

Abstract
Dengue virus is a major human pathogen that infects up to 350 million people annually leading to approximately 500,000 hospitalizations due to severe dengue. Since the only marketed vaccine, Dengvaxia, has recently been shown to increase disease severity in those lacking natural immunity, antivirals to prevent or treat DENV infection represent a large, unmet medical need. Small molecules that target the dengue virus envelope protein, E, on the surface of the virion could act analogously to antibodies by engaging E extracellularly to block infection. Progress towards direct-acting antivirals against E has been hindered by the protein’s lack of a conserved, “druggable” site and a shortage of robust, target-based assays suitable for screening and medicinal chemistry studies. Here we demonstrate that the dengue envelope (E) protein offers a tractable drug target for preventing dengue infection by developing a target-based assay using a recombinantly expressed dengue serotype 2 E protein. We performed a high-throughput screen of ~20,000 compounds followed by secondary assays to confirm target-binding and antiviral activity and counter-screens to exclude compounds with nonspecific activities. These efforts yielded eight distinct chemical leads that inhibit dengue infection by binding to E and preventing E-mediated membrane fusion with potencies equal to or greater than previously described small molecule inhibitors of E. We show that a subset of these compounds inhibit viruses representative of the other three dengue serotypes and Zika virus. This work provides tools for discovery and optimization of direct-acting antivirals against dengue E and shows that this approach may be useful in developing antivirals with broad-spectrum activity against other flavivirus pathogens.
Keywords: antiviral, dengue antiviral, flavivirus antiviral, viral entry inhibitor, target-based HTS
Dengue viruses are a group of related, mosquito-borne viruses within the genus Flavivirus and circulate as four antigenically distinct serotypes, DENV1–4. Over 390 million DENV infections occur annually1. An estimated 500,000 people require hospitalization each year due to severe dengue, and an estimated 2.5% of these patients die due to the infection. We lack effective vaccines and antivirals to combat DENV infection. The only marketed vaccine, Dengvaxia, is effective in boosting natural immunity for those with prior DENV exposure, but actually sensitizes non-immune children to more severe disease and hospitalization if they are subsequently infected2–7. This is due to antibody-dependent enhancement of infection and disease, which has been shown to be directly associated with the presence of pre-existing anti-DENV antibodies in patients8. Antivirals may provide a complementary strategy to reduce viral burden and prevent severe dengue; however, there are currently no approved antivirals to prevent or to treat DENV infection9,10. The success of antivirals targeting the polymerase and protease enzymes of human immunodeficiency virus (HIV) and hepatitis C virus (HCV) has inspired efforts to develop analogous antivirals against DENV11–14; however, no candidate has advanced to clinical trials to date. Alternative antiviral targets and strategies to combat DENV and related flavivirus pathogens are therefore of high interest and need.
The flavivirus envelope protein, E, covers the surface of mature virions as a well-ordered lattice of 90 homodimers and performs essential functions during viral entry. E mediates the initial attachment step by interacting with host factors on the plasma membrane surface15. Following internalization of the virion by a clathrin-dependent process, acidification of the endosomal compartment triggers conformational changes in E that are coupled to fusion of the viral and endosomal membranes. The resulting fusion pore allows escape of the nucleocapsid to the cytoplasm where the viral RNA genome can be expressed. E is an attractive target for direct-acting antivirals due to these essential biochemical functions, which like those of the viral protease and viral polymerase, are well-defined in comparison to those of other flavivirus proteins, most of which are nonstructural. E has no cellular homologue, and the humoral immune response’s success in targeting E’s function in entry provides ample precedent for the effectiveness of targeting E as an antiviral strategy. Unfortunately, viral envelope proteins, including the flavivirus E protein, have generally not been amenable to conventional drug discovery approaches. Rational, structure-based approaches are difficult because, unlike proteases and polymerases, these proteins lack conserved active sites that naturally bind to small molecule substrates. Likewise, conventional, high-throughput screens for inhibitors of E have been limited by the lack of robust, target-based assays for monitoring inhibition of E’s biochemical function(s). Several groups, including our own, have used virtual and/or cell-based screening approaches16–21 to identify small molecules that block DENV entry by targeting E, but the absence of quantitative assays to support target-specific medicinal chemistry optimization efforts has hindered progression of these compounds.
We recently described disubstituted pyrimidines that bind directly to the prefusion, dimeric form of the DENV2 envelope glycoprotein (DENV2 E2) present on mature virions and block viral entry by inhibiting E-mediated membrane fusion21,22. Here we report use of these compounds to establish a competitive, proximity-based luminescence assay and high-throughput screening to identify diverse small molecules that potently inhibit DENV entry by binding to E and blocking membrane fusion. We further show that some small molecules identified in our screen inhibit viruses representative of DENV1, 3, and 4 as well as Zika virus, supporting our hypothesis that this pharmacological strategy may be broadly applicable across multiple flavivirus pathogens. This study provides important tools to discover new small inhibitors of E, to define the structure-activity relationships for antiviral activity mediated by this target, and ultimately to develop small molecule inhibitors of DENV entry as potential anti-DENV therapeutics.
RESULTS AND DISCUSSION
Development of a target-based assay for inhibitors of DENV2 E.
We recently reported the discovery and characterization of 2, 4- and 4,6-disubstituted pyrimidines that inhibit DENV infection by binding directly to the prefusion form of E on the virion surface and preventing E-mediated membrane fusion, an obligate step in viral entry21, 22. Since the phenotypic antiviral assays used to identify and optimize these compounds proved limiting, we sought to establish an amplified luminescent proximity homogeneous assay screen (AlphaScreen) suitable for guiding medicinal chemistry optimization efforts as well as for high-throughput screening to identify other small-molecule inhibitors of E. GNF2 is a 4,6-disubstituted pyrimidine that blocks DENV entry by binding to E and inhibiting E-mediated fusion21, 22 while also affecting a post-entry viral process through allosteric inhibition of Abl kinases21, 23. We recently confirmed through photocrosslinking experiments that a closely related 4,6-disubstituted pyrimidine inhibitor binds in a pocket between domains I and II of DENV E22. To generate an assay probe, we conjugated GNF2 to a linker-modified biotin tag21, 23 to enable immobilization of the resulting probe on streptavidin (SA)-functionalized donor beads. The soluble prefusion E dimer derived from dengue serotype 2 (DENV2 sE2) was expressed as a His-tagged protein and immobilized on acceptor beads. In the absence of competing compounds, binding of GNF2-biotin to DENV2 sE2 brings donor and acceptor beads into close proximity such that energy transfer from excited donor beads to acceptor beads results in the production of a luminescent signal (Figure 1a). Compounds that prevent the interaction of GNF2-biotin with sE2 result in a concentration-dependent loss of luminescence (Figure 1b and S1). We performed cross-titration and order-of-addition experiments to optimize the signal-to-background ratio and competition conditions using 3–110–2220, an established inhibitor of DENV E, and absence of competition with compound JW-068, a negative control compound that does not inhibit DENV2 infection (Figure S1). Although we synthesized and evaluated four other test probes based on other pyrimidine21, 22 or cyanohydrozone20 inhibitors of DENV E, GNF2-biotin consistently exhibited the best probe performance with high signal-to-background ratio and competition signal in pilot assays and was chosen for development of the high-throughput assay (data not shown). Assessing assay performance in 384-well format using GNF2-biotin as the probe and 3–110–22 and DMSO as positive and negative controls, respectively, we observed a Z’ value of 0.53, signal-to-background ratio (S/B) of 11, and coefficient of variation of 3.6% for 3–110–22 and 9.8% for the DMSO control.
Figure 1. Overview of the HTS to identify small molecules that inhibit the fusogenic activity of the dengue virus E protein.

a, Competitive proximity-based luminescence assay using recombinant, soluble prefusion DENV2 E dimer (sE2) or recombinant DI-DII protein and probe GNF2-biotin. b, Concentration-dependent competition of small molecules with GNF2-biotin for binding to sE2 leads to decreasing luminescence. IC50 values in this assay were determined by non-linear regression analysis of data. c, Flowchart of the screening progression followed in this study. d, Schematic representation of the viral infectivity and plaque reduction assays used to assess antiviral activity. For both assays, inhibitor-treatment is limited to pre-incubation of the viral inoculum and the one hour infection of cells, after which all extracellular compound and virus are washed away. In the viral infectivity assay, single-cycle virus yield is taken as a metric of productive viral entry 20–24 hours prior and plotted versus log10 inhibitor concentration to allow determination of antiviral IC90 values by non-linear regression (Tables 1 and 2 and Figure S2). For experiments to assess antiviral activity against other flaviviruses, we followed the same inhibitor treatment protocol but following the washes overlaid with carboxymethylcellulose-containing medium to allow plaque formation. The number of viral plaques is used as a metric reflecting productive viral entry, reported as an empirically determined PRNT50 value (see Table 4) or as a percentage inhibition calculated relative to the number of plaques observed for the DMSO-treated negative control (Figures S4 and Table S1).
We then used this assay as the first step in a high-throughput screening campaign (Figure 1c) designed to identify additional small molecules that can inhibit DENV infection by preventing E-mediated membrane fusion during viral entry. In a primary screen of 21,271 compounds from the Selleck bioactive compound library and the Chemdiv 7 library (see http://Iccb.med.harvard.edu), we observed an intrascreen signal-to-background (S/B) of 3.5 and Z’ values ranging from 0.45 to 0.65. From the primary screen, 218 compounds (1.02% of the total library) were selected based on Z value (−2.5 to −3: medium; < −3: strong).
We culled likely false positives, including (1) known 1O2 scavengers, (2) compounds flagged by computational filters as likely to be associated with pan-assay interference (“PAINS”) properties, (3) compounds with known metabolic liabilities, and (4) compounds for which similar structures were present in the library but were not identified in the screen. This left a set of 50 compounds with reproducible activity in the competitive proximity assay used for HTS. These were purchased from the vendor in larger quantities for further characterization.
Validation of the competitive proximity assay in high-throughput screening for small molecule antivirals that target DENV E.
To measure activity in the target-based assay, we performed dose-response experiments and determined IC50 values by non-linear regression analysis of the data. 35 out of the 50 compounds selected from the primary screen efficiently compete with GNF2-biotin for binding to DENV2 sE2 in the competitive proximity luminescence assay (Figure 1b and Table 1), with IC50 values less than 10 μM. Since GNF2 and other inhibitors of DENV E have been shown by photocrosslinking and site-directed mutagenesis to bind in a pocket located between domains I and II22, we confirmed that the compounds have comparable IC50 values against both sE2 and a recombinant protein corresponding to monomeric domains I and II (DI-DII) and therefore are unlikely to bind elsewhere on prefusion E (data not shown). Based on this finding, we used the recombinant DENV2 DI-DII protein in subsequent secondary screening and SAR studies due to its higher expression yield and 1:1 binding of protein with inhibitor.
Table 1. Summary of activity data for the eight lead compounds.
IC50 values are the concentration of the respective small molecules required to reduce signal in the competitive proximity-based luminescence assay by 50%. These values were measured against recombinant DENV2 DI-DII protein and were determined by non-linear regression analysis. The KD values correspond to equilibrium disassociation constants for small molecule inhibitors bound to DENV2 sE2 and were determined by biolayer interferometry. The IC90 value is defined as the concentration of inhibitor required to reduce single-cycle DENV2 viral yield by 10-fold in the infectivity assay as outlined in Figure 1d. IC90 values against DENV2 NGC were determined by non-linear regression analysis. The CC50 value corresponds to the concentration of inhibitor causing 50% loss of BHK-21 cell viability. The SI50/90 value is a selectivity index, corresponding to CC50/IC90. The IC90, CC50, IC50, and KD values presented here are the average and standard deviation of two or more independent replicates. Compound stability in mouse microsomes was assessed in vitro and is reported as the half-life of the small molecule (T1/2) in minutes. “N.D.” indicates that the value was not determined due to poor signal-to-noise and curve-fits for this compound.
| Compounds | IC50 (DI-DII) /μM | KD/μM | IC90 (DENV2 infectivity assay) /μM | CC50 (Cytotoxicity) /μM | SI50/90 | T1/2 (Microsome stability) |
|---|---|---|---|---|---|---|
| 3–110–22 | 1.1 ± 0.7 | 0.5 ± 0.2 | 2.8 ± 1.3 | 69 ± 15 | 25 | 13.1 |
| S4105 | 0.6 ± 0.2 | 0.9 ± 0.3 | 0.8 ± 0.3 | 50 ± 15 | > 50 | > 120 |
| K786–9739 | 3.0 ± 1.7 | 1.3 ± 0.7 | 3.1 ± 0.1 | 81 ± 13 | 26 | 14.9 |
| C200–5340 | 3.0 ± 1.1 | 7.1 ± 3.8 | 7.2 ± 3.5 | >> 50 | > 10 | 4.2 |
| G199–0398 | 7.0 ± 3.0 | N.D. | 8.8 ± 0.2 | 68 ± 6 | 8 | 70.6 |
| C200–9144 | 2.6 ± 1.3 | 1.2 ± 0.5 | 1.2 ± 0.4 | > 50 | > 40 | 55.6 |
| S7337 | 4.1 ± 1.8 | 1.3 ± 0.5 | 2.7 ± 1.0 | 13 ± 5 | 5 | 3.2 |
| S1633 | 3.1 ±1.0 | 5.2 ± 2.1 | 7.0 ± 2.8 | 50 ± 3 | 7 | 37.7 |
| C066–4182 | 1.4 ± 1.1 | 2.2 ± 1.0 | 17 ± 4.5 | >> 50 | > 2.5 | 5.3 |
To determine if the activity observed in the biochemical assay is associated with antiviral activity, we examined inhibition of dengue virus serotype 2 New Guinea C (DENV2 NGC) in cell culture. To ensure that we identified compounds that inhibit DENV2 by blocking E’s function in viral entry versus compounds that inhibit DENV by interfering with E’s function in viral assembly or via other targets, we made use of an infectivity assay in which compound is pre-incubated with the viral inoculum and present during an initial one hour infection but is otherwise absent during a single-cycle infection (Figure 1d). Quantification of single-cycle viral yield by viral plaque formation assay was then used as a readout of successful viral entry 20–24 hours prior. We initially evaluated antiviral activity at two concentrations (3 and 10 μM) and found that twelve of 35 compounds inhibit DENV2 infectivity by 90% or greater at 10 μM (Figure 1c, Table S1). We then selected the top eight compounds for further characterization, avoiding those that appeared to be non-specific scaffolds or that have chemical groups that are potentially reactive. For these eight, we titrated inhibitor concentration and performed non-linear regression analysis to determine IC90 values - defined as the concentration of inhibitor required to reduce single-cycle DENV2 viral yield by 10-fold in the infectivity assay (Table 1 and Figure S2). The eight lead compounds in this set have chemically diverse structures, and all inhibit DENV infectivity potency equal to or greater than the original disubstituted pyrimidine inhibitors of DENV E (IC90 values ≥ 5 μM)21, 22. With the exception of compound C066–4182, IC50 values measured against DENV2 DI-DII in the competitive proximity assay were well-correlated with antiviral IC90 values measured in the viral infectivity assay (Figure 2b).
Figure 2. Activity in the target-based assay is well-correlated with antiviral activity and binding to sE2.

a, Structures of eight compounds selected for further characterization. b and c, Data from Table 1 are plotted to show correlation of IC50 values measured in the competitive proximity-based luminescence assay with b, the antiviral IC90 values measured in the DENV2 infectivity assay and c, KD of the compounds for DENV2 sE2 protein. C066–4182 was not included in the linear regression analysis. G199–0398 was not included in c because we were unable to measure a KD value for this compound.
We confirmed that antiviral activity for these inhibitors is not due to general cytotoxicity since CC50 values ranged from 3- to more than 50-fold higher than the antiviral IC90 values (Table 1). We additionally showed that the eight compounds have reasonable stability in the presence of mouse microsomes in vitro (Table 1). Then, to demonstrate direct interaction of these inhibitors with the intended target, we used bio-layer interferometry to measure equilibrium dissociation constants (KD) for interaction with recombinant DENV2 sE2. IC50 values measured against DENV2 DI-DII in the competitive proximity assay were well-correlated with KD for DENV2 sE2, again with the exception of compound C066–4182 (Figure 2c). Together, these data validate the use of the competitive proximity-based luminescence assay to identify compounds that inhibit DENV by targeting prefusion E on the virion surface.
Small molecules identified in the target-based screen bind E analogously to GNF2 and block E-mediated fusion.
Implicit in our assay design was the hypothesis that compounds that compete with GNF2-biotin for binding to DENV2 sE2 or DI-DII in the target-based assay would, like GNF2, block E-mediated membrane fusion. To test this hypothesis, we monitored the effects of the inhibitors in an in vitro fusion assay. Briefly, we prepared trypsin-enacpsulating liposomes and mixed these with virions in the presence and absence of inhibitor at both neutral and acidic pH. E-mediated fusion of the viral and liposome membranes at acidic pH creates a fusion pore that renders the viral core protein accessible to the protease; absence of fusion due to neutral pH or small molecule inhibition of E results in protection of core from trypsin digestion (Figure 3a). E on the surface of the virion is never accessible to the protease and serves as an internal control for liposome integrity. We observed that compounds S4105 and C200–9144 protect core from digestion in a concentration-dependent manner (Figure 3b, c), indicating inhibition of E-mediated fusion. Similarly, the other six lead compounds inhibit fusion when present at concentrations corresponding to their respective IC90 values in the viral infectivity assay (Figure 3d, e). The sole exception is C066–4182, a compound for which IC50 in the target-based assay is not well- correlated with the antiviral IC90 value or with KD for sE2 (Figure 2b, c).
Figure 3. Lead compounds identified by HTS inhibit E-mediated fusion of dengue virions with liposomes.

a, Schematic of the capsid protection assay. E-mediated fusion occurs only at acidic pH and results in formation of a fusion pore that enables digestion of the viral core protein by trypsin. Inhibition of E-mediated fusion prevents this, resulting in protection of core. E on the exterior of the virion is never exposed to trypsin and serves as an internal control of liposome integrity. The detergent Triton X-100 causes lysis of both liposomes and virions, resulting in digestion of both E and C. b-c, Western blot analysis of the fusion and control reactions for DENV C and E shows that compounds C200–9144 and S4105 (at 5, 1, and 0.5 μM) inhibit viral fusion in a concentration-dependent manner. d-e, With the exception of C066–4182, the other lead compounds also inhibit E-mediated fusion in this assay at concentrations close to their respective IC90 values (S7337 3 μM, C066–4182 10 μM, S1633 10 μM, C200–5340 10 μM, G199–0398 10 μM, K786–9739 3 μM). Experiments were independently performed two or more times; representative data are shown.
Taken together, these experiments show that the competitive proximity-based luminescence assay identifies antiviral compounds that bind directly to prefusion E and that inhibit E-mediated fusion of the virion with target membranes. While it is most likely that the inhibitors identified using this HTS assay bind at the same site targeted by GNF2 and other pyrimidine inhibitors of E, we cannot formally exclude the possibility that one or more binds at another site on the protein and allosterically prevents binding of GNF2-biotin. In addition to photocrosslinking experiments demonstrating binding of GNF2 and related pyrimidines in the conserved pocket between domains I and II, we have previously determined that a methionine to valine substitution at position 196 (E-M196V) in the pocket confers modest resistance to multiple pyrimidine inhibitors due to decreased binding22. Using the DENV2 E-M196V mutant virus and recombinant, soluble E-M196V protein, we showed that this substitution also confers modest resistance to K786–9739 (Figure 4) and decreases affinity for the recombinant protein target (Figure S3) relative to wildtype DENV2 virus and E protein, respectively.
Figure 4. The E-M196V substitution reduces sensitivity of dengue virus to inhibition by K786–9739.
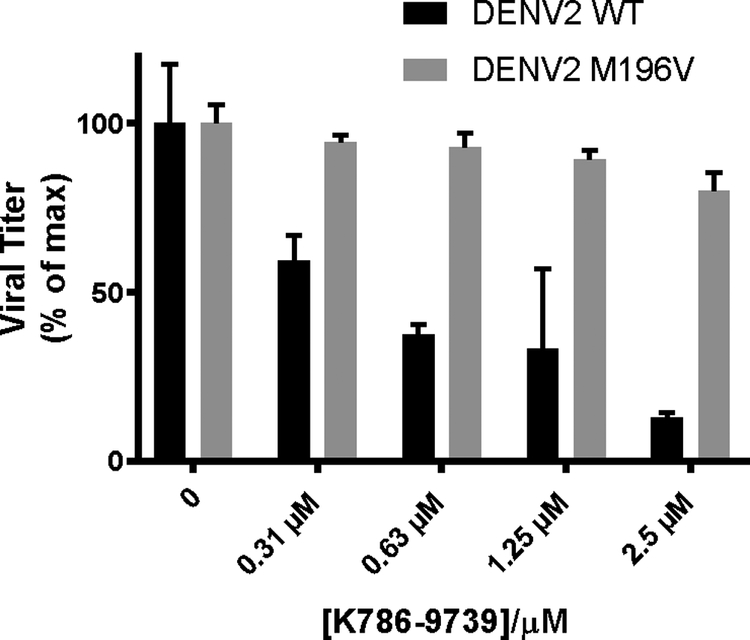
Antiviral activity was measured using the viral infectivity assay as outlined in Figure 1d. Data are normalized to the DMSO-treated negative control samples and are presented as bar plots. Representative data are shown for n ≥ 2 independent experiments. Error bars represent technical replicates in the single experiment.
Empirical evaluation of PAINS properties in lead compounds.
A major challenge in developing selective small molecule inhibitors is the identification and elimination of candidate molecules that have non-specific, off-target activities (i.e., pan-assay interference “PAINS” compounds). Small molecule aggregation and colloid-formation have been identified as a major source of promiscuous activity leading to false positive results in high-throughput screening24,25. These compounds may not be reliably identified via structure-based filters and are here of particular concern given the potential of such aggregates to interact with virions and inhibit infectivity via non-specific mechanisms. To assess the extent to which such non-specific activity may contribute to the antiviral activity of the eight lead compounds, we tested each for detergent-sensitive inhibition of established counter-screening enzymes beta-lactamase (AmpC) and malate dehydrogenase (MDH)24–28. We additionally assessed colloid formation directly using dynamic light scattering to examine particle size of the inhibitors in appropriate assay buffers. All of the lead compounds as well as known DENV E inhibitor 3–110–22 exhibit generally low level inhibition of MDH (IC50 values from 0.3 to > 100 μM) that disappears in the presence of Triton X100 (IC50 values > 100 μM). Several of the compounds also inhibit AmpC in a detergent-sensitive manner and form soluble colloids, as reflected by the formation of larger particle sizes observable at higher concentrations (40 μM) of the compounds in the absence of Tween20 (Table 2). With the exception of compound S4105, the antiviral IC90 values are an order of magnitude lower than the concentrations at which large particle sizes and inhibition of MDH are observed, indicating that these PAINS-like properties are unlikely to contribute appreciably to antiviral activity. This interpretation of the data is also supported by the limited activity of the compounds when tested against an unrelated enveloped virus. Experiments assessing the effect of the compounds on the infectivity of a GFP-expressing vesicular stomatitis virus (VSV-eGFP) detected activity for most of the compounds at 5 or 10 μM; however, the percent inhibition was considerably lower than that observed for DENV2 (Table 2). Compound C066–4182, which has an antiviral IC90 value against DENV2 of 17 μM, likewise exhibits less activity against VSV-eGFP at the tested concentration of 20 μM, suggesting that its modest activity against DENV2 is not due to non-specific mechanisms.
Table 2. Summary of assays examining non-specific sources of antiviral activity.
IC90 values in the viral infectivity assay are reproduced from Table 1 for comparison. IC50 values were measured for inhibition of AmpC and MDH in the presence and absence of Triton-X100. Detergent-sensitive inhibition of one or both enzymes was observed for all eight lead compounds, but this activity is generally at concentrations significantly higher than the antiviral IC90 value. Although many of the compounds appear to form larger particles in the DLS assay, this property does not appear to be correlated with antiviral activity or with detergent-sensitive inhibition of AmpC and MDH. Likewise, modest inhibition of vesicular stomatitis virus (VSV) for the most part occurs at concentrations significantly higher than the concentrations at which the compounds inhibit DENV2 infectivity and does not appear to be correlated with detergent-sensitive inhibition of AmpC or MDH. Representative data from one of two independent experiments are shown for 3–110–22. Other small molecules were tested once in each enzymatic assay. “*” indicates that particle sizes were measured at 20 μM rather than 40 μM due to compound solubility. “**” The smaller particle size observed for 40 μM C066–4182 without detergent versus with detergent may reflect precipitation not visible by eye and loss of the compound during sample preparation. “***” indicates that the percentage inhibition against VSV-eGFP for C066–4182 is reported at 20 μM since the IC90 of this compound against DENV2 is 17 μM.
| Compounds | IC90 (Antiviral) μM |
IC50 μM
MDH no TritonX |
IC50 μM
MDH with TritonX |
IC50 μM
AmpC no TritonX |
IC50 μM
AmpC with TritonX |
Particle size (nm) at 40 μM with Tween20 | Particle size (nm) at 40 μM without Tween20 | Inhibition % vs. VSV-eGFP | |
|---|---|---|---|---|---|---|---|---|---|
| 10 μM | 5 μM | ||||||||
| 3–110–22 | 2.8 ± 1.3 | 44.6 | > 100 | > 100 | > 100 | 7.5 * | 1069 * | 43 | 41 |
| S4105 | 0.8 ± 0.3 | 0.3 | > 100 | 19.5 | > 100 | 13.1 | 82 | 64 | 60 |
| K786–9739 | 3.1 ± 0.1 | 20.1 | > 100 | 36.2 | > 100 | 7.1 | 628 | 63 | 35 |
| C200–5340 | 7.2 ± 3.5 | 42.4 | > 100 | 66.0 | > 100 | 7.5 | 966 | 51 | 20 |
| G199–0398 | 8.8 ± 0.2 | > 100 | > 100 | > 100 | > 100 | 7.2 | 268 | 59 | 33 |
| C200–9144 | 1.2 ± 0.4 | 82.1 | > 100 | 73.5 | > 100 | 8.3 | 1443 | 77 | 76 |
| S7337 | 2.7 ± 1.0 | 86.4 | > 100 | > 100 | > 100 | 8.2 | 144 | 66 | 17 |
| S1633 | 7.0 ± 2.8 | 48.2 | > 100 | 36.6 | > 100 | 9.2 | 823 | 40 | 12 |
| C066–4182 | 17 ± 4.5 | 36.5 | > 100 | 24.3 | > 100 | 63/528 | 0.7** | 49*** | 5 |
Initial SAR demonstrate the potential for further optimization.
Since the absence of structure-activity relationship (SAR) data can severely hinder medicinal chemistry efforts to improve inhibitor activity, we began preliminary examination of SAR for the DENV2 E inhibitors identified by HTS. C200–5340 and K786–9739 were selected for this initial SAR study based on their antiviral potency, binding affinity to recombinant protein, lack of chemical groups of potential concern (e.g., catechol, pentafluorobenzene), lack of PAINS activities, and the accessibility of structural analogs through synthesis or purchase. We collected 34 commercial analogs of lead compound C200–5340 and 26 analogs of K786–9739 (Figure S4a, b) that have diverse substituents on the core heterocycle of each series or that introduce changes to the core structures. We evaluated the activity of these analogs against DENV2 DI-II in the competitive proximity-based luminescence assay to establish SAR trends and then additionally evaluated a subset at two concentrations (5 and 10 μM) in the DENV2 infectivity assay to corroborate SAR in the two assays.
For the C200–5340 series, all analogs with activity comparable to C200–5340 share the 2-sulfonylacetamide-substituted 5-sulfonylpyrimidinone core. Relocation of the chlorine group on the N−phenylacetamide from the meta to the para position (C200–5324, Table 3) is tolerated but removal of the chlorine group (C716–0602, Table 3) ablates activity in both the target-based and the antiviral assays. Activity is tolerant of lipophilic alkyl substituents such as tert−butyl, n−butyl, and methyl groups at the 4-position of the sulfonylbenzene (G605–0654, Table 3; G605–0676 and G605–0163, Figure S4a) but activity is lost with an ethoxy substituent at this position (G605–1076, Table 3). Finally, replacement of the pyrimidinone core with the tautomerized bioisostere 4-aminopyrimidine is associated with loss of potency (E682–0100 and E682–0108, Figure S4a), and various analogs with a pyrimidine core structure (e.g., C716–0055, C200–3730, Y010–0578, Figure S4a) exhibit no inhibitory activity.
Table 3. Initial SAR of C200–5340 and K786–9739.
IC50 values are the concentration of small molecule required to reduce signal in the competitive proximity-based luminescence assay by 50%. Assays were performed against recombinant DENV2 DI-DII protein, and values were determined by non-linear regression analysis. IC50 values for C200–5340 and K786–9739 are the average of three independent replicates. IC50 values for the analogs represent the curve fit for duplicate samples. Activity against DENV2 DI-DII in the target-based assay is in general correlated with inhibition of DENV2 infectivity. Consistent with the parent inhibitors C200–5340 and K786–9739, the analogs show some activity against VSV at 5 and 10 μM but this non-specific activity is less potent than the inhibition of DENV2.
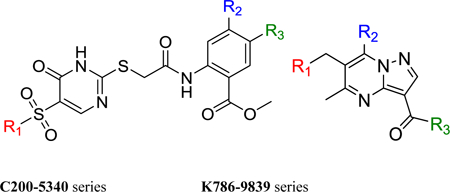
| Compounds | Series | R1 | R2 | R3 | IC50 (DENV2 DI-DII)/ μM | Conc. μM | % Inhibition vs. DENV2 |
% Inhibition vs. VSV-eGFP |
|---|---|---|---|---|---|---|---|---|
| C200–5340 | C200–5340 |  |
−Cl | −H | 3.0 | 5 | > 90 | 50 |
| 10 | > 90 | 80 | ||||||
| C200–5324 | C200–5340 |  |
−H | −Cl | 4.3 | 5 | > 90 | 80 |
| 10 | > 90 | > 90 | ||||||
| C716–0602 | C200–5340 | 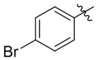 |
−H | −H | > 10 | 5 | No inhibition | 10 |
| 10 | 40 | 20–30 | ||||||
| G605–0654 | C200–5340 | 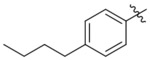 |
−H | −H | 5.2 | 5 | 60 | 10–20 |
| 10 | > 90 | 40–50 | ||||||
| G605–1076 | C200–5340 | 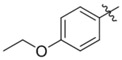 |
−H | −H | > 10 | 5 | No inhibition | No inhibition |
| 10 | < 10 | No inhibition | ||||||
| K786–9739 | K786–9739 | 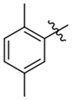 |
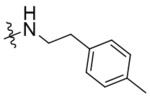 |
−OH | 3.0 | 5 | > 90 | 65 |
| 10 | > 90 | 40 | ||||||
| K784–9103 | K786–9739 | 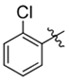 |
 |
−OH | 1.9 | 5 | > 90 | > 90 |
| 10 | > 90 | > 90 | ||||||
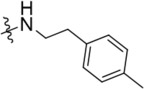
| C218–0512 | K786–9739 | 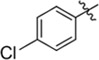 |
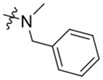 |
−OH | > 10 | 5 | 20 | 10 |
| 10 | 85 | 20–30 | ||||||
| C218–0327 | K786–9739 | 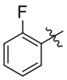 |
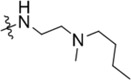 |
−OH | No inhibition | 5 | < 10 | No inhibition |
| 10 | 50 | No inhibition | ||||||
| K785–0399 | K786–9739 | 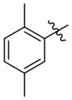 |
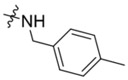 |
−Me | No inhibition | 5 | < 10 | 40 |
| 10 | 50 | > 90 |
For the K786–9739 series, activity generally requires presence of the benzyl group although elimination of this group is tolerated when an additional piperidine substituent is present at the 5-position of the pyrazolopyrimidine core (e.g., C429–0385, Figure S4b). Most analogs with mono- and di-halogen substituents on the benzyl group retain inhibitory activity comparable to the parent compound (e.g., K784–9103 in Table 3 and multiple compounds in Figure S4b. Analogs in which the homobenzyl group has been replaced with cyclic alkyl groups exhibit activities that are also comparable to parent compound K784–9739 (e.g., C218–0301, Figure S4b and K784–9103, Table 3); however, analogs with acyclic alkyl groups lack activity (e.g., C218–0327, Figure S4b). Similarly, simple replacement of the hydroxyl group on the pyrazolopyrimidine core with a methyl group (K785–0399, Table 3 and K784–3392, Figure S4b) or installation of a methyl group on the amide (C218–0512, Table 3) results in significant loss of activity (Figure S4b).
We tested a subset of these C200–5340 and K786–8739 analogs against VSV as a negative control to verify that the antiviral activity we observed is due to specific inhibition of DENV2 and not due to non-specific activity. Although we observed some inhibition of VSV at the concentrations selected, this activity is for the most part less potent than the activity observed against DENV2 (Table 3). Similar values against DENV2 and VSV may reflect a lack of viral specificity but in some cases occurs because the analog’s activity against both viruses was below the detection limit of the experiment.
Targeting the envelope protein as a general strategy for pan-serotype DENV inhibition and other flaviviruses.
Since the four DENV serotypes are hyperendemic in many parts of the world, an additional consideration in prioritizing lead compounds for development of antivirals is broad-spectrum activity against multiple DENV serotypes. Demonstration of activity against multiple serotypes is also important due to the heterogeneity of E sequences across the serotypes due to immune selection. While our HTS was directed against DENV2 E, we have previously observed both serotype-specific and pan-serotype activity for different small molecules originally optimized against DENV2 E20,21 and have demonstrated that some DENV2 E inhibitors can additionally inhibit the entry of Zika and other members of the Flavivirus genus22. We performed plaque reduction assays (Figure 1d) to evaluate activity of the lead compounds against DENV1 WP74, DENV2 NGC, DENV3 THD3, and DENV4 TVP360 viruses as representatives of the four serotypes as well as ZIKV PF-251013–18. As for the viral infectivity assay, we limited inhibitor treatment to a pre-incubation step with the viral inoculum and the initial one hour viral infection; however, instead of quantifying single-cycle viral yield, we instead quantified viral plaque formation directly as a metric of viral infectivity.
A range of serotype specificities was observed, with compounds S4105 and C200–9144 exhibiting potent inhibition of DENV1, DENV2, and DENV4 as well as ZIKV (PRNT50 values ≤ 1μM) and modest activity against DENV3 (PRNT50 ≤ 10 μM) (Table 4 and Figure S5). Compound S7337 likewise exhibits potent inhibition of DENV1, DENV2, and DENV4 (PRNT50 ≤ 1 μM) and modest activity against DENV3 and ZIKV (PRNT50 ≤ 10 μM). Interestingly, compounds K786–9739 and S7337 exhibit broad-spectrum activity against all four dengue serotypes and ZIKV, albeit with lower potencies than observed for S4105 and C200–9144. Noticeably, all of the compounds exhibited relatively limited activity against the strain of DENV3 tested in comparison to activities against the other DENV serotypes and even ZIKV. Rather than reflecting poor interaction of the compounds with the E protein of DENV3 THD3, this instead appears to be correlated with a larger proportion of non-infectious particles (i.e., high particle-to-pfu ratio) for this virus, with non-infectious particles likely acting as a molecular “sponge” competing with infectious particles for binding to the inhibitor (data not shown). More importantly, potent activity against both DENV and ZIKV in these experiments is consistent with the idea that these inhibitors target a site on E that is conserved across the DENV serotypes and ZIKV.
Table 4. Antiviral activity against DENV1, 2, 3, and 4 and Zika viruses.
Antiviral activity against strains representative of DENV1–4 and ZIKV was assessed using the plaque reduction assay depicted in Figure 1d. PRNT50 values, defined as the concentration that causes a fifty percent reduction in the number of viral plaques, were determined empirically and are scored here as follows: ++, PRNT50 ≤ 1 μM; +, PRNT50 ≤ 10 μM; -, PRNT50 > 10 μM. Known DENV inhibitor 2–12–221 was used as a control. Data reflect two or more independent experiments.
| ZIKV | DENV1 | DENV2 | DENV3 | DENV4 | |
|---|---|---|---|---|---|
| 2–12–2 | + | + | + | − | − |
| S4105 | ++ | ++ | ++ | + | ++ |
| K786–9739 | + | + | + | + | + |
| C200–5340 | + | + | + | − | + |
| G199–0398 | − | + | + | − | + |
| C200–9144 | ++ | ++ | ++ | + | ++ |
| S7337 | + | ++ | ++ | + | ++ |
| S1633 | − | + | + | − | + |
| C066–4182 | − | − | + | − | + |
CONCLUSION
Dengue, Zika, and other flavivirus pathogens constitute major challenges due to the illnesses that they cause, the absence of effective vaccines or antivirals, their wide distribution throughout much of the world, and their ability to spread rapidly into non-endemic areas due to transmission by insect vectors. Enhancement of infection and hence disease due to non-neutralizing titers of antibodies has now been demonstrated in humans and limits use of Dengvaxia2–7 to those who have natural immunity due to prior dengue infection. Recent demonstrations that cross-reaction of anti-ZIKV antibodies with DENV can enhance DENV infection and disease and vice versa present an additional set of challenges in developing safe and effective vaccines against these viruses. Small molecules that, like antibodies, target E on the virion can bind extracellularly to block infection, but unlike antibodies, cannot enhance infection or disease. Although viral envelope proteins are attractive targets for direct-acting antivirals due to their well-defined functions in viral entry and their lack of a cellular homolog, the HIV drug enfuvirtide (T20), a peptide, remains the only marketed antiviral drug that acts by preventing membrane fusion mediated by a viral envelope protein. The necessity of delivering therapeutic antibodies and peptides by injection has been a significant limitation in development of these agents as drugs. Small molecule inhibitors that can analogously engage E on the virion surface extracellularly to block viral infection would be attractive due to their potential for delivery by alternative routes of administration (e.g., oral, intradermal). Despite this potential, identification of small molecules that can act by this mechanism has been challenging because these proteins lack a classical active site to which a small molecule can be rationally targeted and because we generally lack the necessary structural models and target-based assays that would enable conventional drug discovery approaches to be applied. Here, we have developed a competitive proximity-based luminescence assay using recombinantly expressed DENV2 sE2 and DI-DII proteins and validated this assay as a tool to identify small molecules that bind directly to prefusion E and inhibit E-mediated membrane fusion. While our current data suggest that the inhibitors identified by the assay directly compete for binding to the conserved pocket targeted by GNF2 and our previously identified inhibitors, additional experiments are needed to exclude definitively the possibility that some bind in another site and allosterically inhibit interaction of the GNF2-biotin probe with E in this assay. Using this assay to conduct a high-throuputput screen, we identified eight lead compounds that inhibit DENV2 infectivity with IC90 values in the single-digit micromolar range. We show that these small molecules all bind to recombinant DENV2 sE2 protein and compete with the probe GNF2-biotin for binding to sE2 in the target-based assays and that this activity is strongly correlated with inhibition of E-mediated membrane fusion. Thus, despite their significantly smaller size compared to antibodies, these small molecules can engage E on the virion surface and effectively block infection via specific inhibition of E’s fusogenic function29. While prior efforts to advance inhibitors of DENV E as antivirals have been hindered by modest potencies and limited tools for target-based improvement of antiviral potency and selectivity16–21, our primary screening effort identified compounds with antiviral potencies equal to or greater than previously optimized inhibitors. These efforts have provided multiple lead compounds suitable for medicinal chemistry optimization of antiviral potency, antiviral spectrum of activity, and resistance profile. We further have demonstrated the utility of the target-based assay as a tool for driving SAR studies and guiding medicinal chemistry optimization of E inhibition. Our observation of cross-inhibition of all four DENV serotypes and ZIKV by a subset of inhibitors suggests that the approach described here may be generalizable to develop inhibitors of other flaviviruses by targeting their respective E proteins and or by elucidating the SAR that enables broad-spectrum inhibition of multiple flaviviruses.
EXPERIMENTAL SECTION
Compound Libraries
High-throughput screening was performed against the Selleck bioactive library (10 mM in DMSO, plates 3651–3657, https://iccb.med.harvard.edu/selleck-bioactive-compound-library) and a subset of ChemDiv library 7 (5 mg/ml in DMSO, plates 3428–3452 and 3501–3550, https://iccb.med.harvard.edu/chemdiv-7) provided by the Harvard Medical School ICCB-Longwood Screening Facility.
All small molecule inhibitors used in secondary screening and structure-activity relationship studies were purchased from Selleck and ChemDiv and characterized by LC-MS.
Chemistry
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. 1H NMR spectra were recorded on 600 MHz (Varian AS600), and chemical shifts are reported in parts per million (ppm, δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), d (doublet), t (triplet), and m (multiplet). Mass spectra were obtained on a Waters Micromass ZQ instrument. Preparative HPLC was performed on a Waters Symmetry C18 column (19 × 50 mm, 5μM) using a gradient of 5–95% methanol in water containing 0.05% trifluoacetic acid (TFA) over 8 min (10 min run time) at a flow rate of 30 mL/min. Purities of assayed compounds were in all cases greater than 95%, as determined by LC-MS analysis.
GNF2-biotin23 and 3–110–2220 were synthesized as previously described. JW-068 was synthesized as in Scheme S1.
N−[4-(4-Methylpiperazin-1-yl)phenyl]-6-phenoxypyrimidin-4-amine (JW-068). 4-chloro-6-phenoxypyrimidine (21 mg, 0.1 mmol, 1 eq.), 4-(4-methylpiperazin-1-yl)aniline (19mg, 0.1 mmol, 1eq.) and trifluoroacetic acid (23 μL, 0.3 mmol, 3 eq.) were dissolved in 2-butanol. The mixture was heated at 110 °C for 4 hours. When the reaction was completed, the reaction mixture was concentrated, and the resulting residue was purified by prep-HPLC to afford the title compound (10.7 mg, 30%). 1H NMR (600 MHz, DMSO-d6) δ 9.36 (s, 1H), 8.31 (d, J = 0.8 Hz, 1H), 7.54 – 7.47 (m, 2H), 7.42 (d, J = 8.5 Hz, 2H), 7.32 (ddt, J = 8.5, 7.3, 1.2 Hz, 1H), 7.26 – 7.18 (m, 2H), 6.98 – 6.93 (m, 2H), 6.03 (d, J = 0.9 Hz, 1H), 3.15 – 3.11 (m, 4H), 2.53 – 2.49 (m, 4H), 2.28 (s, 3H). MS (ESI) m/z 362 (M + H)+.
Cell lines and antibodies
The following cell lines were obtained from the indicated sources and cultured according to procedures previously described22 as indicated: Vero (ATCC), BHK-21 (Eva Harris), and C6/36 cells (ATCC); Sf9 and High Five™ cells (Thermo-Fisher Scientific and Stephen C. Harrison (Harvard Medical School and Boston Childrens Hospital).
Monoclonal antibody 4G2, which recognizes the conserved fusion loop of the DENV E protein, was produced from culture supernatants of hybridoma D1–4G2–4–15 (ATCC HB-112). 4G2 cross-reacts with the fusion loop of ZIKV E and was used for detection of ZIKV E in Western blots performed to characterize virus stocks. Mouse hybridoma producing monoclonal antibody 6F3.1 against DENV2 core protein were generously provided by John Aaskov (Queesnland University of Technology).
Viruses
All work with infectious virus was performed in a biosafety level 2 (BSL2) laboratory using additional safety practices as approved by the Harvard Committee on Microbiological Safety. Dengue virus serotype 2 strain New Guinea C (DENV2 NGC) was donated by Lee Gehrke (MIT). The ZIKV PF-251013–18 strain was isolated from a viremic patient in French Polynesia in 2013 and was shared by Didier Musso (Institut Louis Malardé). Dengue viruses DENV1 WP74, DENV3 THD3, and DENV4 TVP360 were generous gifts from Aravinda De Silva (University of North Carolina Chapel Hill). VSV-eGFP was kindly shared by Sean Whelan (Harvard Medical School). DENV2 NGC E-196V was isolated from resistance selection as previously described22. Viruses were propagated as previously described21, 22 and stored at −80 degrees C.
Quantification of virus titers: DENV1–4 titers were quantified by plaque-formation assay on BHK-21 cells. ZIKV titers were quantified by PFA on Vero cells. Plaque formation assays (PFA) were performed as described before22. Cells were incubated at 37 °C for 2–3 days (ZIKV) or 4 days (DENV1–4).
Characterization of DENV2 (WT and E-196V) stocks for infectivity assays: Since the viral target, E, is present on non-infectious particles that can act as a “sponge” for the inhibitor, we characterized virus stocks to ensure that comparable amounts of target were present in the antiviral assays. DENV2 (both WT and E-M196V) infectivity assays were performed using DENV2 virus stocks with equivalent titers of infectious units (determined by plaque-formation assay as described above), genome equivalents of DENV2 RNA (determined by RT-qPCR), and E (determined by quantitative Western blot) per unit volume of inoculum.22
Characterization of DENV1–4 and ZIKV for plaque reduction assays: For plaque reduction assay experiments performed with DENV1–4 and ZIKV (Table 4 and Figure S5), we performed qualitative Western blots using antibody 4G2 against the conserved fusion loop to confirm comparable amounts of E present per infectious unit of virus. We observed similar signal intensities for E when equivalent numbers of infectious particles of DENV1, 2, 4 and ZIKV were analyzed by Western blot using 4G2. DENV3 THD3 consistently exhibited much higher E signal, indicating a higher particles:infectious unit ratio than for the other viruses (data not shown).
Competitive proximity-based luminescence assay
For the competitive proximity-based luminescence assay, the soluble prefusion DENV2 E dimer (sE2) was used for initial high-throughput screening (HTS) and initial verification of “cherry picks”; recombinant DENV2 DI-DII was used for IC50 determination and structure-activity relationship experiments. 3.5 pmol of DENV2 sE2 or 7 pmol DENV2 DI-DII protein was mixed with AlphaScreen Nickel chelate acceptor beads (15 ng/μL, Perkin Elmer) in 30 μL HEPES buffer (25 mM HEPES, 200 mM NaCl, pH 7.4), in the presence of 10.5 pmol of GNF2-biotin per well. The solution was dispensed into Perkin Elmer standard OptiPlates 384 using a Thermo multidrop Combi machine. For HTS, library compounds (33 nL, 10 mM in DMSO for Selleck bioactive library and 5 mg/ml in DMSO for ChemDiv 7 library) were then transferred to assay plates by Seiko compound pin transfer robot (ICCB-L). For IC50 measurements, individual compound was transferred by HP D300 (ICCB-L) to the assay plates. Assays were incubated at room temperature for 2.5 hours. AlphaScreen streptavidin donor beads (Perkin Elmer) were diluted at 15 ng/μL in HEPES buffer and 5 μL was distributed to the plate by Combi machine followed by incubation at room temperature overnight and measurement of the fluorescence signal (excitation 680 nm, emission 520–620 nm) using a Perkin Elmer EnVision plate reader.
Antiviral activity: viral infectivity and plaque reduction assays
For the viral infectivity assay, virus inocula were diluted in EBSS to achieve a multiplicity of infection (MOI) of 1, and were pre-incubated with the given small molecule at varying concentrations for 45 min at 37 °C. The mixture was then added to cells for 1 hour at 37 °C to allow infection, after which the inoculum was removed and the cells were washed with 1X PBS to remove unbound virus and compound. Cells were overlaid with medium lacking inhibitor and incubated at 37 °C for 20–24 hours, corresponding to a single cycle of infection. Culture supernatants were harvested at this time, and the yield of infectious particles produced was quantified by plaque-formation assay. For initial antiviral screening of HTS “hits,” compounds were tested for activity at 3 and 10 μM (Table S1). For IC90 value determination (Tables 1 and 2 and Figure S2), viral yield (plaque-forming units per milliliter) was plotted versus the log10 of the inhibitor concentration, and non-linear regression analysis of the data (Graphpad Prism) was performed to determine the concentration at which viral yield is reduced 10-fold.
For plaque reduction assays (Table 4 and Figure S5), DENV1, 2, 3, 4 were tested on BHK-21 cells, and ZIKV was tested on Vero cells. Virus inocula were diluted in EBSS to 2500 pfu/ml as the final concentration, and were pre-incubated with different concentrations of small molecule inhibitors (2% DMSO vol/vol final concentration) for 45 min at 37°C, 5% CO2. The mixture (200 μL, 500 pfu of virus) was then added to cells for 1 hour (37 °C, 5% CO2) to allow infection. The inoculum was removed, and the cells were washed with 1X PBS to remove unbound virus and compound. Cells were overlaid with carboxylmethylcellulose and incubated at 37 °C, 5% CO2 to allow the formation of plaques (4 days for DENV1, 2, 3, 4 and 2–3 days for ZIKV). The cells were fixed and plaques visualized by staining of the cell monolayers with crystal violet as previously described21, 22. The number of plaques in inhibitor-treated samples was plotted as a percentage of the number of plaques observed for the DMSO-treated control versus the log10 inhibitor concentration (Figure S5). Empirical analysis was performed to determine the PRNT50 value, defined as the inhibitor concentration needed to reduce plaque formation by 50%. Note that relatively modest inhibition of DENV3 THD3 is consistent with the high particle-to-pfu ratio for this virus resulting in non-infectious particles’ acting as a “sponge” for inhibitor.
Capsid protection assay to monitor fusion of virions with liposomes
Liposomes were made with 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Avanti Polar Lipids), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) (Avanti Polar Lipids), and cholesterol (Sigma-Aldrich) at 1:1:1 molar ratio in TAN buffer (20 mM triethanolamine, 100 mM NaCl, pH 8.0). Lipids were dried down and resuspended thoroughly by vortexing and sonication. Liposomes were prepared by extrusion through a 0.2 micron filter after five freeze/thaw cycles. For trypsin-containing liposomes, 10 mg of trypsin was added to 1 mL of lipids (3 mg each) after the third freeze/thaw cycle, prior to extrusion. Liposomes were separated from unincorporated trypsin by size-exclusion chromatography using a Superdex 200 10/300 Increase column (GE Healthcare Life Sciences) on an Akta fast performance liquid chromatography (FPLC) system. Trypsin-containing liposomes were stored at 4 degrees C and used within 24 hours.
DENV2 virus (~105 infectious particles) was incubated with various concentrations of compounds (0, 0.5, 1, 3, 5, 10 μM in 2% DMSO (vol/vol)) for 45 min at 37 °C in TAN buffer (20mM triethanolamine, 100mM NaCl, pH 8.0) prior to addition of liposomes (15 μL of peak fraction from column) for 10 minutes. After incubation with liposomes, 10 μL 1M sodium acetate (pH 5.0) was added to lower the pH to 5.5 for 10 minutes. Samples were back-neutralized with 10 μL 1.5M Tris (pH 8.8) and incubated for 45 minutes at 37°C to allow trypsin digestion. SDS sample buffer was added, and samples were boiled for 20 minutes before separation by SDS-PAGE on a 12% gel. Proteins were transferred to a nitrocellulose membrane using a semi-dry transfer apparatus. Monoclonal antibodies 4G2 and 6F3.1 were used for detection of DENV envelope and capsid protein, respectively.
Expression, purification, and biotinylation of recombinant, soluble DENV2 envelope proteins
Recombinant DENV2 E wildtype and E-M196V and DI-DII proteins were produced and purified using a Bac-to-Bac baculovirus expression system (Thermo Fisher Scientific)21, 29. Synthetic genes containing the coding sequence for the soluble portion of the DENV2 NGC envelope protein (sE2 or E-WT), the protein carrying E-M196V mutation (sE2−M196V or “E-M196V”), and domains I and II (DI-DII) fused to a N-terminal signal peptide were codon optimized for expression in insect cells. Sf9 and High Five cells were generously donated by Daryl Klein and Stephen C. Harrison (Harvard Medical School and Boston Childrens Hospital. The coding sequence was cloned into a pFastBacTM/CT-TOPO® vector (Thermo Fisher Scientific) to enable expression of protein with a TEV-His6 tag at its C terminus for use in the competitive proximity luminescence assay. The plasmid expressing the wild-type DENV2 E protein was modified to introduce a C-terminal AviTag™21 to allow production of AviTag™ DENV2 sE2 protein for use in bio-layer interferometry experiments.
DENV2 sE2, DENV2 DI-DII, and DENV2 sE2−AviTag™ proteins were purified by Talon resin (Clontech) gravity column followed by size-exclusion chromatography using a Superdex 200 10/300 GL column (GE Healthcare Life Sciences) equilibrated in TAN buffer (20 mM triethanolamine, 100 mM NaCl, pH 8.0). Purified DENV2 sE2 protein carrying the AviTag™, was biotinylated using BirA (Avidity). Excess biotin was removed by size-exclusion chromatography, as mentioned above.
Determination of dissociation constant (KD) values by bio-layer interferometry (BLI)
KD measurements were performed on an Octet RED384 system (ForteBio). Recombinant, soluble, biotinylated DENV2 sE2 protein was immobilized on super-streptavidin (SSA) biosensor tips.22 Equilibrium dissociation constants (KD) values were determined by plotting the local fit maximum response (nm) as a function of small molecule concentrations (μM) using ForteBio software and GraphPad Prism. Titration curves were fit to the following steady-state analysis equation: Response = (Rmax*Conc)/KD + Conc where Rmax is the local fit response maximum; “Conc” is the concentration of small molecule; KD is the equilibrium dissociation constant.
Vesicular stomatitis virus-eGFP (VSV-eGFP) counter screen
Virus inocula were diluted in EBSS to achieve a multiplicity of infection (MOI) of 1, and were pre-incubated with the given small molecule at varying concentrations for 45 min at 37 °C. 100 nM bafilomycin was used as a positive control inhibitor of VSV-eGFP entry. The virus-inhibitor mixture was then added to cells for 1 hour at 37 °C to allow infection, after which the inoculum was removed, and the cells were washed with 1X PBS to remove unbound virus and compound. Cells were overlaid with medium lacking inhibitor and incubated at 37 °C for 6 h, corresponding to multiple cycles of infection. Following removal of the supernatants, the cells were washed with 1X PBS and overlayed with PBS and then imaged. Fluorescence (excitation 488 nm, emission 525 nm) was measured using a Typhoon FLA 9500 (GE Healthcare Life Sciences) and quantified using ImageQuant TL (GE Healthcare Life Sciences).
Non-specific enzyme inhibition assays
AmpC beta-lactamase assay. The AmpC β-lactamase was a kind gift from the Shoichet lab (UCSF). The inhibitor was serially diluted (two-fold dilution series from 100 μM) and pre-incubated with 10 nM enzyme in working buffer (50 mM potassium phosphate, pH 7.0) at room temperature for 5 min. Nitrocefin (100 μM, VWR) was added to the solution and carefully mixed. Absorbance of the final mixture was immediately monitored at 470 nm for 3 min.
Malate dehydrogenase (MDH) assay. Small molecule inhibitors were serially diluted (2-fold dilution series from 100 μM) and were mixed with 200 μM oxaloacetic acid (VWR) and 200 μM NADH (VWR) in working buffer (100 mM potassium phosphate, pH 7.0). Malate dehydrogenase (EMD Millipore) was added to a final concentration of 17.5 nM, and absorbance was immediately monitored at 340 nm for 5 minutes.
For both AmpC and MDH assays, the final concentration of DMSO was 2% for all samples. All assays were repeated in the presence of 0.01% Triton X-100. IC50 values of 3–110–22 presented in Table 2 are representative data from two independent experiments; values for the other lead compounds were measured once for each enzymatic assay.
Dynamic light scattering
Different concentrations of small molecule solutions were prepared in 110 μL of 1X PBS buffer with 2% DMSO (vol/vol). Solutions were centrifuged at 21,130 g for 10 minutes (room temperature). No precipitation by naked eye was observed. Supernatant (100 μL solution) was transferred to a low-volume quartz batch cuvette (ZEN2112, Malvern). Particle size was measured on a Zetasizer Nano instrument (Malvern). The values presented in Table 2 are averages of more than 11 technical replicates.
Cytotoxicity
BHK21 cells (MEM with 2% FBS) were incubated with varying concentrations of inhibitor in a 96-well white plate for 24 hours at 37 degrees C and 5% CO2. CellTiter-Glo (Promega) was used to measure viability following the manufacturer’s instructions. Luminescence was measured using a Biotek Synergy plate reader. Data were plotted versus the log10 inhibitor concentration, and non-linear regression analysis (Graphpad Prism) was used to determine CC50 values, defined as the inhibitor concentration required to cause 50% loss of cell viability. The maximum concentration tested was 100 μM. Values presented in Table 1 are the average of two or more independent experiments.
Supplementary Material
ACKNOWLEDGMENT
Melissanne de Wispelaere, Jared Pitts, and other members of the Yang lab are thanked for scientific discussions. Stephen C. Harrison and Ian Walker are also thanked for technical assistance in expression of recombinant DENV proteins. We thank the following colleagues for sharing reagents: Didier Musso and Nathalie Pardigon for Zika PF13–251013–18; John Aaskov for the hybridoma cell line producing Mab 6F3.1; Lee Gehrke for DENV2 New Guinea C virus; Aravinda de Silva for strains of dengue viruses; Sean Whelan for VSV-eGFP reporter virus; Eva Harris for BHK-21 cells; Stephen C. Harrison for the synthetic DENV2 sE gene and insect cell lines. Kelly Arnett and the Center for Macromolecular Interactions and Jennifer Smith and the ICCB-Longwood Screening Facility provided access to instruments and technical support. We thank Brian Shoichet for help with the PAINS assays utilizing MDH and AmpC. We also thank Liron David and Hao Wu for the instruction of dynamic light scattering experiments. This work was supported by NIH awards R56AI095499, R01AI095499, and U19AI109740. Additional support was provided by The Harvard Clinical and Translational Science Center (National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR001102).
Funding Sources
NIH awards R56AI095499, R01AI095499, and U19AI109740. National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR001102.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental details and additional data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing interests.
REFERENCE
- (1).Bhatt S; Gething PW; Brady OJ; Messina JP; Farlow AW; Moyes CL; Drake JM; Brownstein JS; Hoen AG; Sankoh O; Myers MF; George DB; Jaenisch T; Wint GRW; Simmons CP; Scott TW; Farrar JJ; Hay SI (2013) The global distribution and burden of dengue. Nature 496 (7446), 504–507. DOI: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Capeding MR; Tran NH; Hadinegoro SRS; Ismail HIHM; Chotpitayasunondh T; Chua MN; Luong CQ; Rusmil K; Wirawan DN; Nallusamy R; Pitisuttithum P; Thisyakorn U; Yoon IK; van der Vliet D; Langevin E; Laot T; Hutagalung Y; Frago C; Boaz M; Wartel TA; Tornieporth NG; Saville M; Bouckenooghe A (2014) Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: a phase 3, randomised, observer-masked, placebo-controlled trial. The Lancet 384 (9951), 1358–1365. DOI: 10.1016/S0140-6736(14)61060-6. [DOI] [PubMed] [Google Scholar]
- (3).Sabchareon A; Wallace D; Sirivichayakul C; Limkittikul K; Chanthavanich P; Suvannadabba S; Jiwariyavej V; Dulyachai W; Pengsaa K; Wartel TA; Moureau A; Saville M; Bouckenooghe A; Viviani S; Tornieporth NG; Lang J (2012) Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. The Lancet 380 (9853), 1559–1567. DOI: 10.1016/S0140-6736(12)61428-7. [DOI] [PubMed] [Google Scholar]
- (4).Hadinegoro SR; Arredondo-García JL; Capeding MR; Deseda C; Chotpitayasunondh T; Dietze R; Hj Muhammad Ismail HI; Reynales H; Limkittikul K; Rivera-Medina DM; Tran HN; Bouckenooghe A; Chansinghakul D; Cortés M; Fanouillere K; Forrat R; Frago C; Gailhardou S; Jackson N; Noriega F; Plennevaus E; Wartel TA; Zambrano B; Saville M (2015) Efficacy and long-term safety of a dengue vaccine in regions of endemic disease. N. Engl. J. Med 373 (13), 1195–1206. DOI: 10.1056/NEJMoa1506223. [DOI] [PubMed] [Google Scholar]
- (5).Villar L; Dayan GH; Arredondo-García JL; Rivera DM; Cunha R; Deseda C; Reynales H; Costa MS; Morales-Ramírez JO; Carrasquilla G; Rey LC; Dietze R; Luz K; Rivas E; Miranda Montoya MC; Cortés Supelano M; Zambrano B; Langevin E; Boaz M; Tornieporth N; Saville M; Noriega F (2015) Efficacy of a tetravalent dengue vaccine in children in Latin America. N. Engl. J. Med 372 (2), 113–123. DOI: 10.1056/NEJMoa1411037. [DOI] [PubMed] [Google Scholar]
- (6).Halstead SB (2016) Critique of World Health Organization recommendation of a dengue vaccine. J. Infect. Dis 214 (12), 1793–1795. DOI: 10.1093/infdis/jiw340. [DOI] [PubMed] [Google Scholar]
- (7).Halstead SB; Russell PK (2016) Protective and immunological behavior of chimeric yellow fever dengue vaccine. Vaccine 34 (14), 1643–1647. DOI: 10.1016/j.vaccine.2016.02.004. [DOI] [PubMed] [Google Scholar]
- (8).Katzelnick LC; Gresh L; Halloran ME; Mercado JC; Kuan G; Gordon A; Balmaseda A; Harris E (2017) Antibody-dependent enhancement of severe dengue disease in humans. Science 358 (6365), 929–932. DOI: 10.1126/science.aan6836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Malet H; Massé N; Selisko B; Romette JL; Alvarez K; Guillemot JC; Tolou H; Yap TL; Vasudevan SG; Lescar J; Canard B (2008) The flavivirus polymerase as a target for drug discovery. Antiviral Res. 80 (1), 23–25. DOI: 10.1016/j.antiviral.2008.06.007. [DOI] [PubMed] [Google Scholar]
- (10).Lim SP; Wang Q-Y; Noble CG; Chen Y-L; Dong H; Zou B; Yokokawa F; Nilar S; Smith P; Beer D; Lescar J; Shi PY (2013) Ten years of dengue drug discovery: progress and prospects. Antiviral Res. 100 (2), 500–519. DOI: 10.1016/j.antiviral.2013.09.013. [DOI] [PubMed] [Google Scholar]
- (11).Kuhn RJ; Zhang W; Rossmann MG; Pletnev SV; Corver J; Lenches E; Jones CT; Mukhopadhyay S; Chipman PR; Strauss EG; Baker TS; Strauss JH (2002) Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108 (5), 717–725. DOI: 10.1016/S0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Modis Y; Ogata S; Clements D; Harrison SC (2003) A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci 100 (12), 6986–6991. DOI: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhang X; Ge P; Yu X; Brannan JM; Bi G; Zhang Q; Schein S; Zhou ZH (2013) Cryo-EM structure of the mature dengue virus at 3.5-Å resolution. Nat. Struct. Mol. Biol 20 (1), 105–110. DOI: 10.1038/nsmb.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Krishnan MN; Sukumaran B; Pal U; Agaisse H; Murray JL; Hodge TW; Fikrig E (2007) Rab 5 is required for the cellular entry of dengue and West Nile viruses. J. Virol 81 (9), 4881–4885. DOI: 10.1128/JVI.02210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Harrison SC (2008) Viral membrane fusion. Nat. Struct. Mol. Biol 15 (7), 690–698. DOI: 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou Z; Khaliq M; Suk J-E; Patkar C; Li L; Kuhn RJ; Post CB (2008) Antiviral compounds discovered by virtual screening of small–molecule libraries against dengue virus E protein. ACS Chem. Biol 3 (12), 765–775. DOI: 10.1021/cb800176t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kampmann T; Yennamalli R; Campbell P; Stoermer MJ; Fairlie DP; Kobe B; Young PR (2009) In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses. Antiviral Res. 84 (3), 234–241. DOI: 10.1016/j.antiviral.2009.09.007. [DOI] [PubMed] [Google Scholar]
- (18).Poh MK; Yip A; Zhang S; Priestle JP; Ma NL; Smit JM; Wilschut J; Shi P-Y; Wenk MR; Schul W (2009) A small molecule fusion inhibitor of dengue virus. Antiviral Res. 84 (3), 260–266. DOI: 10.1016/j.antiviral.2009.09.011. [DOI] [PubMed] [Google Scholar]
- (19).Wang Q-Y; Patel SJ; Vangrevelinghe E; Xu HY; Rao R; Jaber D; Schul W; Gu F; Heudi O; Ma NL; Poh MK; Phong WY; Keller TH; Jacoby E; Vasudevan SG (2009) A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother. 53 (5), 1823–1831. DOI: 10.1128/AAC.01148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Schmidt AG; Lee K; Yang PL; Harrison SC (2012) Small-molecule inhibitors of dengue-virus entry. PLOS Pathog. 8 (4), e1002627 DOI: 10.1371/journal.ppat.1002627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Clark MJ; Miduturu C; Schmidt AG; Zhu X; Pitts JD; Wang J; Potisopon S; Zhang J; Wojciechowski A; Hann Chu JJ; Gray NS; Yang PL. (2016) GNF-2 inhibits dengue virus by targeting Abl kinases and the viral E protein. Cell Chem. Biol 23 (4), 443–452. DOI: 10.1016/j.chembiol.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).de Wispelaere M; Lian W; Potisopon S; Li P-C; Jang J; Ficarro S; Clark MJ; Zhu X; Kaplan JB; Pitts JD; Wales TE; Engen JR; Marto J; Gray NS; Yang PL. (2018) Inhibition of flaviviruses by targeting a conserved pocket on the viral envelope protein. Cell Chem. Biol In press. DOI: 10.1016/j.chembiol.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).) Choi Y, Seeliger MA, Panjarian SB, Kim H, Deng X, Sim T, Couch B, Koleske AJ, Smithgall TE, and Gray NS. (2009) N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J. Biol. Chem 284 (42), 29005–29014. DOI: 10.1074/jbc.M109.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Feng BY; Shelat A; Doman TN; Guy RK; Shoichet BK (2005) High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol 1 (3), 146–148. DOI: 10.1038/nchembio718. [DOI] [PubMed] [Google Scholar]
- (25).Irwin JJ; Duan D; Torosyan H; Doak AK; Ziebart KT; Sterling T; Tumanian G; Shoichet BK (2015) An aggregation advisor for ligand discovery. J. Med. Chem 58 (17), 7076–7087. DOI: 10.1021/acs.jmedchem.5b01105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Feng BY; Simeonov A; Jadhav A; Babaoglu K; Inglese J; Shoichet BK; Austin CP (2007) A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem 50 (10), 2385–2390. DOI: 10.1021/jm061317y. [DOI] [PubMed] [Google Scholar]
- (27).McGovern SL; Caselli E; Grigorieff N; Shoichet BK (2002) A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem 45 (8), 1712–1722. DOI: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- (28).Seidler J; McGovern SL; Doman TN; Shoichet BK (2003) Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem 46 (21), 4477–4486. DOI: 10.1021/jm030191r [DOI] [PubMed] [Google Scholar]
- (29).) Klein DE, Choi J,C, Harrison SC. (2009) Structure of a dengue virus envolope protein late-stage fusion intermediate. J. Virol 87 (4), 2287–2293. DOI: 10.1128/JVI.02957-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.