

Abstract
We here determine the influence of mesenchymal stem cell (MSC) therapy on the progression of solid tumors. The influence of MSCs was investigated in human colorectal cancer cells as well as in an immunocompetent rat model of colorectal carcinogenesis representative of the human pathology. Treatment with bone marrow (BM)‐derived MSCs significantly reduced both cancer initiation and cancer progression by increasing the number of tumor‐free animals as well as decreasing the number and the size of the tumors by half, thereby extending their lifespan. The attenuation of cancer progression was mediated by the capacity of the MSCs to modulate the immune component. Specifically, in the adenocarcinomas (ADKs) of MSC‐treated rats, the infiltration of CD68+ monocytes/macrophages was 50% less while the presence of CD3+ lymphocytes increased almost twofold. The MSCs reprogrammed the macrophages to become regulatory cells involved in phagocytosis thereby inhibiting the production of proinflammatory cytokines. Furthermore, the MSCs decreased NK (Natural Killer) and rTh17 cell activities, Treg recruitment, the presence of CD8+ lymphocytes and endothelial cells while restoring Th17 cell activity. The expression of miR‐150 and miR‐7 increased up to fivefold indicating a likely role for these miRNAs in the modulation of tumor growth. Importantly, MSC administration limited the damage of healthy tissues and attenuated tumor growth following radiotherapy. Taken together, we here show that that MSCs have durable action on colon cancer development by modulating the immune component of the tumor microenvironment. In addition, we identify two miRNAs associated with the capacity of MSCs to attenuate cancer growth. stem cells translational medicine 2019;8:285&300
Keywords: Mesenchymal stem cells, Colorectal cancer, Tumor microenvironment, Polarization of immune cells, Cell therapy, Radiotherapy, Pelvic radiation disease
Significance Statement.
Radiotherapy can be accompanied by substantial normal tissue damage. Although mesenchymal stem cells (MSCs) have been used to alleviate radiation damage, uncertainty remains regarding their potential to support growth of residual tumor cells. Administration of MSCs attenuates colorectal cancer progression in immunocompetent animals in the absence or presence of radiotherapy as shown here. Although only transiently present in the colon tissue of treated animals, exogenous MSCs were able to modify the immune profile of the tumor microenvironment even 1 year after the last MSC administration, most likely due to polarization of resident MSCs and immune cells. These results suggest that administration of MSCs may be a safe and innovative therapeutic option to heal normal tissue following cancer radiotherapy.
Introduction
The worldwide number of cancer patients is expected to increase from 14 million in 2012 to more than 19 million in 2025 (http://www.wcrf.org/cancer_statistics/world_cancer_statistics.php). Sixty percent of all cancer patients receive radiation therapy, which is accompanied by detrimental local lesions for at least 500,000 patients. The abdominopelvic area contains major cancer sites as well as highly radiosensitive organs, such as the colon and rectum. Late side effects on the pelvis, that is, pelvic radiation disease (PRD), are mainly characterized by fibrosis and are often lethal 1.
The current treatment of radiotherapy‐induced adverse effects is limited to managing the symptoms; therefore, new strategies are urgently needed 2. No detrimental side effects of treatment with mesenchymal stem cells (MSCs) were reported in a meta‐analysis of 36 clinical trials representing more than 1,000 patients 3.
In contrast, preclinical tumor models treated with exogenous administration of MSCs have resulted in conflicting results concerning their influence on tumor development 4. Depending on the model, it has been suggested that MSCs either inhibit or increase immunity, angiogenesis and apoptosis. One of the reasons for the conflicting results is that most of these studies used local engraftment of human cancer cell lines in mouse models. This system is not representative of the clinical pathology and the tumors are not comparable with human cancers. Another reason is that most of these studies have used immunodeficient animals, in particular mice deficient in T lymphocytes 5, although these cells are known to be important MSC targets for modulation of the tumor microenvironment 6.
In order to evaluate the interest of MSC administration to prevent PRD after cancer radiotherapy, an immunocompetent animal model representative of human cancer development and treatment is needed to address the essential questions approximately mechanism, safety, and efficacy. Besides the cellular component, we have studied miRNAs which play an important role as effectors in inflammation and tumor progression via crosstalk with the MSCs in the tumor microenvironment 8.
We now show that MSC administration to immunocompetent rats treated locally with methylnitronitrosoguanidine (MNNG), a potent carcinogen, decreased tumor progression, and increased overall survival. At early time points, MSCs reversed local acute inflammation by protecting the mucosal structure and function from MNNG. Furthermore, MSC administration was accompanied by a marked modification of the tumor microenvironment which was still apparent 1 year after the last MSC administration. When MSCs were administered after treatment of colorectal cancer (CRC) with fractionated irradiation, MSCs inhibited residual tumor growth and prolonged animal survival. The MSCs also protected healthy tissue from radiation damage by increasing the levels of growth factors, decreasing fibrosis, and mediating intestinal recovery. These results suggest that MSCs induce polarization of resident MSC and immune cells, thereby attenuating CRC development as well as the detrimental side‐effects of radiotherapy on healthy tissues.
Materials and Methods
Isolation and Culture of MSC and CRC Cells
We first used a human coculture model in order to characterize the influence of MSCs on CRC cells. This was followed by in vivo studies to evaluate the influence of bone‐marrow derived MSCs on CRC growth in an immunocompetent rat model (Figs. 1A, 2A, 2B).
Figure 1.
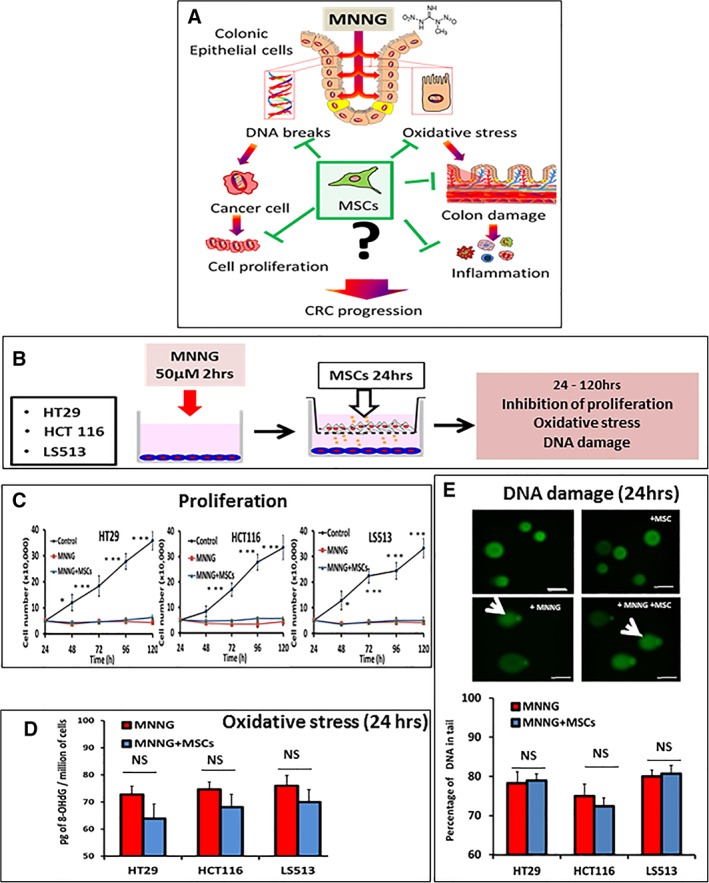
Influence of mesenchymal stem cells (MSCs) on colorectal cancer (CRC) cells. (A): Model illustrating the influence of the alkylating agent methylnitronitrosoguanidine (MNNG) on proliferation, oxidative stress, and DNA damage toward neoplastic and normal CRC cells and tissues. (B): CRC cells were incubated in the presence or absence of MNNG (50 mM) for 2 hours, followed by postincubation in the presence or absence of MSCs in trans‐wells for an additional 24 hours. Then, CRC cells were harvested for determination of adduct formation and DNA damage or, alternatively, postincubated in standard media for 120 hours for determination of proliferation. (C): Proliferation of HT‐29, HCT‐116, and LS513 cells for the indicated times. (D): Presence of 8‐OHdG DNA‐adducts caused by oxidative stress in CRC cells exposed to MNNG in the absence (red columns) or presence (blue columns) of MSCs. (E): DNA damage as determined by single cell electrophoresis (the comet assay). Upper panel, representative images are shown for HT‐29 cells (scale bars = 50 μm). Comet tails are indicated with a white arrow (original magnification ×40). Lower panel, quantitative analysis of fragmented DNA in CRC cells exposed to MNNG in the absence (red columns) or presence (blue columns) of MSCs. The data are representative of three independent experiments and are the mean of triplicate determinations ± SEM. Data represent mean ± SEM; NS, not significant; *, p < .05; **, p < .01; ***, p < .001.
Figure 2.
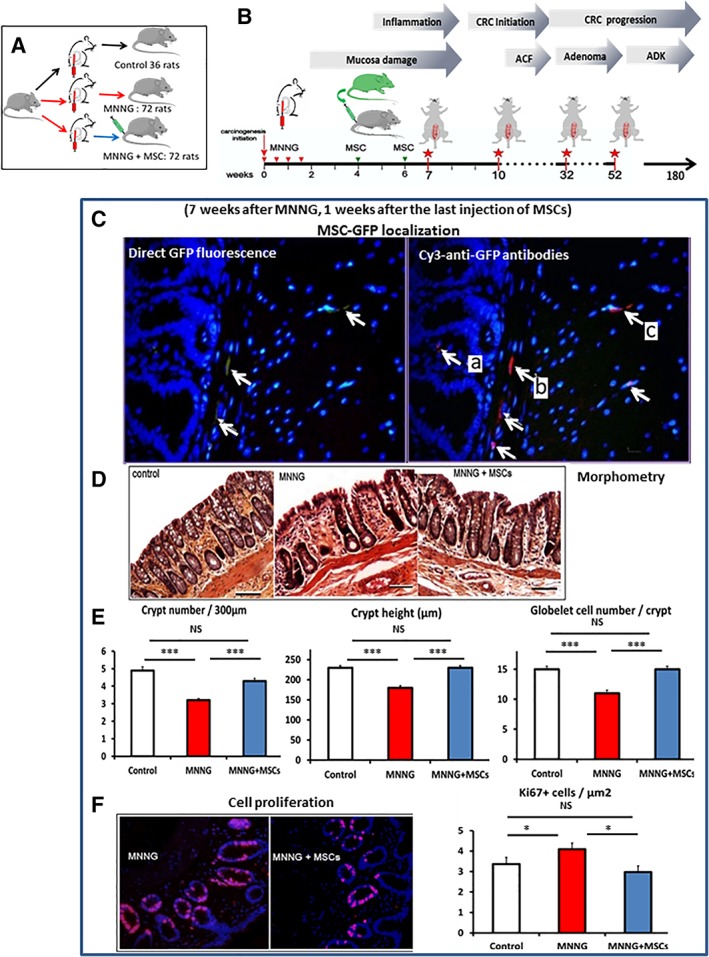
Early effects of methylnitronitrosoguanidine (MNNG) and mesenchymal stem cells (MSCs). (A, B): Protocol used for the animal studies. In a first set of experiment (A, B) Sprague‐Dawley rats (Janvier labs) were divided three groups. The first group was injected with saline solution (control), the second group was injected with MNNG, but not with MSCs, and the third group received MNNG plus MSCs. The study was initiated by local MNNG treatments (two times weekly for 2 weeks) followed by i.v. administration of MSCs (5 million) on weeks 4 and 6. The red stars indicate the times of sampling for data analysis corresponding to 7, 10, 32, and 52 weeks after the start of the experiment. (C): Presence of MSCs in MNNG‐treated colon tissue 1 week after the second injection (7 weeks after MNNG instillation). Exogenous MSCs are detected by direct green fluorescence protein (GFP) fluorescence (left panel, white arrows) or with Cy3‐anti‐GFP antibodies (right panel, white arrows). The presence of MSCs was observed in (A) the lamina propria between the crypts, (B) the muscularis mucosae, and (C) the submucosa. Nuclei are colored by DAPI. Original magnification ×40. (D): Micrographs of the colon mucosa (HE staining) for control animals (left panel) and after treatment with MNNG (middle panel), or with MNNG + MSC (right panel). Original magnification ×10. Bars, 100 μm. (E): Morphometry of the mucosa. Left panel, number of crypts per 300 μm. Middle panel, crypt height. Right panel, number of goblet cells per crypt. White columns, normal mucosa. Red columns, MNNG‐treatment. Blue columns, MNNG + MSC treatment. (F): Proliferating cells as measure by Ki67 immunofluorescence (red) in mucosa treated with MNNG (left) or with MNNG + MSCs (right). Original magnification ×20. Each group of animals was composed of 10 rats Sprague‐Dawley rats and compared using analysis of variance followed by the Mann–Whitney or Dunnett test for group pairwise comparisons. Data represent mean ± SEM; NS, not significant; *, p < .05; **, p < .01; ***, p < .001.
Human CRC cells lines were purchased from European Collection of Cell Cultures (Salisbury, U.K.). MSCs were obtained either from humans or from 7‐week‐old rats. For coculture, CRC cells were incubated with MNNG (50 μM, 2 hours). Then, MSCs and tumor cells were plated in the upper and lower chambers (24 hours), respectively, of trans‐well plates (0.4 mm pore size, Corning) and coincubated for 24 hours followed by collection of cells and culture media. The protocol is further detailed in Figure 1B.
eGFP transgenic rats (strain “green rat CZ‐C04 Tg Act eGFP”) derived from the Sprague‐Dawley strain were obtained by the Institute for Radiological Protection and Nuclear Safety (IRSN) from Pr. Otabe (Osaka University, Osaka, Japan) with Material transfer agreement (MTA) and subsequently bred in the IRSN's animal housing facility. Rat BM cells were isolated from green fluorescence protein (GFP) transgenic Sprague‐Dawley rats via femoral bone aspiration and expanded until the first passage 8. At passage one, cells were detached at 80% confluence and added to 2 ml of phosphate‐buffered saline (PBS) supplemented with 1% bovine serum albumin (Sigma‐Aldrich, St. Louis, MO) and injected intravenously into the tail vein.
To further characterize the alterations observed in vivo in miRNA expression, a coculture model was used (Fig. 3B). CC531 cell line was established from a dimethylhydrazine (DMH)‐induced rat colon ADK. CC531 cell line was plated in the lower chambers of trans‐well plates. Then, MSCs with or without peripheral blood rat mononuclear cells were plated in the upper chambers and coincubated for 24 hours.
Figure 3.
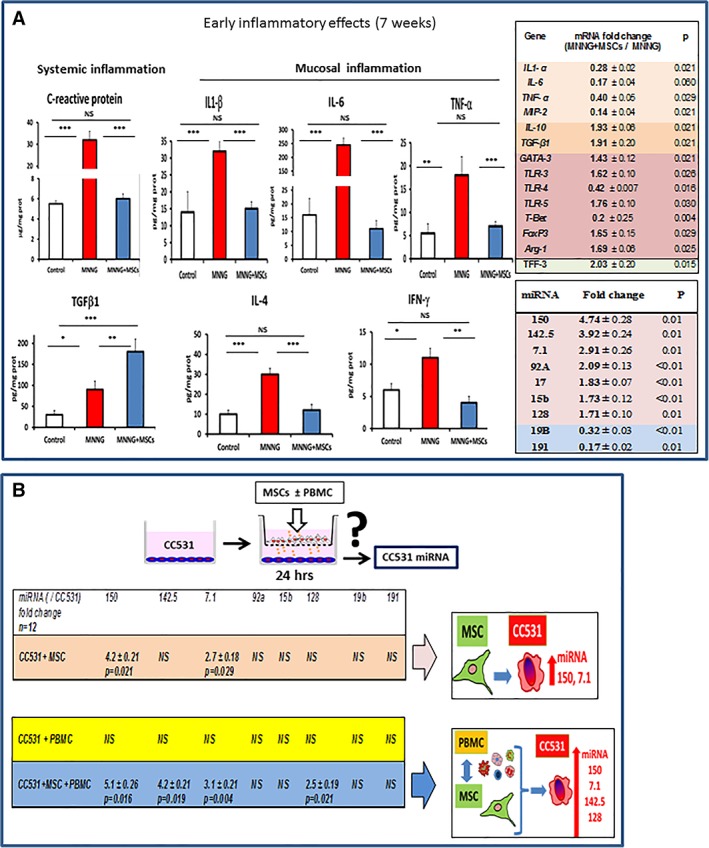
Early inflammatory effects in rats treated with methylnitronitrosoguanidine (MNNG) or with MNNG + mesenchymal stem cells (MSCs). (A): Histograms of serum C‐reactive protein (CRP), an indicator of systemic inflammation, and the local concentrations of IL‐6, IL‐1β, IL‐4, TGFβ, TNF‐1α, and IFNγ proteins 1 week after the second MSC administration. The columns show the protein content in the mucosa of control animals (white columns), after treatment with MNNG (red columns) or with MNNG + MSCs (blue columns). Right panel, expression of mRNA (top) and miRNA (bottom) in colon mucosa in animals treated with MNNG alone or with MNNG + MSCs 1 week after the second MSC administration. The ratio indicates the expression of MNNG + MSCs versus MNNG alone (2−ΔΔCt ± SEM, as determined by the Mann–Whitney test). (B): CC531 colorectal cancer (CRC) cells were incubated with MSCs, with peripheral blood mononuclear cells (PBMC) or with both PBMCs and MSCs in trans‐wells for 24 hours followed by harvesting of the CRC cells for determination of miRNA expression. ***, p < .01. Aberrant crypt foci (ACF), adenoma carcinoma (ADK). Each group of animals was composed of 10 rats Sprague‐Dawley rats and compared using analysis of variance followed by the Mann–Whitney or Dunnett test for group pairwise comparisons. Data represent mean ± SEM; NS, not significant; *, p < .05; **, p < .01; ***, p < .001.
Oxidative Stress
Oxidative DNA damage can trigger tumor initiation. 8‐Hydroxy‐2′‐deoxyguanosine (8‐OHdG) is produced by oxidative damage of DNA by reactive oxygen and nitrogen species, including hydroxyl radical and peroxynitrite. Formation of 8‐OHdG DNA adducts was measured using the Oxidative DNA Damage OxiSelect ELISA Kit (Cell Biolabs Inc., San Diego, CA 92126, USA).
DNA Fragmentation
MNNG is genotoxic toward human CRC cells as shown by single cell electrophoresis under alkaline conditions (the alkaline comet assay). This method permits the detection of DNA strand breaks and alkali‐labile sites in individual cells with the percentage of DNA in the comet tail being proportional to the induction of DNA strand breaks. Genotoxic activity was determined using a comet assay kit (Trevigen 4253‐096‐K). Data are expressed as the percentage of DNA in the tail compared with the total DNA) 9.
Animals
All experiments were performed in compliance with French laws for animal experiments (Act no. 92‐333 of October 2, 2009) and approved by the Ethics Committee of Animal Experimentation “CEEA number 810 (Protocol numbers: P09‐10)”.
In a first set of experiment (Fig. 2A, 2B) Sprague‐Dawley rats (Janvier Labs, Le Genest‐Saint‐Isle, France) were divided into three groups. The first group was injected with a saline solution (control), second group was injected with MNNG, but not with MSCs, and the third group received both MNNG and MSCs.
In a second set of experiments (Fig. 6A) rats were divided into two batches, each with three groups. The first batch was not irradiated while the second batch received fractioned colorectal irradiation for a total dose of 75 Gy. Thirty rats were injected with MNNG, but not with MSCs, and 30 rats received both MNNG and MSCs.
Figure 6.
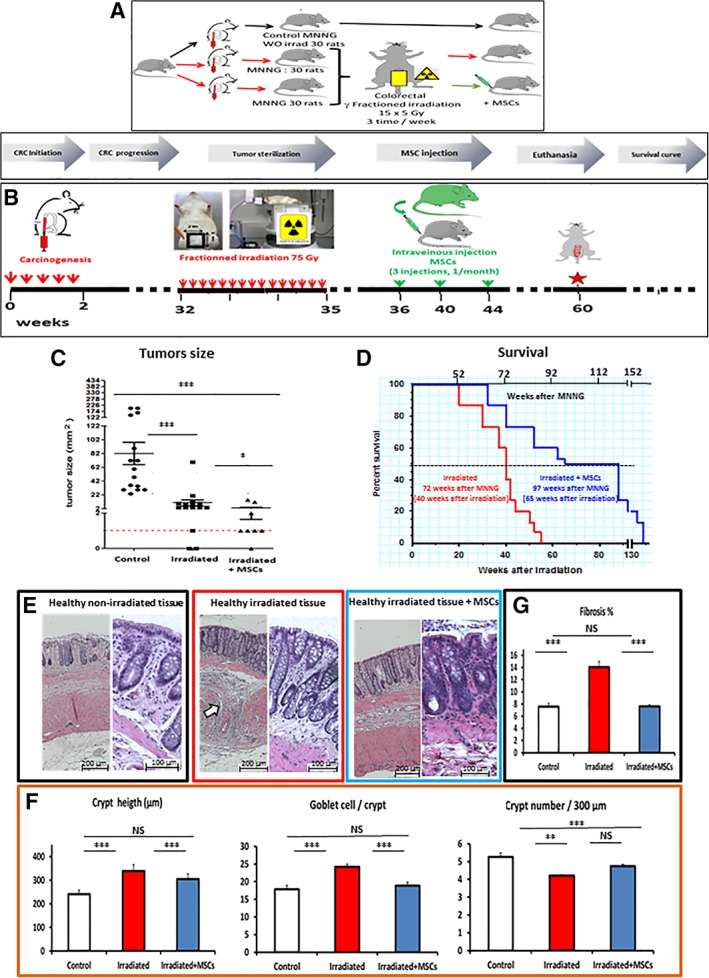
Effects of mesenchymal stem cells (MSCs) administered after fractionated irradiation on tumor size, survival and tissue integrity. (A): Three groups of animals (30 animals/group) were used to determine the influence of MSC administration on tumor size, tissue integrity and overall survival after radiotherapy. (B): Study protocol. the red arrows indicate the various time of fractioned irradiation (15 fractions of 5 Gy each, 3 fractions per week delivered by a 60Co source through a 2 cm × 3 cm window centered on the colorectal region). The green arrows indicate the times of MSCs injection (one injection per month for 3 months, at 36, 40, and 44 weeks after the start of the experiment). The red star indicates the removal of animals (15 animals per group) by 60 weeks for functional and molecular analysis. (C): Tumor size of tumors collected by 60 weeks. (D): Kaplan–Meyer survival curves after irradiation of methylnitronitrosoguanidine (MNNG)‐treated (red line) and MNNG‐treated plus MSC‐treated animals (blue line). (E): Morphometry of the mucosa. Left panels, magnification ×20. Bar 200 μm. Right panels, magnification ×40. Bar 100 μm. The colored boxes illustrate the parameters depicted in (F). White arrows indicate deposition of ECM components. (F): Left, crypt height. Middle, number of goblet cells per crypt. Right, number of crypts per 300 μm. Normal mucosa (white columns), MNNG + irradiation (red columns), MNNG + irradiation + MSCs (blue columns). (G): Scaling of fibrosis. Normal mucosa (white columns), MNNG + irradiation (red columns), MNNG + irradiation + MSCs (blue columns). The number indicates the presence of fibrotic tissue in percentage of total surface. NS, p > .05. Each group of animals was composed of 10 rats Sprague‐Dawley rats and compared using analysis of variance followed by the Mann–Whitney or Dunnett test for group pairwise comparisons. Data represent mean ± SEM; NS, not significant; *, p < .05; **, p < .01; ***, p < .001.
Carcinogenesis and MSC Treatment
MNNG was administered by four intrarectal deposits of 0.5 ml of a 5 mg/ml solution over 2 weeks as described 10. After MNNG treatment, 107 MSCs were injected into the tail vein of anesthetized animals.
The irradiated animals received three intravenous injections of MSCs (one per month) at weeks 36, 40, and 44. Survival, modifications of the mucosa and the development of colonic tumors were determined at 7, 10, 32, and 52 weeks after MNNG treatment and at 60 weeks for the irradiated groups. Tumors collected 52 weeks after MNNG treatment were classified by tumor node metastasis (TNM) after examination at necropsy and by histology.
Radiation and MSC Treatment
Next, we evaluated the influence of MSC treatment on the response to radiotherapy. Thirty weeks after the first MMNG administration, rats were exposed to a γ‐ray source with a radiation (60Co, 1 Gy/minute) field confined to the colorectum (field size: 2 cm × 2.5 cm). Radiation was delivered three times a week in 5‐Gy fractions at a total dose of 75 Gy. One week after the last irradiation, 107 MSCs were injected intravenously into the tail vein of anesthetized animals. The radiation response, animal survival, modification of the mucosa, and the development of colonic tumors were determined 25 days after the last irradiation. The protocol is further detailed in Figure 6B. Fractioned radiation was delivered as previously described 11. One week after the last irradiation, 107 MSCs were injected intravenously. The protocol is further detailed in Figure 6B.
Tissue Sampling and Analysis
Proteins were extracted by tissue disruption in ice‐cold PBS containing protease inhibitors. The protein concentration was assessed using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Villebon‐sur‐Yvette, France). To determine whether MSC administration had an effect on colon cancer development, we used the international TNM classification which is the current clinical standard to estimate the prognosis of CRC patients 12. For this study, an anatomopathological analysis was performed in collaboration with Dr. Betina at the Saint‐Antoine Hospital in Paris, to classify the colon samples collected 52 weeks after carcinogen exposure. To characterize possible tumor regression, the tumor volume (TV) was determined using the World Health Organization criteria as follows. Four intervals were used to describe tumor response: (a) if the tumor regressed below the detection threshold, the response was said to be complete; (b) if the tumor was detectable and the bidimensional measures (height [H] × width [W]), presented a regression of at least 50%, the response was said to be partial, (c) if the regression was between 50% and 25%, it was called stable disease, and finally when growth exceeded 25%, the disease was considered progressive. To evaluate fibrosis, full length longitudinal tissue sections were analyzed. Extracellular matrix (ECM) areas were automatically measured using Histolab software (Microvision Instruments, Lisses, France) to detect Picrosirius stained surfaces (magnification ×40). The percentage of fibrotic area was obtained by dividing the ECM area by the total tissue surface in each field, which was manually measured on Histolab.
Statistical Analysis
Values were expressed as mean ± SEM and compared using analysis of variance followed by the Mann–Whitney or Dunnett test for group pairwise comparisons. Survival curves were compared with the Gehan–Breslow–Wilcoxon test. In the figures, results are expressed as mean ± SEM.
Results
The MSC Secretome Does Not Protect CRC Cells Against Oxidative Stress and DNA Damage After Acute MNNG Exposure
We first determined if MSCs are able to modulate CRC cells directly after acute MNNG exposure. MNNG is a DNA alkylating agent that produces strong oxidative damage followed by DNA strand breaks, genomic instability, inflammation, and proliferation (Fig. 1A) 13. We selected three human CRC cell lines with different defects in DNA damage response and DNA repair. HT‐29 cells show loss of heterozygosity and have mutant p53, HCT‐116 cells have defective mismatch repair while LS513 cells are intrinsic multidrug resistant. The influence of the MSC secretome on CRC cell proliferation (Fig. 1C), oxidative stress (as measured by the presence of oxidative DNA adducts, Fig. 1D), and DNA damage (as measure by DNA strand breaks by the comet assay, Fig. 1E) following MNNG exposure was determined. MNNG exposure was accompanied by the formation of oxidative DNA adducts (Fig. 1D, red columns), induction of DNA strand breaks (Fig. 1E, red columns) and inhibition of proliferation (Fig. 1C, red curves, compared with untreated control cells in black) for the three cell lines (Fig. 1). A similar experiment without MSCs in the upper chamber was used as negative control. In addition, we checked that no MSCs had reached the lower chamber. Coexposure with MSCs decreased the levels of oxidative DNA adducts in a nonsignificant manner (Fig. 1D, blue columns) and had no detectable influence on the levels of DNA strand breaks (Fig. 1E, blue columns) or cell proliferation (Fig. 1C, blue curves). It should be noted, that since a trans‐well assay was used in order to be able to separate CRC and MSC cells following coincubation, the data reflects the influence of the MSC secretome on MNNG‐exposed CRC cells, but not potential heterotypic cell–cell interactions.
Transient Presence of MSCs in the Colon
MSCs were administered twice (5 million by i.v. injection) 4 and 6 weeks after the initiation of MNNG treatment (Fig. 2A, 2B). The first injection was carried out 2 weeks after the last MNNG injection to prevent early damages. The doses of MSCs as well as the repeated injections were chosen according to our previous study which demonstrated a cumulative effect of repeated MSC injections 14, 15, 16. The presence of exogenous MSCs in the colon was verified using an eGFP tag (Fig. 2C, left panel, indicated by white arrows) at 7 weeks after administration. The GFP signal was weak (Fig. 2C, left) and needed amplification in order to permit the visualization of the GFP‐MSCs present in tissue (Fig. 2C, right). Amplification of the signal by use of anti‐GFP antibodies (Fig. 2C, right panel indicated by white arrows) revealed the transient presence of scarce GFP‐labeled cells (indicated in red) localized in (a) the lamina propria, (b) around the bottom of crypts in the muscularis mucosae, and (c) in the submucosa, 1 week after the second MSC injection, but not at later time points. The transient implantation of eGFP‐MSCs was further confirmed by PCR of the eGFP gene in extracts from fixed colon tissue (3.4 ± 1.4 copies of eGFP per 10 ng of DNA, n = 6, data not shown).
MSCs Prevent the Early Damages Induced by MNNG
We decided to study the early effect of MNNG at 7 weeks based on our previous study 10, 12. MNNG induced an alteration of the mucosal structure at 7 weeks after administration (Fig. 2D) as indicated by histological alterations of the mucosa, distortion of the crypts, and immune infiltration (compare controls [left] with MNNG [middle]), which was attenuated by administration of MSCs (right). The number of crypts per micrometre, the height of the crypts as well as the number of goblet cells per crypt were significantly (p < .01) reduced by MNNG and corresponded to 88%, 80%, and 73% of control values, respectively (Fig. 2E, red columns), whereas MSC administration (blue columns) restored the values to the initial levels (white columns). Ki67 expression (in red), reflecting the proliferation of mucosal cells, is shown in Figure 2F (left image, MNNG; right image, MNNG + MSCs). The analysis of labeled cells as a fraction of the total number of epithelial cells is shown in Figure 2F (right). The expression of Ki67 increased significantly (p = .0429) in the MNNG mucosa (4.1 ± 0.2 positive cells/μm2, red column), compared with control animals (3.3 ± 0.2 positive cells/μm2, white column) while MSC administration restored Ki67 expression to normal levels (3.0 ± 0.2 positive cells/μm2, blue column).
MSCs Modulate the Inflammatory Response During the Early Phase of MNNG‐Induced Carcinogenesis
The inflammatory response after MNNG treatment is believed to play a pivotal role in cancer initiation. At the early phase of the experiment (1 week after the second MSC injection), MNNG induced inflammation and mucosal damage, which was attenuated by MSC administration (Figs. 2D–2F, 3A). In addition, MNNG induced a systemic inflammation (p < .001), as evidenced by the serum concentration of C‐reactive protein (CRP) which increased almost sixfold (Fig. 3A, left panel, red column). Locally, the protein levels in the mucosa increased for IL‐6 (17‐fold, p < .001), IL‐1β (2.1‐fold, p < .001), IL‐4 (3.2‐fold, p < .001), TGFβ1 (2‐fold, p = 0,037), TNF‐α (4‐fold, p = .021), and IFNγ (1.8‐fold, p = .030; Fig. 3A, red columns). The presence of MSCs restored the concentration of CRP and mucosal IL‐6, IL‐1β, IL‐4, TNF‐α, and IFNγ to basal levels (Fig. 3A, blue columns compared with control samples, white columns) while TGFβ increased, clearly indicating an anti‐inflammatory activity of the MSCs. These differences were significant (p < .05).
Gene expression analysis of inflammatory modulators present in the mucosa of animals treated with MNNG + MSCs compared with mucosa from animals treated with MNNG alone further confirmed our findings (Fig. 3A, upper table). The mRNA of several proinflammatory genes including IL‐1β (0.3‐fold), IL‐6 (0.2‐fold), TNF‐α (0.4‐fold), and MIP‐2 (0.1‐fold) were significantly downregulated, whereas the mRNA of the anti‐inflammatory genes IL‐10 (1.9‐fold), and TGFβ (1.9‐fold) were upregulated. Immunomodulation of the MSCs was associated with a decrease in the mRNA of T‐Bet (0.2‐fold), TLR4 (0.4‐fold), and an increase in the mRNA of Arginase‐1 (Arg1, 1.7‐fold), GATA3 (1.4‐fold), FoxP3 (1.6‐fold), and TLR5 (1.8‐fold). These differences were significant (p < .05). These findings suggest that the MSCs predominantly stimulated cells of the T cell (GATA3), Treg (IL‐10, FoxP3), and M2 (TNF‐α, Arg1, TLR4) phenotypes. In addition, the increase in the expression of TFF3 (twofold) indicates that the MSCs restored the functionality of the mucosa in agreement with the histological findings.
MicroRNAs are involved in gene regulation and their aberrant expression has been implicated in various pathologies including cancer. To determine miRNA expression early after MNNG treatment, miRNA profiles were analyzed by the Affymetrix GeneChip miRNA Array 2.0. The expression of miRNAs with altered expression was subsequently confirmed by qRT‐PCR. MicroRNA profiling revealed nine microRNAs that were differentially expressed between the mucosa from MNNG and MNNG + MSC animals by 7 weeks, which was confirmed by qRT‐PCR (Fig. 3A). This included seven miRNAs that increased significantly in the mucosa of animals treated with MNNG + MSC, compared with MNNG alone, including miR‐150 (4.7‐fold), miR‐142.5p (3.9‐fold), miR‐7.1 (2.9‐fold), miR‐92A (2.1‐fold), miR‐17 (1.8‐fold), miR‐15b (1.7‐fold), miR‐128 (1.7‐fold), while two miRNAs decreased significantly including (miR‐19b 0.3‐fold), and miR‐191 (0.2‐fold; Fig. 3A, lower table). The increase in IL‐10, miR‐92a, TLR3, and miR‐17 as well as the decrease in TLR4 and IL‐6 transcripts suggest a polarization of resident MSCs into the MSC2 phenotype. These differences were significant (p < .05).
In order to clarify the interaction between MSCs, cancer cells and inflammatory cells, a coculture model was used to characterize the alterations in miRNA expression in the cancer cells without ambiguity (Fig. 3B). As experimental model system, we chose the classic CC531 cell line, originally established from a DMH‐induced rat colon ADK which is a widely used model to study different aspects of tumor growth. A coculture model was used where the CC531 cells were plated in the lower chambers of trans‐well plates. Then, MSCs in the absence or presence of rat peripheral blood mononuclear cells (PBMCs) were plated in the upper chambers and the cells were coincubated for 24 hours. PBMCs were used to test the influence of inflammatory cells on the MSC‐tumor cell crosstalk. The presence of MSCs resulted in increased levels of miR‐150 (4.2‐fold ± 0.2) and miR‐7.1 (2.7‐fold ± 0.2) in CC531 cells compared with CC531 alone. The presence of both MSCs and PBMCs, increased the levels of miR‐150 (5.1‐fold ± 0.3), miR‐142.5 (4.2‐fold ± 0.2), miR‐7.1 (3.1‐fold ± 0.2) and miR‐128 (2.5‐fold ± 0.2) compared with CC531 cells alone. These differences were significant (p < .05). Therefore, the presence of MSCs or MSCs + PMBCs had a significant influence on the expression of miRNA in surrounding CRC cells. Interestingly, the results suggest that the crosstalk between the MSCs and immune cells can influence the tumor cells, since neither MSCs nor PMBCs by themselves had any influence on the expression of miRNA‐142.5 and miR‐128 although their expression was increased when MSCs and PBMCs were together.
MSC Administration Modifies the Phenotype of Immune Cells in the Tumor Microenvironment
One year after the initial MNNG exposure, the tumor microenvironment of animals treated with MNNG alone was very different from that of animals exposed to MNNG + MSCs. In particular, in the tumor, the density of cells expressing the T‐cell marker CD3 was 0.69% ± 0.03% compared with 1.0% ± 0.05% for MNNG and MNNG + MSCs tumors, respectively (p < .001) Therefore, the density of CD3+ cells was 30% higher in the tumors of MSCs‐treated rats (Fig. 4A, upper panels). In contrast, the density of cells expressing the macrophage marker CD68 was 50% lower (p < .001) in tumors from animals treated with MNNG + MSCs (0.8% ± 0.1%) compared with the tumors of animals treated with MNNG alone (1.6% ± 0.1%; Fig. 4B, left panels).
Figure 4.
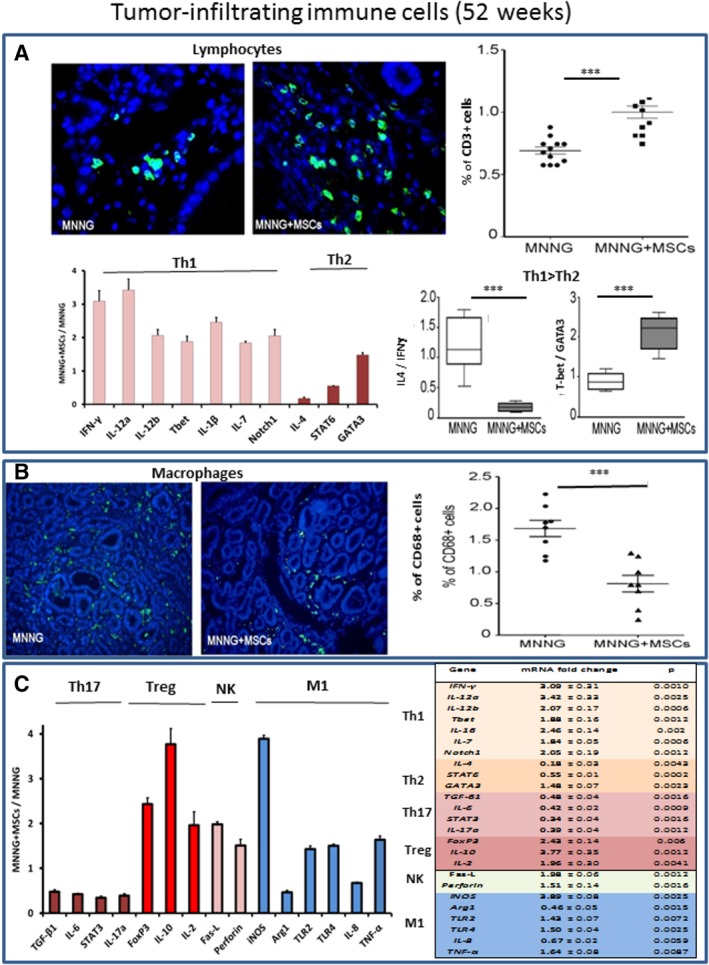
Characteristics of the tumor‐infiltrating immune cells in animals treated with methylnitronitrosoguanidine (MNNG) or with MNNG + mesenchymal stem cells (MSCs) by 52 weeks. (A): Upper panels, CD3‐positive tumor‐infiltrating T lymphocytes (in green, visualization by immunofluorescence, original magnification ×40) in animals treated with MNNG or with MNNG + MSCs. Upper right panel, percentage of CD3‐positive infiltrating cells. Bottom left panel, influence of MSCs on the expression of cytokines and transcription factors as determined by qRT‐PCR. Bottom right panels, the influence of MSC administration on the Th1/Th2 phenotype was determined for animals treated with MNNG alone (white boxes) or with MNNG + MSCs (gray boxes). The initial presence of MSCs reduced the ratios between IL‐4/IFNγ 6.6‐fold (p = .0022) and increased the T‐bet/GATA3 ratio 2.4‐fold (p = .00022). These findings suggest a predominance of the Th1 phenotype in MNNG + MSCs treated tumors. (B): Upper panels, CD68‐positive tumor‐infiltrating macrophages (in green, visualization by immunofluorescence, original magnification ×20) in animals treated with MNNG or with MNNG + MSCs. Upper right panel, percentage of CD68‐positive infiltrating cells in tumors from MNNG treated animals (circles) compared with animals treated with MNNG + MSCs (triangles). (C): Left panel, expression of genes associated with the tumor microenvironment in tumors from MNNG + MSC treated animals compared with animals treated with MNNG alone. Right panel, expression of mRNA in the colon mucosa of animals treated with MNNG or with MNNG + MSCs by 52 weeks. The ratio indicates the expression of MNNG + MSCs versus MNNG (2−ΔΔCt ± SEM). Each group of animals was composed of 10 rats Sprague‐Dawley rats and compared using analysis of variance followed by the Mann–Whitney or Dunnett test for group pairwise comparisons. Data represent mean ± SEM; NS, not significant; *, p < .05; **, p < .01; ***, p < .001.
The expression of cytokines and transcription factors (Fig. 4C, bottom left) showed highly significant differences between tumors from animals treated with MNNG or with MNNG + MSCs compared with nonirradiated animals not exposed to MSCs. The MSC‐mediated increase in mRNA expression between MNNG and MNNG + MSCs tumors was particularly prominent (p < .01) for IFNγ (3.0‐fold ± 0.3), IL‐12a (3.4‐fold ± 0.3), IL‐12b (2.1‐fold ± 0.2), T‐bet (1.9‐fold ± 0.2), IL‐1β (2.5‐fold ± 0.1), IL‐7 (1.8‐fold ± 0.05), and Notch1 (2.1‐fold ± 0.2; Fig. 4A, bottom left). In contrast, MSC administration decreased the mRNA levels for IL‐4 (0.18‐fold ± 0.03) and STAT6 (0.66‐fold ± 0.01), whereas GATA3 levels were slightly increased (1.5‐fold ± 0.07). The ratio between IL‐4 and IFNγ was reduced by a factor of 6.6 (p < .001) while the ratio between T‐bet and GATA3 was increased (p < .001) by a factor of 2.4 (Fig. 4A, lower right). These findings suggest a predominance of the Th1 phenotype in MNNG + MSCs tumors.
The expression of other genes involved in the regulation of the tumor microenvironment is shown in Figure 4C. A comparison of MNNG and MNNG + MSCs tumors reveals that MSC administration was accompanied by a significant decrease (p < .001) in the mRNA levels of TGFβ1 (0.5‐fold ± 0.02), IL6 (0.4‐fold ± 0.02), STAT3 (0.3‐fold ± 0.01) and IL‐17a (0.39‐fold ± 0.04), suggesting a low activity of differentiated Th17 cells. Th17 cells have key functions in the induction of the tissue inflammation and in promoting tumorigenesis. Comparison of genes typical for Treg cells showed that MSC treatment significantly (p < .001) increased the mRNA levels of Foxp3 (2.4‐fold ± 0.1), IL‐10 (3.8‐fold ± 0.4), and IL‐2 (2.0‐fold ± 0.3). The higher levels of IL‐2 in MSC‐treated tumors are coherent with a stimulation of Treg cells. In addition, the expression of M1 macrophage‐related genes including iNOS (3.9‐fold ± 0.08), Arg1 (0.5‐fold ± 0.03), TLR2 (1.4‐fold ± 0.07), and TLR4 (1.6‐fold ± 0.04) increased, whereas the expressions of IL‐8 (0.67‐fold ± 0.02) and TNF‐α (0.64‐fold ± 0.05) decreased (Fig. 4C). The increase in TLR4 and the decrease in TLR3 suggest a polarization of resident MSCs into the MSC2 phenotype.
MSC Administration Limits the Number and the Growth of Colon Tumors Following Carcinogen Treatment
The appearance of the tumors in control animals (without MNNG), in MNNG‐treated animals and in animals treated with MNNG + MSCs by 52 weeks is shown in Figure 5A. Quantitative analysis (Fig. 5B) shows that both the number and frequency of tumors per rat was significantly reduced in animals treated with MSCs at 32 weeks (p = .022) as well as at 52 weeks (p = .0257). In addition, there were significantly (p = .048) more tumor‐free rats in the MNNG + MSC group indicating that the administration of MSCs attenuated tumor initiation thereby decreasing the number of tumors per rat.
Figure 5.
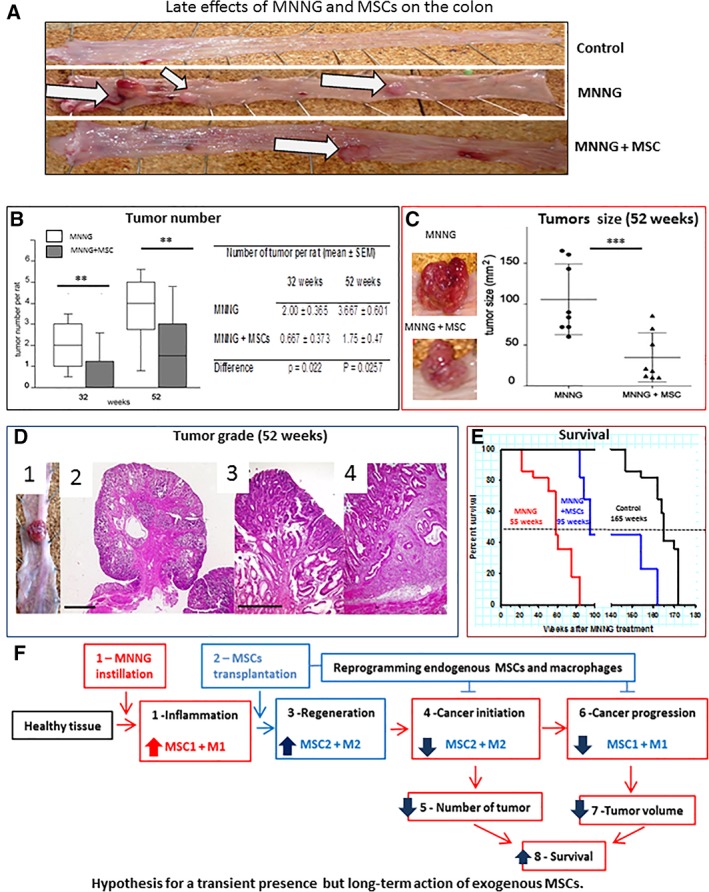
Late effects of methylnitronitrosoguanidine (MNNG) and mesenchymal stem cells (MSCs) on the colon. (A): The appearance of representative colons from untreated control animals and from animals treated with MNNG alone or with MNNG + MSCs by 52 weeks. White arrows, tumor growth. (B): Box plot of the number of tumors by 32 and 52 weeks. White boxes, MNNG alone. Gray boxes, MNNG + MSCs. (C): Analysis of the size of eight representative tumors per group collected by 52 weeks. Circles, MNNG alone; triangles, MNNG + MSCs. (D): (D1, D2): Adenocarcinomas (magnification ×2. Bar = 2,000 μm). (D3): Cancer in situ (magnification ×10). (D4): Adenocarcinoma invading the submucosa (magnification ×10. Bar = 500 μm). (E): Kaplan‐Meyer survival curves of control animals (black line), animals treated with MNNG (red line), or with MNNG + MSCs (blue line). The median survival was 55 weeks for animals treated with MNNG alone, 95 weeks for animals treated with MNNG + MSCs and 165 weeks for control animals. (F): Model for the long‐term effects of exogenous MSCs. (F1): Inflammation is induced in the colon mucosa by MNNG. (F2): Interaction and polarization of resident MSCs and immune cells by the exogenous MSCs. (F3): Polarization of resident MSCs in the colon into MSC2 and of macrophages into M2 will counteract the inflammation and preserve tissue integrity. (F4): Decreased tumor initiation (plain lines) will result in (F5), a lower number of tumors. (F6): The protein and gene expression profile suggests a dominant presence of MSC‐1 and macrophages of a mixed M1/M2 phenotype in the colons of MSC‐treated animals. (F7): Polarization of resident MSCs and immune cells is accompanied by a decreased tumor volume. (F8): The decreased number and size of MNNG‐induced tumors is accompanied by increased life span. Each group of animals was composed of 10 rats Sprague‐Dawley rats and compared using analysis of variance followed by the Mann–Whitney or Dunnett test for group pairwise comparisons. Data represent mean ± SEM; NS, not significant; *, p < .05; **, p < .01; ***, p < .001.
The average size of tumors by 32 weeks was not significantly different for MNNG + MSC animals and animals treated with MNNG alone. However, at 52 weeks, the tumor size was three times less in animals injected with MSCs (34 mm2) compared with animals treated with MNNG alone (106 mm2; p = .0053, Fig. 5C). Similarly, at 52 weeks, the expression of Ki67 mRNA as measured by qRT‐PCR was lower (0.76‐fold ± 0.04) in tumors from the MNNG + MSCs group than for the MNNG group, suggesting a long‐term effect of the MSCs on tumor proliferation.
At 52 weeks, the invasion of tumor cells was limited to the submucosa. In reference to the TNM nomenclature for human tumors, only carcinoma in situ (Tis), Nx (invasion of the regional lymph nodes was not evaluated), M0 (absence of remote metastases), and T1 (tumor less than 2 cm in its largest part) Nx M0 tumors were observed at this stage (Fig. 5D, animal treated with MSCs). In addition, MSC treatment resulted in a nonsignificant decrease in tumor grade.
MSC Administration Significantly Increased the Survival of MNNG‐Treated Animals
The influence of MSC administration on the overall survival is shown in Figure 5D. MNNG treatment significantly (p < .01) reduced the life span of rats (Fig. 5E, red line, median survival 55 weeks), compared with the untreated control animals (black line, 165 weeks). In clear contrast, the injection of MSCs significantly increased (p = .018) the survival of MNNG‐treated animals (blue line, median survival 95 weeks).
MSC Administration Significantly Decreased Tumor Size and Increased Survival in Animals After Fractioned Irradiation
Our experimental protocol was based on a clinical case where patients undergoing radiotherapy for prostate cancer by accident received more than 70 Gy on the rectum. Four patients received repeated intravenous injection of allogenic MSCs resulting in protective effects on proctitis and rectitis 17. One year after radiation, colonoscopy revealed the presence of congested mucosa, telangiectasia and large area of fibrosis. These patients were subsequently treated successfully by intravenous injection of MSCs. A new phase II clinical trial evaluating the efficacy of systemic MSC injections for the treatment of chronic radiotherapy‐induced abdomino‐pelvic complications is scheduled to start this year (ClinicalTrials.gov Identifier: NCT02814864).
To characterize the influence of MSCs on residual tumors after radiotherapy as well as their effect on healthy tissue, we used three groups of MNNG‐treated rats as outlined in Figure 6A. Thirty‐two weeks later, when adenomas had appeared, two groups were irradiated with 75 Gy. The 32 week timepoint was chosen because large carcinomas such as those present at 52 weeks are typically treated with surgery, rather than with radiotherapy, in clinical practice. A dose range of 55–105 Gy was tested and a dose of 75 Gy was retained since higher doses of irradiation strongly increased animal mortality. The dose per fraction (5 Gy) was selected on the basis of irradiator occupancy criteria taking into account the size of the groups of animals. One week after the last irradiation, one group of animals received three intravenous injections of MSCs. At the time fibrosis had developed (26 weeks after irradiation and 60 weeks after the first MNNG treatment), tumors were removed for histological and molecular characterization while the remaining animals were used to determine the influence of the different treatments on the overall survival (Fig. 6B).
Fractionated irradiation reduced the size of the tumors fourfold (20 mm2 ± 2) compared with the nonirradiated control group (82 mm2 ± 20; p < .01). Importantly, MSC administration further decreased the size of the tumors (p = .028) more than twofold (9 mm2 ± 3), compared with irradiation alone (Fig. 6C).
MSC treatment significantly (p < .01) increased the life span of rats after irradiation (Fig. 6D, blue line median survival 65 weeks after irradiation) compared with irradiation alone (red line median survival 40 weeks after irradiation). The results are expressed with two axes the bottom axis represents “weeks after irradiation” while the upper axis corresponds to “weeks after MNNG treatment.”
MSC Administration Decreased Fibrosis and Prevented Tissue Degradation After Fractioned Irradiation
Chronic radiation damage is characterized by an aberrantly repair process associated with chronic inflammation, excessive collagen accumulation, and vascular disorganization resulting in fibrosis, which may lead to organ dysfunction and occlusion 1. Accordingly, fractionated irradiation alone induced an alteration of the mucosal structure as indicated by distortion of the crypts, submucosal edema and immune infiltration and accumulation of components of the ECM (Fig. 6E, compare irradiated tissue [middle panels] with nonirradiated normal tissue [left panels]). These changes were attenuated following MSC exposure (right panels).
Morphometric analysis showed that the height of the crypts per micrometre (348 crypts/μm in average vs. 268 crypts/μm) as well as the number of goblet cells per crypt (24 per crypt vs. 18 per crypt in average) were significantly (p < .001) increased in irradiated colons (Fig. 6F, red columns) compared with nonirradiated controls (white columns) while MSC administration restored basal levels to 288 crypts/μm and the goblet cells to 18 per crypt in average (blue columns). The crypt number per 300 μm was significantly (p < .01) decreased after irradiation (4.1 crypts/300 μm in average, red column) compared with nonirradiated controls (mean 5.2 crypts/300 μm in average, white column) without any detectable effects of the MSCs on the irradiated group (4.2 crypts/300 μm in average).
Fractioned irradiation promoted fibrosis (Fig. 6G, red column, 14% in average) whereas MSC administration significantly decreased fibrosis (blue column, 7.1% in average) to similar levels as the nonirradiated controls (white column, 6.8% in average). Therefore, MSC administration limited fibrosis progression and prevented mucosal damage clearly suggesting an antifibrosis and mucosa‐protective effect of the MSCs against tissue damage induced by fractionated irradiation.
Upregulation of ECM components, ECM degrading enzymes (MMPs: Matrix Metalloproteinases) and their inhibitors (TIMPs: Tissue Inhibitors of Metalloproteinases) are known to cause a defect in ECM turnover inducing fibrosis 18. On the molecular level, fractioned irradiation increased significantly protein levels of TGFβ (2.1‐fold, p < .01) and TIMP1 (2.2‐fold, p < .001; Fig. 7A). MSC treatment also normalized TGFβ compared with control and decreased TIMP1 compared with irradiated (p < .01), but increased the concentration of vascular endothelial growth factor (VEGF) 4.1‐fold (p < .01), compared with basal levels (control), coherent with the influence of MSCs on fibrosis.
Figure 7.
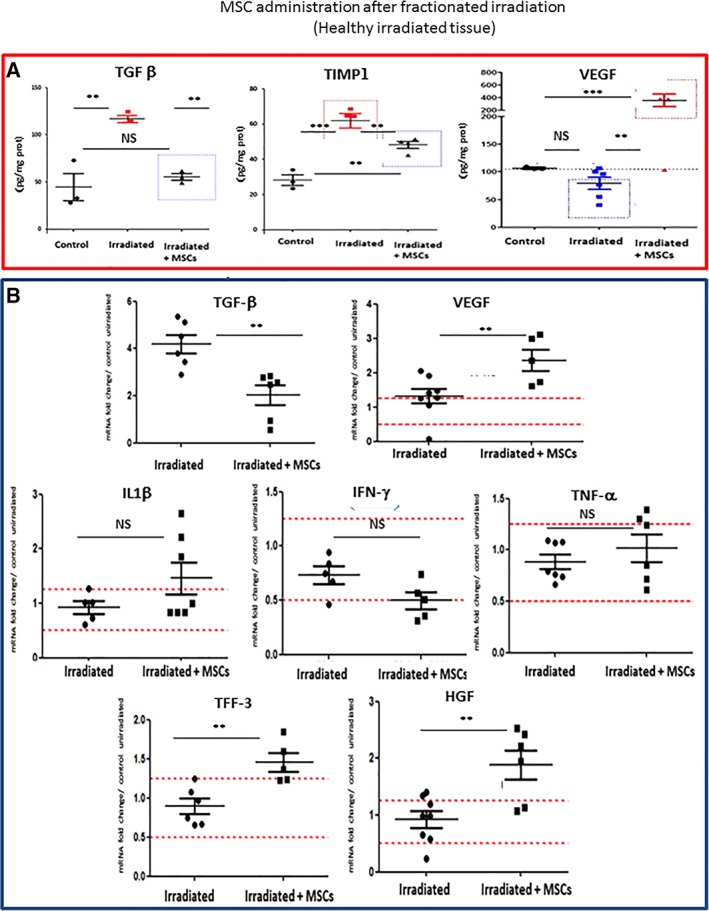
Influence of mesenchymal stem cell (MSC) administration after fractionated irradiation. (A): Protein content of TGFβ, TIMP1, and VEGF in the colon mucosa of control animals and after irradiation followed or not by MSC administration. (B): Expression of inflammatory and regenerative genes in the colon mucosa after irradiation, followed or not by MSC administration. The data illustrates the expression of the indicated genes and are expressed as the ratio of irradiation or irradiation + MSCs, compared with nonirradiated control tissue (2−ΔΔCt ± SEM) as determined by the Mann–Whitney test. **, p < .025. Each group of animals was composed of 10 rats Sprague‐Dawley rats and compared using analysis of variance followed by the Mann–Whitney or Dunnett test for group pairwise comparisons. Data represent mean ± SEM; NS, not significant; *, p < .05; **, p < .01; ***, p < .001.
Gene expression analysis did not show any significant influence of MSC treatment on the expression levels of the proinflammatory genes IL‐1β, IFNγ, and TNF‐α following irradiation (Fig. 7B). In contrast, the expression of TGFβ decreased 1.8‐fold (p < .01) while the expression significantly (p < .01) increased for TFF3 (1.6‐fold), hepatocyte growth factor (HGF; 2‐fold, p < .01), and VEGF (1.6‐fold, p < .01). HGF is a potent antifibrotic protein with pleiotropic activities including immunosuppression and inhibition of TGFβ signaling whereas trefoil factor 3 (TFF3)‐mediates regeneration of an epithelial barrier after mucosal injury 18. These findings indicate that the MSCs restored the functionality of the mucosa while limiting fibrosis progression, in agreement with the histological findings.
Discussion
Radiotherapy is an essential therapeutic option for a wide range of tumor types. However, some patients develop toxic side‐effects due to normal tissue damage. In particular, PRD remains a major problem which is only treated symptomatically 1. The digestive symptoms of PRD are similar to those observed in the so‐called bowel disease as well as for the visceral disorders related to graft versus host disease (GVHD) where administration of MSCs emerge as a promising approach to alleviate the gastrointestinal syndromes 18. Although MSC administration is also able to alleviate tissue damage in irradiated animal models, conflicting data exist concerning their potential influence on residual tumor cells 17. Contradictory results may be explained by the fact that the majority of in vitro studies were carried out in immunodeficient mouse models which are not able to reproduce the immunomodulatory effects of MSCs on resident immune cells. In addition, most studies focused on orthotopic tumor xenografts rather than on endogenous tumor development. We here evaluate the influence of MSCs on tumor growth in an immunocompetent rat model following local exposure to a chemical carcinogen in the presence or absence of fractionated radiotherapy and describe the underlying molecular mechanisms. Specifically, we evaluate the impact of MSCs on tumor development, the inflammatory/immune response and the tumor microenvironment from CRC initiation to its complete development over an entire year. The results show for the first time that intravenous administration of MSCs increase the life span of carcinogen‐exposed rats which display less tumors (tumor initiation) as well as decreased tumor size (tumor progression), indicating that the exogenous MSCs durably inhibited both CRC initiation and progression 19.
To mimic human colon carcinogenesis, which is a long‐term process, we selected an immunocompetent MNNG‐induced CRC rat model. MNNG exposure induced the formation of oxidative DNA adducts and DNA strand breaks. The MNNG model is relevant for human carcinogenesis since rectal administration of MNNG mimic the detrimental action of alimentary nitroso‐compounds 20 that are closely associated with the induction of CRC with a physiopathology similar to that observed in humans 21. Furthermore, intrarectal administration of MNNG act only on the colon mucosa 22 in contrast to carcinogenesis induced by 1,2‐dimethylhydrazine or azoxymethane which also induce tumors in other tissues 23.
The most prominent early effect of MNNG is induction of inflammation which has been closely linked to tumor initiation 12. Inflammation is accompanied by mucosal lesions as evidenced by a decrease in the number of mucus‐secreting cells as well as a decrease in crypt number, crypt height, and the number of goblet mucus‐secreting cells which may result in colon dysfunction. The influence of MSC administration is summarized in Figure 5F. MSCs seem to decrease CRC initiation via the immune system rather than by a direct effect on the tumor cells. Specifically, the MSCs showed only marginal effect on MNNG‐mediated oxidative stress, DNA damage and cell proliferation in isolated CRC cells.
MSCs modulate the MNNG‐induced inflammation, at least in part, by lowering the local IL‐6 concentrations, since IL‐6 is implicated in tumor initiation in inflammatory bowel disease 24. Furthermore, MSC treatment protected the structure of the colon epithelium by reducing the ulcerations caused by MNNG 12, 25. These early effects of MSCs, which counteract the deleterious action of MNNG, are likely associated with TGFβ‐ and/or TFF3‐dependent tissue repair in agreement with the results presented here. Indeed, these two regulatory peptides can stimulate the restitution on the basolateral (TGFβ) and apical (TFF) sides of the colon mucosa via independent pathways 25. Finally, the administration of MSCs restored gut morphology and function in agreement with previous findings 26, 27, 28.
We then characterized the expression of miRNAs that have been implicated in modulating tumor initiation. The microRNA expression profile identified nine microRNAs that were differentially expressed. To clarify the interplay between tumor cells, MSCs and immune cells, an in vitro model was used. The results show that the presence of MSCs increased the levels of miR‐150 and miR‐7.1 in the tumor cells. Interestingly, the joint presence of MSCs and inflammatory cells specifically increased tumor cell expression of miR‐142.5 and miR‐128 pointing to the importance of crosstalk between MSCs and inflammatory cells. The inhibition of MNNG‐induced inflammation by the MSCs was reflected by an increase in miR‐150 and miR‐7.1 that are known to inhibit inflammation in sepsis (miR‐150) and gastritis (miR‐7.1), and a decrease in miR‐19b, that has been associated with increased inflammation in rheumatoid arthritis 29. Similarly, MSCs induced an increase in antioncogenic miRNAs (miRs 150, 142‐5p, 7.1, 15b, 128, and 17) and a decrease in pro‐oncogenic miRNAs (miRs 19b and 191]. Interestingly, miR‐17 is produced by MSC2s and negatively regulates expression of Smurf1 30, a potential oncogene that has been implicated in CRC 31. Taken together, the results indicate that MSCs inhibited tumor initiation by preventing MNNG‐mediated inflammation thereby protecting the colon and decreasing the number of tumors. The inhibition of tumor initiation by MSCs is in agreement with a previous report in a rat model treated with a different carcinogen 19, 32, although the cited study did not include a characterization of local inflammation or epithelial function.
Next, we characterized the long‐term influence of MSCs on the tumor microenvironment, a known participant in tumor development 33 and the composition of tumor‐infiltrating immune cells which may become a prognostic tool 34. Indeed, a comparison of tumors induced by MNNG alone with those developed after treatment with both MNNG and MSCs showed important differences.
By 52 weeks, MNNG‐induced tumors treated with MSCs (MNNG + MSCs) were infiltrated with more CD3+ lymphocytes compared with MNNG‐induced tumors. MSCs directed the lymphocyte response toward a Th1/Treg. The increased expression of IL‐10, TGFβ, Foxp3, and IL‐1β suggests an important Treg population activated by the high level of IL‐2 in the microenvironment. FoxP3+ Tregs have been associated with a better prognosis in CRC while the Th1 response has been shown to be anticarcinogenic in CRC 35. Decrease expression of IL‐17a suggested a decrease of Th17 cell activity. This result is in agreement with Yoshida et al. who suggested that a decreased of Th17 expression may improve the survival of CRC patients 36. Human colon carcinoma tumors infiltrated with more CD3+ cells, presenting a high Th1 adaptive cluster have a better prognosis. Similarly, patients with high levels of T‐Bet and FoxP3 showed better survival while no correlation was observed with respect to the Th2 gene cluster 35.
In comparison, the number of infiltrating CD68+ macrophages decreased in MNNG + MSCs tumors. High levels of TAMs (tumor‐associated macrophages) are generally correlated with a nonfavorable outcome in various cancers including CRC 37. The lower levels of CD68‐positive cells may explain the decreased expression of IL‐8 mRNA (which is implicated in the carcinogenic process) 38. It is generally accepted that macrophages may be activated into M1 (classical) or M2 (alternative) subtypes. A switch from M1 to M2 macrophages has been described during tumor progression 39. We found an increase in iNOs, TLR2, and TLR4 expression in MNNG + MSCs tumors, suggesting a differentiation toward the M1 subtype 40. In agreement, MNNG + MSCs tumors exhibited low levels of STAT6, Arg1, and IL‐4 suggesting a low degree of M2 differentiation 41. Furthermore, the inhibition of tumor growth observed in MNNG + MSCs‐treated animals was associated with an increased expression of perforin (secreted by NK and cytotoxic T cells), FasL, and iNOs (which contribute to tumor cell killing). Expression levels of IL‐6 and STAT3 were low in MNNG + MSCs tumors. A systematic review and meta‐analysis showed that STAT3 may have oncogenic activities 42. In addition, high levels of STAT3 and IL‐6 have been associated with CRC progression 43. Therefore, our results are in full agreement with the current understanding of the IL‐6/STAT3 pathway.
PRD is characterized by severe gastrointestinal disorders. Radiation‐induced colorectal damage involves severe mucosal damage, chronic inflammation and activation of the profibrotic transforming growth factor beta (TGFβ)/Smad pathway, which is a major contributor to the fibrogenic process 44. In this study we selected a rat model of fractionated γ‐irradiation 11 which is considered to be representative of the colonic irradiation damage observed in patients 14. Importantly, administration of MSCs after radiotherapy decreased the size of residual tumors and protected colon integrity, thereby improving animal survival. This model of colorectal irradiation generates histopathological lesions similar to those observed clinically during the severe chronic phase. In this model, MSCs promoted intestinal regeneration after irradiation in agreement with the multiple therapeutic effects of MSCs including stimulation of proliferation, inhibition of apoptosis, stimulation of angiogenesis and a large range of anti‐inflammatory activities 18. Our results highlights a dynamic progression of ECM remodeling with a balance toward fibrogenesis in irradiated tissues which is counteracted by MSC administration coherent with previous findings 15, 16. The regenerative capacity of MSCs was mediated by secretion of HGF and VEGF. HGF and TSG‐6 are key effectors of MSC‐mediated inhibition of fibrosis by suppressing TGFβ signaling and regulating the polarization of macrophages. TGFβ signaling has pleiotropic activities on the immune system, tissue regeneration and fibrosis 44 and can thus play multiple roles during tumor progression dependent on the context. Taken together, these data support the capacity of MSCs to establish a normal microenvironment after irradiation by acting on endogenous tissue cells.
Interestingly, the presence of exogenous MSCs was observed only at early time points after injection. In the tumor microenvironment, administered MSCs interact with resident cells and tumor cells resulting in a complex crosstalk. Tunneling nanotube‐mediated intercellular crosstalk and mitochondria transfer between MSCs, cancer cells and resident immune cells may also modulate tumor response and the inflammatory environment 45. MSCs can polarize into two distinct phenotypes. TLR4‐primed MSCs secrete proinflammatory mediators while TLR3‐primed MSCs express immunosuppressive effectors. Thus, MSCs can be classified in proinflammatory and immunocompetent MSC1 and immunosuppressive MSC2 46. Therefore, the durable antitumor effects of the MSCs observed is most likely explained by “reprogramming” of resident immune cells by the MSCs via epigenetic mechanisms, in agreement with the “hit and run” mechanism suggested by Von Bahr et al. 7.
Our results suggest that at early time points, MSCs promotes an anti‐inflammatory response via polarization of resident MSCs into anti‐inflammatory MSC2s decreasing tumor initiation. At later time points, we observed polarization of resident macrophages into the M1 subtype in agreement with the findings of Eggenhofer et al. 48 who reported that MSCs are able to induce M1 polarization of resident macrophages, thereby inducing a microenvironment unfavorable to tumor progression.
Conclusion
The results presented here show that administration of MSCs increase the life span of carcinogen‐exposed rats by attenuating both CRC initiation and progression. Considering the transient presence of the MSCs this is likely mediated by polarization of resident immune cells which in turn interferes with tumor growth. After fractionated irradiation, MSCs inhibited residual tumor growth, protected healthy tissue and prolonged animal survival. These findings support the use of MSC administration for treatment of severe radiation damage in patients.
Author Contributions
A.C., A.K.L., and M.‐E.F.‐L.: conception, experimental design, data collection, data analysis and interpretation, manuscript writing; B.L.H., B.U., and S.F.: technical support; S.F., B.L.H., and B.U.: animal assistance; S.F. and B.U.: study concept; L.D. and M.B.: manuscript revision; S.F., B.U., M.‐E.F.‐L., B.L.H., M.B., L.D., N.‐C.G., A.K.L., and A.C.: final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
We thank Bettina Fabiani (Hôpital Saint‐Antoine, Paris, France) for tumor analysis, Christian Gespach (Hôpital Saint‐Antoine, Paris, France) for helpful discussions, and Pierre P. Levy (Hôpital Tenon, Paris, France) for fruitful discussions on statistical analysis. We also thank Thi Cam Ha Che, Sandrine Bouchet, Fahd Hached, Djaber Benaoumeur, Yoann Ristic, and Marion Baumann (IRSN, PRP‐HOM, SDE, LDRI, Fontenay, France), GSEA (IRSN, PRP‐HOM, SRBE, GSEA, Fontenay, France), for technical support. This work was supported by IRSN, INSERM, and by a collaborative research grant from Centre de Recherche Saint‐Antoine (CRSA).
Contributor Information
Annette K. Larsen, Email: annette.larsen@sorbonne-universite.fr
Alain Chapel, Email: alain.chapel@irsn.fr.
References
- 1. Andreyev HJ, Wotherspoon A, Denham JW et al. Defining pelvic‐radiation disease for the survivorship era. Lancet Oncol 2010;11:310–312. [DOI] [PubMed] [Google Scholar]
- 2. Benderitter M, Caviggioli F, Chapel A et al. Stem cell therapies for the treatment of radiation‐induced normal tissue side effects. Antioxid Redox Signal 2014;21:338–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lalu MM, McIntyre L, Pugliese C et al. Safety of cell therapy with mesenchymal stromal cells (SafeCell): A systematic review and meta‐analysis of clinical trials. PLoS ONE 2012;7:e47559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee RH, Yoon N, Reneau JC et al. Preactivation of human MSCs with TNF‐α enhances tumor‐suppressive activity. Cell Stem Cell 2012;11:825–835. [DOI] [PubMed] [Google Scholar]
- 5. Zhao S, Wehner R, Bornhauser M et al. Immunomodulatory properties of mesenchymal stromal cells and their therapeutic consequences for immune‐mediated disorders. Stem Cells Dev 2010;19:607–614. [DOI] [PubMed] [Google Scholar]
- 6. Lazennec G, Richmond A. Chemokines and chemokine receptors: New insights into cancer‐related inflammation. Trends Mol Med 2010;16:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Von Bahr L, Batsis I, Moll G et al. Analysis of tissues following mesenchymal stromal cell therapy in humans indicates limited long‐term engraftment and no ectopic tissue formation. Stem Cells 2012;30:1575–1578. [DOI] [PubMed] [Google Scholar]
- 8. Whiteside TL. Exosome and mesenchymal stem cell cross‐talk in the tumor microenvironment. Semin Immunol 2018;35:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. François S, Eder V, Belmokhtar K et al. Synergistic effect of human bone morphogenic protein‐2 and mesenchymal stromal cells on chronic wounds through hypoxia‐inducible factor‐1 α induction. Sci Rep 2018;8:6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Soares DG, Escargueil AE, Poindessous V et al. Replication and homologous recombination repair regulate DNA double‐strand break formation by the antitumor alkylator ecteinascidin 743. Proc Natl Acad Sci USA 2007;104:13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maurin N, Forgue‐Lafitte ME, Levy P et al. Progression of tumors arising from large ACF is associated with the MUC5AC expression during rat colon MNNG carcinogenis. Int J Cancer 2007;120:477–483. [DOI] [PubMed] [Google Scholar]
- 12. Gremy O, Benderitter M, Linard C. Acute and persisting Th2‐like immune response after fractionated colorectal γ‐irradiation. World J Gastroenterol 2008;14:7075–7085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Che TC, Francois S, Bouchet S et al. Early lesions induced in rat colon epithelium by N‐methyl‐N′‐nitro‐N‐nitrosoguanidine. Tissue Cell 2010;42:190–194. [DOI] [PubMed] [Google Scholar]
- 14. Lowe EL, Crother TR, Rabizadeh S et al. Toll‐like receptor 2 signaling protects mice from tumor development in a mouse model of colitis‐induced cancer. PLoS ONE 2010;5:e13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity 2010;32:593–604. [DOI] [PubMed] [Google Scholar]
- 16. Ji K, Zhang M, Chu Q et al. The role of p‐STAT3 as a prognostic and clinicopathological marker in colorectal cancer: A systematic review and meta‐analysis. PLoS ONE 2016;9:e0160125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bessout R, Demarquay C, Moussa L et al. TH17 predominant T‐cell responses in radiation‐induced boweldisease are modulated by treatment with adipose‐derived mesenchymal stromal cells. J Pathol 2015;237:435–446. [DOI] [PubMed] [Google Scholar]
- 18. Koi M, Umar A, Chauhan DP et al. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces W‐methyl‐W‐nitro‐A′‐nitrosoguanidine tolerance in colon tumor cells with homozygous hMLHl mutation. Cancer Res 1994;54:4308–4312. [PubMed] [Google Scholar]
- 19. Bessout R, Sémont A, Demarquay C et al. Mesenchymal stem cell therapy induces glucocorticoid synthesis in colonic mucosa and suppresses radiation‐activated T cells: New insights into MSC immunomodulation. Mucosal Immunol 2014;7:656–669. [DOI] [PubMed] [Google Scholar]
- 20. Linard C, Busson E, Holler V et al. Repeated autologous bone marrow‐derived mesenchymal stem cell injections improve radiation‐induced proctitis in pigs. Stem Cells Translational Medicine 2013;2:916–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Voswinkel J, Francois S, Simon JM et al. Use of mesenchymal stem cells (MSC) in chronic inflammatory fistulizing and fibrotic diseases: A comprehensive review. Clin Rev Allergy Immunol 2013;45:180–192. [DOI] [PubMed] [Google Scholar]
- 22. Moussa L, Usunier B, demarquay C et al. Bowel radiation injury: Complexity of the pathophysiology and promise of cell and tissue engineering. Cell Transplant 2016;25:1723–1746. [DOI] [PubMed] [Google Scholar]
- 23. Katsuno T, Ochi M, Tominaga K et al. Mesenchymal stem cells administered in the early phase of tumorigenesis inhibit colorectal tumor development in rats. J Clin Biochem Nutr 2013;53:170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li SK, Martin A. Mismatch repair and colon cancer: Mechanisms and therapies explored. Trends Mol Med 2016;22:274–289. [DOI] [PubMed] [Google Scholar]
- 25. Narisawa T, Sato T, Hayakawa M et al. Carcinoma of the colon and rectum of rats by rectal infusion of N‐methyl‐N′‐nitro‐N‐nitrosoguanidine. Gan 1971;62:231–234. [PubMed] [Google Scholar]
- 26. Reddy BS, Weisburger JH, Narisawa T. Wynder EL colon carcinogenesis in germ‐free rats with 1,2‐dimethylhydrazine and N‐methyl‐N′‐nitro‐N‐nitrosoguanidine. Cancer Res 1974;34:2368–2372. [PubMed] [Google Scholar]
- 27. Schmahl D, Danisman A, Habs M et al. Experimental investigations on the influence upon the chemical carcinogenesis. III communication: Studies with 1,2‐dimethylhydrazine. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol 1976;86:89–94. [DOI] [PubMed] [Google Scholar]
- 28. Grivennikov S, Karin E, Terzic J et al. IL‐6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis‐associated cancer. Cancer Cell 2009;15:103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sémont A, Demarquay C, Bessout R et al. Mesenchymal stem cell therapy stimulates endogenous host progenitor cells to improve colonic epithelial regeneration. PLoS ONE 2013;8:e70170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sturm A, Dignass AU. Epithelial restitution and wound healing in inflammatory bowel disease. World J Gastroenterol 2008;14:348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sémont A, Francois S, Mouiseddine M et al. Mesenchymal stem cells increase self‐renewal of small intestinal epithelium and accelerate structural recovery after radiation injury. Adv Exp Med Biol 2006;585:19–30. [DOI] [PubMed] [Google Scholar]
- 32. Sémont A, Mouiseddine M, Francois A et al. Mesenchymal stem cells improve small intestinal integrity through regulation of endogenous epithelial cell homeostasis. Cell Death Differ 2010;17:952–961. [DOI] [PubMed] [Google Scholar]
- 33. Gantier MP, Stunden HJ, McCoy CE et al. A miR‐19 regulon that controls NF‐kappaB signaling. Nucleic Acids Res 2012;40:8048–8058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y, Liu W, Hu C et al. MiR‐17 modulates osteogenic differentiation through a coherent feed‐forward loop in mesenchymal stem cells isolated from periodontal ligaments of patients with periodontitis. Stem Cells 2011;29:1804–1816. [DOI] [PubMed] [Google Scholar]
- 35. Xie P, Zhang M, He S et al. The covalent modifier Nedd8 is critical for the activation of Smurf1 ubiquitin ligase in tumorigenesis. Nat Commun 2014;5:3733. [DOI] [PubMed] [Google Scholar]
- 36. Nasuno M, Arimura Y, Nagaishi K et al. Mesenchymal stem cells cancel azoxymethane‐induced tumor initiation. Stem Cells 2014;32:913–925. [DOI] [PubMed] [Google Scholar]
- 37. Bissell MJ, Hines WC. Why don't we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med 2011;17:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tosolini M, Kirilovsky A, Mlecnik B et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 2011;71:1263–1271. [DOI] [PubMed] [Google Scholar]
- 39. Salama P, Phillips M, Grieu F et al. Tumor‐infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 2009;27:186–192. [DOI] [PubMed] [Google Scholar]
- 40. Yoshida N, Kinugasa T, Miyoshi H et al. A High RORγT/CD3 ratio is a strong prognostic factor for postoperative survival in advanced colorectal cancer: Analysis of helper T cell lymphocytes (Th1, Th2, Th17 and regulatory T cells). Ann Surg Oncol 2016;23:919–927. [DOI] [PubMed] [Google Scholar]
- 41. Peddareddigari VG, Wang D, Dubois RN. The tumor microenvironment in colorectal carcinogenesis. Cancer Microenviron 2010;3:149–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Waugh DJ, Wilson C. The interleukin‐8 pathway in cancer. Clin Cancer Res 2008;14:6735–6741. [DOI] [PubMed] [Google Scholar]
- 43. Schmieder A, Michel J, Schonhaar K et al. Differentiation and gene expression profile of tumor‐associated macrophages. Semin Cancer Biol 2012;22:289–297. [DOI] [PubMed] [Google Scholar]
- 44. De Simone V, Franzè E, Ronchetti G et al. Th17‐type cytokines, IL‐6 and TNF‐α synergistically activate STAT3 and NF‐kB to promote colorectal cancer cell growth. Oncogene 2015;34:3493–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Usunier B, Benderitter M, Tamarat R et al. Management of fibrosis: The mesenchymal stromal cells breakthrough. Stem Cells Int 2014;2014:340257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matula Z, Németh A, Lőrincz P et al. The role of extracellular vesicle and tunneling nanotube‐mediated intercellular cross‐talk between mesenchymal stem cells and human peripheral T cells. Stem Cells Dev 2016;25:1818–1832. [DOI] [PubMed] [Google Scholar]
- 47. Waterman RS, Henkle SL, Betancourt AM. Mesenchymal stem cell 1 (MSC1)‐based therapy attenuates tumor growth whereas MSC2‐treatment promotes tumor growth and metastasis. PLoS ONE 2012;7:e45590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eggenhofer E, Luk F, Dahlke MH et al. The life and fate of mesenchymal stem cells. Front Immunol 2014;5:148. [DOI] [PMC free article] [PubMed] [Google Scholar]