

Abstract
Associations between Sternorrhyncha insects and intracellular bacteria are common in nature. Mealybugs are destructive pests that seriously threaten the production of agriculture and forestry. Mealybugs have evolved intimate endosymbiotic relationships with bacteria, which provide them with essential amino acids, vitamins, and other nutrients. In this study, the divergence of five mealybugs was analyzed based up the sequences of the mitochondrial cytochrome oxidase I (mtCOI). Meanwhile, the distinct regions of the 16S rRNA gene of primary symbionts in the mealybugs were sequenced. Finally, high‐throughput sequencing (HTS) techniques were used to study the microbial abundance and diversity in mealybugs. Molecular phylogenetic analyses revealed that these five mealybugs were subdivided into two different clusters. One cluster of mealybugs (Dysmicoccus neobrevipes, Pseudococcus comstocki, and Planococcus minor) harbored the primary endosymbiont “Candidatus Tremblaya princeps,” and another cluster (Phenacoccus solenopsis and Phenacoccus solani) harbored “Ca. Tremblaya phenacola.” The mtCOI sequence divergence between the two clusters was similar to the 16S rRNA sequence divergence between T. princeps and T. phenacola. Thus, we concluded that the symbiont phylogeny was largely concordant with the host phylogeny. The HTS showed that the microbial abundance and diversity within P. solani and P. solenopsis were highly similar, and there was lower overall species richness compared to the other mealybugs. Among the five mealybugs, we also found significant differences in Shannon diversity and observed species. These results provide a theoretical basis for further research on the coevolution of mealybugs and their symbiotic microorganisms. These findings are also useful for research on the effect of symbiont diversity on the pest status of mealybugs in agricultural systems.
Keywords: coevolution, high‐throughput sequencing, mealybugs, symbiont diversity
1. INTRODUCTION
Hemiptera insects, such as aphids, scales, and whiteflies, feed solely on plant sap throughout their life (Baumann & Baumann, 2005). However, phloem is rich in carbohydrates and deficient in amino acids (Douglas, 1998). Therefore, mealybugs require the presence of microorganisms to provide additional nutrients (Downie & Gullan, 2004, 2005; Moran, Plague, Sandström, & Wilcox, 2003). In terms of their inseparability from insect survival, endosymbionts are further classified as primary endosymbionts (Abb. P‐endosymbiont) and secondary symbionts (Abb. S‐symbiont here) (Engel & Moran, 2013; Wernegreen & Wheeler, 2009). The dominant bacteria in bacteriocytes (generally, P‐endosymbionts) rarely experience interference from the external environment and can be transmitted vertically by hosts. Thus, the insects are able to precisely control the location and abundance of the endosymbionts (Gruwell, Hardy, Gullan, & Dittmar, 2010). S‐symbionts, which can be horizontally or vertically transferred, are not restricted only to bacteriocytes. Apart from their important roles in supplementing essential nutrients to insect, some endosymbionts can also play crucial roles in protecting hosts from damage by producing toxins and regulating the reproductive and immune systems of the insect (Moran, McCutcheon, & Nakabachi, 2008).
Mealybugs (Hemiptera: Pseudococcidae), a large family of scale insects (Coccoidea) including 259 genera and 1997 species (García Morales et al., 2016), cause serious threats to the production of agriculture and forestry (Wu, Ma, Hu, & Zeng, 2015). However, the morphological identification of mealybug species is difficult and limited by a high degree of similarity and polymorphism (especially in nymphs or eggs), which pose challenges in the study and management of these insects (Pacheco da Silva et al., 2014). The diagnostic PCR assay developed here provides a quick, simple, and reliable molecular technique for identifying and monitoring mealybugs, and this assay will be useful for intercepting and blocking the further spread of invasive mealybugs (Beltrà, Soto, & Malausa, 2012; Gruwell et al., 2010; Malausa et al., 2011). The mitochondrial cytochrome oxidase I (COI) gene has been demonstrated to be effective in studying the phylogenetic relationships of insects, for example, Bemisia tabaci (De Barro, Liu, Boykin, & Dinsdale, 2011; Rao, Luo, Zhang, Guo, & Devine, 2011). Likewise, this method has also been applied to the scale insects to construct biological and phylogenetic models, providing a theoretical basis for pest control (Abd‐Rabou et al., 2012; Correa, Germain, Malausa, & Zaviezo, 2012; Park, Suh, Hebert, Oh, & Hong, 2011; Saccaggi, Krüger, & Pietersen, 2008). This sequencing method has been widely used in biological species identification and genetic analysis. A recent study proposed that certain endosymbiotic Flavobacteria, which are considered to be primary endosymbionts of scale insects, codiversified with their Monophlebidae hosts (Von Dohlen, Kohler, Alsop, & McManus, 2001; Gruwell et al., 2010; Rosenblueth, Sayavedra, Samano‐Sanchez, Roth, & Martinez‐Romero, 2012). The primary endosymbionts, “Candidatus Tremblaya princeps,” are members of the β‐subdivision of the Proteobacteria, which are found in nearly all mealybug species and have a monophyletic origin (Baumann & Baumann, 2005; Downie & Gullan, 2005). The research showed that certain Phenacoccus species harbor another β‐proteobacterial endosymbiont, “Candidatus Tremblaya phenacola.” T. phenacola is a sister clade of T. princeps, suggesting that the Pseudococcidae share a common and single evolutionary source of the endosymbionts (Downie & Gullan, 2005; Gruwell et al., 2010), whereas the secondary γ‐proteobacterial endosymbionts, including Moranella endobia (Mccutcheon & Dohlen, 2011), are of polyphyletic evolutionary origins. The S‐symbiont clusters of 12 representative mealybug species were distinct from each other and from other insect‐associated bacteria (Thao, Gullan, & Baumann, 2002). A number of mealybugs harbor secondary γ‐proteobacterial endosymbionts, which are contained within the cell of the β‐proteobacteria, T. princeps (Von Dohlen et al., 2001). This unusual nested relationship between the β‐proteobacteria and γ‐proteobacteria has been well studied in recent research (Gatehouse, Sutherland, Forgie, Kaji, & Christeller, 2011; Kono, Koga, Shimada, & Fukatsu, 2008; Sergio, Amparo, Manuel, Andrés, & Rosario, 2013; Von Dohlen et al., 2001). For example, T. princeps in Planococcus citri (Risso) harbors the γ–proteobacterium “Candidatus Moranella endobia” (Lopez‐Madrigal et al., 2014). Through the intensive study of this nested structure, genome information revealed that M. endobia appears to be responsible for the biosynthesis of the majority of the cellular constituents and the energy supply, as well as the regulation of most informational courses for this special consortium, while T. princeps reserves the genes involved in indispensable informational functions (Lopez‐Madrigal et al., 2013).
Additionally, the β‐endosymbiotic genome has experienced a tremendous reduction due to the high incidence of gene drift and the relaxation of gene purification options (Sabatermuñoz, Toft, Alvarez‐Ponce, & Fares, 2017) Therefore, P‐endosymbionts are likely to lack the genes associated with the metabolic functions required by insects, but studies have also shown that these genes can be obtained from S‐symbionts that are nested within P‐endosymbionts (Wernegreen & Wheeler, 2009).
The 16S ribosomal RNA (especially the V3–V4 variable region) gene has been verified to be an accurate, reliable, and repeatable marker for symbiont identification and phylogenetic analysis, since the sequence is highly conserved and universally distributed (Degnan & Ochman, 2012). In addition, with the continuous development of high‐throughput sequencing platforms, HTS is considered to be a valid tool for researching microbial communities, which do not depend on previous knowledge about the diversity of the bacterial communities that are under investigation (Goodrich et al., 2014). These methods are based on the deep sequencing of PCR‐amplified bacterial 16S rRNA gene fragments, and the sequence data generated by HTS techniques are usually grouped into the same operational taxonomic unit (OTU) based on sequence similarity (≥97%), enabling the detection of all bacterial species existing in various samples. OTU is an effective tool for exploring bacterial communities, such as the symbiosis in aphids and psyllidae (Gauthier, Outreman, Mieuzet, & Simon, 2015; Hao & Chen, 2012; Jousselin et al., 2016; Overholt, Diaz, Rosskopf, Green, & Overholt, 2015). Therefore, HTS techniques are the first choice for studying the diversity of microbial communities (Degnan & Ochman, 2012).
In this study, the phylogenies of five species of mealybugs Phenacoccus solenopsis (Tinsley), Phenacoccus solani (Ferris), Dysmicoccus neobrevipes (Beardsley), Pseudococcus comstocki (Kuwana), and Planococcus minor (Maskell) were studied base on the divergence of the mtCOI gene. Meanwhile, regions of the 16S rRNA genes of the P‐endosymbionts from the five mealybugs were sequenced to explore the phylogenetic congruence of the P‐endosymbionts and their hosts. In addition, to gain insight into the microbial abundance and diversity in mealybugs, the polymorphism of the bacteria present in the mealybugs was investigated using high‐throughput sequencing (HTS) techniques (Wang et al., 2018). The findings presented in this study will be helpful for future research on the coevolution of mealybugs and their endosymbionts.
2. MATERIALS AND METHODS
2.1. Insects and DNA extraction
The mealybug samples used in the study were collected from different areas. Phenacoccus solenopsis were collected from eggplant Solanum melongena in Linan, Zhejiang Province. Pseudococcus comstocki were collected from vineyard in Xianju, Zhejiang Province. Dysmicoccus neobrevipes were provided by Shanghai Customs and Planococcus minor were provided by Zhoushan Customs. Both of their host plants are pineapple Ananas comosus. P. solani was provided by Hangzhou Customs and its host plant is succulent plant Senecio. All mealybug samples were collected individually in separate microfuge tubes containing 75% ethanol and were stored at −20°C for further study. The out‐groups, Icerya purchasi (Maskell), were collected from Citrus medica in Xiamen, Fujian Province. Asiacornococcus kaki (Kuwana) was collected from Diospyros in Heze, Shandong Province. For mtCOI gene amplification and endosymbionts identification, an individual insect DNA (three replicates per species) was extracted. For microbiome analysis, the mixed five individuals were extracted (three replicates per species). All DNA was extracted using the E.Z.N.A.® Insect DNA Kit (Omega Bio‐Tek, Norcross, GA, USA).
2.2. Genetic identification of Mealybugs samples
The mtCOI gene of insects (each species were repeated three times) was amplified using forward primer‐M2183 (5′‐CAACATTTATTTTTGATTTTTTGG‐3′) and reverse primer‐M2568 (5′‐GCWACWACRTAATAKGTATCATG‐3′) (Gullan et al., 2003). The gene was amplified with TaKaRa Taq™. The reaction conditions were 95°C for 5 min thereafter by 35 cycles of 95°C for 1 min, 52°C for 1 min and 72°C for 1.5 min with a final elongation step for 5 min at 72°C. The PCR products were sent to Sangon Biotech for sequencing (Shanghai, China).
2.3. Endosymbionts identification of Mealybugs samples
The 16S rRNA gene was amplified using the universal primers 16 s‐27F (5' AGAGTTTGATCMT GGCTCAG‐3') and 16 s‐1495R (5'‐CTACGGCTACCTTGTTACGA‐3') (Barzanti et al., 2007). The reaction conditions were five cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 2 min followed by 25 cycles of 95°C for 30 s, 50°C for 30 s, and 72°C for 2 min and 72°C 10 min. The PCR product was purified and then was cloned using pEASY‐T1 Cloning Kit according to the manufacturer's protocol. The sequencing process was performed by Sangon Biotech (Shanghai, China).
2.4. Molecular phylogenetic and evolutionary analyses
All sequences were aligned with Clustal X by MEGA (Version 6.06). The evolutionary divergence of different samples based on mtDNA CO1 used the Kimura 2‐parameter model. The phylogenetic trees were carried out by using the neighbor‐joining (NJ) method. Bootstrap values were calculated from 1,000 bootstrap replicates. The trees were rooted with a distinct out‐group. The genetic distance for these samples was calculated in the same software.
2.5. Microbiome identification of the mealybugs with distinct regions of 16S rRNA gene amplification and sequencing
The 16S ribosomal RNA (rRNA) V3‐V4 gene was amplified to assess the microbial diversity based on the Illumina HiSeq sequencing platform (Novogene Bioinformatics Technology Co., Ltd.). All amplicons with bright main strip were in the size range of 400–450 bp, and sequencing libraries were produced using TruSeq® DNA PCR‐Free Sample Preparation Kit (Illumina, USA). After the library quality was assessed (only high‐quality sequences were remained), double‐ended sequencing was performed by using the method of paired‐end and paired‐end reads (250 bp) were produced. Paired‐end reads were merged using FLASH (Version 1.2.7) (Magoc & Salzberg 2011), and then, the newly synthesized sequences were retained as raw tags. In order to get the clean tags with the high quality, the QIIME (Version 1.7.0) quality controlled process was applied to quality filtering on the raw tags (Bokulich et al., 2013). Then, the tags were compared with Gold database using UCHIME algorithm (Edgar, Haas, Clemente, Quince, & Knight, 2011) to detect and removed chimera sequences. Finally, the obtained “Effective Tags” were for subsequent analysis (Haas et al., 2011).
2.6. Data analyses of HTS
2.6.1. OTU cluster and species annotation
All Effective Tags of samples were clustered using Uparse software (Version 7.0.1001) (Edgar, 2013). The sequences with ≥97% identity were clustered into the same Operational Taxonomic Units (OTUs). At the same time, the highest frequency sequence of OTUs is selected as the representative sequence of OTUs according to its algorithm principle, which was conducted by Classifier algorithm (Wang, Garrity, Tiedje, & Cole, 2007). The MUSCLE (Version 3.8.31) software was used to perform rapid multiple sequence alignments to obtain the phylogenetic relationships of all OTUs representative sequences (Edgar, 2004). Finally, the data of each sample are homogenized, and then, the subsequent analysis was all based on the data after normalization. In order to further explore the phylogenetic relationships of different OTUs, and to compare the divergence of dominant species between different samples, we performed multiple sequence alignments. The phylogenetic trees were also carried out by neighbor‐joining (NJ) method using the programs MEGA (Version 6.06).
2.6.2. Alpha diversity
The Estimated‐species (observed species), ACE, Chao1, Simpson, Shannon, and Goods‐coverage indices were calculated using the QIIME software and the dilution curve, rank abundance curve and species cumulative curve were plotted using the R software, which was applied to analyze the difference of alpha diversity index between the groups. A box‐plot chart of abundance estimators were also generated by the QIIME toolkit. One‐way ANOVA (SPSS.18.0) was implemented to evaluate the alpha diversity among groups.
2.6.3. Beta diversity
Beta diversity analysis was service as an important tool to evaluate the distinction of all three samples in species complexity. The Unifrac distance was calculated using QIIME software to build the Unweighted Pair‐group Method with Arithmetic Means (UPGMA) clustering tree. Then, Principal component analysis (PCA) and Principal Coordinate Analysis (PCoA) graphs were carried out by using the R software. The LEfSe software was applied to analyze effect size, the default setting LDA score screening value of four. Two‐sided Student's t test was used for significance test of beta diversity difference between sample groups. Finally, to highlight differences in bacterial communities, the QIIME toolkit was chosen to construct a box‐plot of the dominant bacterial genus between different sample groups. And the composition of dominant bacterial genera among different groups was evaluated using the R statistical software (permutation test) at the level of Species (White, Nagarajan, & Pop, 2009).
3. RESULTS
3.1. Phylogenetic analysis of mealybugs and their endosymbionts
According to the phylogenetic tree, P. solenopsis was closely related to P. solani, sharing an identity of 95.03% (on average). P. comstocki shared a similarity of 94.38% (on average) with D. brevipes and 92.43% with P. minor. D. neobrevipes and D. brevipes were clustered in one clade. The results revealed that the intraspecies average genetic distances of the five mealybugs were 0.28% for P. solenopsis, 0.93% for P. solani, 0.35% for P. comstocki, 0.48% for P. minor, and 0.5% for D. neobrevipes (Figure 1).
Figure 1.

The phylogenetic tree based on mtDNA COI sequences divergence of the mealybugs and the divergence of the 16s rRNA sequences of their P‐endosymbionts. The phylogenetic trees were constructed by neighbor‐joining method using the programs MEGA (Version 6.06).Left, Relationship among mtDNA COI sequences of mealybugs and out‐group. The red diamond on behalf of five mealybugs (the orange dotted box region represents Dysmicoccus neobrevipes, the purple ones represent P. comstocki,the blue one represents P. minor,the red one represents P. solenopsis, and green one represents P. solani) sequenced in our study and the blue one represent the out‐groups sequenced in our study. Right, relationship of 16S rRNA sequences of their primary endosymbionts (the color of dotted line box are the same with the left). The red triangle on behalf of the P‐endosymbionts of five mealybugs sequenced in our study and the blue one represents out‐group sequenced in our study. The Information concerning the mealybugs used in this study and the accession numbers of the 16S rRNA of the P‐endosymbionts were shown in Supporting information Table S2
Molecular phylogenetic analyses revealed that the β‐proteobacterial sequences of P. comstocki, P. minor and D. brevipes were placed within the clade of T. princeps. A fragment of approximately 1,500 bp within the 16S rRNA gene was sequenced in the endosymbionts, and these 16S rRNA sequences showed the highest sequence similarity (97.77%, on average) to that of P. comstocki, P. minor, and D. brevipes (Figure 1). Nevertheless, the P‐endosymbionts in P. solenopsis and P. solani were T. phenacola, which were 0.67% divergent from each other. Simultaneously, as shown in Figure 1, there was an 18.88% sequence divergence between T. princeps and T. phenacola. In addition, a fragment of approximately 1,500 bp within the 16S rRNA gene was also sequenced in the S‐symbiont in P. comstocki and D. brevipes, and the results are shown in Figure 2. Further information about the mealybugs used in this study and the accession numbers are listed in Supporting information Table S2.
Figure 2.

Dominant OTUs were inserted into a precompiled phylogenetic tree using Mega 6.06. The phylogenetic trees were also constructed by neighbor‐joining method using the programs MEGA. The P‐ endosymbionts and the S‐symbionts of five mealybugs (Phenacoccus solenopsis, P. solani, P. comstocki, P. minor, and D. brevipes) and out‐group (Icerya purchasi) shown in this figure was sequenced in our study and others were downloaded in NCBI. The information of OTUs was the same with that in Figure 3
3.2. Basic statistics of V3–V4 16S rRNA gene sequences by HTS
A total of 1,357,058 V3–V4 16S rRNA effective sequences reads were acquired from the total DNA extracts of seven species (three replicates, respectively), with an average of 64,622 valid sequences reads (the minimum of a sample was 57,895 sequences reads, and the maximum was 72,513 sequences reads). The average length of all valid sequences reads was 420 bp (Supporting information Table S3). The sparse curves of all samples showed that the database constructed by the target gene sequences had substantial abundance, and thus, sufficient depth in the analysis can be obtained from the data and information concerning the diversity of microbial community (Supporting information Figure S1).
The bacterial diversity and relative abundance of all samples in the various taxonomies are presented in Supporting information Figure S2. Five phylogenetic groups, α‐, β‐, and γ‐proteobacteria, Flavobacteria, and Chloroplast, were identified as the major bacterial taxa of the scales bacterial community in all samples. The later probably came from stomach food. Based on relative abundance, the Proteobacteria (primarily β‐proteobacteria) was the most abundant bacterial phylum in the Phenacoccus populations, with an average relative abundance of over 93%. The γ‐proteobacteria had a higher relative abundance compared with other classes. The taxonomic distribution of each sample at the genus level is shown in Figure 3. The microbiome of P. minor was highly distinct from those of all other tested mealybugs, in which the dominant taxa were more abundant. The P. comstocki microbiome contained dominant taxa that were closely related to the dominant taxa in D. brevipes, while the microbiome of P. solani was similar to the dominant taxa in P. solenopsis. The bacteria from the Candidatus genus T. princeps (OTU3) were the most abundant bacteria in P. comstocki and D. brevipes, and the second most abundant bacteria were those them from Enterobacteriaceae family (OTU4, OTU30, and OTU247). Thus, these four OTUs represented between 83.81 and 95.28% of rRNA gene amplicon sequences from each sample. Similarly, 1–3 dominant OTUs within the Phenacoccus clade were detected, among which OTU1 was highly enriched in Phenacoccus (P. solani and P. solenopsis). In addition, all core bacteria in P. minor, such as OTU2, OTU5, OTU8, OTU10, OTU13, and others, found in our study were rarely detected in the Phenacoccus populations (Figure 3a). Moreover, the bacteria Wolbachia (OTU5) were detected in P. minor, with an average relative abundance of >4%.
Figure 3.

The relative abundances of enteric bacteria at the species (a) and phylum (b) level in five species mealybugs and two species out‐group was assessed using 16S rRNA high‐throughput sequencing. Microbial community composition for each microbiome analyzed. Dominant OTUs from each species. Only taxa with a relative abundance ≥1% in at least one sample were considered. Taxonomic classification includes genus when possible. OTUs from each dominant clade are colored with the same hue, and in all cases these dominant clades represent most of the total community
3.3. Characterization of the microbiome of mealybugs
To assess the bacterial community structure succession, the corresponding results revealed an apparent trend in the relative abundance of distinct bacterial taxa (Figure 3b). In five mealybugs, the Proteobacteria was the dominant phylum, whereas in the two out‐groups, Bacteroidetes and Firmicutes were the dominant phyla (Figure 3).
The box‐plot of the dominant bacterial genera constructed among the different sample groups (Figures 4 and 5) indicated that the bacterial community compositions were further confirmed by the clear clustering of the dominant bacterial genus and species corresponding to different populations in the heat map, as shown in Supporting information Figures S3 and S4. Three alpha diversity measurements were calculated, including the Shannon diversity index, observed species (observed OTUs), and Chao1 (Estimated Richness) (Table 1). For observed species comparison, P. minor and E. kaki had a significantly higher number of observed and estimated (Chao1) OTUs compared with the other species (p = 0.007, F = 4.786, Figure 4a). We also found significant differences between P. minor and the other four mealybugs (p = 0.000, F = 18.729, Figure 4b) in Shannon diversity. Moreover, the two indicators of P. comstocki and D. brevipes were significantly higher than that of P. solenopsis. Meanwhile, no significant differences in richness were observed between P. solenopsis and P. solani.
Figure 4.
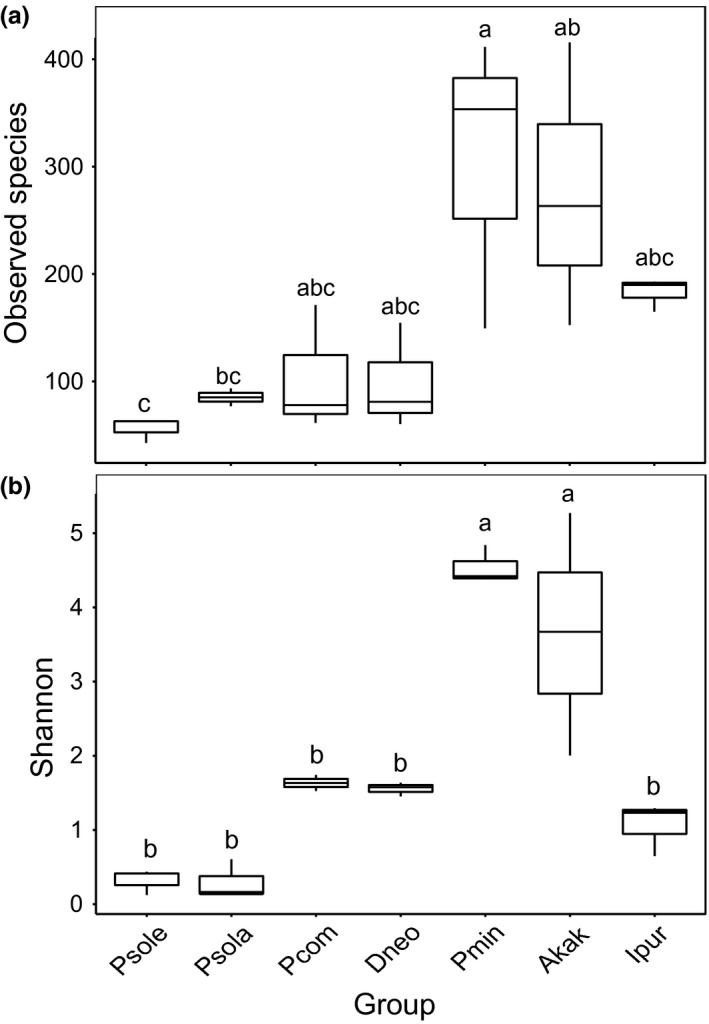
Differences in bacterial community diversity, richness and structure among seven samples. A box‐plot of richness estimators observed species (a) and Shannon diversity index (b) were examined by 16S high‐throughput sequencing was constructed using the QIIME toolkit. Alpha diversities were further tested by comparing the alpha diversity indexes between groups using one‐way ANOVA (SPSS. 18.0), n = 3 per group. Statistically significant differences are indicated. For observed species comparison, Planococcus minor and Asiacornococcus kaki had significantly higher number than others (p = 0.007, F = 4.786, Figure 4a). We also found significant difference in Shannon diversity between P. minor and other four mealybugs (p = 0.000, F = 18.729, Figure 4b). The top and bottom boundaries of each box indicate the 75th and 25th quartile values, respectively, and lines within each box represent the 50th quartile (median) values
Figure 5.
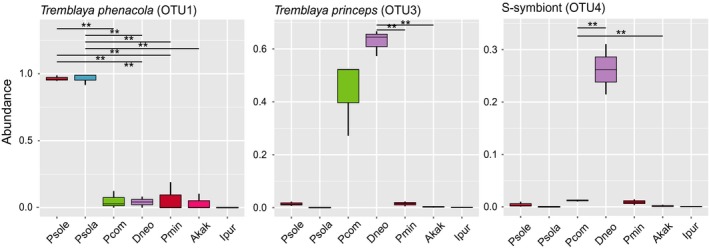
The relative abundances of P‐ and S‐ bacteria at the phylum, genus and species level in the five species mealybugs and two out‐groups species. Metalats analysis using the R software (Version 2.15.3) at the classification level, make the permutation test between groups, get p value, and then use Benjamini and Hochberg False Discovery Rate method for correcting the value of p, q value obtained. Finally, according to the q value marked the significance, to find significant differences in symbiotic bacteria (**q < 0.01)
Table 1.
HTS Sequencing summary of microbiomes from mealybugs
| Sample ID | Sequences Retrieved | Number of OTUs | Estimated Richness | Shannon | Num OTU > 1% Abundance | GC content |
|---|---|---|---|---|---|---|
| Psole01 | 66,640 | 62 | 69.814 | 0.438 | 3 | 49.90 |
| Psole02 | 64,341 | 42 | 50.141 | 0.124 | 1 | 49.72 |
| Psole03 | 61,525 | 61 | 68.501 | 0.390 | 2 | 49.85 |
| Psola01 | 70,960 | 91 | 106.853 | 0.606 | 2 | 49.68 |
| Psola02 | 59,937 | 83 | 98.810 | 0.134 | 1 | 49.49 |
| Psola03 | 61,027 | 75 | 85.041 | 0.150 | 1 | 49.52 |
| Pcom01 | 66,907 | 76 | 91.430 | 1.743 | 7 | 52.94 |
| Pcom02 | 68,025 | 60 | 78.713 | 1.525 | 6 | 52.97 |
| Pcom03 | 58,235 | 166 | 190.675 | 1.634 | 5 | 51.65 |
| Dneo01 | 72,513 | 79 | 95.985 | 1.576 | 4 | 53.90 |
| Dneo02 | 66,011 | 59 | 65.401 | 1.637 | 4 | 53.58 |
| Dneo03 | 57,895 | 150 | 218.779 | 1.452 | 3 | 54.05 |
| Pmin01 | 62,919 | 342 | 360.022 | 4.379 | 13 | 51.22 |
| Pmin02 | 63,763 | 398 | 422.432 | 4.407 | 9 | 51.13 |
| Pmin03 | 64,609 | 145 | 152.072 | 4.840 | 15 | 53.67 |
| Ekak01 | 59,155 | 402 | 424.079 | 5.273 | 11 | 54.02 |
| Ekak02 | 61,178 | 255 | 264.810 | 3.671 | 13 | 53.14 |
| Ekak03 | 68,386 | 148 | 164.381 | 2.004 | 7 | 53.14 |
| Ipur01 | 69,644 | 187 | 224.176 | 0.648 | 2 | 45.67 |
| Ipur02 | 72,743 | 185 | 242.068 | 1.294 | 2 | 46.44 |
| Ipur03 | 60,645 | 160 | 194.825 | 1.247 | 2 | 46.14 |
Although similar microbial richness was observed between mealybugs, the community structures were significantly different among the microbiomes of all tested organisms, of which P. solani and P. solenopsis shared a high similarity. Additionally, the results confirmed that two species of mealybugs had low overall species richness with 42–91 detected OTUs and housed 1–3 dominant taxa (Table 1). As a result, only 1–3 OTUs had >1% abundance in populations from P. solani and P. solenopsis, while 4–5 OTUs with >1% abundance were found in other mealybugs (Table 1). The Shannon diversity indices ranged from 0.124 to 0.606 in P. solani and P. solenopsis, and the microbial communities sampled from P. minor, P. comstocki, and D. brevipes were significantly more diverse than those from P. solani and P. solenopsis (Table 1). The Estimated Richness and diversity in E. kaki were higher compared with the others. According to the GC content, P. solani and P. solenopsis belong to Phenacoccus, with a GC content of approximately 50%, whereas the GC content of the others species was slightly higher (except the out‐group I. purchasi, with an average GC content of 46%). The OTU number and alpha diversity metrics for P. minor were higher compared to those from the other four mealybugs (Table 1).
A Venn diagram was used to compare the similarities and differences between the communities in the samples. The P. solenopsis, P. solani, P. comstocki, D. brevipes, and P. minor communities had 40 OTUs in common (Supporting information Figure S5A), with unique OTUs totaling 12, 46, 47, 33, and 330 in the P. solenopsis, P. solani, P. comstocki, D. brevipes, and P. minor communities, respectively (Figure S5B).
3.4. Clustering patterns of different species of scales
According to the unweighted unifrac PCoA, the samples were all clustered into three distinct clusters (Figure 6a). On the basis of principal coordinate 1 (PC1) and PC2 analyses (18.14% and 11.46% of variance explained, respectively), the microbial communities of P. minor were separated from the microbial communities of the four other mealybugs and clustered into the clades of out‐groups. The separation between samples across different species and the similarity between four mealybugs (P. solani, P. solenopsis, P. comstocki, and D. brevipes) were more notable on the weighted unifrac‐based plot. According to the weighted unifrac PCoA, all of the samples were grouped into two distinct clusters based on PC1 and PC2 analyses (49.79% and 23.67% of variance explained, respectively; Figure 6b). Four species of mealybugs formed a unique cluster, separate from the other samples. However, as shown in Figure 6, sample P.min 3 clustered separately from samples in the same group and showed a different pattern of relative abundance (this might have been caused by sampling error or poor sample quality).
Figure 6.

Two‐dimensional principal coordinates analysis (PCoA) plot of unweighted (abundance is ignored) (a) and weighted (abundance is considered) (b) unifrac distance matrices for seven samples
4. DISCUSSIONS
4.1. Phylogenetic analysis of mealybugs and their endosymbionts
The phylogenetic tree showed that the β‐proteobacterial sequences from P. comstocki, P. minor, and D. brevipes were placed within the clade of T. princeps, while the P‐endosymbionts in P. solenopsis and P. solani were identified as T. phenacola. A number of mealybugs harbor secondary γ‐proteobacterial endosymbionts, which are contained within the cell of the β‐proteobacteria T. princeps (Von Dohlen et al., 2001). The unusual nested relationship between the β‐proteobacteria and γ‐proteobacteria has been well studied in recent research (Gatehouse et al., 2011; Kono et al., 2008; Sergio et al., 2013). According to previous studies, mealybugs, especially the mealybugs, display a unique nested model where in the β‐proteobacteria T. princeps contains secondary bacteria (Gatehouse et al., 2012; Von Dohlen et al., 2001). For example, T. princeps in Planococcus citri harbors a γ‐proteobacterium, “Candidatus Moranella endobia.” Similar to P. citri, the results showed that the primary endosymbionts of P. comstocki, P. minor, and D. brevipes were the β‐proteobacteria T. princeps. In addition, we also detected two distinct secondary bacteria in P. comstocki and D. brevipes by sequencing the 16S rRNA gene. Based on the phylogenetic tree analysis, we can speculate that the secondary bacteria of P. comstocki and D. brevipes were probably also present in β‐proteobacteria, suggesting that these two mealybugs also have the same nested structure.
From many previous studies, we found that various insects, such as the whitefly, mealybugs, and others, exhibited distinct but stable symbiont systems and a close‐knit host–symbiont connection over evolutionary time, implying that the development of symbiotic systems generally mirror the phylogenetic relationships of the host insects (Clark et al., 2000; Thao et al., 2000; Thao & Baumann, 2004; Rao et al., 2015). This phenomenon is similar to the findings on “Ca. Tremblaya” in mealybugs in this study. The horizontal transfer of genes influences the endosymbionts of insects in the Phenacoccinae, which clearly reveal the mealybugs’ two typical endosymbiotic systems of historical evolution (Lopez‐Madrigal et al., 2014). Interestingly, the previous study also revealed that the theory of host–symbiont symbiosis can also be applied to gut symbionts. The stable relationship between host and symbiont implies the significant biological role of the symbiont in the host insects, which is preserved by generations in a vertical transfer manner. The findings of Kikuchi and Hosokawa strongly confirmed that the intracellular or extracellular environment had little effect on symbiont genomic evolutionary change (Kikuchi et al., 2009). As we expected, our results are consistent with the assumption of co‐diversification of “Ca. Tremblaya” and the host mealybug.
4.2. Basic statistics of V3–V4 16S rRNA gene sequences by HST
Interestingly, P. comstocki and D. neobrevipes harbor a distinct dominant Enterobacteriaceae group. The OTU4 and OTU247, which belong to Enterobacteriaceae, were abundant in P. comstocki and D. neobrevipes. OTU4 was closer to γ‐proteobacteria in other mealybugs, which means that OTU4 was nested in β‐proteobacteria (Figure 3). However, this pattern is different from the nested endosymbiotic pattern of P. comstocki and D. neobrevipes, consisting primarily of the β‐proteobacterial T. princeps that contains the γ‐proteobacterium (Figure 3). The abundance of OTU4 in D. neobrevipes was higher than in other species (Figure 5). Whether the common origin of T. princeps and T. phenacola had already been connected with the γ‐proteobacterium or not might have influenced their genome structure. The bacteria Staphylococcus (OTU7) and Bacillus (OTU6) are common members of the gut microbial communities in insects such as whitefly Bemisia tabaci and Apriona germari, and these bacteria have been reported for their role in reducing the pathogenic gut microbe population by maintaining the gut pH (Takatsuka & Kunimi, 2000). Moreover, the association with Bacillus, Staphylococcus, and Micrococcus at all B. tabaci developmental stages indicates a symbiosis and, indeed, suggests the detoxification of toxic substances (Genta, Dillon, Terra, & Ferreira, 2006; Lauzon, Potter, & Prokopy, 2003). Previous studies have reported the role of bacterial symbionts in insecticide resistance, confirming that bacterial symbionts of the genus Burkholderia in insects could degrade fenitrothion and thus enhance insect resistance to the insecticide. At the same time, we speculated that the insecticide‐degrading symbiotic bacteria could transfer horizontally through different pests of the same species (Kikuchi et al., 2009). The mealybugs in this study also contain endosymbionts of the genus Burkholderia, and some of them harbor Staphylococcus (OTU7) and Bacillus (OTU6), which may play a role in conferring insecticide resistance to mealybugs. These symbiotic bacteria may be obtained by insects from environmental soil or plants (Li et al., 2017). Vibrionaceae (OTU8) is a family of γ‐proteobacteria, which is typically found as symbionts in deep‐sea creatures and was also abundant in P. minor (average relative abundance was approximately 11.90%) in our study. Certain members of the genus Vibrio cause diarrhea or gastroenteritis in humans (Lin, Kumagai, Baba, Mekalanos, & Nishibuchi, 1993). Other members of the genus may cause a rapidly progressive hemorrhagic septicemia that can account for high mortalities among marine animals (Singer, Choe, Schmidt, & Makula, 1992). At the same time, this symbiont can also generate neurotoxins, such as tetrodotoxin (Simidu, Noguchi, Hwang, Shida, & Hashimoto, 1987), which protect itself from the predation of its natural enemies (Johnson et al., 2018).
4.3. Characterization of the microbiome of mealybugs
Alpha diversity was applied in analyzing the complexity of species diversity, and a Beta diversity analysis was used to assess discrepancies of samples in species integrality. Through these analyses, we observed that P. minor harbors a rich microbiome. The OTU number and alpha diversity metrics for P. minor were higher compared with the samples collected from the other four mealybugs. Moreover, the GC content data are also listed in Table 1. The measurement of GC content has an intimate correlation with the amino acid content of proteins, codon usage in messenger ribonucleic acid, and other properties of biology fields. The greatest potential of GC analysis may be its usefulness as a marker for classification that can be used to differentiate microorganisms with similar phenotypes (Mesbah, Premachandran, & Whitman, 1989). Previous research showed that the 16S rRNA gene sequences of T. princeps were GC‐rich (39%–46% AT) in comparison with those of free‐living β‐proteobacteria (43%–47% AT) (Baumann, Thao, Hess, Johnson, & Baumann, 2002; Mccutcheon & Dohlen, 2011). By contrast, the 16S rRNA gene sequences of T. phenacola were AT‐rich (49%–54% AT) (Gruwell et al., 2010). This study showed that the GC content of P. solani and P. solenopsis is approximately 50%, but that of P. minor, P. comstocki and D. neobrevipes are higher (Table 1). For amino acid synthesis, T. princeps differed from the other endosymbionts in that there were more amino acid‐encoding regions with a high GC content compared to the P‐endosymbiont in aphid and P‐endosymbiont in phylloxera (Clark, Baumann, Thao, Moran, & Baumann, 2001). These results correlated with the GC contents of the DNAs, which encode these proteins (Baumann et al., 2002). However, so far, there have been few studies on the relationship between inducing pathogens and insect invasion potentials, especially in the case of phytophagous insects, including invasive insects, which contain large amounts of symbiotic bacteria (Douglas, 2009). Some studies have proposed that there is a connection between the intracellular lifestyle of eukaryotic symbionts and pathogens with a low GC content. Interestingly, the bacterial communities associated with the living host environment are essential factors shaping the microbiota of insects (Linnenbrink et al., 2013). As a result, we concluded that certain groups within the microbiota and the bacterial richness in the insect gut may be affected by the host diet, habitat, and developmental stage (Yun et al., 2014). Certainly, the diversity of plant nutrient sources may also affect the abundance of bacteria in the host insects that feed on plants (Francis & Currie, 2003; Parkinson, Gobin, & Hughes, 2016).
4.4. Clustering patterns of different species of scales
Phenacoccus solenopsis is an invasive pest in a wide array of host plants, which was first reported to be found in Guangzhou, China, and has spread to other provinces since then (Wu et al., 2015). In the study, all of the mtCOI data from the P. solenopsis samples from China had a closer distant phylogenetic relationship with those from Pakistan and other Asian countries (Figure 1). These experimental results are consistent with previously reported results (Wu et al., 2015; Zhe et al., 2013). A geographical population analysis was conducted on another four kinds of mealybugs. The results of that study were similar to those of the present study, in that D. neobrevipes had a close relationship with the species from Vietnam. To adapt to new conditions, mealybugs may face changes that may be inherited by invasive species (Wu et al., 2015). Therefore, mealybugs of different geographical populations have various adaptations to different environments and thus affect endosymbionts indirectly (Ross, Shuker, Normark, & Pen, 2012). In this study, we demonstrated that the P. solani and P. solenopsis have a simple endosymbiotic system involving T. phenacola, which belongs to the β‐proteobacteria, contained within phenacoccinae mealybugs. The stable host–symbiont association is implicit in important biological roles of the endosymbionts for the host insect and rigorous vertical transmission of the symbiont through host generations. In addition, the results of HTS revealed that the observed species and Shannon diversity indices of P. solenopsis were lower than those of three native species (P. comstocki, E. kaki and I. purchasi) and two exotic species (D. brevipes and P. minor). Meanwhile, no significant differences in richness were observed between P. solenopsis and P. solani. Thus, the coexistence of endosymbionts may contribute to the invasiveness of exogenous harmful insects, which may be due to the presence of a single P‐endosymbiont and less abundant S‐symbionts in P. solenopsis. Genetic reductions in invasive species have been widely documented. For example, the loss of endosymbiotic bacteria in invasive populations is common in ant species (Rey et al., 2013; Yang et al., 2010). Another study found that the geographic invasion of mosquitoes is related to their symbiotic microbiome communities. Invasive species have a significantly reduced diversity of microbial populations in their hosts compared to native species (Minard et al., 2015). An important reason for the reduction in the diversity of endosymbiotic bacteria in the invasive species may be that the invasive species can gradually replace the native species. In future studies, it will be valuable to collect more samples of mealybugs from different parts of China and analyze the genetic structure of mealybugs using different and diverse molecular markers. This will help us to further understand the sources of invasive mealybugs and the relation between their invasion and endosymbiotic bacteria.
CONFLICT OF INTERESTS
The author(s) declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
QR and HMW conceived and designed the experiments. DL, LZ, WDS, and XLL performed experiments. LD, QR, and XYL analyzed the data. LD and QR drafted the manuscript. All authors read and approved the final manuscript.
Supporting information
ACKNOWLEDGMENTS
This work was supported by National Key R&D Program of China (Grant No. 2017YFD0202000). We thank Dr. Zhihong Xu for morphological identification of the mealybugs; Dr. Yunlin Su for revising and improving the paper.
lin D, Zhang L, Shao W, et al. Phylogenetic analyses and characteristics of the microbiomes from five mealybugs (Hemiptera: Pseudococcidae). Ecol Evol. 2019;9:1972–1984. 10.1002/ece3.4889
Contributor Information
Huiming Wu, Email: wuhm@zafu.edu.cn.
Qiong Rao, Email: qiong.rao@zafu.edu.cn.
DATA ACCESSIBILITY
Novel nucleotide sequences have been deposited in the GenBank nucleotide database (Accession nos.: partial mtCOI sequences of mealybugs, MF966987‐MF966993; 16S rRNA sequences of primary endosymbionts, MF939352‐MF939360). All other data are presented in the Supporting information.
REFERENCES
- Abd‐Rabou, S. , Shalaby, H. , Germain, J. F. , Ris, N. , Kreiter, P. , & Malausa, T. (2012). Identification of mealybug pest species (Hemiptera: Pseudococcidae) in egypt and france, using a dna barcoding approach. Bulletin of Entomological Research, 102(5), 515–523. 10.1017/S0007485312000041 [DOI] [PubMed] [Google Scholar]
- Barzanti, R. , Ozino, F. , Bazzicalupo, M. , Gabbrielli, R. , Galardi, F. , Gonnelli, C. , & Mengoni, A. (2007). Isolation and characterization of endophytic bacteria from the nickel hyperaccumulator plant Alyssum bertolonii. Microbial Ecology, 53, 306–316. 10.1007/s00248-006-9164-3. [DOI] [PubMed] [Google Scholar]
- Baumann, L. , & Baumann, P. (2005). Cospeciation between the primary endosymbionts of mealybugs and their hosts. Current Microbioloy, 50, 84–87. 10.1007/s00284-004-4437-x [DOI] [PubMed] [Google Scholar]
- Baumann, L. , Thao, M. L. , Hess, J. M. , Johnson, M. W. , & Baumann, P. (2002). The genetic properties of the primary endosymbionts of mealybugs differ from those of other endosymbionts of plant sap‐sucking insects. Applied and Environmental Microbiology, 68, 3198–3205. 10.1128/AEM.68.7.3198-3205.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrà, A. , Soto, A. , & Malausa, T. (2012). Molecular and morphological characterisation of Pseudococcidae surveyed on crops and ornamental plants in Spain. Bulletin of Entomological Research, 102, 165–172. 10.1017/S0007485311000514 [DOI] [PubMed] [Google Scholar]
- Bokulich, N. A. , Subramanian, S. , Faith, J. J. , Gevers, D. , Gordon, J. I. , Knight, R. , … Caporaso, J. G. (2013). Quality‐filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nature Methods, 10, 57–59. 10.1038/nmeth.2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, M. A. , Baumann, L. , Thao, M. L. , Moran, N. A. , & Baumann, P. (2001). Degenerative minimalism in the genome of a psyllid endosymbiont. Journal of Bacteriology, 183, 1853–1861. 10.1128/JB.183.6.1853-1861.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, M. A. , Moran, N. A. , Baumann, P. , & Wernegreen, J. J. (2000) Cospeciation between bacterial endosymbionts (Buchnera) and a recent radiation of aphids (Uroleucon) and pitfalls of testing for phylogenetic congruence. Evolution, 54, 517–525. 10.1554/0014-3820(2000)054[0517:CBBEBA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Correa, M. C. , Germain, J. F. , Malausa, T. , & Zaviezo, T. (2012). Molecular and morphological characterization of mealybugs (Hemiptera: Pseudococcidae) from chilean vineyards. Bulletin of Entomological Research, 102(5), 524–530. 10.1017/S0007485312000053 [DOI] [PubMed] [Google Scholar]
- De Barro, P. J. , Liu, S. S. , Boykin, L. M. , & Dinsdale, A. B. (2011). Bemisia tabaci: A statement of species status. Annual Review of Entomology, 56, 1–19. 10.1146/annurev-ento-112408-085504 [DOI] [PubMed] [Google Scholar]
- Degnan, P. H. , & Ochman, H. (2012). Illumina‐based analysis of microbial community diversity. ISME Journal, 6, 183 10.1038/ismej.2011.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas, A. E. (1998). Nutritional interactions in insect‐microbial symbioses: Aphids and their symbiotic bacteria buchnera. Annual Review of Entomology, 43, 17–37. 10.1146/annurev.ento.43.1.17 [DOI] [PubMed] [Google Scholar]
- Douglas, A. E. (2009). The microbial dimension in insect nutritional ecology. Functional Ecology, 23, 38–47. 10.1111/j.1365-2435.2008.01442.x [DOI] [Google Scholar]
- Downie, D. A. , & Gullan, P. J. (2004). Phylogenetic analysis of mealybugs (Hemiptera: Coccoidea: Pseudococcidae) based on DNA sequences from three nuclear genes, and a review of the higher classification. Systematic Entomology, 29, 238–260. 10.1111/j.0307-6970.2004.00241.x [DOI] [Google Scholar]
- Downie, D. A. , & Gullan, P. J. (2005). Phylogenetic congruence of mealybugs and their primary endosymbionts. Journal of Evolutionary Biology, 18, 315–324. 10.1111/j.1420-9101.2004.00834.x [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research, 32, 1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2013). UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nature Methods, 10, 996–998. 10.1038/nmeth.2604 [DOI] [PubMed] [Google Scholar]
- Edgar, R. C. , Haas, B. J. , Clemente, J. C. , Quince, C. , & Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics, 27, 2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel, P. , & Moran, N. A. (2013). The gut microbiota of insects ‐ diversity in structure and function. FEMS Microbiology Reviews, 37, 699–735. 10.1111/1574-6976.12025 [DOI] [PubMed] [Google Scholar]
- Francis, A. P. , & Currie, D. J. (2003). A globally consistent richness‐climate relationship for angiosperms. American Naturalist, 161, 523–536. 10.1086/368223 [DOI] [PubMed] [Google Scholar]
- García Morales, M. , Denno, B. D. , Miller, D. R. , Miller, G. L. , Ben‐Dov, Y. , & Hardy, N. B. (2016). ScaleNet: A literature‐based model of scale insect biology and systematics. Database, 2016, 1–5. 10.1093/database/bav118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatehouse, L. N. , Sutherland, P. , Forgie, S. A. , Kaji, R. , & Christeller, J. T. (2011). Molecular and histological characterisation of primary (β‐proteobacteria) and secondary (γ‐proteobacteria) endosymbionts of three species of mealybugs, (Pseudococcidae, Hemiptera) and prediction of interactions of their GroEL homologs with Grape Leaf Roll‐associated Virus‐3. Fertility and Sterility, 99, 533–542. 10.1128/AEM.06340-11 [DOI] [Google Scholar]
- Gatehouse, L. N. , Sutherland, P. , Forgie, S. A. , Kaji, R. , & Christeller, J. T. (2012). Molecular and histological characterization of primary (betaproteobacteria) and secondary (gammaproteobacteria) endosymbionts of three mealybug species. Applied and Environmental Microbiology, 78(4), 1187–1197. 10.1128/AEM.06340-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier, J. P. , Outreman, Y. , Mieuzet, L. , & Simon, J. C. (2015). Bacterial communities associated with host‐adapted populations of pea aphids revealed by deep sequencing of 16S ribosomal DNA. PLoS ONE, 10, e0120664 10.1371/journal.pone.0120664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genta, F. A. , Dillon, R. J. , Terra, W. R. , & Ferreira, C. (2006). Potential role for gut microbiota in cell wall digestion and glucoside detoxification in Tenebrio molitor larvae. Journal of Insect Physiology, 52, 593–601. 10.1016/j.jinsphys.2006.02.007 [DOI] [PubMed] [Google Scholar]
- Goodrich, J. K. , Di Rienzi, S. C. , Poole, A. C. , Koren, O. , Walters, W. A. , Caporaso, J. G. , … Ley, R. E. (2014). Conducting a microbiome study. Cell, 158, 250–262. 10.1016/j.cell.2014.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruwell, M. E. , Hardy, N. B. , Gullan, P. J. , & Dittmar, K. (2010). Evolutionary relationships among primary endosymbionts of the mealybug subfamily phenacoccinae (Hemiptera: Coccoidea: Pseudococcidae). Applied and Environmental Microbiology, 76, 7521–7525. 10.1128/AEM.01354-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullan, P. J. , Downie, D. A. , & Steffan, S. A. (2003) A new pest species of the Mealybug Genus Ferrisia Fullaway (Hemiptera: Pseudococcidae) from the United State. Annals of the Entomological Society of America, 96, 723–737. 10.1603/0013-8746(2003)096[0723:ANPSOT]2.0.CO;2 [DOI] [Google Scholar]
- Haas, B. J. , Gevers, D. , Earl, A. M. , Feldgarden, M. , Ward, D. V. , Giannoukos, G. , … Birren, B. W. (2011). Chimeric 16S rRNA sequence formation and detection in Sanger and 454‐pyrosequenced PCR amplicons. Genome Research, 21, 494–504. 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, X. , & Chen, T. (2012). OTU analysis using metagenomic shotgun sequencing data. PLoS One, 7, e49785 10.1371/journal.pone.0049785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, P. T. J. , Calhoun, D. M. , Stokes, A. N. , Susbilla, C. B. , McDevitt‐Galles, T. , Briggs, C. J. , … de Roode, J. C. (2018). Of poisons and parasites: The defensive role of tetrodotoxin against infections in newts. Journal of Animal Ecology, 87, 1192–1204. 10.1111/1365-2656.12816 [DOI] [PubMed] [Google Scholar]
- Jousselin, E. , Clamens, A. L. , Galan, M. , Bernard, M. , Maman, S. , Gschloessl, B. , … Coeur d'acier, A., (2016). Assessment of a 16S rRNA amplicon Illumina sequencing procedure for studying the microbiome of a symbiont‐rich aphid genus. Molecular Ecology Resources, 16, 628–640. 10.1111/1755-0998.12478 [DOI] [PubMed] [Google Scholar]
- Kikuchi, Y. , Hosokawa, T. , Nikoh, N. , Meng, X. Y. , Kamagata, Y. , & Fukatsu, T. (2009). Host‐symbiont co‐speciation and reductive genome evolution in gut symbiotic bacteria of acanthosomatid stinkbugs. BMC Biology, 7, 2 10.1186/1741-7007-7-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono, M. , Koga, R. , Shimada, M. , & Fukatsu, T. (2008). Infection dynamics of coexisting beta‐ and gammaproteobacteria in the nested endosymbiotic system of mealybugs. Applied and Environmental Microbiology, 74, 4175–4184. 10.1128/AEM.00250-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauzon, C. R. , Potter, S. E. , & Prokopy, R. J. (2003). Degradation and Detoxification of the Dihydrochalcone Phloridzin by Enterobacter agglomerans, a Bacterium Associated with the Apple Pest, Rhagoletis pomonella (Walsh) (Diptera: Tephritidae). Environmental Entomology, 32, 953–962. 10.1603/0046-225X-32.5.953 [DOI] [Google Scholar]
- Li, S. J. , Ahmed, M. Z. , Lv, N. , Shi, P. Q. , Wang, X. M. , Huang, J. L. , & Qiu, B. L. (2017). Plantmediated horizontal transmission of Wolbachia between whiteflies. The ISME Journal, 11, 1019–1028. 10.1038/ismej.2016.164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Z. , Kumagai, K. , Baba, K. , Mekalanos, J. J. , & Nishibuchi, M. (1993). Vibrio parahaemolyticus has a homolog of the Vibrio cholerae toxRS operon that mediates environmentally induced regulation of the thermostable direct hemolysin gene. Journal of Bacteriology, 175, 3844–3855. 10.1128/jb.175.12.3844-3855.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnenbrink, M. , Wang, J. , Hardouin, E. A. , Künzel, S. , Metzler, D. , Baines, J. F. , & Miriam, L. (2013). The role of biogeography in shaping diversity of the intestinal microbiota in house mice. Molecular Ecology, 22, 1904–1916. 10.1111/mec.12206. [DOI] [PubMed] [Google Scholar]
- Lopez‐Madrigal, S. , Balmand, S. , Latorre, A. , Heddi, A. , Moya, A. , & Gil, R. (2013). How does Tremblaya princeps get essential proteins from its nested partner Moranella endobia in the Mealybug Planoccocus citri? PLoS One, 8, e77307 10.1371/journal.pone.0077307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Madrigal, S. , Beltra, A. , Resurreccion, S. , Soto, A. , Latorre, A. , Moya, A. , & Gil, R. (2014). Molecular evidence for ongoing complementarity and horizontal gene transfer in endosymbiotic systems of mealybugs. Front in Microbiology, 5, 449 10.3389/fmicb.2014.00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoc, T. , & Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics, 27, 2957–2963. 10.1093/bioinformatics/btr507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malausa, T. , Fenis, A. , Warot, S. , Germain, J. F. , Ris, N. , Prado, E. , … Kreiter, P. (2011). DNA markers to disentangle complexes of cryptic taxa in mealybugs (Hemiptera: Pseudococcidae). Journal of Applied Entomology, 135, 142–155. 10.1111/j.1439-0418.2009.01495.x [DOI] [Google Scholar]
- Mccutcheon, J. P. , & Dohlen, C. D. V. (2011). An interdependent metabolic patchwork in the nested symbiosis of mealybugs. Current Biology, 21, 1366–1372. 10.1016/j.cub.2011.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesbah, M. , Premachandran, U. , & Whitman, W. B. (1989). Precise measurement of the G+C content of deoxyribonucleic acid by high‐performance liquid chromatography. International Journal of Systematic Bacteriology, 39, 159–167. 10.1099/00207713-39-2-159 [DOI] [Google Scholar]
- Minard, G. , Tran, F. H. , Van Tran Van, C. G. , Bellet, C. , Lambert, G. , Kim, K. L. H. , & Moro, C. V. (2015). French invasive Asian tiger mosquito populations harbor reduced bacterial microbiota and genetic diversity compared to Vietnamese autochthonous relatives. Front in Microbiology, 6, 970 10.3389/fmicb.2015.00970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran, N. A. , McCutcheon, J. P. , & Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annual Review of Genetics, 42, 165–190. 10.1146/annurev.genet.41.110306.130119 [DOI] [PubMed] [Google Scholar]
- Moran, N. A. , Plague, G. R. , Sandström, J. P. , & Wilcox, J. L. (2003). A genomic perspective on nutrient provisioning by bacterial symbionts of insects. Proceedings of the National Academy of Sciences, 100, 14543–14548. 10.1073/pnas.2135345100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholt, W. A. , Diaz, R. , Rosskopf, E. , Green, S. J. , & Overholt, W. A. (2015). Deep characterization of the Microbiomes of Calophya spp. (Hemiptera: Calophyidae) gall‐inducing psyllids reveals the absence of plant pathogenic bacteria and three dominant endosymbionts. PLoS One, 10, e0132248 10.1371/journal.pone.0132248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco da Silva, V. C. , Bertin, A. , Blin, A. , Germain, J. F. , Bernardi, D. , Rignol, G. , … Thibaut, M. (2014). Molecular and morphological identification of mealybug species (Hemiptera: Pseudococcidae) in brazilian vineyards. PLoS One, 9(7), e103267 10.1371/journal.pone.0103267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, D. S. , Suh, S. J. , Hebert, P. D. , Oh, H. W. , & Hong, K. J. (2011). Dna barcodes for two scale insect families, mealybugs (hemiptera: Pseudococcidae) and armored scales (Hemiptera: Diaspididae). Bulletin of Entomological Research, 101(4), 429–434. 10.1017/S0007485310000714 [DOI] [PubMed] [Google Scholar]
- Parkinson, J. F. , Gobin, B. , & Hughes, W. O. (2016). Heritability of symbiont density reveals distinct regulatory mechanisms in a tripartite symbiosis. Ecology and Evolution, 6, 2053–2060. 10.1002/ece3.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, Q. , Luo, C. , Zhang, H. , Guo, X. , & Devine, G. J. (2011). Distribution and dynamics of Bemisia tabaci invasive biotypes in central China. Bulletin of Entomological Research, 101, 81–88. 10.1017/S0007485310000428 [DOI] [PubMed] [Google Scholar]
- Rao, Q. , Rollat‐Farnier, P. A. , Zhu, D. T. , Santos‐Garcia, D. , Silva, F. J. , Moya, A. , … Wang, X. W. (2015). Genome reduction and potential metabolic complementation of the dual endosymbionts in the whitefly Bemisia tabaci. BMC Genomics, 16, 226 10.1186/s12864-015-1379-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, O. , Estoup, A. , Facon, B. , Loiseau, A. , Aebi, A. , Duron, O. , & Foucaud, J. (2013). Distribution of endosymbiotic reproductive manipulators reflects invasion process and not reproductive system polymorphism in the little fire ant wasmannia auropunctata. PLoS One, 8, e58467 10.1371/journal.pone.0058467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblueth, M. , Sayavedra, L. , Samano‐Sanchez, H. , Roth, A. , & Martinez‐Romero, E. (2012). Evolutionary relationships of flavobacterial and enterobacterial endosymbionts with their scale insect hosts (Hemiptera: Coccoidea). Journal of Evolutionary Biology, 25, 2357–2368. 10.1111/j.1420-9101.2012.02611.x [DOI] [PubMed] [Google Scholar]
- Ross, L. , Shuker, D. M. , Normark, B. B. , & Pen, I. (2012). The role of endosymbionts in the evolution of haploid‐male genetic systems in scale insects (Coccoidea). Ecology and Evolution, 2, 1071–1081. 10.1002/ece3.222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabater-Muñoz, B. , Toft, C. , Alvarez-Ponce, D. , & Fares, M. A. (2017). Chance and necessity in the genome evolution of endosymbiotic bacteria of insects. The ISME Journal, 11, 1291–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccaggi, D. L. , Krüger, K. , & Pietersen, G. (2008). A multiplex PCR assay for the simultaneous identification of three mealybug species (Hemiptera: Pseudococcidae). Bulletin of Entomological Research, 98, 27–33. 10.1017/S000748530700538X [DOI] [PubMed] [Google Scholar]
- Sergio, L. M. , Amparo, L. , Manuel, P. , Andrés, M. , & Rosario, G. (2013). Mealybugs nested endosymbiosis: Going into the ‘matryoshka’ system in planococcus citri in depth. BMC Microbiology, 13, 74 10.1186/1471-2180-13-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simidu, U. , Noguchi, T. , Hwang, D. F. , Shida, Y. , & Hashimoto, K. (1987). Marine bacteria which produce tetrodotoxin. Applied & Environmental Microbiology, 53, 1714–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, J. T. , Choe, W. , Schmidt, K. A. , & Makula, R. A. (1992). Virulence plasmid pJM1 prevents the conjugal entry of plasmid DNA into the marine fish pathogen Vibrio anguillarum 775. Journal of General Microbiology, 138, 2485–2490. 10.1099/00221287-138-12-2485 [DOI] [PubMed] [Google Scholar]
- Takatsuka, J. , & Kunimi, Y. (2000). Intestinal bacteria affect growth of Bacillus thuringiensis in larvae of the oriental tea tortrix, Homona magnanima diakonoff (Lepidoptera: Tortricidae). Journal of Invertebrate Pathology, 76, 222–226. 10.1006/jipa.2000.4973 [DOI] [PubMed] [Google Scholar]
- Thao, M. L. , & Baumann, P. (2004). Evolutionary relationships of primary prokaryotic endosymbionts of whiteflies and their hosts. Applied and Environmental Microbiology, 70, 3401–3406. 10.1128/AEM.70.6.3401-3406.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao, M. L. , Gullan, P. J. , & Baumann, P. (2002). Secondary ( ‐Proteobacteria) endosymbionts infect the primary ( ‐Proteobacteria) endosymbionts of mealybugs multiple times and coevolve with their hosts. Applied and Environmental Microbiology, 68, 3190–3197. 10.1128/AEM.68.7.3190-3197.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao, M. L. , Moran, N. A. , Abbot, P. , Brennan, E. B. , Burckhardt, D. H. , & Baumann, P. (2000). Cospeciation of psyllids and their primary prokaryotic endosymbionts. Applied and Environmental Microbiology, 66, 2898–2905. 10.1128/AEM.66.7.2898-2905.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Dohlen, C. D. , Kohler, S. , Alsop, S. T. , & McManus, W. R. (2001). Mealybug β‐proteobacterial endosymbionts contain γ‐proteobacterial symbionts. Nature, 412, 433–436. 10.1038/35086563 [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261–5267. 10.1128/AEM.00062-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S. , Tang, J. Y. , Ma, J. , Li, X. D. , & Li, Y. H. (2018). Moss habitats distinctly affect their associated bacterial community structures as revealed by the high‐throughput sequencing method. World Journal Microbiology and Biotechnology, 34, 58 10.1007/s11274-018-2436-5 [DOI] [PubMed] [Google Scholar]
- Wernegreen, J. J. , & Wheeler, D. E. (2009). Remaining flexible in old alliances: Functional plasticity in constrained. DNA and Cell Biology, 28, 371–382. 10.1089/dna.2009.0872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, J. R. , Nagarajan, N. , & Pop, M. (2009). Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Computational Biology, 5, e1000352 10.1371/journal.pcbi.1000352.g001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, F. Z. , Ma, J. , Hu, X. N. , & Zeng, L. (2015). Homology difference analysis of invasive mealybug species Phenacoccus solenopsis Tinsley in Southern China with COI gene sequence variability. Bulletin of Entomological Research, 105, 32–39. 10.1017/S0007485314000674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, C. C. , Yu, Y. C. , Valles, S. M. , Oi, D. H. , Chen, Y. C. , Shoemaker, D. , & Shih, C. J. (2010). Loss of microbial (pathogen) infections associated with recent invasions of the red imported fire ant Solenopsis invicta. Biological Invasions, 12, 3307–3318. 10.1007/s10530-010-9724-9 [DOI] [Google Scholar]
- Yun, J.‐H. , Roh, S. W. , Whon, T. W. , Jung, M.‐J. , Kim, M.‐S. , Park, D.‐S. , … Bae, J.‐W. (2014). Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Applied and Environmental Microbiology, 80, 5254 10.1128/AEM.01226-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhe, C. , Jiang, Z. , Hangfei, F. , Zhengzheng, X. , Kunzheng, D. , & Jiayong, Z. (2013). On the validity of the species Phenacoccus solenopsis based on morpho‐logical and mitochondrial COI data, with the description of a new body color variety. Biodiversity Science, 20, 443–450. 10.3724/sp.j.1003.2012.08202 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Novel nucleotide sequences have been deposited in the GenBank nucleotide database (Accession nos.: partial mtCOI sequences of mealybugs, MF966987‐MF966993; 16S rRNA sequences of primary endosymbionts, MF939352‐MF939360). All other data are presented in the Supporting information.