

Abstract
Human induced pluripotent stem cells (hiPSCs) are an important tool for research and regenerative medicine, but their efficient cryopreservation remains a major challenge. The current gold standard is slow‐rate freezing of dissociated colonies in suspension, but low recovery rates limit immediate post‐thawing applicability. We tested whether ultrafast cooling by adherent vitrification improves post‐thawing survival in a selection of hiPSCs and small molecule neural precursor cells (smNPCs) from Parkinson's disease and controls. In a dual‐center study, we compared the results by immunocytochemistry (ICC), fluorescence‐activated cell sorting analysis, and RNA‐sequencing (RNA‐seq). Adherent vitrification was achieved in the so‐called TWIST substrate, a device combining cultivation, vitrification, storage, and post‐thawing cultivation. Adherent vitrification resulted in preserved confluency and significantly higher cell numbers, and viability at day 1 after thawing, while results were not significantly different at day 4 after thawing. RNA‐seq and ICC of hiPSCs revealed no change in gene expression and pluripotency markers, indicating that physical damage of slow‐rate freezing disrupts cellular membranes. Scanning electron microscopy showed preserved colony integrity by adherent vitrification. Experiments using smNPCs demonstrated that adherent vitrification is also applicable to neural derivatives of hiPSCs. Our data suggest that, compared to the state‐of‐the‐art slow‐rate freezing in suspension, adherent vitrification is an improved cryopreservation technique for hiPSCs and derivatives. stem cells translational medicine 2019;8:247&259
Significance Statement.
Efficient cryopreservation of human induced pluripotent stem cells (hiPSCs) is a prerequisite for future stem cell‐based research. The current gold standard slow‐rate freezing of dissociated colonies in suspension limit immediate post‐thaw applicability due to low survival rates after thawing. These data suggest that ultra‐fast cooling by adherent vitrification in the TWIST substrate improves direct post‐thaw applicability of hiPSCs and neural derivatives. The TWIST technique provides ready‐to‐use samples and might therefore enable large‐scale vitrification of adherent cell systems. This advanced cryopreservation method can be transferred to current stem cell research and can make large‐scale experiments more efficient and comparable.
Introduction
The isolation of human embryonic stem cells (hESCs) in 1998 and the derivation of human‐induced pluripotent stem cells (hiPSCs) by reprogramming of somatic cells via overexpression of the pluripotency‐specific transcription factors OCT3/4, SOX2, KLF4, and c‐MYC in 2007 have marked the beginning of a new era in translational research. Since then, hiPSCs proved to be a powerful tool for basic scientific research, disease modeling, compound testing, and regenerative medicine 1. One major limiting factor for the experimental reproducibility and translational applicability of hiPSCs is efficient cryopreservation. HiPSCs and hESCs are highly sensitive to freezing and thawing procedures, resulting in spontaneous differentiation, low reattachment, and low recovery rates 2, 3, 4. HiPSCs are typically frozen as dissociated cell clumps with slow cooling rates of approximately −1°C/minute. Resuspension in cryomedia containing various concentrations of cryoprotective agents (CPAs) like 10% dimethylsulfoxide (DMSO) minimizes the freezing damage to the cells 5, 6. However, cryoinjury during slow‐rate freezing due to intracellular/extracellular freezing, toxicity of CPAs, and osmotic imbalances between the intracellular and extracellular environment endanger post‐thawing viability of pluripotent stem cells 6. The disruption of cell–cell contacts renders hiPSCs vulnerable and results in apoptosis or anoikis (detachment‐induced apoptosis) after thawing 4, 7, 8.
It has been shown in hESCs, that loss of E‐cadherin dependent cell–cell adhesion triggers cell death by Rho/Rho‐associated protein kinase (ROCK) and myosin hyperactivation 9. A chemical‐based method to improve the survival of dissociated pluripotent stem cells is the addition of ROCK inhibitor (RI, Y27632) to the medium during the first 24 hours and thereby inhibiting dissociation‐induced cell death 10, 11, 12, 13, 14, 15. However, it has been shown that morphological and metabolic changes can occur in human pluripotent stem cells after RI treatment 16, 17.
In contrast to slow‐rate freezing, vitrification overcomes intracellular and extracellular ice crystallization and thus minimizes osmotic effects. Temporarily high concentrations of CPAs and ultrafast cooling rates by immersion into liquid nitrogen inhibit ice formation and transform the cytoplasm of the cells into a glasslike structure 18. Successful vitrification of pluripotent stem cells was originally performed in straws 2. Thus far, disadvantages of vitrification are the complex handling and freezing procedure, and the highly limited number of cells that can be vitrified at the same time 2, 3. A major advance for vitrification of human pluripotent stem cells was made by further developing a surface‐based approach 19, 20, 21. Adherent vitrification has been successfully shown for hESCs, using a special disposable allowing cultivation, cryopreservation, storage, and post‐thawing cultivation in a single device (Fig. 1A) 20. The ultra‐thin bottom of the so‐called TWIST substrate enables adherent vitrification of pluripotent stem cells without direct contact to the potentially nonsterile liquid nitrogen. Regarding the influence of cryopreservation methods to human pluripotent stem cells on genetic level, only little knowledge has been reported so far 4, 22. Previous data suggest that cell‐line‐specific variations in DNA methylation and gene expression are more prevalent in hiPSCs than in hESCs that might affect reproducibility of experiments 23. The availability of protocols for a minimal or even noninvasive cryopreservation of hiPSCs in their physiological state is a prerequisite for hiPSC‐based research and future application.
Figure 1.
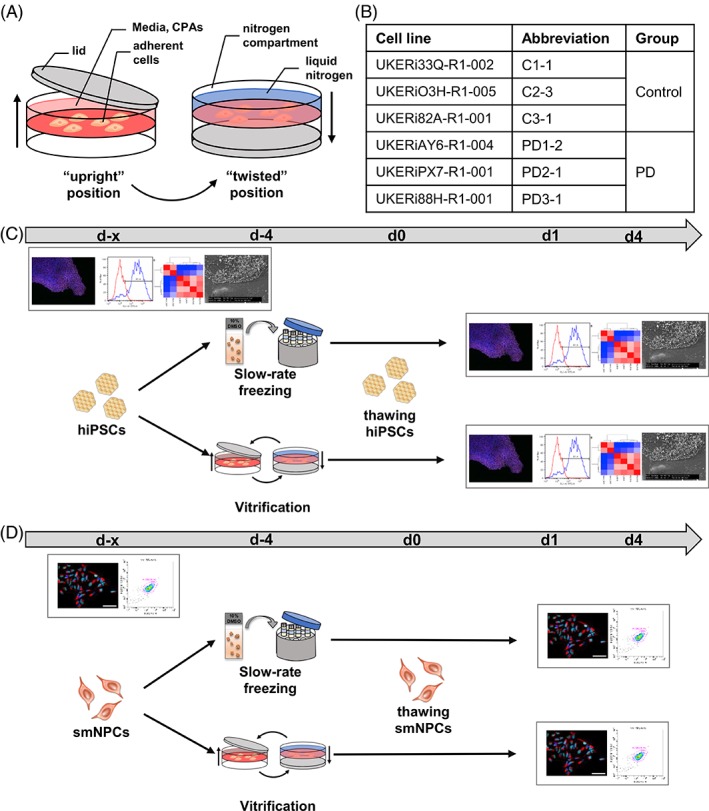
Experimental method and paradigm. (A): Vitrification process. (B): Control and patient cell lines used for all experiments. (C and D): hiPSCs (C) and smNPCs (D) were cryopreserved via slow‐rate freezing in suspension and adherent vitrification in TWIST substrate. Experiments were performed for unfrozen cells and cryopreserved cells 1 day (d1) and 4 days (d4) after rapid thawing using immunocytochemistry, fluorescence‐activated cell sorting analysis, RNA‐sequencing, and scanning electron microscopy. Abbreviations: C, control; CPA, cryoprotective agents; hiPSCs, human‐induced pluripotent stem cells; PD, Parkinson's disease; smNPCs, small molecule neural precursor cells.
The aim of this study was to provide a detailed comparison of adherent vitrification to the standard technique applied in human stem cell research by using a cohort of hiPSCs and neural derivatives in a dual‐center approach. A selection of hiPSCs and derived small molecule neural precursor cells (smNPCs) from healthy individuals and patients with Parkinson's disease (PD) were used (Fig. 1B). We compared unfrozen and cryopreserved cells at day 1 and day 4 after thawing. Our data demonstrate that adherent vitrification is an improved and scalable cryopreservation method for hiPSCs and neural derivatives. Dissociation‐induced cell death is prevented and thus possible clonal selection during freeze–thaw cycles is reduced.
Materials and Methods
Generation and Cultivation of hiPSCs
Human fibroblasts were reprogrammed by retroviral transduction with SOX2, KLF4, c‐MYC, and OCT3/4, and screened for pluripotency and stable karyotype as previously described 24. HiPSCs were cultivated in mTeSR1 medium (STEMCELL Technologies, Vancouver) with 1% Penicillin/Streptavidin (Thermo Fisher Scientific, Waltham, MA) on geltrex‐coated (Gibco® Thermo Fisher Scientific) or Matrigel‐coated (Corning, NY) six‐well plates (Nunc™ Thermo Fisher Scientific). Cells were passaged using Gentle Cell Dissociation Reagent (GCDR) (STEMCELL Technologies) or 0.5 M EDTA (Thermo Fisher Scientific) treatment for 6 minutes at RT or 5 minutes at 37°C, respectively.
Generation and Cultivation of smNPCs
HiPSCs were differentiated into smNPCs as previously described 25. In short, hiPSCs were transferred to ultra‐low attachment six‐well plates (Corning) to allow formation of embryoid bodies using collagenase IV (Gibco® Thermo Fisher Scientific) treatment. For this step, mTeSR1 supplemented with 1 μM LDN, 10 μM SB431542, 3 μM CHIR99021, 0.5 μM purmorphamine (PMA) (all from Tocris, Bristol) was used for 2 days. The medium was then changed to N2B27 medium (50% DMEM/F12, 50% neurobasal medium, 1:200 N2, 1:100 B27 (all from Thermo Fisher Scientific) supplemented with the same small molecules. On day 4, the medium was changed to smNPC medium (N2B27 medium supplemented with 3 μM CHIR99021, 0.5 μM PMA and 150 μM ascorbic acid (Sigma‐Aldrich, St. Louis, MO). Cell colonies were replated on geltrex‐coated 12‐well plates in smNPC medium supplemented with RI Y27532 (Tocris) after two more days of cultivation. SmNPCs were passaged once a week as single cells in a ratio of 1:6 to 1:9 using accutase (Thermo Fisher Scientific).
Cryopreservation Procedure
For slow‐rate freezing, cells were detached using GCDR/EDTA and accutase for hiPSCs and smNPCs, respectively. The cells were resuspended in knockout serum replacement (Thermo Fisher Scientific) with 10% DMSO (Carl Roth, Karlsruhe), transferred to cryovials (Greiner Bio‐One, Frickenhausen), and placed in a freezing container (Nalgene® Mr. Frosty, Thermo Fisher Scientific). Cells were frozen to −80°C overnight and stored for 3 days in the vapor phase of liquid nitrogen. For rapid thawing of the cells, cryovials were immersed in a water bath at 37°C. The cells were replated in cultivation medium supplemented with 10 μM RI for 24 hours.
Cryopreservation via adherent vitrification was achieved using the TWIST technique (Fig. 1A). Prior to vitrification of hiPSCs and smNPCs, the cells were cultivated in the TWIST substrate. Cells were incubated with precooled vitrification solution 1 (VS1) (cultivation medium with 10% DMSO and 10% ethylene glycol [Carl Roth]) at RT for 5 or 1 minute for vitrification of hiPSCs and smNPCs, respectively. VS1 was replaced with precooled vitrification solution 2 (VS2) (cultivation medium with 20% DMSO, 20% ethylene glycol, and 30% 1 M sucrose (Carl Roth) in cultivation medium), and incubated for 5 seconds at RT. After aspiration of VS2, the cultivation compartment was closed with the lid to maintain sterility and twisted. Liquid nitrogen was filled into the nitrogen compartment of the TWIST substrate and the cells were stored in the vapor phase of liquid nitrogen 4 days. The cells were thawed by incubation with 37°C prewarmed warming solution 1 (WS1) (cultivation medium with 20% 1 M sucrose) for 1 minutes at RT. WS1 was replaced with warming solution 2 (WS2) (cultivation medium with 10% 1 M sucrose) and incubated for 5 minutes at RT. After thawing, WS2 was replaced with cultivation medium and the cells were placed in an incubator (37°C, 5% CO2).
Cell Counting and Cell Viability
Cell counting and viability analysis was performed with the LUNA‐FL™ Dual Fluorescence Cell Counter (Logos Biosystems, Anyang‐si) or NucleoCounter NC‐200 (Chemometec, Allerod), following manufacturer's instructions. Viability of cells was measured by acridine orange (AO) and propidium iodide (PI) or 4′,6‐diamidino‐2‐phenylindole (DAPI) staining.
Confluency Analysis
Bright‐field images were taken with an Olympus SZX16 stereomicroscope and analyzed using ImageJ (version 1.51) or NIS‐Elements AR (Nikon, Tokio, version 4.60.00).
Flow Cytometry Analysis
For flow cytometry analysis of hiPSCs, cells were detached using accutase or TrypLE (Thermo Fisher Scientific) treatment (3–5 minutes, 37°C, 5% CO2) and washed with FACS‐PBS (PBS, 2% FCS (Gibco® Thermo Fisher Scientific), 0.01% NaN3 (Sigma‐Aldrich)). For surface staining, cells were incubated with saturating amounts of fluorescently labeled antibodies for 15–30 minutes at 4°C in the dark. Cells were washed and collected in 300–350 μl FACS‐PBS.
For flow cytometry of smNPCs, cells were detached using accutase treatment (5 minutes, 37°C, 5% CO2). 1 × 106 cells were fixed with BD Cytofix/Cytoperm (BD Biosciences, San Jose, CA) for 20 minutes at RT and permeabilized with 0.3% Triton X‐100 (Sigma‐Aldrich) in PBS for 5 minutes at RT. The cells were incubated with saturating amounts of fluorescently labeled antibodies for 15 minutes at RT in the dark. Afterwards, the cells collected in BD PermWash buffer.
Flow cytometry was performed using Cytoflex (Beckman Coulter, Brea, CA) or BD FACS Aria III (BD Biosciences) and analyzed using FlowJo software (LLC) and CytExpert (Beckman Coulter) or BD FACS Diva Software (BD Biosciences).
The following antibodies were used for flow cytometry: anti‐Tra‐1‐60‐FITC (#330614, BioLegend, San Diego, CA), anti‐Sox2‐PE (#560291, BD Biosciences), anti‐Nestin‐PerCP‐CY5.5 (#561231, BD Biosciences), anti‐PE‐Mouse‐IgG2a isotype control (#554648, BD Biosciences), anti‐PerCP‐Cy5.5‐Mouse‐IgG1 isotype control (#550795, BD Biosciences), and anti‐Mouse‐IgM isotype control (#401617, BioLegend).
Immunocytochemistry and Image Analysis
HiPSCs and smNPCs were fixed with 4% PFA (Carl Roth) or BD Cytofix (BD Biosciences) for 10 minutes at RT or 30 minutes at 4°C. Fixed cells were blocked for 1 hour at RT. Primary antibodies were diluted in blocking solution (1% bovine serum albumin and 0.2% Tween in PBS, Sigma‐Aldrich) and incubated overnight at 4°C. The next day, cells were incubated with fluorescently labeled secondary antibodies for 1 hour at RT, followed by an incubation with DAPI (10 μg/ml, Thermo Fisher Scientific) for 1 minute at RT to the stain the nuclei. Stained cells were mounted on microscope slides using 10 μl of mounting solution (Aqua‐Poly/Mount, Polysciences, Warrington, PA). After drying overnight at RT in the dark, the object glasses were stored at 4°C in the dark. Images were taken with Axio Observer.Z1 fluorescence microscope (Carl Zeiss, Oberkochen) or Leica DMI6000 (Leica, Wetzlar) and analyzed with respective software or ImageJ software (version 1.51).
The following primary antibodies were used: anti‐Nanog (#AF2729, R&D Systems, Minneapolis, MN, 1:300), anti‐Oct3/4 (#Sc‐5,279, Santa Cruz, Santa Cruz, CA, 1:300), anti‐Sox2 (#D6D9, Cell Signaling, Danvers, MA, 1:200), and anti‐Nestin (#Sc‐21,247, Santa Cruz, 1:300). The following secondary antibodies were used: anti‐goat 546 (#A11056, Life Technology, Carlsbad, CA, 1:700), anti‐rabbit 488 (#A21206, Life Technology, 1:700), and anti‐mouse 546 (#A10036, Life Technology, 1:700).
Scanning Electron Microscopy
The samples were washed in PBS and fixed with 2% glutaraldehyde on 0.1 M cacodylate buffer (both Agar Scientific, Essex, UK). The following preparation steps were performed as previously described 26. The postfixation implemented in consecutive steps composed of incubation in 2% osmium tetroxide (Carl Roth), 1% tannic acid (Sigma‐Aldrich), and 1% uranyl acetate (Ted Pella, Redding, CA) solutions. Subsequently, dehydration in increased ethanol series and drying in hexamethyldisilazane (Sigma‐Aldrich) ensued according to Katsen‐Globa et al. 27. The prepared samples were investigated at a 5 kV accelerating voltage and work distance of 10 mm in Philips FESEM XL30 (FEI, Eindhoven, Netherlands).
RNA Sequencing
Libraries for next‐generation sequencing were prepared with total RNA isolated from unfrozen and thawed hiPSCs using the TruSeq Stranded mRNA kit (Illumina, San Diego, CA) according to the manufacturer's instructions. Quality control was performed with a Bioanalyzer® system (Agilent, Santa Clara, CA). Samples were sequenced at 100 bp paired‐end at a depth of >40,000,000 reads. After sequencing with the HiSeq 2,500 sequencer (Illumina), samples files were converted to .fastq files.
FastQ files were aligned to the human genome (hg19) using STAR under previously published conditions 28. Aligned sam‐files were annotated with gencode.v19 and features counted using SubReads featureCounts. DESeq2 was used to determine differential expression. Stringent criteria were set to determine significantly dysregulated genes: adjusted p value below .05 and log2 fold change (log2FC) of greater than one. To reduce false positives due to high variability of lowly expressed transcripts, only genes with a mean expression value of greater than one reads per kilobase per million mapped reads (RPKM) throughout the dataset were considered. Hierarchical clustering was generated using the seaborn package in python. Principal component analysis (PCA) plots were performed in R using DESeq2.
Statistical Analysis
The results of this study were obtained from three hiPSC lines of PD patients and three hiPSC lines of controls unless stated differently. Three independent experiments were performed with each hiPSC line. All statistical analyses were conducted with Prism 5 (GraphPad Software, La Jolla, CA). Significance level was assumed at p value <.05. Differences between two groups were analyzed by one‐way‐ANOVA followed by Bonferroni post hoc test. When more than two groups were compared, differences were analyzed with two‐way ANOVA followed by Sidak's post hoc test.
Results
Adherent Vitrification Preserves Confluency, Cell Numbers, and Cell Viability of hiPSCs
For adherent vitrification, cells were cultivated and incubated with CPAs prior to vitrification in the “upright” position of the device and vitrified in the “twisted” position by filling liquid nitrogen into the nitrogen compartment (Fig. 1A). To compare the efficiency of adherent vitrification of hiPSCs in the TWIST substrate to conventional slow‐rate freezing, six hiPSC and smNPC lines were used. Respective fibroblasts were previously reprogrammed from controls and patients suffering from PD (Fig. 1B) 29. HiPSCs and smNPCs were cryopreserved via slow‐rate freezing in suspension and adherent vitrification in the TWIST substrate and analyzed after thawing (Fig. 1C, 1D). Rapid thawing was applied for both freezing methods as previously described 19, 20. Experiments were performed for unfrozen control cells and cryopreserved cells 1 day (d1) and 4 days (d4) after thawing using ICC, FACS analysis, RNA‐Seq, and scanning electron microscopy (SEM) (Fig. 1C, 1D). The experimental design was identical at centers 1 and 2, except for few variations (Supporting Information Tables S1 and S2).
First, we assessed the cryopreservation efficiency of adherent vitrification for hiPSCs and therefore analyzed the confluency in the TWIST substrates (Fig. 2A). We used bright‐field images of the TWIST cultivation surface, allowing tracing of the identical hiPSC colonies before and at d1 after vitrification (Fig. 2B). Quantification revealed preserved confluency of hiPSCs after adherent vitrification on the TWIST substrate (mean confluency before freezing 63.3% ± 7.2% vs. d1 after freezing 57.8% ± 4.4%) (Fig. 2C). No differences between experimental centers and control/disease hiPSC lines were detectable, indicating reproducibility of adherent vitrification on the TWIST substrate. In a second step, we compared the absolute cell numbers and cell viability of slow‐rate frozen and vitrified hiPSCs at d1 after thawing to unfrozen cells of the identical cell lines (Fig. 2D–2G). Slow‐rate freezing in suspension resulted in a significant cell loss of average 69% ± 7% (Fig. 2D), whereas adherent vitrification in the TWIST substrate did not reduce absolute cell numbers (Fig. 2E). Furthermore, the viability of thawed hiPSCs was reduced at d1 after slow‐rate freezing in suspension, as measured by AO for total population and PI or DAPI for nonviable cells (viability before freezing 81.0% ± 4.5% vs. d1 after thawing 67.7% ± 6.2%) (Fig. 2F). In contrast, the viability of vitrified hiPSCs was even increased after thawing to 93.14% ± 2.3% (Fig. 2G). The total recovery of hiPSCs was 26% ± 13% and 110% ± 24% after slow‐rate freezing and adherent vitrification, respectively. In summary, adherent vitrification in the TWIST substrate resulted in preserved confluency, cell numbers, a higher cell viability, and higher cell recovery compared to conventional slow‐rate freezing.
Figure 2.
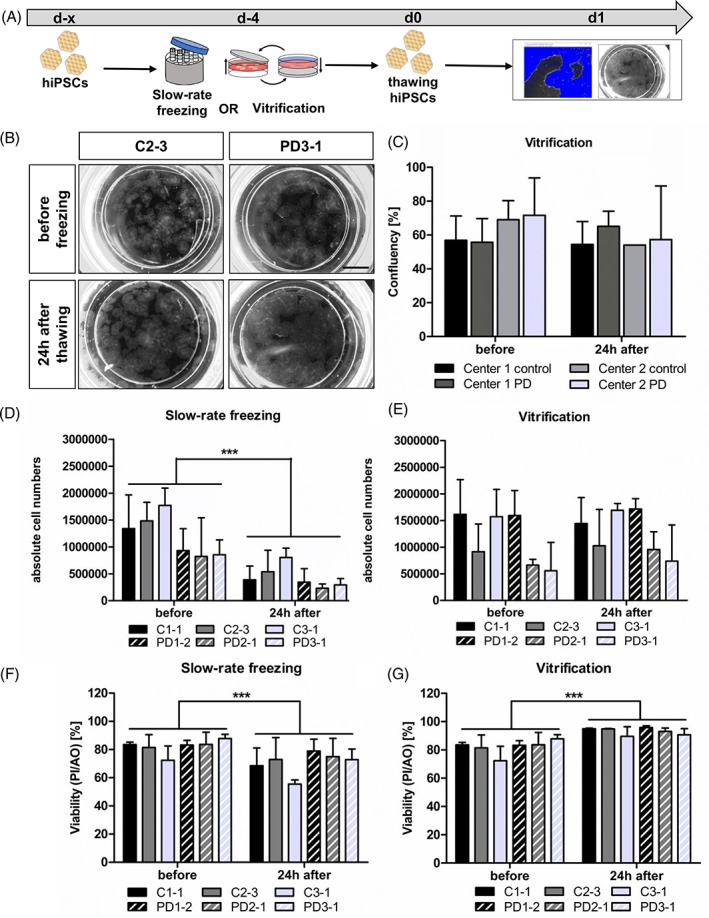
Preservation of confluency, cell numbers, and cell viability by adherent vitrification of hiPSCs. (A): Experimental paradigm. (B): Representative bright‐field images of C and PD hiPSCs in TWIST substrate before and after vitrification. Scale bar 5 mm. (C): Quantification of hiPSC confluency before and after adherent vitrification showed no significant changes. Results are shown as mean ± SD (n = 4 for disease/control group). Statistical analysis: two‐way ANOVA followed by Bonferroni post hoc test. (D–G): Absolute cell numbers and viability of hiPSCs were determined before and after cryopreservation via slow‐rate freezing in suspension (D, F) and adherent vitrification (E, G), as measured by PI and AO staining. Slow‐rate freezing resulted in decreased cell numbers and viability 24 hours after thawing (D). adherent vitrification had no effect on the absolute cell numbers and cell viability (E,G). Results are shown as mean ± SD. Three independent experiments were performed for each hiPSC line (n = 3 per control/disease group). **p value < .005, ***p value < .0001 by two‐way ANOVA followed by Bonferroni post hoc test. see also Supporting Information Figure S1. Abbreviations: AO, acridine orange; C, control; hiPSCs, human‐induced pluripotent stem cells; PD, Parkinson's disease; PI, propidium iodide.
Furthermore, we analyzed the influence of RI on thawing efficiency after slow‐rate freezing and adherent vitrification (Supporting Information Fig. S1). When we thawed slow‐rate frozen hiPSCs without supplementing RI to the medium for the first 24 hours, we detected almost no surviving hiPSCs at d1. Conversely, addition of RI to vitrified hiPSCs had no additional effect on surviving colonies after thawing. This experiment shows that RI is crucial for slow‐rate frozen hiPSCs, but redundant for thawing of hiPSCs after adherent vitrification.
Reduction of Tra‐1‐60‐Positive hiPSCs after Slow‐Rate Freezing in Suspension
Next, we examined the effect of cryopreservation on the pluripotency state of hiPSCs (Fig. 3A). ICC for the pluripotency markers NANOG (green) and OCT3/4 (red) revealed strong expression of both markers in the majority of cells before and at d1/ d4 after cryopreservation for both freezing methods (Fig. 3B and Supporting Information Fig. S2). All groups showed typical compact morphology of pluripotent stem cell colonies with clear borders and no indications for differentiating cells, whereas slow‐rate frozen hiPSCs at d1 were missing borders and the cells were scattered throughout the cultivation dish (Fig. 3B). Next, we performed FACS analysis for the surface protein and pluripotent stem cell marker Tra‐1‐60 (Fig. 3C). At d1 after slow‐rate freezing in suspension, Tra‐1‐60‐positive hiPSCs were significantly decreased (before freezing 86.0% ± 3.4% vs. d1 70.0% ± 3.4% vs. d4 86.8% ± 5.1%). Experiments at center 1 and center 2 were comparable for the decrease of Tra‐1‐60‐positive hiPSCs. There were no significant differences between disease and control cell lines. After 4 days of cultivation, the percentage of Tra‐1‐60‐positive hiPSCs was not different from the level of unfrozen cells any more (Fig. 3D). Adherent vitrification of hiPSCs on the TWIST substrate had no effect on the percentage of Tra‐1‐60‐positive hiPSCs at d1 and d4 after thawing (before freezing 86.0% ± 3.4% vs. d1 87.23% ± 1.3% vs. d4 88.0% ± 4.3%) (Fig. 3E). These results show that the expression of the intracellular pluripotency markers NANOG and OCT3/4 is unchanged in all groups after cryopreservation, indicative for preserved pluripotency. The decreased number of the cell surface marker Tra‐1‐60 might be attributed to loosened colony morphology or shifts in membrane at d1 after thawing, indicative for the impairment of cell–cell contacts at d1.
Figure 3.
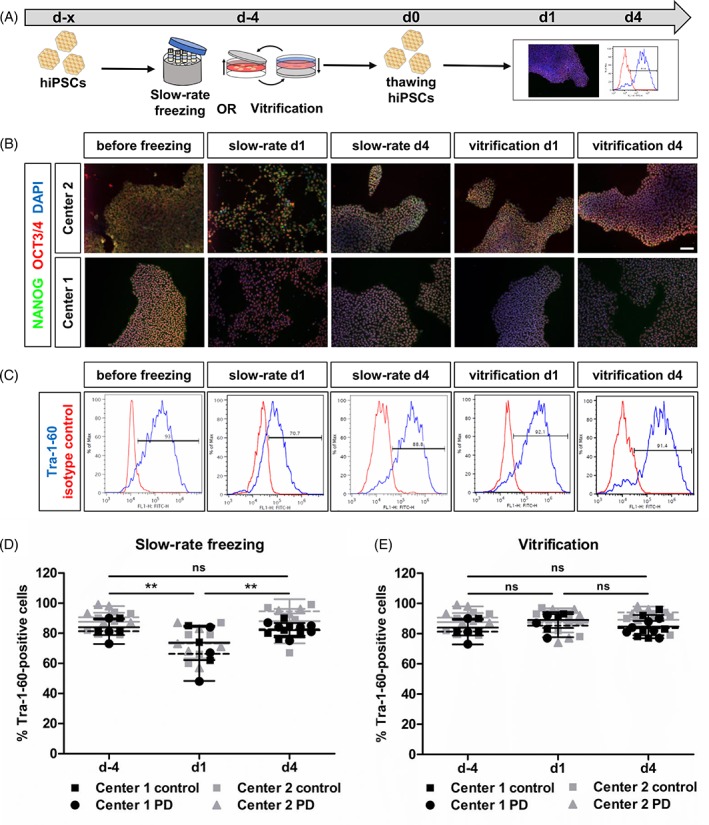
Reduction of Tra‐1‐60‐positive hiPSCs after slow‐rate freezing in suspension. (A): Experimental paradigm. (B): Representative ICC images of pluripotency markers OCT3/4 (red) and NANOG (green) staining before and after cryopreservation. Scale bar 100 μm. See also Supporting Information Figure S2. (C): Representative FACS plots for Tra‐1‐60 expression in hiPSCs before and after freezing. (D and E): Quantification of Tra‐1‐60 FACS analysis of hiPSCs after slow‐rate freezing in suspension (D) and adherent vitrification (E). The percentage of Tra‐1‐60‐positive hiPSCs was significantly decreased after slow‐rate freezing (d1). Adherent vitrification of hiPSCs had no influence on the percentage of Tra‐1‐60‐positive cells. Results are shown as mean ± SD (n = 4 per control/disease group). **p < .0001 by two‐way ANOVA followed by Bonferroni post hoc test. Abbreviations: hiPSCs, human‐induced pluripotent stem cells; ICC, immunocytochemistry; PD, Parkinson's disease.
Unchanged Gene Expression of hiPSCs after Cryopreservation
To elucidate whether the different cryopreservation technologies induce global and long‐lasting transcriptome changes, we performed RNA‐seq of hiPSCs prior to freezing and at days 1 and 4 after thawing (Fig. 4A). Hierarchical clustering of Pearson correlation of the whole expressed transcriptome (mean RPKM > 16,560 genes) clustered the samples according to the hiPSC line. However, the different cryopreservation techniques did not play a significant role (Fig. 4B). This finding was validated by using PCA. Two clusters were identified that diverge in a group with the hiPSC lines C1‐1 and C3‐1, and a second one containing PD1‐1 samples (Fig. 4D). No difference due to cryopreservation was observed (Fig. 4C). This diversification might be due to the different sex of the donors (C1‐1 and C3‐1 are female while PD1‐2 is male). To determine if single gene expression levels are altered, we performed differential expression analysis using DESeq2 and compared each time point and freezing method to the cells prior to freezing. Even this sensitive method revealed no differences in gene expression using our stringency thresholds (adjusted p value <.05, log2 fold change >1, and mean RPKM >1). These unexpected results suggest that cryopreservation of hiPSCs—independent of the used method—does not change the stem cell transcriptome.
Figure 4.
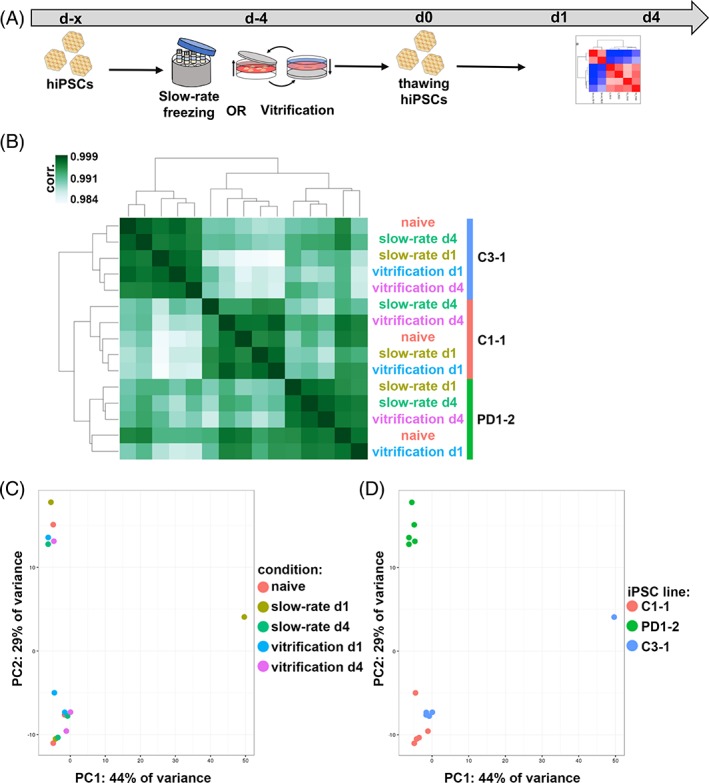
Cryopreservation does not alter global transcriptome of hiPSCs. (A): Experimental paradigm. (B): Hierarchical clustering of Pearson correlation coefficients of transcripts with mean reads per kilobase per million > 1. (C and D): Transcriptome‐wide PC analysis highlighting experimental conditions (C) and cell lines (D). Abbreviations: hiPSCs, human‐induced pluripotent stem cells; PC, principal component; PD, Parkinson's disease.
Preserved hiPSC Colony Integrity by Adherent Vitrification
Since we observed a significant decrease of Tra‐1‐60‐positive hiPSCs after slow‐rate freezing but no differences in global transcriptome and expression of pluripotency markers NANOG and OCT3/4, we analyzed whether physical damage during freezing results in decreased number of the surface marker Tra‐1‐60. Therefore, we performed SEM of hiPSC colonies before and after cryopreservation via slow‐rate freezing and adherent vitrification (Fig. 5A and Supporting Information Figure S3). Prior to freezing, hiPSC colonies showed intact cell–cell contacts (Fig. 5B and Supporting Information Fig. S3A: D2/4, arrows) with the cell surface either smooth or covered with short microvilli located at the cell borders (Supporting Information Fig. S3A: B2/4, C2/4, D2/4). Small cells (5‐7 μm in diameter) (Supporting Information Fig. S3A, B1/3) or cell fragments with intact (Supporting Information Fig. S3A: B1/3, asterisks) or damaged cell membrane (Supporting Information Fig. S3A: B/D1 and B/D3, double asterisks) were only observed at the edges of hiPSC colonies (Supporting Information Fig. S3A: A1/3, dash‐dots quadrats). Strikingly, at day 1 after cryopreservation, adherent vitrification on the TWIST substrate resulted in intact hiPSC colonies comparable to unfrozen hiPSCs (Fig. 5B and Supporting Information Fig. S3B: A3‐4). Cells exhibiting numerous long microvilli were observed after adherent vitrification (Fig. 5B and Supporting Information Fig. S3B: B3‐4, C3‐4, D3‐4). In contrast, at day 1 after slow‐rate freezing, only small hiPSC colonies and enormous amounts of damaged cell material and artifacts with the extracellular matrix (ECM) coating were observed (Supporting Information Fig. S3B: A1‐2). Moreover, large holes in the plasmatic membrane were visible after slow‐rate freezing (Fig. 5B; S3B: C1‐2, D1‐2, arrows). At day 4 after thawing, large intact colonies with numerous microvilli were observed for both freezing methods (Fig. 5B and Supporting Information Fig. S3C: A2‐4, D1‐4). Small hiPSC colonies of C2‐3 are most likely the result of a pipetting or centrifugation error (Supporting Information Fig. S3C: A1). Furthermore, round cells with or without damaged membrane were detected for both freezing methods (Supporting Information Fig. S3C: B2‐4, C1‐2, undamaged marked with asterisks, damaged marked with double asterisks). Gel‐like formations were detected, indicating artifacts of damaged cells and disturbances with the underlying ECM that was used for coating and were disrupted during sample preparation for electron microscopy. Due to their electrical charge, they appear brighter in the images (Supporting Information Fig. S3C: B1, quadrats). In summary, SEM analysis of hiPSC colonies before and after cryopreservation revealed preservation of intact cell–cell contacts by adherent vitrification.
Figure 5.
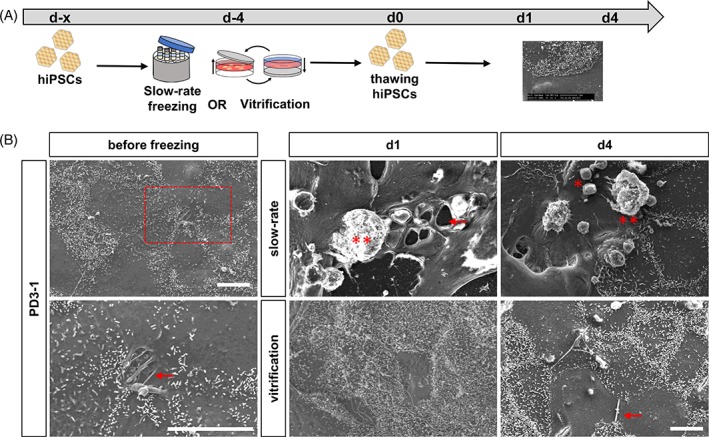
Scanning electron microscopy (SEM) revealed preservation of colony integrity of hiPSCs by adherent vitrification. (A): Experimental paradigm. (B): Representative SEM images of hiPSCs before and after cryopreservation via slow‐rate freezing and vitrification. Prior to freezing intact cell–cell contacts were visible (arrows) and hiPSCs displayed numerous microvilli. After thawing of slow‐rate frozen hiPSCs, large holes within colonies (arrow) and damaged cells were detected (asterisks). Adherent vitrification preserved cell–cell adhesions (arrows) and hiPSCs were covered with numerous microvilli. Scale bars 10 μm. See also Supporting Information Figure S3. Abbreviations: hiPSCs, human‐induced pluripotent stem cells; PD, Parkinson's disease.
Preserved Cell Numbers and Cell Viability of smNPCs by Adherent Vitrification
To examine if adherent vitrification in the TWIST substrate is also feasible for neural precursor cells, we vitrified smNPCs. The smNPCs were generated using an adapted version of a published protocol for a dual‐SMAD‐based generation of midbrain neurons from hiPSCs (Fig. 6A) 25, 29. The cryopreservation experiments were designed using the same groups and freezing conditions as for the hiPSCs (Fig. 1D). First, we analyzed if absolute cell numbers and viability of smNPCs are affected by slow‐rate freezing in suspension or adherent vitrification, in comparison to unfrozen cells of the identical cell lines (Fig. 6B). Slow‐rate freezing of smNPCs resulted in a significant cell loss of average 51% after thawing (Fig. 6C), whereas after vitrification the absolute numbers of plated smNPCs was not reduced (Fig. 6D). Furthermore, the viability of smNPCs was decreased after slow‐rate freezing (before freezing 84.3% ± 8.8% vs. d1 after thawing 66.4% ± 18.7%) (Fig. 6E). Adherent vitrification in the TWIST substrate had no influence on the viability of smNPCs (before freezing 84.3% ± 8.8% vs. d1 after thawing 81.0% ± 5.2%) (Fig. 6F). The total recovery of smNPCs was 41% ± 23% and 142% ± 45% after slow‐rate freezing and adherent vitrification, respectively. In summary, our data indicate that adherent vitrification is applicable for neural derivatives of hiPSCs.
Figure 6.
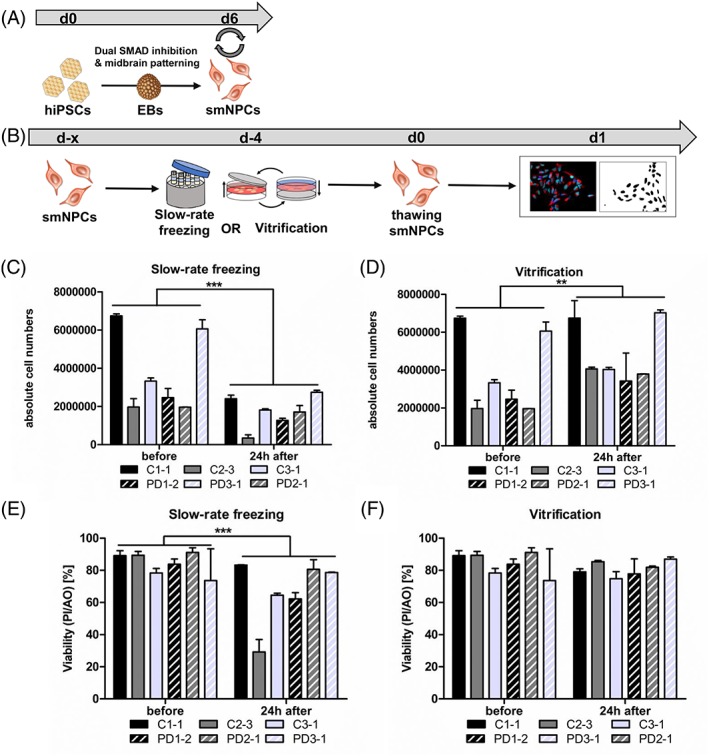
Adherent vitrification preserves cell numbers and cell viability of smNPCs. (A): Scheme of midbrain differentiation of hiPSCs into smNPCs via EBs intermediate state. (B): Experimental paradigm. (C–F): Absolute cell numbers and viability of smNPCs were determined before and after cryopreservation via slow‐rate freezing in suspension (C, E) and adherent vitrification (D, F), as measured by PI and AO staining. Slow‐rate freezing resulted in significant cell loss and reduced cell viability (C, E). Adherent vitrification preserved absolute cell numbers and viability of smNPCs. Results are shown as mean ± SD. Two independent experiments were performed for each smNPC line (n = 3 per control/disease line). **p value = .005, ***p value < .0001 by two‐way ANOVA followed by Bonferroni post hoc test. Abbreviations: AO, acridine orange; C, control; EBs, embryoid bodies; hiPSCs, human‐induced pluripotent stem cells; PD, Parkinson's disease; PI, propidium iodide; smNPCs, small molecule neural precursor cells.
Unchanged Neural Identity of smNPCs after Cryopreservation
For further characterization of smNPCs after cryopreservation, we performed ICC and FACS analysis for respective neural markers (Fig. 7A). Staining for the neural identity markers SOX2 (green) and NESTIN (red) revealed strong expression in the majority of cells before and after cryopreservation via slow‐rate freezing and vitrification. All groups displayed colony‐like morphology, while slow‐rate frozen smNPCs at d1 after thawing were dispersed throughout the cultivation surface (Fig. 7B). Moreover, we performed FACS analysis for the neural identity markers SOX2 and NESTIN (Fig. 7C). Quantification revealed no significant change in the percentage of double positive smNPCs after slow‐rate freezing in suspension (before freezing 89.0% ± 6.1% vs. d1 81.5% ± 6.4% vs. d4 85.7% ± 6.5%) (Fig. 7D) and adherent vitrification on the TWIST substrate (before freezing 89.0% ± 6.1% vs. d1 84.6% ± 7.5% vs. d4 79.5% ± 5.4%) (Fig. 7E). Taken together, both cryopreservation methods preserve the neural identity of smNPCs.
Figure 7.
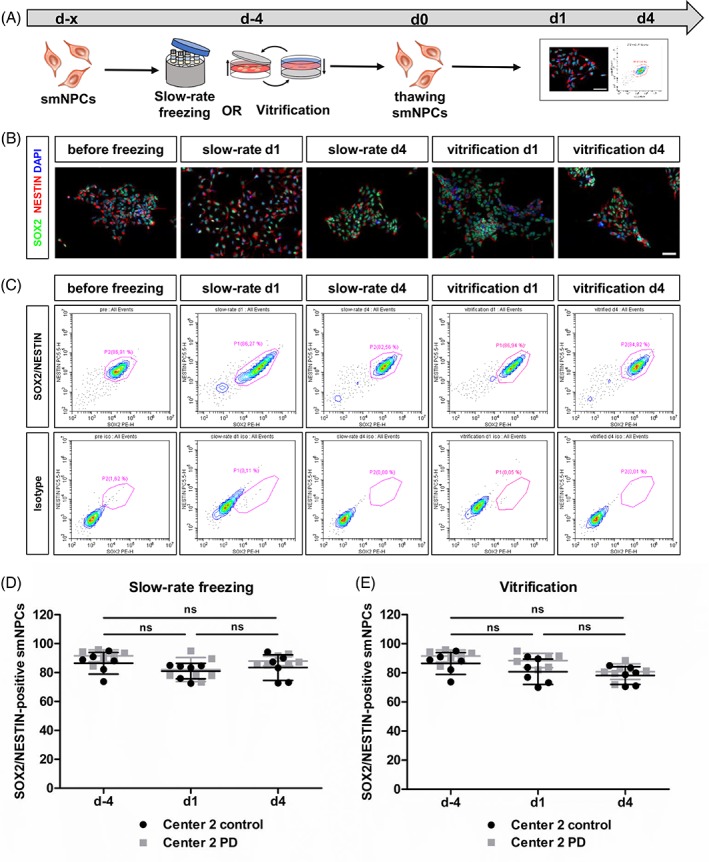
Unchanged neural identity of smNPCs after cryopreservation.(A): Experimental paradigm. (B): Representative immunocytochemistry images of neural identity markers SOX2 (green) and NESTIN (red) staining before and after cryopreservation. Scale bar 50 μm. (C): Representative FACS plots for SOX2/NESTIN expression of smNPCs before and after cryopreservation. (D and E): Quantification of SOX2/NESTIN FACS analysis of smNPCs after slow‐rate freezing in suspension (D) and adherent vitrification (E) revealed no effect of both cryopreservation methods on the number of SOX2/NESTIN‐positive smNPCs. Results are shown as mean ± SD. Two independent experiments were performed for each smNPC line (n = 3 per control/disease group). Statistical analysis: one‐way ANOVA followed by Bonferroni post hoc test. Abbreviations: PD, Parkinson's disease; smNPCs, small molecule neural precursor cells.
Discussion
A prerequisite of hiPSC‐based research is the availability of high‐quality human pluripotent stem cells. Efficient cryopreservation of hiPSCs and derivatives can impact reproducibility, robustness, and simplicity of this application. This dual‐center study compared a new cryopreservation approach (adherent vitrification) for hiPSCs and neural derivatives of control and PD human cell lines to the standard procedure. Adherent vitrification of hiPSCs and smNPCs in the TWIST substrate resulted in higher confluency, cell numbers, and viability compared to conventional slow‐rate freezing of dissociated colonies in suspension. Moreover, cell–cell adhesions of hiPSC colonies were preserved by adherent vitrification. The global transcriptomic profile of hiPSCs was unchanged upon cryopreservation for both freezing methods.
With our adherent vitrification approach, we could retain confluent hiPSC growth in the TWIST substrate immediately after thawing. Moreover, absolute cell numbers of hiPSCs and smNPCs were preserved after adherent vitrification compared to conventional slow‐rate freezing. Cell viability 24 hours after thawing was significantly higher for vitrified hiPSCs and smNPCs than for slow‐rate frozen cells, where we observed a viability loss of average 13% and 20% for hiPSCs and smNPCs, respectively. Conventional slow‐rate freezing in suspension is the current gold standard for cryopreservation of large quantities of hiPSCs and neural derivatives. Our data confirm previously published data of low survival rates of human pluripotent stem cells 2, 3, 4, 30, 31. For instance, Wagh et al. reported a decreased colony area of hESCs after slow‐rate freezing and the lowest cell viability 24 hours after thawing. In our study the timepoint of 24 hours after thawing, at which hiPSCs showed morphological changes, revealed no changes in the transcriptome of hiPSCs using RNA‐seq. The study of Wagh et al. found differences in hESCs at different timepoints after cryopreservation using microarrays in one single cell line 4. As our study was performed with various lines and replicates, our study identified that there are no reproducible changes when looking at a larger number of lines. Moreover, Wagh et al. showed that the hESCs were more similar to unfrozen stem cells 24 hours after thawing than at the other timepoints. Interestingly, a single‐cell RNA‐seq based study found also no transcriptomic changes of different cryopreserved cell types and primary tissues 32. This outcome differs from previous RNA‐seq studies of fresh and frozen cells and provides new insight for future stem cell‐based studies.
Furthermore, human pluripotent stem cells are highly sensitive to the disruption of cell–cell/cell‐matrix adhesions and colony dissociation 9, 33, 34. The method of choice to inhibit cell death after slow‐rate freezing is currently the interference of the Rho/ROCK signaling pathway 11, 14, 35. However, it has been shown recently that the RI Y‐27632 alters the metabolism of hiPSCs by reducing glycolysis, glutaminolysis, and the citric acid cycle 17. Hence, one of the major advantages of adherent vitrification is that ROCK inhibition is not necessary. By avoiding cell detachment prior to cryopreservation, we maintain confluency on the cultivation surface, contributing to higher survival rates, and avoid possible side effects of RI on hiPSCs, for example, actin bundling and disruption of colony formation 36.
Previous studies showed that various cell adhesion molecules regulate self‐renewal, pluripotency, and survival of pluripotent stem cells 37, 38, 39, 40. Even though current protocols for pluripotent stem cells avoid complete dissociation by detaching, passaging, and freezing cells as clumps, conventional slow‐rate freezing in suspension can lead to cell death of dissociated cells 8. After adherent vitrification on the TWIST substrate, we observed preserved compact colony morphology, intact cell–cell‐adhesions, and numerous microvilli indicating induction of proliferation shortly after thawing. In contrast, SEM revealed major cryoinjury of hiPSC colonies after slow‐rate freezing in suspension. Strikingly, the number of Tra‐1‐60‐exhibiting hiPSCs was significantly reduced at d1 after slow‐rate freezing and unchanged after adherent vitrification. Tra‐1‐60, classically used as a pluripotency marker, is located on the membrane of hiPSCs and hence more susceptible to physical damage occurring during freezing procedure. Our results indicate that adherent vitrification might prevent cryoinjury of hiPSCs and neural derivatives, maybe by reducing osmotic stress or ice crystallization and therefore protects cell–cell/cell‐matrix adhesions during the freezing process as it has been postulated, for example, for bone marrow cells 41. For example, disruption of cell–cell adhesions mediated by the epithelial cadherin (E‐cadherin) might induce Rho/ROCK hyperactivation and trigger detachment‐induced cell death after slow‐rate freezing, as previously described 9, 34, 42. Thus, by avoiding cell detachment prior to cryopreservation, the adherent vitrification approach maintains the physical integrity of the hiPSC membrane and results in high cell survival.
In this study, we used standard containers with controlled cooling rates of approximately −1°C/minute for unadherent colonies, as detachment of colonies to either single cells or clumps is still a standard procedure, commonly used in academic research institutes and operating biobanks for stock keeping (e.g. HipSci, WiCell). However, the standard slow‐freezing method consists of additional steps that will influence the cells: the effect of detachment, of freezing, and the formation of extracellular ice. The procedure of vitrification prevents ice formation but puts osmotic damages at risk due to necessary high concentrations of CPAs. While there are fundamental differences in both approaches, our comparative study compared adherent vitrification to the state of the art technique. Our data show that adherent vitrification exceeds by far the recovery of hiPSCs and neural derivatives in all six of the tested lines and provides ready‐to‐use cells immediately after thawing.
While slow‐rate freezing in suspension is still the method of choice for biobanking and large‐scale experiments, adherent vitrification in the TWIST substrates offers a sterile vitrification of adherent cells, highly compatible with clinical‐grade cell lines. Additionally, vitrified cells are ready‐to‐use for therapeutic application immediately after thawing, for example, differentiated cells for transplantation on demand. In this study, 35 mm cell culture dishes (TWIST) were used. This size is not yet ready for large‐scale biobanking. But further development of large‐scale, society for biomolecular screening‐compliant formats like 24‐ or even 96‐well plates, mapping the underlying principles that are compatible with automated cell processing, can realize the transfer of the TWIST technique to large‐scale cell managing and biobanking. We suggest that the adherent vitrification approach can be transferred to any adherent cell system to make large‐scale experiments more efficient and comparable. Avoiding time‐consuming seeding and differentiation processes after thawing makes the TWIST technique also applicable for neural derivatives and potentially neurons.
Conclusion
The findings of our dual‐center study suggest that compared to standard methods adherent vitrification is an advanced cryopreservation method for hiPSCs and neural derivatives. Adherent vitrification in the TWIST substrate resulted in higher absolute cell numbers and viability of hiPSCs and smNPCs at day 1 after thawing. Nonetheless, we observed no changes in expression of pluripotency markers and global transcriptome after both freezing methods. Using SEM, we could show that the slow‐rate freezing procedure possibly damages cellular membranes of hiPSCs, whereas adherent vitrification in the TWIST substrate preserved cell–cell and cell‐matrix adhesions.
Author Contributions
J.K.: collection and/or assembly of data, data analysis and interpretation, manuscript writing—original draft, final approval of manuscript; I.M., conception and design, data analysis and interpretation, manuscript writing—review & editing, final approval of manuscript; J.M., collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; A.S.: conception and design, collection and/or assembly of data, data analysis and interpretation, final approval of manuscript; F.K.: data analysis and interpretation, final approval of manuscript; A.K.: collection and/or assembly of data, data analysis and interpretation; J.W: provision of study material or patients, financial support, final approval of manuscript; H.Z.: conception and design, financial support, final approval of manuscript; J.N.: conception and design, financial support, final approval of manuscript; B.W.: conception and design, financial support, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supplemental Figure 1: Rho‐associated protein kinase (ROCK) inhibitor (RI) is redundant after cryopreservation of human induced pluripotent stem cells (hiPSCs) via adherent vitrification.
HiPSCs were cryopreserved via slow‐rate freezing and vitrification and thawed with or without RI. We observed that RI inhibitor is crucial for thawing of slow‐rate frozen hiPSCs, but redundant for thawing after freezing via adherent vitrification. Scale bars 500 μm. Related to Figure 2.
Supplemental Figure 2: Strong expression of pluripotency markers in human induced pluripotent stem cells (hiPSCs) before and after cryopreservation.
Respective immunocytochemistry (ICC) images of pluripotency markers OCT3/4 (red) and NANOG (green) staining for both freezing methods. ICC revealed strong expression of both markers in the majority of the cells. Scale bar 100 μm. Related to Figure 3.
Supplemental Figure 3: Scanning electron microscopy (SEM) revealed preservation of cell–cell contacts of human induced pluripotent stem cells (hiPSCs) by adherent vitrification.
(A) SEM images of hiPSCs before cryopreservation. Cells within colonies displayed numerous microvilli and intact cell–cell adhesions (regions of interest, arrows). Round and damaged cells were only detected at colony borders (asterisks). (B) SEM images at day 1 after thawing. Slow‐rate frozen hiPSC colonies were decreased in size (regions of interest), showed large holes and disruption of colony integrity (arrows). Round cells with undamaged and damaged membrane were detected (asterisks and double asterisks, respectively). Adherent vitrification maintained large hiPSC colonies. Cells were covered with numerous microvilli (regions of interest). (C) SEM images at day 4 after thawing. Slow‐rate frozen hiPSCs increased in size, displayed microvilli and few round detached or damaged cells were detected (asterisks). Artifacts of the extracellular matrix (ECM) coating were visible. Vitrified hiPSCs showed intact cell–cell adhesions and only few round detached and damaged cells (asterisks). Related to Figure 5.
Appendix S1: Supporting Information Table 1
Appendix S2: Supporting Information Table 2
Acknowledgments
This work has been funded by the German Federal Ministry of Education and Research BMBF (grant 01EK1609A) and by the Bavarian Ministry of Economic Affairs, Energy and Technology (grant PROMIB). J.K. is an associated member of Research Training Grant GRK2162 of the Deutsche Forschungsgemeinschaft. Additional support came from the Bavarian Ministry of Education and Culture, Science and the Arts in the framework of the Bavarian Research Network Induced Pluripotent Stem Cells for ForIPS, the German Federal Ministry of Education and Research (BMBF: 01GQ113, 01GM1520A), the DFG funded research training group GRK2162 (B.W., J.W.) and DRG grant 410/45‐1 FUGG, and the Interdisciplinary Centre for Clinical Research (University Hospital of Erlangen E25). F.K. received funding from the Bavarian California Technology Center (BaCaTec).
Contributor Information
Julia C. Neubauer, Email: julia.neubauer@ibmt.fraunhofer.de
Beate Winner, Email: beate.winner@fau.de.
References
- 1. Singh VK, Kalsan M, Kumar N et al. Induced pluripotent stem cells: Applications in regenerative medicine, disease modeling, and drug discovery. Front Cell Dev Biol 2015;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Reubinoff BE, Pera MF, Vajta G et al. Effective cryopreservation of human embryonic stem cells by the open pulled straw vitrification method. Hum Reprod 2001;16:2187–2194. [DOI] [PubMed] [Google Scholar]
- 3. Richards M, Fong C‐Y, Tan S et al. An efficient and safe xeno‐free cryopreservation method for the storage of human embryonic stem cells. Stem Cells 2004;22:779–789. [DOI] [PubMed] [Google Scholar]
- 4. Wagh V, Meganathan K, Jagtap S et al. Effects of cryopreservation on the transcriptome of human embryonic stem cells after thawing and culturing. Stem Cell Rev Rep 2011;7:506–517. [DOI] [PubMed] [Google Scholar]
- 5. Cohen RI, Thompson ML, Schryver B et al. Standardized cryopreservation of pluripotent stem cells. Curr Protoc Stem Cell Biol 2014;28:1C.14.1–1C.14.10. [DOI] [PubMed] [Google Scholar]
- 6. Heng BC, Kuleshova LL, Bested SM et al. The cryopreservation of human embryonic stem cells. Biotechnol Appl Biochem 2005;41:97. [DOI] [PubMed] [Google Scholar]
- 7. Heng BC, Ye CP, Liu H et al. Loss of viability during freeze–thaw of intact and adherent human embryonic stem cells with conventional slow‐cooling protocols is predominantly due to apoptosis rather than cellular necrosis. J Biomed Sci 2006;13:433–445. [DOI] [PubMed] [Google Scholar]
- 8. Xu X, Cowley S, Flaim CJ et al. The roles of apoptotic pathways in the low recovery rate after cryopreservation of dissociated human embryonic stem cells. Biotechnol Prog 2010;26:827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ohgushi M, Matsumura M, Eiraku M et al. Molecular pathway and cell state responsible for dissociation‐induced apoptosis in human pluripotent stem cells. Cell Stem Cell 2010;7:225–239. [DOI] [PubMed] [Google Scholar]
- 10. Baharvand H, Salekdeh GH, Taei A et al. An efficient and easy‐to‐use cryopreservation protocol for human ES and iPS cells. Nat Protoc 2010;5:588–594. [DOI] [PubMed] [Google Scholar]
- 11. Claassen DA, Desler MM, Rizzino A. ROCK inhibition enhances the recovery and growth of cryopreserved human embryonic stem cells and human induced pluripotent stem cells. Mol Reprod Dev 2009;76:722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li X, Meng G, Krawetz R et al. The ROCK inhibitor Y‐27632 enhances the survival rate of human embryonic stem cells following cryopreservation. Stem Cells Dev 2008;17:1079–1086. [DOI] [PubMed] [Google Scholar]
- 13. Martín‐Ibáñez R, Strömberg AM, Hovatta O et al. Cryopreservation of dissociated human embryonic stem cells in the presence of ROCK inhibitor. Curr. Protoc. Stem Cell Biol. 2009;Chapter 1:Unit 1C.8. [DOI] [PubMed] [Google Scholar]
- 14. Mollamohammadi S, Taei A, Pakzad M et al. A simple and efficient cryopreservation method for feeder‐free dissociated human induced pluripotent stem cells and human embryonic stem cells. Hum Reprod 2009;24:2468–2476. [DOI] [PubMed] [Google Scholar]
- 15. Narumiya S, Ishizaki T, Uehata M. Use and properties of ROCK‐specific inhibitor Y‐27632. Methods Enzymol 2000;325:273–284. [DOI] [PubMed] [Google Scholar]
- 16. Heng BC. Effect of Rho‐associated kinase (ROCK) inhibitor Y‐27632 on the post‐thaw viability of cryopreserved human bone marrow‐derived mesenchymal stem cells. Tissue Cell 2009;41:376–380. [DOI] [PubMed] [Google Scholar]
- 17. Vernardis SI, Terzoudis K, Panoskaltsis N et al. Human embryonic and induced pluripotent stem cells maintain phenotype but alter their metabolism after exposure to ROCK inhibitor. Sci Rep 2017;7:42138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wowk B. Thermodynamic aspects of vitrification. Cryobiology 2010;60:11–22. [DOI] [PubMed] [Google Scholar]
- 19. Beier SJC, Dörr D et al. Effective surface‐based cryopreservation of human embryonic stem cells by vitrification. Cryobiology 2011;63:175–185. [DOI] [PubMed] [Google Scholar]
- 20. Beier AF, Schulz JC, Zimmermann H. Cryopreservation with a twist—Towards a sterile, serum‐free surface‐based vitrification of hESCs. Cryobiology 2013;66:8–16. [DOI] [PubMed] [Google Scholar]
- 21. Ji L, de Pablo JJ, Palecek SP. Cryopreservation of adherent human embryonic stem cells. Biotechnol Bioeng 2004;88:299–312. [DOI] [PubMed] [Google Scholar]
- 22. Chatterjee A, Saha D, Niemann H et al. Effects of cryopreservation on the epigenetic profile of cells. Cryobiology 2017;74:1–7. [DOI] [PubMed] [Google Scholar]
- 23. Bock C, Kiskinis E, Verstappen G et al. Reference maps of human ES and iPS cell variation enable high‐throughput characterization of pluripotent cell lines. Cell 2011;144:439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Havlicek S, Kohl Z, Mishra HK et al. Gene dosage‐dependent rescue of HSP neurite defects in SPG4 patients' neurons. Hum Mol Genet 2014;23:2527–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reinhardt P, Glatza M, Hemmer K et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One 2013;8:e59252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Katsen AD, Vollmar B, Mestres‐Ventura P et al. Cell surface and nuclear changes during TNF‐α‐induced apoptosis in WEHI 164 murine fibrosarcoma cells. Virchows Arch 1998;433:75–83. [DOI] [PubMed] [Google Scholar]
- 27. Katsen‐Globa A, Puetz N, Gepp MM et al. Study of SEM preparation artefacts with correlative microscopy: Cell shrinkage of adherent cells by HMDS‐drying. Scanning 2016;38:625–633. [DOI] [PubMed] [Google Scholar]
- 28. Kapeli K, Pratt GA, Vu AQ et al. Distinct and shared functions of ALS‐associated proteins TDP‐43, FUS and TAF15 revealed by multisystem analyses. Nat Commun 2016;7:12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sommer A, Maxreiter F, Krach F et al. Th17 lymphocytes induce neuronal cell death in a human iPSC‐based model of Parkinson's disease. Cell Stem Cell 2018;23:123–131.e6. [DOI] [PubMed] [Google Scholar]
- 30. Ha SY, Jee BC, Suh CS et al. Cryopreservation of human embryonic stem cells without the use of a programmable freezer. Hum Reprod 2005;20:1779–1785. [DOI] [PubMed] [Google Scholar]
- 31. Lee JY, Lee JE, Kim DK et al. High concentration of synthetic serum, stepwise equilibration and slow cooling as an efficient technique for large‐scale cryopreservation of human embryonic stem cells. Fertil Steril 2010;93:976–985. [DOI] [PubMed] [Google Scholar]
- 32. Guillaumet‐Adkins A, Rodríguez‐Esteban G, Mereu E et al. Single‐cell transcriptome conservation in cryopreserved cells and tissues. Genome Biol 2017;18:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Watanabe K, Ueno M, Kamiya D et al. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat Biotechnol 2007;25:681–686. [DOI] [PubMed] [Google Scholar]
- 34. Xu Y, Zhu X, Hahm HS et al. Revealing a core signaling regulatory mechanism for pluripotent stem cell survival and self‐renewal by small molecules. Proc Natl Acad Sci U S A 2010;107:8129–8134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li X, Krawetz R, Liu S et al. ROCK inhibitor improves survival of cryopreserved serum/feeder‐free single human embryonic stem cells. Hum Reprod 2008;24:580–589. [DOI] [PubMed] [Google Scholar]
- 36. Maldonado M, Luu RJ, Ramos MEP et al. ROCK inhibitor primes human induced pluripotent stem cells to selectively differentiate towards mesendodermal lineage via epithelial‐mesenchymal transition‐like modulation. Stem Cell Res 2016;17:222–227. [DOI] [PubMed] [Google Scholar]
- 37. Chen T, Yuan D, Wei B et al. E‐Cadherin‐mediated cell‐cell contact is critical for induced pluripotent stem cell generation. Stem Cells 2010;28:1315–1325. [DOI] [PubMed] [Google Scholar]
- 38. Prokhorova TA, Rigbolt KTG, Johansen PT et al. Stable isotope labeling by amino acids in cell culture (SILAC) and quantitative comparison of the membrane proteomes of self‐renewing and differentiating human embryonic stem cells. Mol Cell Proteomics 2009;8:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rowland TJ, Miller LM, Blaschke AJ et al. Roles of integrins in human induced pluripotent stem cell growth on matrigel and vitronectin. Stem Cells Dev 2010;19:1231–1240. [DOI] [PubMed] [Google Scholar]
- 40. Son YS, Seong RH, Ryu CJ et al. Brief report: L1 cell adhesion molecule, a novel surface molecule of human embryonic stem cells, is essential for self‐renewal and pluripotency. Stem Cells 2011;29:2094–2099. [DOI] [PubMed] [Google Scholar]
- 41. Kuleshova LL, Gouk SS, Hutmacher DW. Vitrification as a prospect for cryopreservation of tissue‐engineered constructs. Biomaterials 2007;28:1585–1596. [DOI] [PubMed] [Google Scholar]
- 42. Harb N, Archer TK, Sato N. The Rho‐Rock‐Myosin signaling axis determines cell‐cell integrity of self‐renewing pluripotent stem cells. PLoS One 2008;3:e3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Rho‐associated protein kinase (ROCK) inhibitor (RI) is redundant after cryopreservation of human induced pluripotent stem cells (hiPSCs) via adherent vitrification.
HiPSCs were cryopreserved via slow‐rate freezing and vitrification and thawed with or without RI. We observed that RI inhibitor is crucial for thawing of slow‐rate frozen hiPSCs, but redundant for thawing after freezing via adherent vitrification. Scale bars 500 μm. Related to Figure 2.
Supplemental Figure 2: Strong expression of pluripotency markers in human induced pluripotent stem cells (hiPSCs) before and after cryopreservation.
Respective immunocytochemistry (ICC) images of pluripotency markers OCT3/4 (red) and NANOG (green) staining for both freezing methods. ICC revealed strong expression of both markers in the majority of the cells. Scale bar 100 μm. Related to Figure 3.
Supplemental Figure 3: Scanning electron microscopy (SEM) revealed preservation of cell–cell contacts of human induced pluripotent stem cells (hiPSCs) by adherent vitrification.
(A) SEM images of hiPSCs before cryopreservation. Cells within colonies displayed numerous microvilli and intact cell–cell adhesions (regions of interest, arrows). Round and damaged cells were only detected at colony borders (asterisks). (B) SEM images at day 1 after thawing. Slow‐rate frozen hiPSC colonies were decreased in size (regions of interest), showed large holes and disruption of colony integrity (arrows). Round cells with undamaged and damaged membrane were detected (asterisks and double asterisks, respectively). Adherent vitrification maintained large hiPSC colonies. Cells were covered with numerous microvilli (regions of interest). (C) SEM images at day 4 after thawing. Slow‐rate frozen hiPSCs increased in size, displayed microvilli and few round detached or damaged cells were detected (asterisks). Artifacts of the extracellular matrix (ECM) coating were visible. Vitrified hiPSCs showed intact cell–cell adhesions and only few round detached and damaged cells (asterisks). Related to Figure 5.
Appendix S1: Supporting Information Table 1
Appendix S2: Supporting Information Table 2