

Abstract
We describe a novel therapeutic approach for cirrhosis using mesenchymal stem cells (MSCs) and colony‐stimulating factor‐1‐induced bone marrow‐derived macrophages (id‐BMMs) and analyze the mechanisms underlying fibrosis improvement and regeneration. Mouse MSCs and id‐BMMs were cultured from mouse bone marrow and their interactions analyzed in vitro. MSCs, id‐BMMs, and a combination therapy using MSCs and id‐BMMs were administered to mice with CCl4‐induced cirrhosis. Fibrosis regression, liver regeneration, and liver‐migrating host cells were evaluated. Administered cell behavior was also tracked by intravital imaging. In coculture, MSCs induced switching of id‐BMMs toward the M2 phenotype with high phagocytic activity. In vivo, the combination therapy reduced liver fibrosis (associated with increased matrix metalloproteinases expression), increased hepatocyte proliferation (associated with increased hepatocyte growth factor, vascular endothelial growth factor, and oncostatin M in the liver), and reduced blood levels of liver enzymes, more effectively than MSCs or id‐BMMs monotherapy. Intravital imaging showed that after combination cell administration, a large number of id‐BMMs, which phagocytosed hepatocyte debris and were retained in the liver for more than 7 days, along with a few MSCs, the majority of which were trapped in the lung, migrated to the fibrotic area in the liver. Host macrophages and neutrophils infiltrated after combination therapy and contributed to liver fibrosis regression and promoted regeneration along with administered cells. Indirect effector MSCs and direct effector id‐BMMs synergistically improved cirrhosis along with host cells in mice. These studies pave the way for new treatments for cirrhosis. stem cells translational medicine 2019;8:271&284
Keywords: Mesenchymal stem cells, Induced bone marrow‐derived macrophages, Cirrhosis, Combination cell therapy, Intravital imaging
Significance Statement.
Cirrhosis is a life‐threatening condition. Thus, the development of novel therapeutic approaches for liver fibrosis regression and regeneration is urgently needed. The authors focused on two cell types; that is, bone marrow‐derived mesenchymal stem cells (MSCs) and colony‐stimulating factor‐1‐induced bone marrow‐derived macrophages (id‐BMMs), previously reported to be effective for cirrhosis, and showed that combination therapy with the two cell types synergistically improves liver function and fibrosis (over monotherapy with MSCs or id‐BMMs), in part by enhancing host endogenous regenerative responses. The authors also, for the first time, succeeded in tracing the detailed behavior of administered cells in the liver by intravital imaging technology and believe that these studies pave the way for new treatments.
Introduction
Liver cirrhosis is a life‐threatening condition mediated by hepatocyte loss and accumulation of excess extracellular matrix (ECM). Recently, evidence from studies using antiviral therapy indicated that liver fibrosis and function are reversible. However, even after treatment, some patients with decompensated cirrhosis progress to irreversible end‐stage liver disease 1, 2, 3.
Since 2003, autologous bone marrow cell infusion therapy, which improves liver fibrosis and induces liver regeneration, has been applied in experimental clinical trials for cirrhosis 4; however, many fundamental issues for cell therapy in this context require further study. First, the most effective cells among the heterogeneous bone marrow cells should be identified. Second, the detailed behavior of the administered cells remains unknown. Third, the mechanisms underlying liver fibrosis improvement and promotion of liver regeneration has not been shown. Because cell therapy involves multiple factors, this leads to difficulty in analyzing mechanisms. In this study, we focused on these unresolved points.
Macrophages and mesenchymal stem cells (MSCs) have attracted attention as cells that improve liver fibrosis and promote regeneration in cirrhosis. Macrophages present diverse phenotypes and plasticity 5, with two macrophage phenotypes having been defined: “classically activated” (M1) and “alternatively activated” (M2). Following liver injury, M1 macrophages contribute to the induction of inflammation and formation of fibrosis through activation of hepatic stellate cells and myofibroblasts. In contrast, when liver injury stabilizes, M2 macrophages contribute to the inactivation of inflammation and fibrosis regression 6. Furthermore, macrophages induce the differentiation and fate decision of hepatic stem/progenitor cells by producing oncostatin M (OSM) and Wnt3A 7, 8. Macrophages derived from the bone marrow and cultured with colony‐stimulating factor‐1 (CSF‐1) induce regression of CCl4‐induced liver fibrosis in mice 9. Here, the phenotype of culture‐induced macrophages was not polarized as M1 or M2, and theoretically could polarize from M1 to M2 or vice versa in vivo 5. The therapeutic effect of these CSF‐1‐induced bone marrow‐derived macrophages (id‐BMMs) in cirrhotic mice is directly and indirectly mediated by (a) factors related to fibrosis (matrix metalloproteinases; MMPs), (b) anti‐inflammatory molecules (IL‐10), (c) chemoattractants (CCl2, CXCL1, and CXCL2), and (d) proregeneration factors (OSM). However, migration and engraftment of administered macrophages and their contribution to the phagocytosis of debris and fibrosis regression in the liver has not been detected using intravital imaging.
MSCs can expand easily and, after induction, can differentiate into osteoblasts, adipocytes, and chondrocytes 10. MSCs represent a heterogeneous population that presents species‐dependent specific surface markers such as CD105 and CD73 10. MSCs secrete various cytokines, chemokines, and growth factors as well as exosomes, which help tissue repair indirectly and remotely 11. Importantly, MSCs regulate trophic factors, which indirectly mediate antiapoptosis, antioxidant, antifibrosis, angiogenic, and immunosuppressive effects 11. Owing to their multiple functions, ready expansion, and low antigenicity, not only autologous but also allogeneic MSCs have been used in more than 700 clinical trials in a variety of diseases. There are approximately 50 trials using MSCs targeting chronic and acute liver diseases.
The functions of macrophages and MSCs are clearly different; however, both have therapeutic effects on cirrhosis. Thus, we focused on the different characteristics of these cells; their behavior in the body especially in the liver, and their interactions and mechanisms supporting their effects on liver fibrosis and liver function. We showed the differences and interactions between id‐BMMs and MSCs finding that combination therapy using both id‐BMMs and MSCs had synergistic effects including effects on host cells compared with those of therapy using each cell type alone in a mouse model of cirrhosis. Furthermore, we succeeded for the first time in tracing the detailed behavior of id‐BMMs and MSCs using intravital imaging with two‐photon excitation microscopy 12. The detailed analysis of the mechanisms underlying the effects of MSCs and id‐BMMs using intravital imaging of the liver is a new concept in cell‐based therapies directed at liver diseases.
Materials and Methods
Mice
Animals were housed in a specific pathogen‐free environment and kept under standard conditions with a 12‐hour day/night cycle and access to food and water ad libitum. All animal experiments were conducted in compliance with regulations and approved by the Institutional Animal Care and Committee at the Niigata University and Osaka University. C57BL/6 male mice were purchased from Charles River (Yokohama, Japan). Green fluorescent protein (GFP)‐transgenic mice (Tg[CAG‐enhanced green fluorescent protein (EGFP)]C14‐Y01‐FM131Osb) were kindly provided by Professor Masahito Ikawa, Osaka University. DsRed‐transgenic mice (Tg[CAG‐DsRedMST1]Nagy/J) were purchased from Jackson Laboratory (Bar Harbor, ME).
Preparation of the Cirrhosis Mouse Model and Cell Transplantation
Male mice were intraperitoneally (i.p.) injected with 1.0 ml/kg carbon tetrachloride (CCl4; Wako, Osaka, Japan) to induce cirrhosis. CCl4 was dissolved in corn oil (Wako) at 1:10 ratio. The mice received CCl4 i.p. over a 12‐week period, twice weekly. At eight weeks, 1 × 106 MSCs (MSC100) or id‐BMMs (id‐BMM100), or 5 × 105 MSCs and 5 × 105 id‐BMMs (50/50; cells were mixed immediately prior to injection) were injected into CCl4‐induced mice through the tail vein.
Intravital Imaging
For intravital imaging of the liver in mice, mice were anesthetized with isoflurane, the median lobe of the liver was surgically exposed, and the internal surface was observed using inverted two‐photon excitation microscopy. The imaging system was composed of a two‐photon inverted microscope (A1R‐MP, Nikon, Tokyo, Japan) driven by a laser (Chameleon Vision Ti: Sapphire, Coherent) tuned to 880 nm, with a water multi‐immersion objective lens (Plan Fluor; N.A., 0.75; Nikon). Fluorescence signals were detected through bandpass emission filters at 525/50 nm (for EGFP) and 575/25 nm (for DsRed). Raw imaging data were processed using Imaris software (Bitplane, Zurich, Switzerland). To estimate the amount of administered cells distributed in the liver, lung, and spleen, the number of GFP‐positive cells or DsRed‐positive cells in 40 fields of each organ (1 field = 400 μm × 400 μm × 10 μm) was counted, and this number was extrapolated for each organ using the total volume of the organ. Based on the number of administered cells (1 × 106), the frequency of populating cells was calculated for each organ.
Cell Culture and MSC and id‐BMM Isolation
For MSC and id‐BMM isolation, 8‐ to 10‐week‐old male C57BL6 mice were euthanized by cervical dislocation and their limbs were removed. Bone marrow precursors were flushed from the medullary cavities of tibiae and femurs. MSCs were cultured at 37°C, under 5% O2 and 5% CO2 in specific dishes (Collagen Type I‐Coated Microplate, Iwaki, Shizuoka, Japan) and medium (MesenCult MSC basal medium, mouse; Stem Cell Technologies, Vancouver, BC, Canada) containing stimulatory supplements (MesenCult Stem Cell Stimulatory Supplements, Mouse; Stem Cell Technologies), 0.1 μl/ml supplements (MesenPure ×1,000, mouse; Stem Cell Technologies), and 10 μg/ml fibroblast growth factor 2 (FGF‐2; human FGF‐2; Miltenyi Biotec, Bergisch Gladbach, Germany). MSCs were cultured for 2 weeks with the medium changed twice weekly. Approximately 2 × 106 MSCs were obtained from one mouse, and cultured cells were administered without passage or used to monitor the phenotype (Supporting Information Fig. S1).
The id‐BMMs were cultured at 37°C under 5% CO2 in specific dishes (ultra‐low attachment flasks; Corning, Armonk, NY) and medium (Dulbecco's modified Eagle's medium/F12 media; Thermo Fisher Scientific, Waltham, MA) containing 20 ng/ml CSF‐1 (Recombinant Murine M‐CSF; Peprotech Inc., Rocky Hill, NJ). The id‐BMMs were cultured for 7 days with the medium changed twice weekly. Approximately 5 × 106 id‐BMMs were obtained from one mouse, and cultured cells were administered without passage or used to monitor the phenotype (Supporting Information Fig. S1).
Polymerase Chain Reaction
Total RNA was reverse‐transcribed using a QuantiTect reverse transcription kit (Qiagen, Hilden, Germany). Gene expression analysis was achieved by using prevalidated QuantiTech primers (Supporting Information Table S1) with QuantiTech SYBR reagent (Qiagen). Real‐time PCR was conducted by using a Step One Plus Real‐time PCR System (Applied Biosystems, Foster City, CA). The results were obtained from at least three separate samples. Gapdh was used as an internal control. The fold change in relative gene expression from the control was calculated by using the ΔΔCt method.
Immunostaining
For immunohistochemistry, 10% formalin‐fixed tissue was cut into 3‐μm‐thick sections. When necessary, heat‐mediated antigen retrieval was performed in 10 mM sodium citrate buffer at pH 6.0. For 3,3′‐diaminobenzidine (DAB) staining, sections were blocked using 3% hydrogen peroxide for 10 minutes at room temperature. Primary antibodies (Supporting Information Table S2) were incubated overnight at 4°C. Species‐specific anti‐IgG biotinylated antibodies were used for detection. Slides were then stained using a Vectastain ABC kit (Vector Laboratories, Inc., Burlingame, CA) and DAB substrate (Muto Pure Chemicals, Tokyo, Japan). Nuclei were stained using hematoxylin (Vector Laboratories, Inc.). Photographs were acquired using an HS all‐in‐one fluorescence microscope (BZ‐9000; Keyence, Osaka, Japan).
Statistical Analysis
Statistical analysis was performed by using GraphPad Prism5 software (GraphPad Software Inc., La Jolla, CA) and Microsoft Excel (Microsoft, Redmond, WA). Data are presented as the means ± SD; all data analyzed were normally distributed. The results were assessed using the Student's t test. Differences between groups were analyzed by one‐way analysis of variance. Differences were considered significant when p < .05. Furthermore, description of the experimental procedures used is provided in Supporting Information.
Results
Cultured MSCs and id‐BMMs Express a Variety of Unique Factors and Show Altered Characteristics After Addition of Serum from Mice with CCl4‐Induced Liver Damage
Before assessing the therapeutic effects of MSCs and id‐BMMs on cirrhosis in vivo, we performed a comprehensive analysis of factors expressed by these two populations by microarray (Supporting Information Fig. S2). We compared several factors related to fibrosis (MMP tissue inhibitor of metalloprotease [TIMP]), chemoattractants (C‐X‐C chemokine receptor 4 [CXCR4] and stromal cell‐derived factor 1 [SDF‐1]), proregeneration factors (OSM and hepatocyte growth factor [HGF]), inflammation/anti‐inflammation factors (IL‐10 and IL‐6), macrophage phenotype markers (INOS and YM‐1), and others (PGE2 and TSG‐6). Cultured macrophages express many factors that affect cirrhosis treatment directly or indirectly 9. In the present study, the expression of factors related to fibrosis (MMP‐8, ‐12, and ‐13), chemoattractant factors (CXCR4), anti‐inflammation factors (IL‐10), and proregenerative factors (OSM) was stronger in id‐BMMs than in MSCs, suggesting that id‐BMMs play a pivotal role in the improvement of liver fibrosis and regeneration. Cultured MSCs expressed different factors from those of id‐BMMs such as the antifibrosis factor MMP‐2 and the angiogenic factor vascular endothelial growth factor (VEGF). We further evaluated other factors related to immunomodulation, especially those affecting macrophage polarization. PGE2, TSG‐6, and IL‐13 are factors that change the macrophage phenotype toward M2 and are secreted from MSCs only when MSCs are stimulated by inflammatory stimuli. The mRNA expression of PGE2, TSG‐6, and IL‐13 in simple cultured MSCs was not as strong as previously reported 11.
MSCs and macrophages change their characteristics in response to environmental signals. Furthermore, once MSCs are stimulated by inflammatory stimuli, they produce factors such as PGE2 and IL‐13, thereby switching the macrophage phenotype toward M2 13. Because the serum from mice with CCl4‐induced liver damage contains various factors, we suspected that after injection into these mice, the characteristics of both MSCs and id‐BMMs would change. Thus, to mimic the change in the characteristics of MSCs and id‐BMMs in vivo, cultured cells were incubated for 72 hours with serum from mice with CCl4‐induced liver damage and changes in mRNA levels were analyzed in vitro. In MSCs, after addition of serum, mRNA levels of the antifibrotic factor MMP‐13 (p < .001), the chemoattractant factor SDF‐1 (p < .001), the anti‐inflammatory factor IL‐10 (p < .001), the M2 macrophage‐inducing factors IL‐13 (p < .001, and PGE2 (p = .021) were upregulated, whereas, the mRNA expression of the metalloproteinase inhibitor TIMP3 (p < .001) was downregulated (Fig. 1A). In id‐BMMs, which secreted a high level of MMP‐13 after addition of serum from mice with CCl4‐induced liver damage in vitro, although Mmp13 (p = .048) mRNA levels remained high, mRNA expression of the antifibrosis factors MMP‐8 (p < .001) and MMP‐9 (p < .001), the anti‐inflammatory factor IL‐10 (p < .001), the angiogenic factor VEGF (p < .001), the SDF‐1 receptor CXCR4 (p = .001), the proregenerative factor OSM (p < .001), the macrophage and neutrophil chemoattractant factors CXCL1 (p < .001), CXCL2 (p < .001) and CCL2 (p < .001), the M2 macrophage markers YM‐1 (p = .018) and FIZZ‐1 (p < .001), and the mannose receptor (MR; p < .001) was upregulated. In contrast, mRNA expression of the M1 macrophage markers IL‐6 (p < .001), INOS (p = .029), and TNF‐α (p < .001) was downregulated (Fig. 1B), suggesting that trophic factors in the serum changed the id‐BMM characteristics toward the M2 phenotype. These results revealed that both MSCs and id‐BMMs were affected by the serum from liver‐damaged mice and their characteristics were changed.
Figure 1.
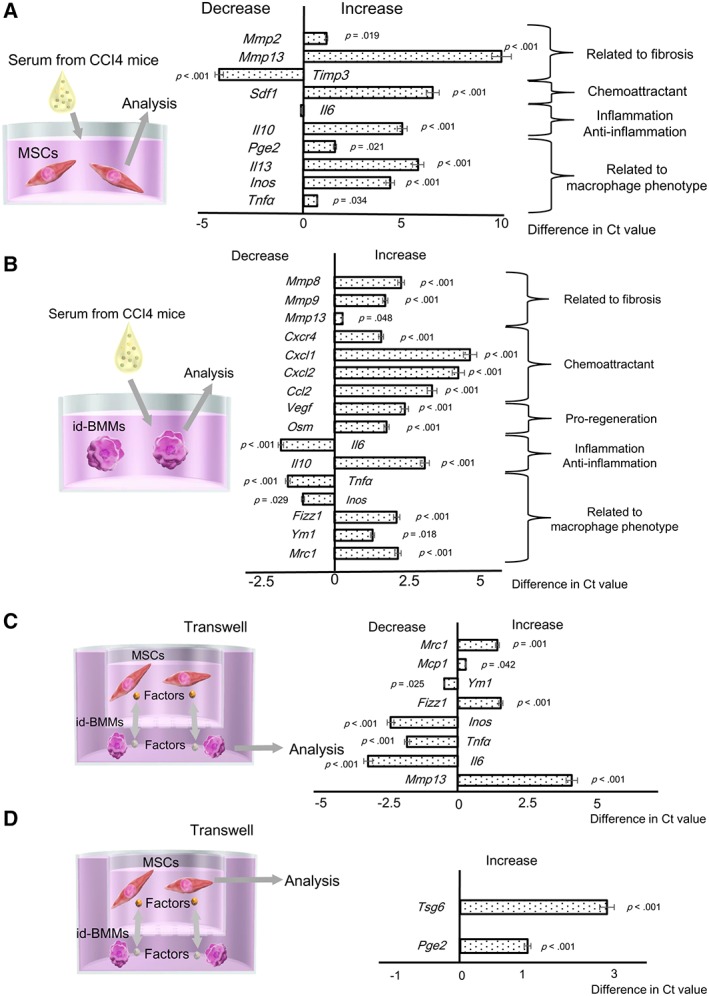
Analysis of mRNA expression changes in mesenchymal stem cells (MSCs) and induced bone marrow‐derived macrophages (id‐BMMs) after addition of serum from mice with CCl4‐induced liver damage to mimic the in vivo microenvironment after cell administration, and after coculture of MSCs and id‐BMMs. Data are presented as the means ± SD, n = 12 in each experiment. (A): mRNA expression changes in MSCs after addition of serum from liver‐damaged mice. Mmp2 (p = .019), Mmp13 (p < .001), Sdf1 (p < .001), Il10 (p < .001), Pge2 (p = .021), Il13 (p < .001), Inos (p < .001), Tnfa (p = .034); upregulated. Timp3 (p < .001); downregulated. (B): mRNA expression changes in id‐BMMs after addition of serum from liver‐damaged mice. Mmp8 (p < .001), Mmp9 (p < .001), Mmp13 (p = .048), Cxcr4 (p < .001), Cxcl1 (p < .001), Cxcl2 (p < .001), Ccl2 (p < .001), Vegf (p < .001), Osm (p < .001), Il10 (p < .001), Fizz1 (p < .001), Ym1 (p = .018), Mrc1 (mannose receptor; p < .001); upregulated. Il6 (p < .001), Tnfa (p < .001), Inos (p = .029); downregulated. (C): mRNA expression changes in id‐BMMs after coculture. Mrc1 (p = .001), Mcp1 (p = .042), Fizz1 (p < .001), Mmp13 (p < .001); upregulated. Ym1 (p = .025), Inos (p < .001), Tnfa (p < .001), Il6 (p < .001); downregulated. (D): mRNA expression change in MSCs after coculture. Tsg6 (p < .001), Pge2 (p < .001); upregulated.
MSCs Change id‐BMM Characteristics Toward M2 Macrophages, Which Gain High Phagocytic Activity
To further show the interaction between MSCs and id‐BMMs mediated by trophic factors, we cocultured MSCs and id‐BMMs in a noncontact transwell culture system. After 72 hours of coculture changes in mRNA expression in id‐BMMs in the presence or absence of MSCs and those in MSCs in the presence or absence of id‐BMMs were compared. In id‐BMMs cultured in presence of MSCs the expression of the M2 macrophage markers FIZZ‐1 (p < .001) MR (p = .001) and that of the antifibrosis factor MMP‐13 (p < .001) increased, whereas, that of the M1 macrophage markers INOS (p < .001) TNF‐α (p < .001) and IL‐6 (p < .001) decreased (Fig. 1C) suggesting that MSC trophic factors induced a switch toward the M2 phenotype. In MSCs cultured in the presence of id‐BMMs, the expression of PGE2 (p < .001) and TSG‐6 (p < .001) that induced the switch toward the M2 macrophage phenotype (Fig. 1D) increased suggesting that id‐BMM trophic factors affected the characteristics of MSCs as M2 macrophage inducers. These results revealed that by utilizing MSCs, the characteristics of id‐BMMs were changed toward the M2 phenotype. Furthermore, to determine whether the phagocytic activity of M2‐polarized id‐BMMs changed, we compared the number of macrophages that phagocytosed fluorescent particles (zymosan) between id‐BMMs cultured in the presence and absence of MSCs in vitro. The phagocytic activity in M2‐polarized id‐BMMs was 1.23‐fold stronger (p = .008) than that in nonpolarized id‐BMMs revealing that macrophages polarized toward the M2 phenotype by MSCs had a higher phagocytic activity (Supporting Information Fig. 3A, 3B).
Combination Therapy Using MSCs and id‐BMMs (50/50) Has a Stronger Therapeutic Effect in Cirrhotic Mice than MSCs or id‐BMMs Alone
Altogether, the results obtained in vitro suggested that combination therapy using MSCs and id‐BMMs could improve liver fibrosis and regeneration. To show the therapeutic effect of MSCs and id‐BMMs on cirrhosis, 1 × 106 MSCs (MSC 100), 1 × 106 id‐BMMs (id‐BMM 100), a combination of 5 × 105 MSCs and 5 × 105 id‐BMMs (50/50; total 1 × 106 cells), or phosphate buffered solution control were administered to cirrhotic mice (Fig. 2A). Four weeks after cell administration, Sirius red staining indicated reduced liver fibrosis in the MSC100 (5.40%; p = .201), id‐BMM100 (18.8%, p = .036), and 50/50 groups (27.3%; p = .002) compared with the control group. However, the improvement of fibrosis was stronger in the 50/50 group than in the MSC100 and id‐BMM100 groups (Fig. 2B), and quantitative analysis of hydroxyproline, which is the major component of collagen, also indicated reduced liver fibrosis in the MSC100 (2.43%; p = .315), id‐BMM100 (19.8%; p = .006), and 50/50 groups (37.2%; p < .001) compared with that in the control group (Fig. 2C). Decreased serum ALT levels were observed in the id‐BMM100 (p = .001) and 50/50 groups (p < .001), whereas decreased serum ALP levels were observed in all groups (MSC100; p = .036, id‐BMM100; p = .029, 50/50; p = .012), and serum levels of total bilirubin were decreased in the id‐BMM100 and 50/50 groups (id‐BMM100; p = .014, 50/50; p = .016; Fig. 2D). These results revealed that combination therapy using MSCs and id‐BMMs (50/50) had the strongest therapeutic effect for improvement of liver fibrosis.
Figure 2.
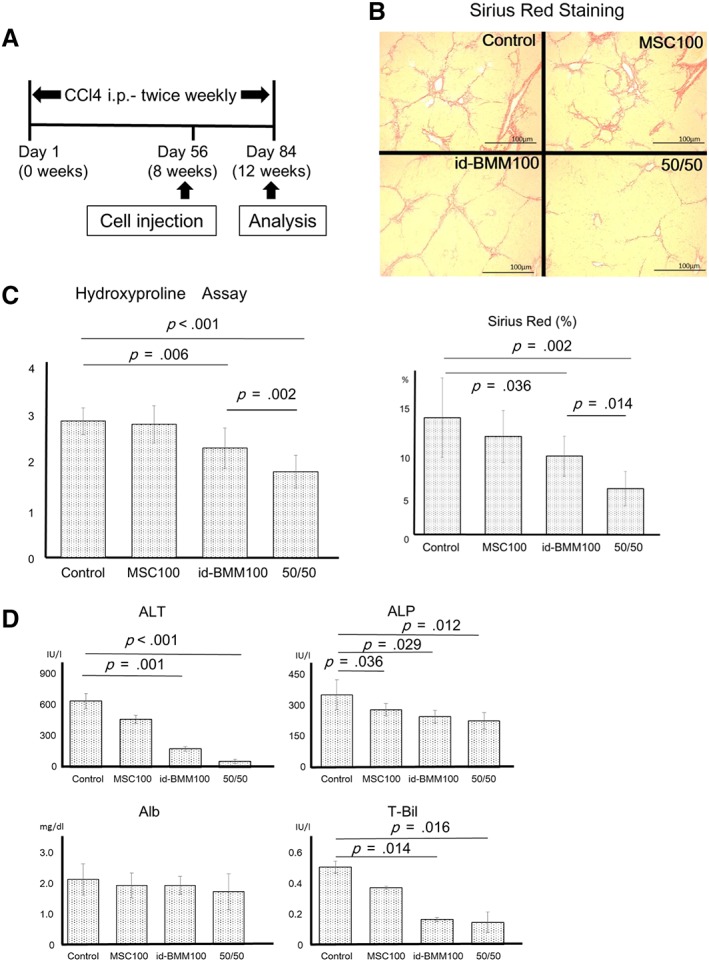
Therapeutic effect of mesenchymal stem cells (MSCs) and induced bone marrow‐derived macrophages (id‐BMMs) on CCl4‐induced cirrhosis in mice. (A): The schematic diagram represents the experimental design regarding the induction of fibrosis, cell administration, and analysis in this study. (B, C): Sirius red staining and hydroxyproline assay showing the degree of fibrosis in mice from the control, MSC100, id‐BMM100, and 50/50 groups. Data are presented as the means ± SD, n = 12 mice in each group, p = .315 (hydroxyproline, MSC100), p = .201 (Sirius red, MSC100), p = .006 (hydroxyproline, id‐BMM100), p = .036 (Sirius red, id‐BMM100), p < .001 (hydroxyproline, 50/50), p = .002 (Sirius red, 50/50), compared with control group. (D): Serum levels of ALT, ALP, albumin, and total bilirubin 4 weeks after cell injection. Data are presented as the means ± SD, n = 12 mice in each group, p = .001 (ALT, id‐BMM100), p < .001 (ALT, 50/50), p = .036 (ALP, MSC100), p = .029 (ALP, id‐BMM100), p = .012 (ALP, 50/50), p = .014 (T‐Bil, id‐BMM100), p = .016 (T‐Bil, 50/50), compared with the control group. Scale bar: 100 μm.
Combination Therapy Leads to Increased Expression of Antifibrosis Markers Followed by Proregenerative Factors
To evaluate the mechanisms underlying the effects of combination therapy shortly after cell administration, liver tissue was analyzed 1, 3, and 7 days after cell administration. On day 28 after cell injection, the mRNA changes in combination therapy group were not significantly different compared with the no cell injection or monotherapy groups (data not shown). Regarding fibrosis‐related factors, Mmp8 (p < .001; day 3, compared with control), Mmp9 (p < .001; day 3, compared with control), and Mmp13 (p < .001; day 3, compared with control) mRNA levels in the combination therapy group were upregulated, whereas those of TIMP‐1 (p < .001; day 3, compared with control) and TIMP‐3 (p < .001; day 3, compared with control; antagonist of MMP) were markedly downregulated at day 3 in the combination therapy group (Fig. 3A). Regarding the proregenerative factors, mRNA levels of HGF (p < .001; day 7, compared with control), OSM (p < .001; day 7, compared with control), and VEGF (p < .001; day 7, compared with control), especially those of VEGF and OSM, were upregulated in the combination therapy group at day 7 (Fig. 3B). We further evaluated the regeneration of hepatocytes by liver weight and body weight ratio (LW/BW) and proliferating cell nuclear antigen (PCNA) staining. Although there was no significant difference in LW/BW, the largest number of dividing hepatocytes were present in the 50/50 group at day 7 (p = .001 compared with day 1, p = .003 compared with day 3; Fig. 3C). These results revealed that after combination therapy, the expression of MMPs, which are antifibrosis markers, increased, followed by an increase in proregenerative factors, leading to liver regeneration.
Figure 3.
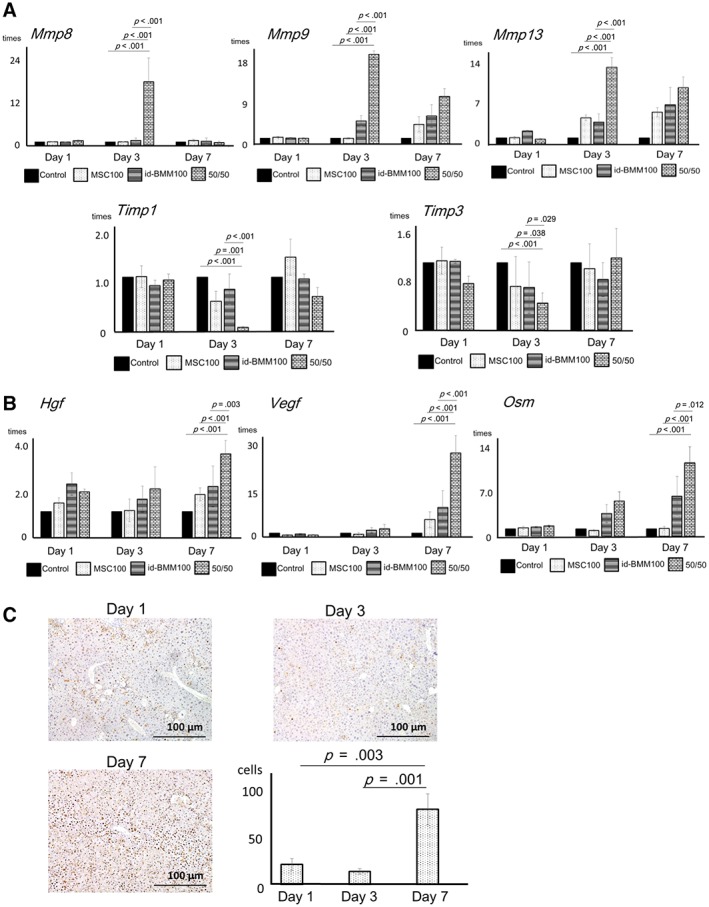
Analysis of mRNA expression changes for factors related to fibrosis and proregenerative factors at 1, 3, and 7 days after cell administration in the liver, and analysis of the proliferating hepatocytes. (A): mRNA expression changes of factors related to fibrosis (matrix metalloproteinases [MMP] and tissue inhibitor of metalloprotease [TIMP]). Data are presented as the means ± SD, n = 12 in each experiment. Representatively, in the 50/50 group, mRNA levels of MMP‐8 (p < .001; day 3, compared with control, p < .001; day 3, compared with MSC100, p < .001; day 3, compared with id‐BMM100), MMP‐9 (p < .001; day 3, compared with control, p < .001; day 3, compared with MSC100, p < .001; day 3, compared with id‐BMM100), and MMP‐13 (p < .001; day 3, compared with control, p < .001; day 3, compared with MSC100, p < .001; day 3, compared with id‐BMM100) are upregulated, whereas in 50/50 group, mRNA levels of TIMP‐1 (p < .001; day 3, compared with control, p = .001; day 3, compared with MSC100, p < .001; day 3, compared with id‐BMM100), and TIMP‐3 (p < .001; day 3, compared with control, p = .038; day 3, compared with MSC100, p = .029; day 3, compared with id‐BMM100) are downregulated. (B): mRNA expression changes of proregenerative factors (hepatocyte growth factor [HGF], vascular endothelial growth factor [VEGF], and oncostatin M [OSM]). Data are presented as the means ± SD, n = 12 in each experiment. Representatively, in the 50/50 group, mRNA levels of HGF (p < .001; day 7, compared with control, p < .001; day 7, compared with MSC100, p = .003; day 7, compared with id‐BMM100), VEGF (p < .001; day 7, compared with control, p < .001; day 7, compared with MSC100, p < .001; day 7, compared with id‐BMM100), and OSM (p < .001; day 7, compared with control, p < .001; day 7, compared with MSC100, p = .012; day 7, compared with id‐BMM100), are upregulated. (C): Analysis of the proliferating hepatocytes by PCNA staining at day 7 after cell administration. Data are presented as the means ± SD, n = 12 in each experiment, p = .003 (day 7) compared with day 1, p = .001 (day 7) compared with day 3. Scale bar: 100 μm.
A Large Number of id‐BMMs and Few MSCs Migrate to the Fibrotic Area in the Liver and Combination Therapy Increases the Frequency of id‐BMM Engraftment
Although combination therapy using MSCs and id‐BMMs (50/50) was the most effective for improvement of liver fibrosis and regeneration, the behavior of each cell type and the mechanisms underlying the effect of combination therapy needed to be shown. We first evaluated the detailed behavior of the administered cells using intravital imaging in mice with CCl4‐induced liver damage using two‐photon excitation microscopy. Previous studies reported the engraftment of administered cells indirectly using still images or an in vivo imaging system (IVIS) 14. However, it was difficult to distinguish dead hepatocytes from the administered cells using such techniques. Additionally, the detailed area of cell engraftment and the behavior of the cells in the engrafted area were not fully shown. Intravital imaging using two‐photon excitation microscopy overcame these problems. The administered fluorescently labeled cells were able to be distinguished from dead cells. Moreover, we confirmed their location and behavior in the intravital tissue. Furthermore, migration or phagocytosis in the specific area was observed by intravital imaging. To detect administered cells, id‐BMMs derived from GFP knock‐in mice and MSCs derived from DsRed knock‐in mice were administered to cirrhotic mice (Supporting Information Fig. S4). The liver, spleen, and lung were observed by using two‐photon excitation microscopy (liver, Fig. 4A; spleen and lung, Supporting Information Fig. S5A), which enabled intravital cell imaging of tissues at days 1, 3, and 7 after cell administration. On day 28, most of the administered cells had disappeared (data not shown). Among the cells administered, the majority of id‐BMMs migrated to the liver (42.6% ± 5.5%), especially near the fibrotic area, or to the lung (47.7% ± 7.9%) from day 1 and they remained for 7 days (Fig. 4B). In contrast, most administered MSCs migrated to the lung (84.4% ± 6.1%) and were rarely detected in the liver and spleen at day 1 (6.2% ± 2.0%). MSCs gradually disappeared and were not detected in the liver, spleen, and lung at day 7 after cell administration (Fig. 4C). Similar results were observed when the mice received the combination therapy. However, the frequency of migration of id‐BMMs into the liver was significantly higher (1.70‐fold at day 1, 1.26‐fold at day 3, and 1.50‐fold at day 7) than that in mice receiving id‐BMM monotherapy (Supporting Information Fig. S5B). These results, together with the results showing that MSCs changed the polarity of macrophages toward M2 in coculture, suggest that id‐BMMs are the direct effectors against liver fibrosis and induce liver regeneration, whereas MSCs are indirect modulators that affect the behavior and characteristics of macrophages.
Figure 4.

Localization of administered mesenchymal stem cells (MSCs) and induced bone marrow‐derived macrophages (id‐BMMs) at 1, 3, and 7 days after cell injection in intravital imaging analysis, and analysis of changes in id‐BMM characteristics. (A): Intravital imaging was performed with two‐photon excitation microscopy of the liver, 3 days after cell administration in the MSC100 (left panels), id‐BMM100 (middle panels), and 50/50 (right panels) groups. Green cells represent administered id‐BMMs, and red cells are administered MSCs. Nuclei were stained with DAPI (blue), and the dense blue area composed of blue fibers represents fibrosis, white dotted lines represent the area of fibrotic liver damage, and white spots represent hepatocyte debris (scale bar: 100 μm). (B, C): Behavior of administered id‐BMMs and MSCs in the liver, lung, and spleen at 1, 3, and 7 days after combination cell administration, n = 12 mice in each group. (D): Representative intravital time‐lapse images of phagocytosis by id‐BMMs. Green cells represent administered id‐BMMs, nuclei are stained with DAPI (blue), the dense blue area composed of blue fibers represents fibrosis, and white spots represent hepatocyte debris (scale bar: left panel, 50 μm; magnified image of left panel, 10 μm), n = 4 mice in each group. (Di): Start point of intravital imaging. (Dii): Three minutes after starting the video, id‐BMMs approached debris. (Diii, Div): After 9–16 minutes, id‐BMMs surrounded, phagocytosed, and digested the debris (phagocytosis activity). (Dv, Dvi): After 21–30 minutes, id‐BMMs reapproached and phagocytosed residual debris. (E): Analysis of changes in id‐BMM characteristics after phagocytosis of hepatocyte debris. mRNA expression changes in id‐BMMs after phagocytosis of hepatocyte debris. Data are presented as the means ± SD, n = 12 in each experiment (Vegf; 3.18‐fold, p < .001, and Osm; 4.25‐fold, p < .001, compared with mRNA levels before phagocytosis).
Administered id‐BMMs Migrate to the Fibrotic Area in the Liver and Phagocytose the Debris near the Fibrotic Area
We showed that id‐BMMs migrated to the liver and were directly involved in the improvement of liver fibrosis and regeneration. However, the behavior of injected id‐BMMs in the fibrotic area is unknown. Time‐lapse intravital cell imaging was performed by using two‐photon excitation microscopy to investigate the behavior of id‐BMMs in the fibrotic area. During the time‐lapse intravital cell imaging, we succeeded in recording the phagocytosis of hepatocyte debris by administering id‐BMMs for the first time. The id‐BMMs migrated and surrounded the hepatocyte debris near the fibrotic area and gradually phagocytosed the hepatocyte debris in 30 minutes (Supporting Information Video S1 and Fig. 4D). Phagocytosing macrophages change their characteristics 8. Thus, to evaluate the changes in id‐BMM characteristics after phagocytosis of debris, hepatocyte debris was added to macrophages cultured in vitro. Vegf and Osm mRNA levels increased after phagocytosis, suggesting that the injected id‐BMMs that phagocytose hepatocyte debris promote liver regeneration (Vegf; 3.18‐fold, p < .001 and Osm; 4.25‐fold, p < .001, compared with mRNA levels before phagocytosis; Fig. 4E). Altogether, our results suggest that the administered id‐BMMs modulated by MSCs present high phagocytic activity, which promotes the healing of damaged tissues and liver regeneration.
Combination Therapy Induces the Migration of Host Macrophages and Neutrophils to the Damaged Liver Where these Host Cells Express Antifibrotic MMPs
During macrophage therapy, endogenous macrophages and neutrophils that migrate into the damaged liver contribute to improve resolution of liver fibrosis 9. From our in vivo intravital imaging results, the therapeutic effect could not be solely explained by direct effects of the injected cells. Thus, we next investigated the contribution of host cells. The damaged liver expresses the chemoattractant SDF‐1 15. Moreover, our macrophages cultured with or without serum from mice with CCl4‐induced liver damage expressed high levels of chemoattractants such as CXCL1, CXCL2, CCL2, and CXCR4. Thus, we suspected that the concentration of chemoattractants is high in the injured liver. In addition, we evaluated the change of mRNA expression levels of chemoattractants in the liver over time after cell administration, confirming that mRNA expression levels of CXCL1 (p < .001; day 3, compared with control) and CXCL2 (p < .001; day 3, compared with control), chemoattractants for neutrophils, in the liver were upregulated in the 50/50 group. Cxcl2 mRNA expression was upregulated not only in the 50/50 group but also in the id‐BMM group (Supporting Information Fig. S6). These results suggest that administration of the combination therapy or id‐BMMs alone supports the migration of host macrophages and neutrophils into the liver. To test this hypothesis, immunohistochemistry for F4/80 (macrophage marker) and Ly‐6G (neutrophil marker) was performed, revealing that the number of macrophages and neutrophils in the liver of mice in the id‐BMM100 and 50/50 groups at day 3 after cell administration was higher than that in the control group, and the number of these cells at day 3 in the 50/50 group was highest (1.61‐fold increase in macrophages; p < .001 and 5.30‐fold increase in neutrophils; p < .001 in the 50/50 group compared with the control group; Fig. 5A, 5B). Furthermore, to strengthen our findings, the frequency of macrophages and neutrophils among whole blood cells in the liver was analyzed by flow cytometry. The frequency of F4/80+/CD11b+ cells, which was the macrophage population showing a significant increase in the id‐BMM100 and 50/50 groups compared with the control group, was highest in the 50/50 group (Fig. 6A; 1.56‐fold increase in the id‐BMM100 group; p < .001, 2.11‐fold increase in the 50/50 group; p < .001, compared with the control group). F4/80+/CD11b+ cells comprised host macrophages in the liver and administered id‐BMMs that migrated to the liver. Because the administered id‐BMMs expressed the GFP fluorescent protein, host macrophages (GFP‐negative cells) and administered id‐BMMs (GFP‐positive cells) could be distinguished by FACS. The content of GFP‐positive id‐BMMs was extremely low, suggesting that the increase in the number of macrophages mainly resulted from the migration of host macrophages. The frequency of Ly‐6G+/CD11b+ cells, which represent the neutrophil population 16, also increased significantly in the id‐BMM100 and 50/50 groups compared with that in the control group, and was highest in the 50/50 group (Fig. 6B; 1.62‐fold increase in the id‐BMM100 group; p = .004, 4.58‐fold increase in the 50/50 group; p < .001 compared with the control group). Our in vitro study confirmed that the interaction between MSCs and id‐BMMs resulted in a change in the macrophage phenotype toward the M2 phenotype. Thus, we investigated the polarization of macrophages in the liver after cell administration by flow cytometry. The frequency of F4/80+/CD11b+/CD206+ cells, the M2 macrophage population, increased in the MSC100 and 50/50 groups compared with that in the control group (Supporting Information Fig. S7). Altogether, our results indicate that after combination therapy with MSCs and id‐BMMs, host macrophages and neutrophils migrated to the damaged liver, and the frequency of M2 macrophages increased in the liver, resulting in an increase of Mmps as antifibrotic factors in the liver at day 3. Moreover, id‐BMMs modulated by MSCs had a high phagocytosis activity, which promoted the healing of damaged tissues and liver regeneration at day 7 after cell administration.
Figure 5.
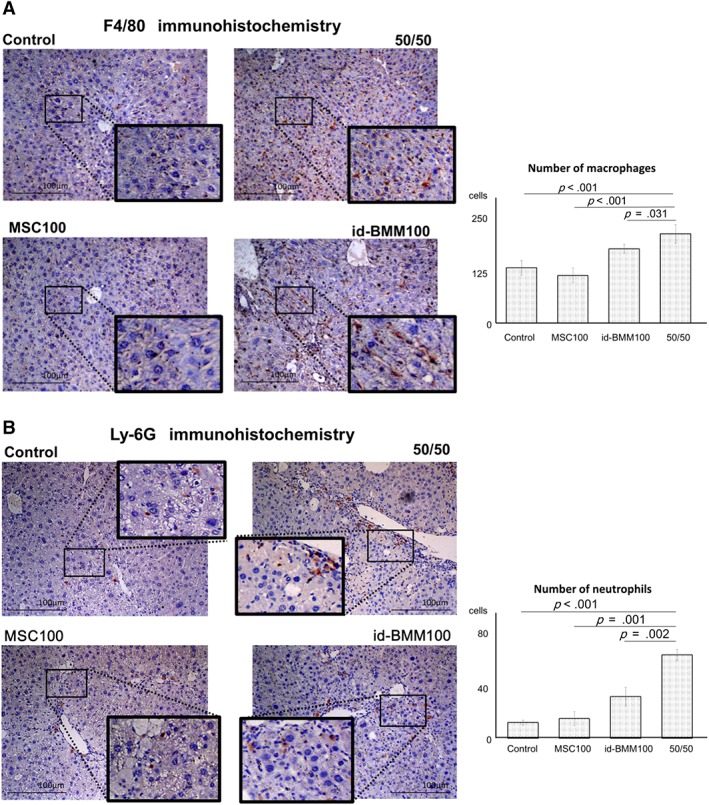
Involvement of host macrophages and neutrophils during improvement of liver fibrosis and regeneration. (A, B): Immunostaining for F4/80 and Ly‐6G in the liver, n = 12 for each group. Data are presented as the means ± SD, p < .001 (F4/80, 50/50 compared with control), p < .001 (F4/80, 50/50 compared with MSC100), p = .031 (F4/80, 50/50 compared with id‐BMM100), p < .001 (Ly‐6G, 50/50 compared with control), p = .001 (Ly‐6G, 50/50 compared with MSC100), p = .002 (Ly‐6G, 50/50 compared with id‐BMM100). Scale bar: 100 μm.
Figure 6.

Host macrophages and neutrophils in the liver. (A, B): Flow cytometric analysis of macrophages (F4/80) and neutrophils (Ly‐6G) in 1 × 106 hematopoietic cells in the liver. The macrophage population contained administered induced bone marrow‐derived macrophages (id‐BMMs; GFP positive cells). Data are presented as the means ± SD, n = 12 mice in each group, p < .001 (macrophages, 50/50 compared with control), p < .001 (macrophages, 50/50 compared with MSC100), p = .004 (macrophages, 50/50 compared with id‐BMM100), p < .001 (neutrophils, 50/50 compared with control), p < .001 (neutrophils, 50/50 compared with MSC100), p = .001 (neutrophils, 50/50 compared with id‐BMM100).
Discussion
MSCs and macrophages are effective for liver fibrosis regression and regeneration. However, the differences in the characteristics of these cells, their detailed behavior, and the mechanism underlying the improvement of liver fibrosis and the promotion of liver regeneration have not been fully shown. This study demonstrates that combination therapy using MSCs and id‐BMMs synergistically improves liver fibrosis and regeneration. Furthermore, we showed the behavior of MSCs and id‐BMMs using intravital imaging and partially showed the mechanisms underlying liver fibrosis improvement and liver regeneration.
In vitro data in simple culture and serum‐added culture revealed that MSCs may not directly affect liver fibrosis regression and liver regeneration as strongly as id‐BMMs. Based on these in vitro experiments, in vivo studies were performed to show the most effective cells and cell populations for the treatment of cirrhosis. The therapeutic effect of MSCs alone was relatively low confirming our in vitro data. However, the combination therapy significantly improved liver fibrosis.
Next, we focused on the detailed behavior of MSCs and id‐BMMs after administration using intravital imaging. First, by using two‐photon excitation microscopy, we showed that id‐BMMs abundantly migrated to the liver, whereas MSCs rarely migrated to the liver. Furthermore, we clearly detected the engraftment of id‐BMMs near the fibrotic area and distinguished the id‐BMMs from dead cells. Finally, we succeeded in capturing the migrating and phagocytosing id‐BMMs, which is difficult using conventional methods such as immunohistochemistry and IVIS 14. Most of the administered MSCs migrated to the lung, were rarely detected in the liver, and disappeared from the lung, liver, and spleen by 7 days after cell administration. Given that combination therapy greatly improved liver fibrosis and regeneration and that id‐BMMs engrafted near the fibrotic area in the liver, expressing antifibrosis factors, we speculated that MSCs, as indirect effectors, and id‐BMMs, as direct effectors, synergistically improved liver fibrosis and regeneration. We further speculated that owing to the cell sizes of MSCs, they were trapped in the lung, whereas macrophage migration to specific places in particular organs was probably due to the actions of chemoattractants and adhesion molecules. Nevertheless, these cells synergistically improved cirrhosis. The location and direction of MSC effects on macrophages require further analyses.
Thus, in this study, we comprehensively analyzed the characteristics and relationship of MSCs and id‐BMMs. We assessed the relationship between MSCs and id‐BMMs comprehensively. A recent report suggested that MSCs polarize macrophages toward the M2 phenotype 13, 17, 18. Thus, we evaluated the interaction between MSCs and id‐BMMs by coculture in transwell dishes. The mRNA expression of PGE2 and TSG‐6, which can polarize macrophages toward the M2 phenotype, was upregulated in MSCs after coculture, and id‐BMMs increasingly showed the M2 phenotype after coculture with MSCs. These results suggest that by utilizing MSCs, the characteristics of id‐BMMs were changed toward the M2 phenotype.
Additionally, the phagocytic activity of M2‐polarized id‐BMMs after coculture with MSCs increased. A change in the expression of various factors is observed in macrophages after phagocytosis 8. In our id‐BMMs, mRNA levels of proregenerative factors (OSM and VEGF) were upregulated after phagocytosis of hepatocyte debris, suggesting that liver regeneration is accelerated after phagocytosis of hepatocytes by id‐BMMs. From these results, we considered that M2 polarization of macrophages prior to injection, or drug treatment to induce M2‐polarized macrophages might represent strategies for future cell therapies.
Finally, we determined whether host cells were recruited to the damaged site during fibrosis regression and liver regeneration. id‐BMMs express several chemoattractants to recruit host macrophages and neutrophils 9. Furthermore, the damaged liver itself expresses chemoattractants such as SDF‐1. Thus, the host cells are likely involved the therapeutic effect. In our study, after cell administration, especially after combination therapy, host macrophages and neutrophils migrated to the damaged liver. These results revealed that the combination therapy triggered the migration of macrophages and neutrophils and their involvement in fibrosis regression and liver regeneration. In addition, the polarization of macrophages in the whole liver was analyzed after cell administration. After combination therapy, the frequency of M2 macrophages, including host and administered macrophages, increased in the liver. Time‐lapse changes in mRNA expression after cell administration indicated the upregulation of antifibrotic factors such as MMPs followed by the upregulation of proregenerative factors such as OSM and VEGF, suggesting that fibrosis regression occurs first, followed by liver regeneration. Altogether, these results suggest that both cell types are necessary to improve liver fibrosis and regeneration and that MSCs, which contributed to therapy remotely and indirectly, and id‐BMMs, which contributed directly, acted together to synergistically improve liver fibrosis and regeneration (Fig. 7).
Figure 7.
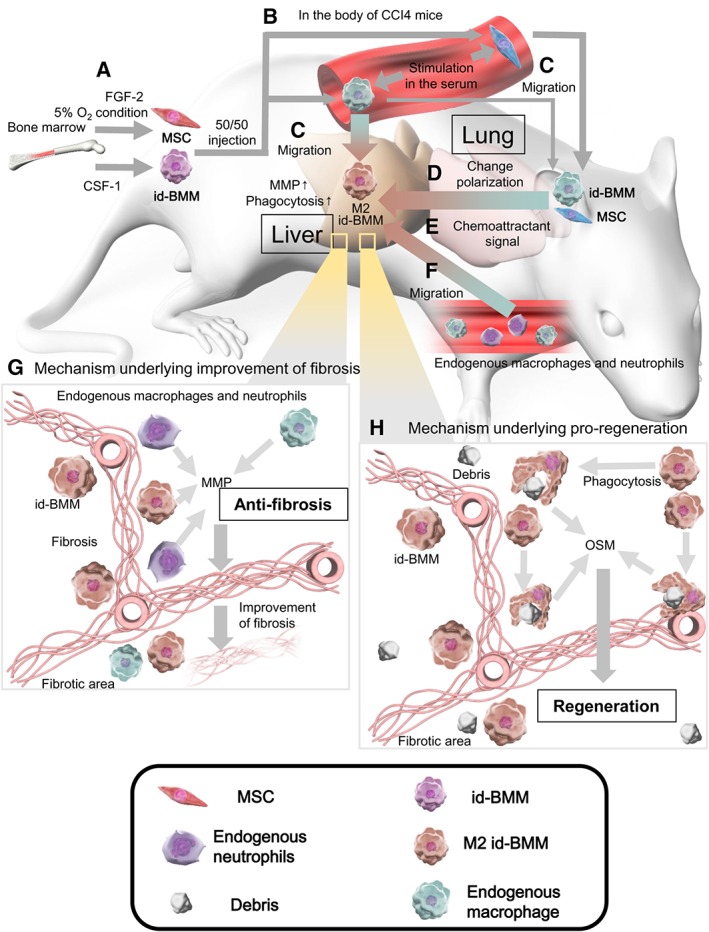
Schematic representation of the mechanisms underlying the improvement of liver fibrosis and regeneration. (A): Highly enriched mesenchymal stem cells (MSCs) and induced bone marrow‐derived macrophages (id‐BMMs) were cultured from mouse bone marrow in the presence of FGF‐2 and 5% O2 condition, or CSF‐1. (B): After MSCs and id‐BMMs were administered to the mice, the administered cells were activated by cytokines and chemokines in the serum from liver‐damaged mice. (C): Most MSCs migrated to the lung, whereas many id‐BMMs migrated to the liver, and id‐BMMs engrafted near the fibrotic area in the liver. (D): Signals from MSCs that migrated to the lung changed id‐BMMs toward the M2 phenotype, promoting an anti‐inflammatory environment in the liver. (E): Activated id‐BMMs secreted chemoattractant signals in the liver. (F): Endogenous macrophages and neutrophils were recruited from the peripheral blood to the liver by chemoattractant signals. (G): Matrix metalloproteinases secreted from administered id‐BMMs and endogenous cells that migrated to the liver induced regression of fibrosis (antifibrosis). (H): M2 id‐BMMs that gained high phagocytic activity phagocytosed hepatocyte debris more effectively and expressed proregenerative factors (proregeneration).
The activities of MSCs, but not synergistic effects with id‐BMMs, have been previously reported. MSCs affect various events in the fibrotic cascade, which represents a series of processes leading to the production of ECM as follows: hepatocyte injury, inflammation, activation of myofibroblasts, and then ECM production. MSCs reduce hepatocyte damage and liver injury 19, suppress inflammation in the liver 20, and inhibit ECM production by inactivating hepatic stellate cells 21. Recently, it has been reported that milk fat globule‐EGF factor 8 (MFG‐E8) related to the inactivation of hepatic stellate cells suppresses ECM production 21. Because the expression of this factor was high in the microarray analysis of our MSCs, this mechanism may also affect the suppression of liver fibrosis. In this manner, MSCs affected the fibrotic cascade indirectly and suppressed fibrosis formation. In our study, the majority of MSCs were trapped in the lung, in which increased Il10 mRNA levels and decreased Il6 mRNA levels were detected (data not shown). Thus, we speculate that MSCs support a systemic anti‐inflammatory state and help to change host and administered macrophages to the M2 phenotype.
Limitations of our studies included our use of the CCl4‐induced cirrhosis model mouse, which differs from human cirrhosis wherein excess fibrosis occurs over the long‐term. Furthermore, direct intra‐arterial MSC injection to the liver performed in some human clinical trials 22. Our studies did not show the effect of direct injection of the cell combination to the liver.
Conclusion
In conclusion, our results indicate that MSCs indirectly affect the improvement of liver fibrosis, whereas M2‐polarized id‐BMMs, together with host macrophages and neutrophils, directly improve liver fibrosis and regeneration. We consider that these studies are important for future cell therapies. Our developed studies of combination therapy might serve as an alternative therapy and fulfill unmet needs for decompensated cirrhosis. Because MSCs can be obtained from medical waste such as adipose tissue, umbilical cord tissue, and dental pulp and exhibit low antigenicity, they can be applied for allogeneic administration on demand 22, 23, 24, 25. Furthermore, recent research advances suggest the possibility of cell‐free therapy. Notably, MSCs express various cytokines, chemokines, and growth factors as well as exosomes, which assist with tissue repair both indirectly and remotely 11. Accordingly, some studies have reported that only conditioned medium or exosomes provide effects similar to those of cell therapies 26, 27. Moreover, because macrophages can be obtained from peripheral blood using apheresis, it is possible to obtain a sufficient number of macrophages repeatedly, although some adverse effects may occur during apheresis of patients with cirrhosis 28, 29, and owing to their antigenicity, allogeneic administration of macrophages may be difficult. Overall, combination therapy constitutes a useful tool to show the mechanisms underlying liver fibrosis regression and liver regeneration and paves the way to develop future therapies for decompensated cirrhosis.
Author Contributions
Y.W., A.T., and ST: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; S.S., Y.K., Y.K., S.I., H.K., and S.Y.: collection and/or assembly of data; L.P.J.S., W.Y.L., and J.K.: conception and design, supervision of the study; S.J.F., M.I., and S.T.: conception and design, supervision of the study, manuscript writing.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Appendix S1: Supporting Information
Supplemental Figure 1. Characterization of mouse bone marrow‐derived mesenchymal stem cells (MSCs) and induced bone marrow‐derived macrophages (id‐BMMs). (A) Hematoxylin staining of id‐BMMs. (Scale bars: 100 μm.) (B) Flow cytometric analysis for the cell surface markers of id‐BMMs. (C) Microscopic analysis of MSCs. (Scale bars: 100 μm.) (D) Flow cytometric analysis for the cell surface markers of MSCs. (E) Tri‐lineage differentiation potential of MSCs. Chondrogenic (left panel), adipogenic (middle panel), and osteogenic (right panel) differentiation was confirmed by Alcian blue staining, Oil red O staining, and Alizarin red staining, respectively. (Scale bars: 100 μm.)
Supplemental Figure 2. Gene expression in cultured MSCs and id‐BMMs analyzed by microarray. Heatmaps of 5 major categories are shown. mRNA expression of genes related to fibrosis, chemoattractants, pro‐regeneration, inflammation/anti‐inflammation, and macrophage phenotypes is shown.
Supplemental Figure 3. Phagocytic ability of id‐BMMs determined by zymosan phagocytosis assay. Data are presented as the means ± SD, n = 6 in each experiment, p = 0.038 (co‐culture compared to simple culture), p = 0.041 (addition of serum compared to simple culture), p = 0.008 (co‐culture and addition of serum compared to simple culture), p = 0.036 (co‐culture and addition of serum compared to co‐culture), p = 0.032 (co‐culture and addition of serum compared to addition of serum). (Scale bar: 50 μm.)
Supplemental Figure 4. Schematic representation of the experimental design for live imaging to show the detailed behavior of administered MSCs (DsRed; red) and id‐BMMs (GFP; green). The id‐BMMs derived from GFP knock‐in mice and MSCs derived from DsRed knock‐in mice were administered to the mice with CCl4‐induced liver damage via the tail vein. (Scale bar: 100 μm.)
Supplemental Figure 5. Localization of administered MSCs and id‐BMMs at 1, 3, and 7 days after cell injection in intravital imaging analysis. (A) Intravital imaging using two‐photon excitation microscopy of the lung (upper panels), and spleen (lower panels) 3 days after cell administration in the MSC100 (left panels), id‐BMM100 (middle panels), and 50/50 (right panels) groups. Green cells represent administered id‐BMMs, red cells are administered MSCs. Nuclei are stained with DAPI (blue), the dense blue area composed of blue fibers represents fibrosis, and white spots represent debris of hepatocytes. (Scale bar: 100 μm.) (B) Comparison of localization of administered id‐BMMs between the 50/50 and id‐BMM100 groups at 1, 3, and 7 days, n = 12 mice in each group.
Supplemental Figure 6. Cxcl1 and Cxcl2 mRNA levels in the id‐BMM100 and 50/50 groups were markedly upregulated at 3 days after cell administration. Data are presented as the means ± SD, n = 12 in each experiment. Representatively, in the 50/50 group, mRNA levels of CXCL1 (p < 0.001; day 3, compared to control, p < 0.001; day 3, compared to MSC 100, p < 0.001; day 3, compared to id‐BMM100) and CXCL2 (p < 0.001; day 3, compared to control, p < 0.001; day 3, compared to MSC 100, p = 0.086; day 3, compared to id‐BMM100) are upregulated.
Supplemental Figure 7. Flow cytometric analysis of CD206‐positive M2 polarized macrophages. The values represent the frequency of F4/80+/CD11b+/CD206+ cells (M2 macrophages) among all macrophages. Data are presented as the means ± SD, n = 12 mice in each group.
Supplemental Table 1. List of primers used for real‐time PCR. The names of primers, catalog numbers, species origin, and company are provided.
Supplemental Table 2. List of antibodies used for immunostaining. The names of antibodies, clones, species origin, company names, dilution, antigen retrieval, and heating time are provided.
Supplemental Table 3. List of antibodies used for flow cytometry. The names of antibodies, clones, species origin, and company names are provided.
Supplemental Video 1. Intravital two‐photon imaging of id‐BMMs phagocytizing debris in the liver. Green cells are the administered id‐BMMs, nuclei are stained with DAPI (blue), the dense blue area composed of blue fibers represents fibrosis, and white spots represent hepatocyte debris. Three minutes after starting the video, id‐BMMs approached debris. After 9–16 minutes, id‐BMMs surrounded and phagocytized the debris, and digested it(Phagocytosis activity). After 21–30 minutes, id‐BMMs re‐approached and phagocytized residual debris. Scale bar, 50 μm. Playback speed = 100×.
Acknowledgments
This research was supported by a Grant‐in‐Aid for Scientific Research (B; 26293175) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, by the Highway Program for Realization of Regenerative Medicine from the Japan Agency for Medical Research and Development (AMED; 16bm0504003h0206), and by the Research Program on Hepatitis from AMED to Shuji Terai (17fk0210101h0001).
Contributor Information
Atsunori Tsuchiya, Email: atsunori@med.niigata-u.ac.jp.
Shuji Terai, Email: terais@med.niigata-u.ac.jp.
References
- 1. Curry MP, O'Leary JG, Bzowej N et al. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med 2015;373:2618–2628. [DOI] [PubMed] [Google Scholar]
- 2. Terai S, Tsuchiya A. Status of and candidates for cell therapy in liver cirrhosis: Overcoming the “point of no return” in advanced liver cirrhosis. J Gastroenterol 2017;52:129–140. [DOI] [PubMed] [Google Scholar]
- 3. Tsuchiya A, Kojima Y, Ikarashi S et al. Clinical trials using mesenchymal stem cells in liver diseases and inflammatory bowel diseases. Inflamm Regener 2017;37:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Terai S, Ishikawa T, Omori K et al. Improved liver function in patients with liver cirrhosis after autologous bone marrow cell infusion therapy. Stem Cells 2006;24:2292–2298. [DOI] [PubMed] [Google Scholar]
- 5. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest 2012;122:787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Duffield JS, Forbes SJ, Constandinou CM et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005;115:56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamiya A, Kinoshita T, Ito Y et al. Fetal liver development requires a paracrine action of oncostatin M through the gp130 signal transducer. EMBO J 1999;18:2127–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boulter L, Govaere O, Bird TG et al. Macrophage‐derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med 2012;18:572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thomas JA, Pope C, Wojtacha D et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology 2011;53:2003–2015. [DOI] [PubMed] [Google Scholar]
- 10. Zhou BO, Yue R, Murphy MM et al. Leptin‐receptor‐expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 2014;15:154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang Y, Chen X, Cao W et al. Plasticity of mesenchymal stem cells in immunomodulation: Pathological and therapeutic implications. Nat Immunol 2014;15:1009–1016. [DOI] [PubMed] [Google Scholar]
- 12. Stoll S, Delon J, Brotz TM et al. Dynamic imaging of T cell‐dendritic cell interactions in lymph nodes. Science 2002;296:1873–1876. [DOI] [PubMed] [Google Scholar]
- 13. Eggenhofer E, Hoogduijn MJ. Mesenchymal stem cell‐educated macrophages. Transplant Res 2012;1:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haga J, Wakabayashi G, Shimazu M et al. In vivo visualization and portally repeated transplantation of bone marrow cells in rats with liver damage. Stem Cells Dev 2007;16:319–328. [DOI] [PubMed] [Google Scholar]
- 15. Liepelt A, Tacke F. Stromal cell‐derived factor‐1 (SDF‐1) as a target in liver diseases. Am J Physiol Gastrointest Liver Physiol 2016;311:G203–G209. [DOI] [PubMed] [Google Scholar]
- 16. Leon B, Martinez del Hoyo G, Parrillas V et al. Dendritic cell differentiation potential of mouse monocytes: Monocytes represent immediate precursors of CD8− and CD8+ splenic dendritic cells. Blood 2004;103:2668–2676. [DOI] [PubMed] [Google Scholar]
- 17. Kim J, Hematti P. Mesenchymal stem cell‐educated macrophages: A novel type of alternatively activated macrophages. Exp Hematol 2009;37:1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maggini J, Mirkin G, Bognanni I et al. Mouse bone marrow‐derived mesenchymal stromal cells turn activated macrophages into a regulatory‐like profile. PLoS ONE 2010;5:e9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forbes SJ, Newsome PN. New horizons for stem cell therapy in liver disease. J Hepatol 2012;56:496–499. [DOI] [PubMed] [Google Scholar]
- 20. Han Z, Jing Y, Zhang S et al. The role of immunosuppression of mesenchymal stem cells in tissue repair and tumor growth. Cell Biosci 2012;2:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. An SY, Jang YJ, Lim HJ et al. Milk fat globule‐EGF factor 8, secreted by mesenchymal stem cells, protects against liver fibrosis in mice. Gastroenterology 2017;152:1174–1186. [DOI] [PubMed] [Google Scholar]
- 22. Suk KT, Yoon JH, Kim MY et al. Transplantation with autologous bone marrow‐derived mesenchymal stem cells for alcoholic cirrhosis: Phase 2 trial. Hepatology 2016;64:2185–2197. [DOI] [PubMed] [Google Scholar]
- 23. Peng L, Xie DY, Lin BL et al. Autologous bone marrow mesenchymal stem cell transplantation in liver failure patients caused by hepatitis B: Short‐term and long‐term outcomes. Hepatology 2011;54:820–828. [DOI] [PubMed] [Google Scholar]
- 24. Wang L, Han Q, Chen H et al. Allogeneic bone marrow mesenchymal stem cell transplantation in patients with UDCA‐resistant primary biliary cirrhosis. Stem Cells Dev 2014;23:2482–2489. [DOI] [PubMed] [Google Scholar]
- 25. Trounson A, McDonald C. Stem cell therapies in clinical trials: Progress and challenges. Cell Stem Cell 2015;17:11–22. [DOI] [PubMed] [Google Scholar]
- 26. Watanabe S, Arimura Y, Nagaishi K et al. Conditioned mesenchymal stem cells produce pleiotropic gut trophic factors. J Gastroenterol 2014;49:270–282. [DOI] [PubMed] [Google Scholar]
- 27. Li T, Yan Y, Wang B et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev 2013;22:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stutchfield BM, Antoine DJ, Mackinnon AC et al. CSF1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology 2015;149:1896.e14–1909.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Newsome PN, Fox R, King AL et al. Granulocyte colony‐stimulating factor and autologous CD133‐positive stem‐cell therapy in liver cirrhosis (REALISTIC): An open‐label, randomised, controlled phase 2 trial. Lancet Gastroenterol Hepatol 2018;3:25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information
Supplemental Figure 1. Characterization of mouse bone marrow‐derived mesenchymal stem cells (MSCs) and induced bone marrow‐derived macrophages (id‐BMMs). (A) Hematoxylin staining of id‐BMMs. (Scale bars: 100 μm.) (B) Flow cytometric analysis for the cell surface markers of id‐BMMs. (C) Microscopic analysis of MSCs. (Scale bars: 100 μm.) (D) Flow cytometric analysis for the cell surface markers of MSCs. (E) Tri‐lineage differentiation potential of MSCs. Chondrogenic (left panel), adipogenic (middle panel), and osteogenic (right panel) differentiation was confirmed by Alcian blue staining, Oil red O staining, and Alizarin red staining, respectively. (Scale bars: 100 μm.)
Supplemental Figure 2. Gene expression in cultured MSCs and id‐BMMs analyzed by microarray. Heatmaps of 5 major categories are shown. mRNA expression of genes related to fibrosis, chemoattractants, pro‐regeneration, inflammation/anti‐inflammation, and macrophage phenotypes is shown.
Supplemental Figure 3. Phagocytic ability of id‐BMMs determined by zymosan phagocytosis assay. Data are presented as the means ± SD, n = 6 in each experiment, p = 0.038 (co‐culture compared to simple culture), p = 0.041 (addition of serum compared to simple culture), p = 0.008 (co‐culture and addition of serum compared to simple culture), p = 0.036 (co‐culture and addition of serum compared to co‐culture), p = 0.032 (co‐culture and addition of serum compared to addition of serum). (Scale bar: 50 μm.)
Supplemental Figure 4. Schematic representation of the experimental design for live imaging to show the detailed behavior of administered MSCs (DsRed; red) and id‐BMMs (GFP; green). The id‐BMMs derived from GFP knock‐in mice and MSCs derived from DsRed knock‐in mice were administered to the mice with CCl4‐induced liver damage via the tail vein. (Scale bar: 100 μm.)
Supplemental Figure 5. Localization of administered MSCs and id‐BMMs at 1, 3, and 7 days after cell injection in intravital imaging analysis. (A) Intravital imaging using two‐photon excitation microscopy of the lung (upper panels), and spleen (lower panels) 3 days after cell administration in the MSC100 (left panels), id‐BMM100 (middle panels), and 50/50 (right panels) groups. Green cells represent administered id‐BMMs, red cells are administered MSCs. Nuclei are stained with DAPI (blue), the dense blue area composed of blue fibers represents fibrosis, and white spots represent debris of hepatocytes. (Scale bar: 100 μm.) (B) Comparison of localization of administered id‐BMMs between the 50/50 and id‐BMM100 groups at 1, 3, and 7 days, n = 12 mice in each group.
Supplemental Figure 6. Cxcl1 and Cxcl2 mRNA levels in the id‐BMM100 and 50/50 groups were markedly upregulated at 3 days after cell administration. Data are presented as the means ± SD, n = 12 in each experiment. Representatively, in the 50/50 group, mRNA levels of CXCL1 (p < 0.001; day 3, compared to control, p < 0.001; day 3, compared to MSC 100, p < 0.001; day 3, compared to id‐BMM100) and CXCL2 (p < 0.001; day 3, compared to control, p < 0.001; day 3, compared to MSC 100, p = 0.086; day 3, compared to id‐BMM100) are upregulated.
Supplemental Figure 7. Flow cytometric analysis of CD206‐positive M2 polarized macrophages. The values represent the frequency of F4/80+/CD11b+/CD206+ cells (M2 macrophages) among all macrophages. Data are presented as the means ± SD, n = 12 mice in each group.
Supplemental Table 1. List of primers used for real‐time PCR. The names of primers, catalog numbers, species origin, and company are provided.
Supplemental Table 2. List of antibodies used for immunostaining. The names of antibodies, clones, species origin, company names, dilution, antigen retrieval, and heating time are provided.
Supplemental Table 3. List of antibodies used for flow cytometry. The names of antibodies, clones, species origin, and company names are provided.
Supplemental Video 1. Intravital two‐photon imaging of id‐BMMs phagocytizing debris in the liver. Green cells are the administered id‐BMMs, nuclei are stained with DAPI (blue), the dense blue area composed of blue fibers represents fibrosis, and white spots represent hepatocyte debris. Three minutes after starting the video, id‐BMMs approached debris. After 9–16 minutes, id‐BMMs surrounded and phagocytized the debris, and digested it(Phagocytosis activity). After 21–30 minutes, id‐BMMs re‐approached and phagocytized residual debris. Scale bar, 50 μm. Playback speed = 100×.