Soc. 1993, 115 (2), 803. 10.1021/ja00055a070. [DOI] [Google Scholar]
- Versluis C.; van der Staaij A.; Stokvis E.; Heck A. J.; de Craene B. J. Am. Soc. Mass Spectrom. 2001, 12 (3), 329. 10.1016/S1044-0305(00)00227-0. [DOI] [PubMed] [Google Scholar]
- Sinelnikov I.; Kitova E. N.; Klassen J. S. J. Am. Soc. Mass Spectrom. 2007, 18 (4), 617. 10.1016/j.jasms.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Sharon M. J. Am. Soc. Mass Spectrom. 2010, 21 (4), 487. 10.1016/j.jasms.2009.12.017. [DOI] [PubMed] [Google Scholar]
- Benesch J. L. P.; Robinson C. V. Curr. Opin. Struct. Biol. 2006, 16 (2), 245. 10.1016/j.sbi.2006.03.009. [DOI] [PubMed] [Google Scholar]
- Jurchen J. C.; Williams E. R. J. Am. Chem. Soc. 2003, 125 (12), 2817. 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciuto S. V.; Liu J.; Konermann L. J. Am. Soc. Mass Spectrom. 2011, 22 (10), 1679. 10.1007/s13361-011-0205-x. [DOI] [PubMed] [Google Scholar]
- Beardsley R. L.; Jones C. M.; Galhena A. S.; Wysocki V. H. Anal. Chem. 2009, 81 (4), 1347. 10.1021/ac801883k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobott F.; Robinson C. V. Curr. Opin. Struct. Biol. 2002, 12 (6), 729. 10.1016/S0959-440X(02)00400-1. [DOI] [PubMed] [Google Scholar]
- Jones C. M.; Beardsley R. L.; Galhena A. S.; Dagan S.; Cheng G.; Wysocki V. H. J. Am. Chem. Soc. 2006, 128 (47), 15044. 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- Wysocki V. H.; Joyce K. E.; Jones C. M.; Beardsley R. L. J. Am. Soc. Mass Spectrom. 2008, 19 (2), 190. 10.1016/j.jasms.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbelt J. S. Chem. Soc. Rev. 2014, 43 (8), 2757. 10.1039/c3cs60444f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim M. A.; Girod M.; MacAleese L.; Lemoine J.; Antoine R.; Dugourd P. J. Am. Soc. Mass Spectrom. 2016, 27 (3), 474. 10.1007/s13361-015-1297-5. [DOI] [PubMed] [Google Scholar]
- Joly L.; Antoine R.; Broyer M.; Dugourd P.; Lemoine J. J. Mass Spectrom. 2007, 42 (6), 818. 10.1002/jms.1222. [DOI] [PubMed] [Google Scholar]
- Ko B. J.; Brodbelt J. S. J. Am. Soc. Mass Spectrom. 2011, 22 (1), 49. 10.1007/s13361-010-0016-5. [DOI] [PubMed] [Google Scholar]
- Madsen J. A.; Kaoud T. S.; Dalby K. N.; Brodbelt J. S. Proteomics 2011, 11 (7), 1329. 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly J. P. Mass Spectrom. Rev. 2009, 28 (3), 425. 10.1002/mas.20214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort K. L.; Dyachenko A.; Potel C. M.; Corradini E.; Marino F.; Barendregt A.; Makarov A. A.; Scheltema R. A.; Heck A. J. R. Anal. Chem. 2016, 88 (4), 2303. 10.1021/acs.analchem.5b04162. [DOI] [PubMed] [Google Scholar]
- Mcluckey S. A.; Goeringer D. E. J. Mass Spectrom. 1997, 32, 461.. [DOI] [PubMed] [Google Scholar]
- Dyachenko A.; Wang G.; Belov M. E.; Makarov A.; de Jong R. N.; van den Bremer E. T. J.; Parren P. W. H. I.; Heck A. J. R. Anal. Chem. 2015, 87 (12), 6095. 10.1021/acs.analchem.5b00788. [DOI] [PubMed] [Google Scholar]
- Hall Z.; Hernández H.; Marsh J. A.; Teichmann S. A.; Robinson C. V. Structure 2013, 21 (8), 1325. 10.1016/j.str.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose R.; Damoc E.; Denisov E.; Makarov A.; Heck A. Nat. Methods 2012, 9 (11), 1084–1086. 10.1038/nmeth.2208. [DOI] [PubMed] [Google Scholar]
- Van Dongen W. D.; Heck A. J. R. Analyst 2000, 125 (4), 583. 10.1039/a907957b. [DOI] [Google Scholar]
- Zhou M.; Dagan S.; Wysocki V. H. Analyst 2013, 138 (5), 1353. 10.1039/c2an36525a. [DOI] [PubMed] [Google Scholar]
- Lermyte F.; Williams J. P.; Brown J. M.; Martin E. M.; Sobott F. J. Am. Soc. Mass Spectrom. 2015, 26, 1068. 10.1007/s13361-015-1124-z. [DOI] [PubMed] [Google Scholar]
- Crespo-Otero R.; Mardykov A.; Sanchez-Garcia E.; Sander W.; Barbatti M. Phys. Chem. Chem. Phys. 2014, 16 (35), 18877. 10.1039/C4CP02518K. [DOI] [PubMed] [Google Scholar]
- O’Brien J. P.; Li W.; Zhang Y.; Brodbelt J. S. J. Am. Chem. Soc. 2014, 136 (37), 12920. 10.1021/ja505217w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.; Hernández H.; Marsh J. A.; Teichmann S. A.; Robinson C. V. Structure 2013, 21 (8), 1325. 10.1016/j.str.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B. Y.; Bewley M. C.; Creamer L. K.; Baker H. M.; Baker E. N.; Jameson G. B. Biochemistry 1998, 37 (40), 14014. 10.1021/bi981016t. [DOI] [PubMed] [Google Scholar]
- Kükrer B.; Barbu I. M.; Copps J.; Hogan P.; Taylor S. S.; van Duijn E.; Heck A. J. R. J. Am. Soc. Mass Spectrom. 2012, 23 (9), 1534. 10.1007/s13361-012-0441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S.; Mohanty D.; Surolia A. Biophys. J. 2009, 96 (1), 21. 10.1529/biophysj.108.134601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer-Hartl M.; Bracher A.; Hartl F. U. Trends Biochem. Sci. 2016, 41 (1), 62. 10.1016/j.tibs.2015.07.009. [DOI] [PubMed] [Google Scholar]
- Gruber R.; Horovitz A. Chem. Rev. 2016, 116 (11), 6588. 10.1021/acs.chemrev.5b00556. [DOI] [PubMed] [Google Scholar]
- Xu Z.; Horwich A. L.; Sigler P. B. Nature 1997, 388 (6644), 741. 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- Jones C. M.; Beardsley R. L.; Galhena A. S.; Dagan S.; Cheng G.; Wysocki V. H. J. Am. Chem. Soc. 2006, 128 (47), 15044. 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- Clare D. K.; Bakkes P. J.; van Heerikhuizen H.; van der Vies S. M.; Saibil H. R. J. Mol. Biol. 2006, 358 (3), 905. 10.1016/j.jmb.2006.02.033. [DOI] [PubMed] [Google Scholar]
- Hunt J. F.; van der Vies S. M.; Henry L.; Deisenhofer J. Cell 1997, 90 (2), 361. 10.1016/S0092-8674(00)80343-8. [DOI] [PubMed] [Google Scholar]
- Belov M. E.; Damoc E.; Denisov E.; Compton P. D.; Horning S.; Makarov A. a; Kelleher N. L. Anal. Chem. 2013, 85 (23), 11163. 10.1021/ac4029328. [DOI] [PubMed] [Google Scholar]
- van der Vies S. M. Methods Mol. Biol. 2000, 140, 51. 10.1385/1-59259-061-6:51. [DOI] [PubMed] [Google Scholar]
- Quaite-Randall E.; Joachimiak A. Methods Mol. Biol. 2000, 140, 41. 10.1385/1-59259-061-6:41. [DOI] [PubMed] [Google Scholar]
- Zabrouskov V.; Senko M. W.; Du Y.; Leduc R. D.; Kelleher N. L. J. Am. Soc. Mass Spectrom. 2005, 16 (12), 2027. 10.1016/j.jasms.2005.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H.ggplot2 - Elegant Graphics for Data Analysis; Springer: New York, 2009. [Google Scholar]
- Ihaka R.; Gentleman R. J. Comput. Graph. Stat. 1996, 5 (3), 1991. [Google Scholar]
- Cavallo L. Nucleic Acids Res. 2003, 31 (13), 3364. 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.