

SUMMARY
The gut microbiota plays a significant role in the progression of fatty liver disease; however, the mediators and their mechanisms remain to be elucidated. Comparing metabolite profile differences between germ-free and conventionally raised mice against differences between mice fed a low- and high-fat diet (HFD), we identified tryptamine and indole-3-acetate (I3A) as metabolites that depend on the microbiota and are depleted under a HFD. Both metabolites reduced fatty-acid- and LPS-stimulated production of pro-inflammatory cytokines in macrophages and inhibited the migration of cells toward a chemokine, with I3A exhibiting greater potency. In hepatocytes, I3A attenuated inflammatory responses under lipid loading and reduced the expression of fatty acid synthase and sterol regulatory element-binding protein-1c. These effects were abrogated in the presence of an aryl-hydrocarbon receptor (AhR) antagonist, indicating that the effects are AhR dependent. Our results suggest that gut microbiota could influence inflammatory responses in the liver through metabolites engaging host receptors.
In Brief
Dysbiosis of the intestinal microbiota is an emerging factor contributing to the progression of fatty liver disease. Krishnan et al. utilize metabolomics and biochemical assays in conjunction with animal and cell culture models to identify microbiota-dependent metabolites that engage a host receptor to affect liver inflammatory responses under lipid loading.
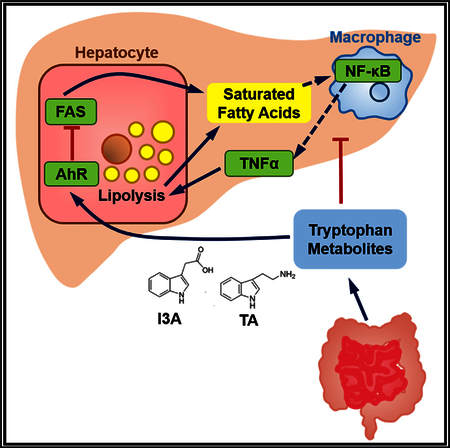
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease in Western countries, and its incidence strongly correlates with obesity and the metabolic syndrome (Day, 2011). It encompasses a spectrum of disorders from simple steatosis, by itself a benign condition, to non-alcoholic steatohepatitis (NASH), characterized by intrahepatic inflammation, hepatocellular ballooning, and progressive fibrosis (Loomba and Sanyal, 2013). Sustained steatohepatitis can lead to cirrhosis and liver cancer (Buzzetti et al., 2016).
The mechanisms driving NAFLD progression are not well understood, although it is generally agreed that there are at least two components. First, cellular lipid deposits accumulate in the liver. This is then followed by activation of immune cells and production of pro-inflammatory cytokines (Dowman et al., 2010). Nutritional, genetic, and epigenetic factors each play a role in determining whether an individual with steatosis develops the more severe pathologies of fatty liver disease (Eslam et al., 2018). Another emerging factor contributing to NAFLD pathogenesis is dysbiosis of the intestinal microbiota. Germ-free (GF) mice fed a high-fat diet (HFD) and inoculated with bacterial isolates from feces of hyperglycemic mice developed steatohepatitis, whereas GF mice fed the same HFD but inoculated with bacteria from normoglycemic mice only showed mild steatosis (Le Roy et al., 2013). A study comparing intestinal microbiomes of children found subtle, yet significant differences between children with obesity and NASH (Zhu et al., 2013).
Several hypotheses have been put forth regarding the role of dysbiosis (Schippa and Conte, 2014). One possibility is that dysbiosis disrupts production of microbial metabolites that are utilized by the intestinal epithelial cells for maintaining barrier integrity, which could elevate bacterial endotoxins in circulation, and trigger a pro-inflammatory cytokine cascade in the liver. Another hypothesis is that dysbiosis leads to increased generation of toxic metabolic byproducts in the intestine and elevates the chemical burden on the liver. A third hypothesis is that dysbiosis leads to aberrant metabolism of dietary residues (e.g., choline) and/or endogenous metabolites (e.g., bile acids), which imapirs the export of lipids or elevates lipogenesis in the liver. A common denominator of these hypotheses is that progression toward steatohepatitis is linked to alterations in the metabolic outputs of the intestinal microbiota. Recent findings have shown that the microbiota produces bioactive metabolites that engage host cellular pathways to modulate immune cell fate and function (Sridharan et al., 2014) and lipid absorption in the intestine (Ghazalpour et al., 2016). Microbial metabolites have been detected in circulation (Wikoff et al., 2009), and the liver (Vernocchi et al., 2016) and several of these metabolites (e.g., indoxyl sulfate) are ligands for host receptors, such as the aryl hydrocarbon receptor (AhR). Studies in transgenic animal models suggest that the AhR plays a significant role in regulating lipid and fatty acid metabolism (Lee et al., 2010). In mice, AhR activation negatively regulates several lipogenesis enzymes, including fatty acid synthase (Fasn), and the cholesterol metabolism regulator sterol regulatory element-binding protein-1c (SREBP-1c) (Tanos et al., 2012). In humans, exposure to the AhR ligand TCDD disrupts lipid and fatty acid metabolism (Pelclova´ et al., 2002).
The present study investigates the hypothesis that gut microbiota dysbiosis perturbs the balance of immunomodulatory microbiota metabolites, and increases the susceptibility of the liver to inflammation. We utilize a metabolomics approach to identify microbiota-dependent metabolites that activate the AhR and characterize their effects on liver inflammatory responses and metabolic function.
RESULTS
Microbiota-Dependent Metabolites in the Intestine Include Tryptophan-Derived AhR Ligands
Cecal contents and fecal material from GF and conventionally raised (CONV-R) mice were analyzed using both targeted and untargeted liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiments. Hierarchical clustering of detected features showed qualitative differences between the two groups (Figure 1A). The majority of the differentially present features (60% of total) were reduced in GF samples, suggesting that these features include products that depend on microbiota metabolic activity (Figure 1B). In the fecal material, a majority of differentially present features (36% of total) were elevated in GF samples (Figure 1C), suggesting that these features include dietary resides that could have been catabolized by the intestinal microbiota or host intestinal enzymes activated by the microbiota.
Figure 1. Results from an Untargeted Analysis of Samples from GF and CONV-R (Female C57BL/6N) Mice.
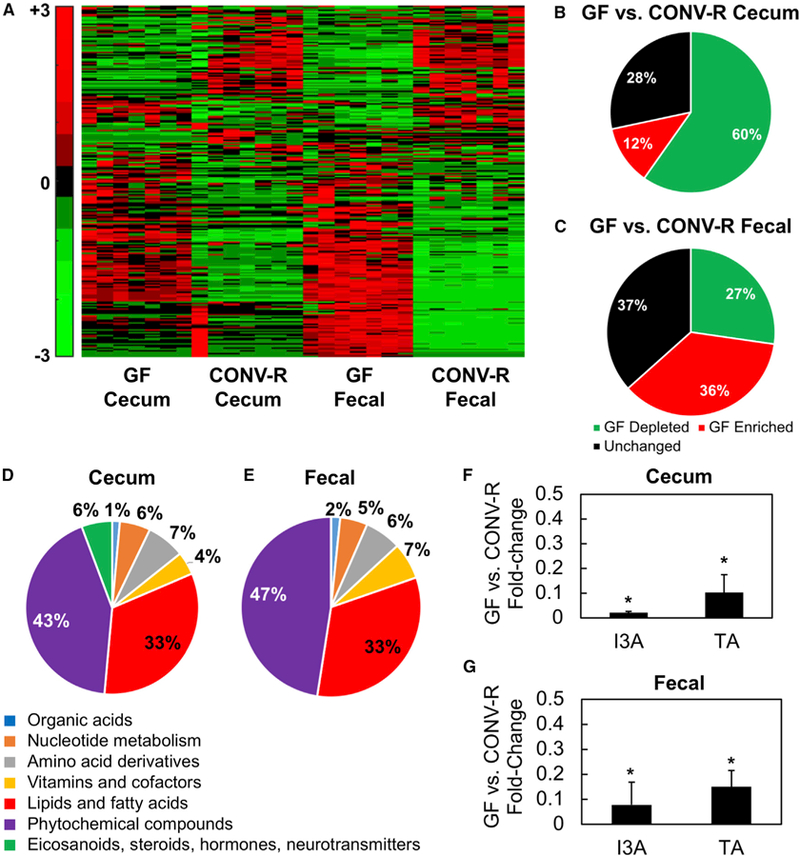
(A) Heatmaps of hierarchically clustered LC-MS/MS features.
(B and C) Features from fecal material and cecal contents were separately scaled prior to clustering using the Pareto method. Fraction of detected features that are depleted, elevated, or not significantly different in cecal contents (B) and fecal material (C) between GF and CONV-R mice.
(D–G) Putatively identified metabolites that depend on the gut microbiota. Features depleted in cecal contents (D) and fecal material (E) from GF mice were annotated and mapped to metabolic pathways in Kyoto Encyclopedia of Genes and Genomes (KEGG). Statistical tests on fold changes identified several metabolites that are consistently depleted in both fecal material and cecal contents from GF mice. I3A and TA are both significantly reduced in cecal contents (F) and fecal material (G) from GF mice.
Data shown are averages of n = 7 mice. Error bars represent 1 SD. *p < 0.05 using Wilcoxon rank-sum test.
Of the 1,444 LC-MS/MS features depleted in both cecal contents and fecal material from GF mice, we annotated 156 features with putative identities. The majority of these metabolites are associated with phytochemical and lipid metabolism pathways (Figures 1D and 1E). Other major categories are amino acid derived compounds, nucleotides, vitamins, and cofactors. Notably, these depleted metabolites include bioactive molecules, such as indole-3-acetate (I3A) and tryptamine (TA) (Figures 1F and 1G), which we previously found (Sridharan et al., 2014) to activate the AhR in a breast cancer cell line.
HFD Alters the Levels of Microbiota-Dependent Metabolites in Cecum, Serum, and Liver
Next, we investigated whether administering a HFD, which can induce steatosis in mice following a prolonged feeding period (Reid and Eksteen, 2015), alters the levels of microbiota-dependent metabolites. The duration of HFD feeding was kept relatively short to avoid substantially altering host physiology. Using qRT-PCR, we confirmed that an 8-week HFD significantly perturbed the microbiota. The ratio of Firmicutes to Bacteroidetes was significantly higher in fecal material from HFD-fed mice (8.62) compared to mice fed a low-fat diet (LFD) (0.82) (Figure S1A). No significant differences in liver triglyceride levels were observed (data not shown).
Untargeted analysis of metabolites in the cecum, serum, and liver (Figure S1B) did not show any obvious qualitative differences between HFD- and LFD-fed mice. However, a more detailed examination of the LC-MS/MS data using hypothesis testing for equal medians (Wilcoxon rank-sum test) followed by feature annotation revealed significant differences in several classes of metabolites (Figure 2A). The major classes are phytochemicals, lipids, and amino-acid-derived compounds (Figure S1C). Intersecting the list of metabolites differentially present in HFD and LFD mice with the list of metabolites differentially present in GF and CONV-R mice identified 19 microbiota-dependent metabolites that are consistently altered by the HFD in at least two of the three tissue compartments (Figure 2C). This panel of 19 metabolites was further narrowed to eliminate small effect sizes (fold change < 2 between HFD and LFD mice). Finally, searching the narrowed list for potential AhR ligands identified three tryptophan metabolites: I3A, TA, and xanthurenic acid (XA).
Figure 2. Liver, Cecum, and Serum Samples from LFD- and HFD-Fed CONV-R Mice (Male C57BL/6J) Were Analyzed Using Untargeted LC/MS-MS Experiments.
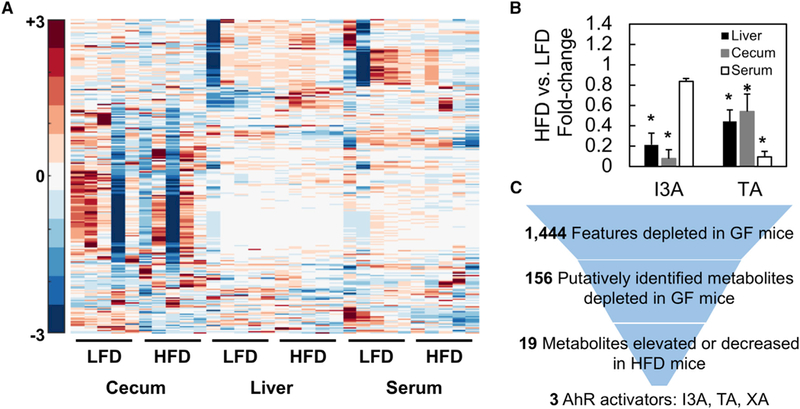
(A) Significantly different features were Pareto scaled and clustered along with features of interest based on annotation of GF and CONV-R samples.
(B) TA and I3A were quantified using targeted analysis to confirm depletion of these metabolites in HFD samples. Metabolite levels were normalized to sample weights and plotted as fold changes comparing HFD to LFD.
(C) Data shown are averages of n = 5 mice. Error bars represent 1 SD. *p < 0.05. Metabolite selection process based on features differentially present in GF versus CONV-R samples and HFD versus LFD samples. Final selections are suspected AhR ligands.
Targeted LC-MS/MS analysis for I3A and TA confirmed significant depletion of both metabolites in the liver and cecum of HFD mice compared to LFD mice (Figure 2B). The decrease in TA varied from 2- (cecum) to 10-fold (liver). Similarly, I3A decreased between 3- (liver) and 10-fold (cecum). The concentrations of TA and I3A in the LFD liver are 2 and 0.03 mM/g tissue, respectively.
HFD Increases Levels of Free Fatty Acids and Alters the Composition of Primary Bile Acid Pool
Targeted analysis for major free fatty acid (FFA) species confirmed significant increases in both saturated and unsaturated fatty acids in liver, cecum, and serum of HFD mice compared to LFD mice (Figure 3). In the liver, palmitic and stearic acids were increased 4-fold. In the cecum, palmitic acid was increased 10-fold, while stearic acid was unchanged (Figure 3B). In serum, feeding with the HFD increased almost every major FFA species (Figure 3C). The increases in serum FFAs largely correlated with the composition of the HFD. The quantitatively dominant FFAs in the diet are palmitic, oleic, linoleic, and stearic acids (Figure 3D); these are also the four most abundant FFAs in all three tissue compartments examined in this study.
Figure 3. Comparison of Free Fatty Acid (FFA) Profiles in HFD and LFD Mice.
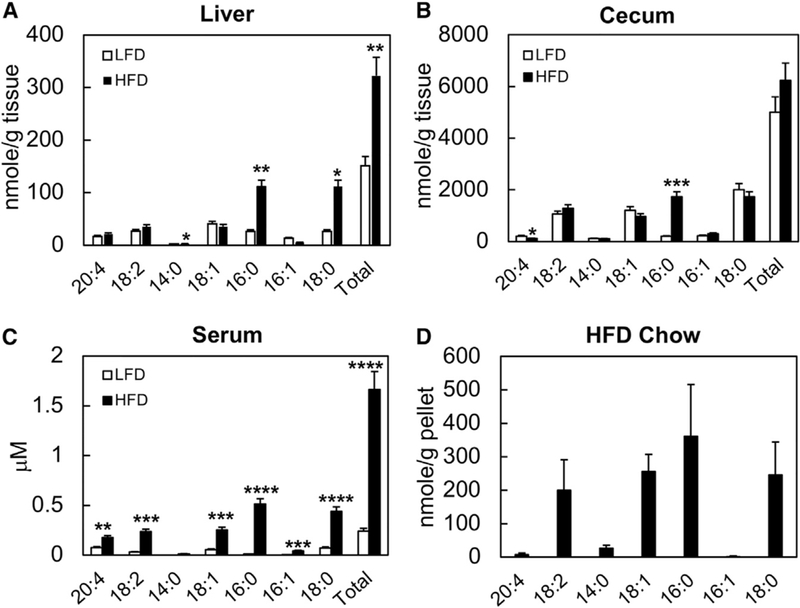
(A–C) Major FFA species were quantified in liver (A), cecal luminal content (B), and serum (C) samples from HFD- and LFD-fed mice.
(D) The same FFAs were quantified in the HFD pellets (D).
Data shown are averages of n = 5 mice. Error bars represent 1 SD. *p < 0.05, **p < 0.01, ***p < 0.001, ***p < 0.0005.
In addition to increasing the FFA levels, HFD also altered the composition of the primary bile acid pools in the liver, cecum, and serum. Targeted analysis for cholic acid (CA) and chenodeoxycholic acid (CDCA) found a significant (p = 0.013) decrease in the relative pool size of CA in the liver of HFD mice (Figure S1D).
I3A and TA Attenuate Pro-inflammatory Cytokine Expression in Macrophages
In a previous study (Bansal et al., 2010), we found that the tryptophan-derived bacterial metabolite indole attenuates indicators of inflammation in epithelial cells. As inflammation is a key secondary insult driving NASH, we investigated whether the tryptophan metabolites decreased in HFD could attenuate inflammation. Cultured macrophages were first exposed to palmitic acid and then lipopolysaccharide (LPS) to mimic the sequence of two key insults in NAFLD. Changes in the expression of tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and monocyte chemoattractant protein-1 (MCP-1) in the presence of I3A and TA were determined (Figures 4A and 4B). Pretreating macrophages with I3A or TA prior to palmitate and LPS exposure significantly attenuated the increase in the mRNA levels of TNF-α, IL-1β, and MCP-1. Of the two metabolites, I3A exhibited greater potency, reducing the expression of all three cytokines in a dose-dependent manner (Figure 4A). While TA pretreatment resulted in similar trends, only IL-1β expression was significantly attenuated (Figure 4B). Moreover, significant toxicity was observed at TA concentrations greater than 500 μM. The trends for cytokine secretion were largely consistent with gene expression (Figure 4C).
Figure 4. I3A and TA Reduce Pro-inflammatory Cytokine Production at the mRNA and Protein Levels and Chemotactic Migration in Macrophages Exposed to Palmitate and LPS.
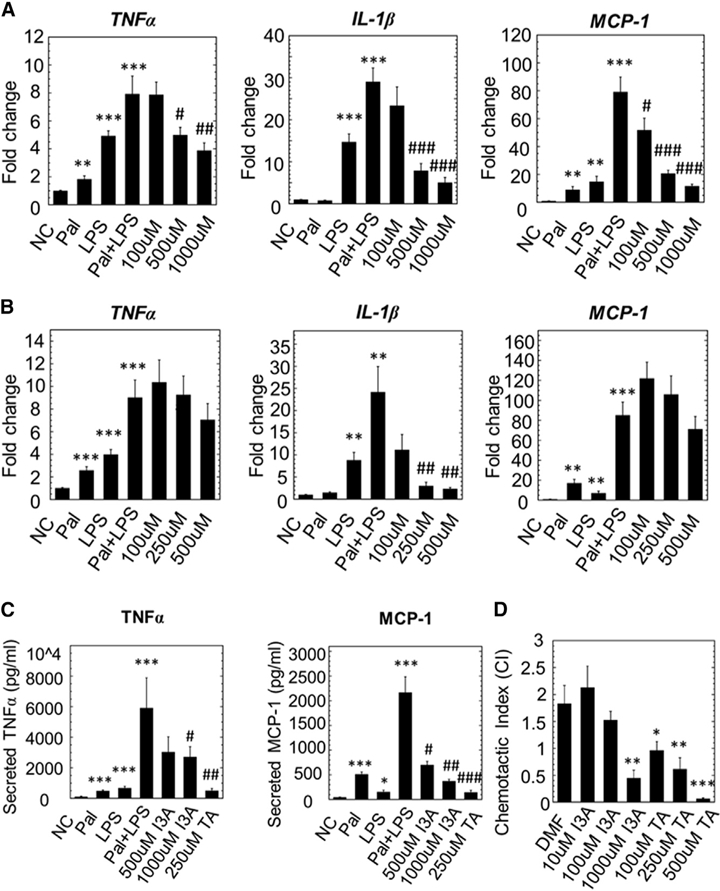
(A and B) RAW 264.7 cells were stimulated with palmitic acid (Pal) followed by LPS, with or without addition of varying doses of I3A (A) or TA (B). The negative control (NC) group was only treated with the vehicle, dimethylformamide (DMF). Changes in the expression of TNF-α, IL-1β, and MCP-1were determined using qRT-PCR.
(C) Culture supernatants were analyzed for secreted cytokines using a bead-based assay. Data shown are averages of 3 independent experiments with 3 biological replicates. Error bars represent one standard error of the mean. *p < 0.05, **p < 0.01, and ***p < 0.001 compared to NC; #p < 0.05, ##p < 0.01, and ###p < 0.001 compared to Pal+LPS group.
(D) I3A and TA inhibit BMDM migration toward MCP-1. BMDMs were incubated with MCP-1 in a transwell with vehicle (DMF) or different doses of I3A or TA. The chemotactic index (CI) for a treatment condition was calculated as the ratio of average number of migrated cells in the treatment group relative to the control group (incubated in medium only). Data shown are averages of three independent experiments with three biological replicates. Error bars represent 1 SEM. *p < 0.05, **p < 0.01, and ***p < 0.001.
I3A and TA Inhibit Macrophage Migration to MCP-1
In addition to activation of resident macrophages, progression of steatohepatitis is marked by increased infiltration of macrophages into the liver. Macrophage infiltration is promoted by MCP-1, a chemokine that is increased in serum and liver of NASH patients (Haukeland et al., 2006). Thus, we investigated if I3A or TA alters migration of macrophages toward MCP-1. Pretreatment of bone-marrow-derived macrophages (BMDMs) with I3A or TA prior to stimulation with MCP-1 significantly decreased migration in a dose-dependent manner. At the highest concentrations tested in this study, I3A (1 mM) and TA (500 μM) both completely abolished BMDM migration toward MCP-1 (Figure 4D).
I3A Attenuates Effects of TNF-α and Fatty Acids in Hepatocytes
Based on the above observation that I3A significantly attenuated the production of inflammatory mediators in macrophages, we examined whether I3A also modulated the metabolic response of hepatocytes. We first confirmed that culturing AML12 cells in medium supplemented with palmitic and oleic acids at a high, but sub-toxic (Malhi et al., 2006), dose (500 μM) led to accumulation of visible lipid droplets within 24 to 48 hr (Figures 5A–5C). A similar trend was observed in HepG2 cells, a human liver cell line (Figures S2A–S2C), which accumulated noticeably larger lipid droplets. We followed the fatty acid supplementation with TNF-α treatment, taking this cytokine as a representative pro-inflammatory signal produced by macrophages in the liver.
Figure 5. Micrographs of AML12 Hepatocytes Obtained Using CARS Microscopy.
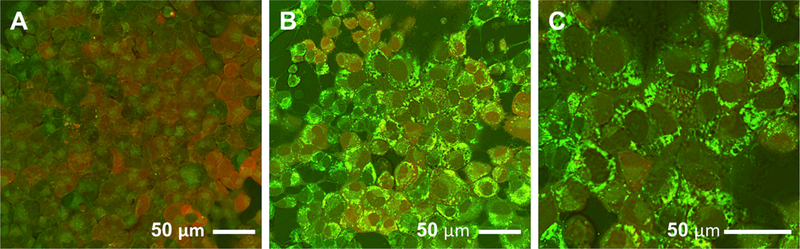
(A–C) Micrographs were recorded after the cells had been treated with either (A) vehicle or (B and C) FFAs (500 μM palmitate + 500 μM oleate) for 48 hr. (C) A close-up of (B). All images are composites of forward- (green) and epi-CARS (red) signals. Lipid inclusion bodies are primarily detected by the stronger forward signal.
Targeted LC-MS/MS analysis of major fatty acid species showed that the AML12 cells cultured in FFA-supplemented medium contained 5-fold greater levels of intracellular palmitic and oleic acids (Figures 6A–6C), suggesting that not all of the FFAs taken up by the cells were stored into droplets. A similar trend was observed for the HepG2 cells (Figures S2D–S2F). In both AML12 and HepG2 cells, exposure to TNF-α increased the intracellular levels of both palmitic and oleic acids, even without fatty acid preconditioning. Combining the treatments did not lead to an additive effect. Pretreating the cells with I3A prior to TNF-α exposure significantly decreased the levels of palmitic and oleic acids. Similarly, I3A treatment attenuated the effects of fatty acid preconditioning, even when it was combined with TNF-α.
Figure 6. I3A Attenuates the Effects of TNF-a in Cultured AML12 Cells Preconditioned with FFAs.
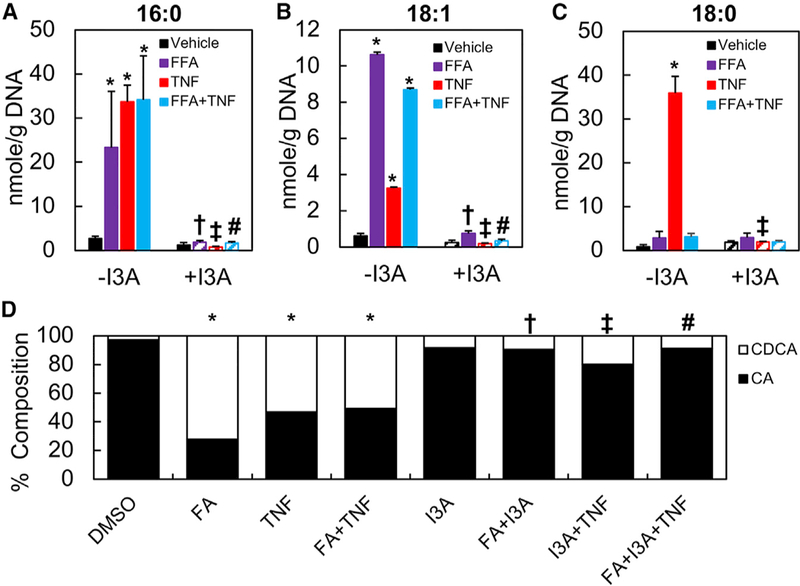
(A–C) Palmitic (A), oleic (B), and stearic acids (C) were quantified using targeted LC-MS/MS experiments.
(D) CA and CDCA were quantified and represented as a percentage of the contribution to the combined pool of primary bile acids.
Data shown are averages of 4 independent experiments with 6 biological replicates. Error bars represent 1 SD. *p < 0.05, **p < 0.01, ***p < 0.001 compared to cells in the negative control group exposed to vehicle (DMSO) only; yp < 0.05 compared to the FFA group that was not treated with TNF-a; zp < 0.05 compared to the TNF group that was not preconditioned with FFAs; #p < 0.05 compared to the FFA+TNF group that was pre-conditioned with FFAs and treated with TNF-a.
Since a hallmark of liver inflammation in fatty liver disease is dysregulation of bile acid metabolism (Chow et al., 2017), we examined the effect of I3A on bile acid production. Targeted LC-MS/MS analysis showed that fatty acid preconditioning and TNF-α exposure independently and together increased the fraction of CDCA by more than 30% in AML12 cells (Figure 6D). A similar trend was observed for the HepG2 cells (Figure S2G). By itself, I3A also increased the CDCA fraction, but when combined with the other treatments, I3A reduced the CDCA fraction by 20% compared to the conditions where the cells were exposed to either FFAs or TNF-α. However, I3A treatment had no significant impact on cultures exposed to both TNF-α with FFA. Similar trends were observed in HepG2 cell cultures (Figure S2G); however, CA accounted for a larger share of the bile acid pool in the control cultures and TNF-α exposure increased the CDCA fraction by 40%. In HepG2 cells, I3A treatment had a significant impact on bile acid levels even when both FFA and TNF-α were present.
I3A Attenuates Hepatocyte Response to TNF-α
Based on the observation that I3A attenuates the both palmitic acid- and TNF-α-stimulated fatty acid level increase, we more broadly profiled the effects of I3A on hepatocyte metabolite levels using untargeted LC-MS/MS analysis. Hierarchical clustering (Figure S3A) suggested that I3A pretreatment was more effective in reversing the metabolic alterations due to TNF-α exposure compared to fatty acid preconditioning. A similar trend was observed when the metabolite profiles were compared using partial least-squares discriminant analysis (PLS-DA) (Figures S3B and S3C). The profiles of cells cultured in fatty-acid-supplemented medium formed a distinct cluster independent of I3A treatment, whereas the cells exposed to TNF-α following I3A pretreatment clustered closely with vehicle control.
I3A Modulates the Expression of Fasn and SREBP-1c via Activation of the AhR
To investigate the impact of I3A on lipid and fatty acid pathways, we measured the expression of a key lipogenesis enzyme, Fasn, and a transcription factor regulating cholesterol/bile acid biosynthesis, SREBP-1c. By itself, I3A treatment significantly reduced the expression of Fasn in AML12 cells. This effect was also observed when I3A treatment was combined with fatty acid preconditioning and/or TNF-α exposure (Figure 7A). The effect of I3A on Fasn at the protein level was consistent with the gene expression data (Figure 7B). Treatment with TNF-α independently or in conjunction with FFA increased Fasn protein levels by 4-fold while independent treatment with FFA did not significantly alter Fasn. Treatment with I3A significantly attenuated the effects of TNF-α on Fasn by 2-fold. While fatty acid supplementation increased SREBP-1c gene expression, TNF-α exerted a weaker effect (Figure 7C). Combining both treatments did not have a significant effect compared to vehicle control.
Figure 7. I3A Attenuates the Effects of TNF-a on AhR and Its Target Genes in Cultured AML12 Cells Preconditioned with FAs.
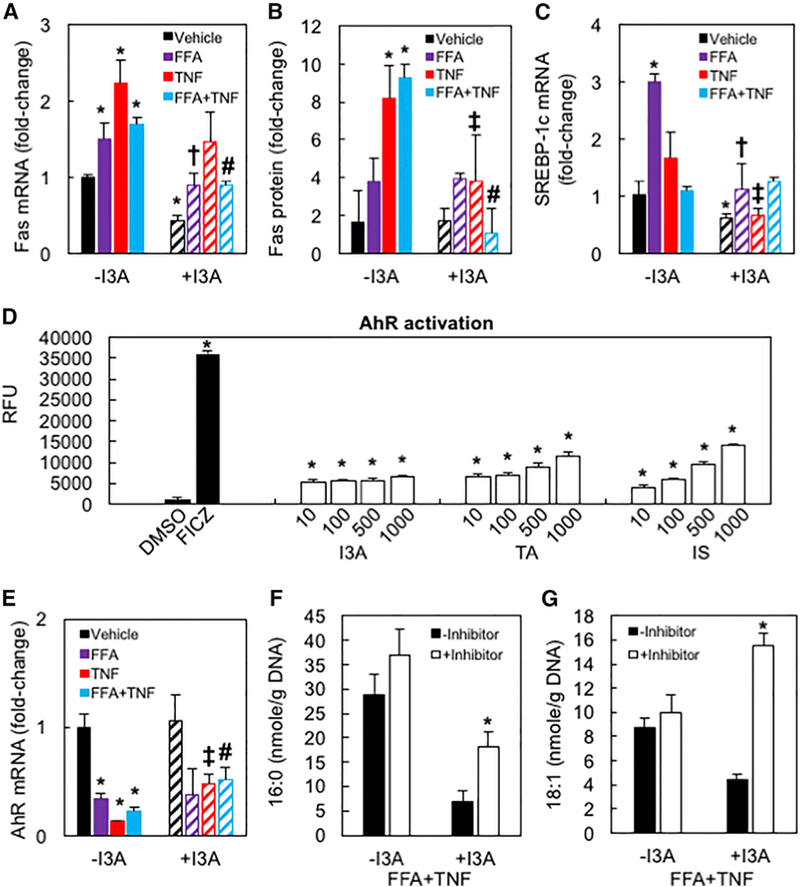
(A and B) Gene expression (A) and relative abundance of Fasn protein (B).
(C) Gene expression of SREBP-1c.
(D) AhR activation by different doses of I3A, TA, and IS. FICZ and DMSO were used as positive and negative controls (NC), respectively. ***p < 0.005, ****p < 0.001 compared to NC.
(E) Gene expression of AhR. Data shown are averages of 4 independent experiments with six biological replicates. Error bars represent 1 SD. *p < 0.05, **p < 0.01, and ***p < 0.001 compared to NC; †p < 0.05 compared to FA group, ‡p < 0.05 compared to TNF group; #p < 0.05 compared to FA+TNF group.
(F and G) Palmitic (F) and oleic acids (G) in cultures treated with AhR antagonist CH-223191. *p < 0.05 compared to corresponding culture without CH-223191.
Treatment with I3A again attenuated the effect of TNF-α or fatty acid supplementation. We also tested the effects of I3A on AhR gene expression and activation, which negatively regulates both Fasn and SREBP-1c. Fatty acid supplementation and TNF-α exposure, either individually or in combination, significantly reduced AhR gene expression compared to vehicle control. Treatment with I3A attenuated the TNF-α-induced reduction in AhR expression, regardless of fatty acid supplementation (Figure 7D). Treatment with I3A by itself had no significant impact on AhR gene expression, suggesting that the effects of I3A on AhR target genes are mediated through ligand activation of the nuclear receptor. Activation of AhR by I3A was confirmed in a H4IIE murine liver cell line expressing a stable enhanced green fluorescent protein (EGFP) reporter regulated by a minimal promoter containing xenobiotic response elements. At 10 μM or higher doses, I3A increased EGFP expression by over 5-fold compared to vehicle control (Figure 7E). To determine if the observed effects of I3A are AhR-dependent, AML12 cells were incubated with CH-223191, a potent antagonist of AhR (Zhao et al., 2010), prior to I3A treatment. In the presence of the inhibitor, I3A only weakly attenuated the TNF-α- or fatty acid supplementation-induced increases in palmitic (Figure 7F) and oleic acids (Figure 7G). In the case of oleic acid, incubation with CH-223191 completely abrogated the effects of I3A.
DISCUSSION
A number of studies have shown that the gut microbiota composition is altered in a HFD regimen, suggesting that this dysbiosis could drive various phenotypic changes and progression of HFD-associated diseases, including fatty liver disease (Murphy et al., 2015). By co-analyzing the differences in the metabolite profiles between HFD- and LFD-fed mice alongside the differences between CONV-R and GF mice, we identified metabolites that depend on the gut microbiota and are significantly depleted under HFD. The metabolite profile comparisons were only performed within the same sub-strain of mice (C57BL/6J or C57BL/6N) to examine the effect of a single factor, either diet or colonization.
The untargeted LC-MS/MS experiments detected a large number of features differentially present in GF and CONV-R mice (Figures 1A–1C). A subset of these features (26%) is consistently depleted in both cecal luminal contents and fecal material from GF mice and thus could be products of gut microbiota metabolism. A common practice for annotating untargeted LC-MS/MS data is to match the MS/MS spectra of the detected features to spectral libraries of reference databases. Unfortunately, the coverage of bacterial metabolites in these databases remains incomplete. Moreover, different databases often return conflicting results. To address these issues, we searched multiple databases and supplemented these searches with in silico fragmentation analyses. A putative identity was assigned only if two or more of the data sources agreed. The identification rate achieved in the present study (156/1,444) is comparable to other global profiling studies, with the identified LC-MS/MS features broadly representing known categories of microbiota metabolites (Figures 2D–2E).
In order to determine which of the microbiota-dependent intestinal metabolites could be altered in fatty liver disease, we compared the metabolite profiles of liver, cecum, and serum from HFD-fed mice against corresponding profiles from LFD-fed mice. The HFD model has been extensively used to study phenotypes associated with fatty liver disease in mice (Murphy et al., 2015). It is generally accepted that a HFD typically leads to simple steatosis and that steatohepatitis and fibrosis require additional challenges, such as fructose supplementation (Asgharpour et al., 2016). We limited the HFD to 8 weeks based on previous reports (Kim et al., 2012) showing that this duration is sufficient to significantly modify the gut microbiome, while also elevating circulating levels of inflammatory cytokines, without producing overt liver dysfunction. Targeted LC-MS/MS experiments confirmed the impact of the HFD on cecum, serum, and liver FFA profiles, in good agreement with previous reports on similar HFD models (Kim et al., 2012; Pang et al., 2016).
Of the 156 putatively identified metabolites depleted in GF mice, 19 were also depleted by the HFD in at least two of the three tissue compartments. Querying this panel of metabolites against the literature for previously reported bioactivity, specifically ligand activation of the AhR, further narrowed the panel to three metabolites (Figure 2C): XA, TA, and I3A. Enzymes for producing TA (Williams et al., 2014) and I3A (Sridharan et al., 2014) from tryptophan have been characterized in the gut microbiota, whereas such enzymes have not yet been identified for XA. Therefore, we focused on TA and I3A as likely microbiota metabolites. The doses of TA and I3A were based on concentrations measured in the liver, estimated to be 2 and 0.03 mM, respectively, assuming a tissue density of 1 g/ml.
Multiple triggers can activate liver resident macrophages and induce hepatic inflammation. One well-known trigger is exposure to LPS. Increased bacterial translocation from the digestive tract has been observed in NASH patients (Miele et al., 2009). Another potential trigger is activation of toll-like receptors (e.g., TLR4) by elevated FFAs (Shi et al., 2006). Treating the cells with both palmitate and LPS synergistically induced the production of several pro-inflammatory cytokines. This finding is consistent with an earlier study reporting that palmitate amplifies the production of inflammatory cytokines (IL-6 and IL-8) upon LPS stimulation (Schwartz et al., 2010) of in human THP-1 monocytes. A similar observation was reported by Wen et al. (2011), who showed palmitate induction of IL-1β secretion in LPS primed macrophages. The synergistic effects of LPS and palmitate could involve production of ceramides (Schilling et al., 2013), which strongly induce lipotoxicity (Pagadala et al., 2012), or inflammasome activation through AMP-activated protein kinase (Wen et al., 2011). Both palmitate and LPS have been shown to activate macrophages through TLR4 and nuclear factor-κB (NF-κB) signaling (Shi et al., 2006), suggesting that signaling from both stimuli could converge onto this classical inflammation pathway. The addition of I3A or TA to the culture medium significantly decreased palmitate and LPS induced cytokine production in a dose-dependent manner (Figures 4A–4C). Interestingly, TA shows greater potency than I3A in decreasing TNF-α and MCP-1 secretion, but is less potent in decreasing their gene expression. One possible explanation for this observation is that I3A and TA affect cytokine production at different points.
Another source of hepatic inflammation in NASH is increased infiltration of monocytes to the liver. Activated Kupffer cells produce chemokines, including MCP-1, which recruit multiple immune cells including monocytes (Tacke and Zimmermann, 2014). Accumulation of monocytes in the liver depends on signaling between C-C chemokine receptor type 2 (CCR2) and MCP-1 (Tacke and Zimmermann, 2014), and blocking this axis improves NASH in a murine model (Baeck et al., 2012). Our results show that I3A and TA both inhibit BMDM migration toward MCP-1 in a dose-dependent manner; however, the mechanism for this inhibition remains to be elucidated. While both I3A and TA exhibit the ability to attenuate inflammation in macrophages, only I3A significantly attenuated both gene expression and secretion of cytokines (Figure 4D). Moreover, TA exhibited toxicity at higher doses. Based on these observations, we focused on I3A for further in vitro testing in hepatocytes.
Fatty acid supplementation has been used to mimic diet-derived lipid accumulation in hepatocytes (García-Ruiz et al., 2015), although these previous studies did not report visual evidence for lipid droplets that characterizes steatosis. Here, we used an imaging technique to confirm the presence of lipid droplets in both murine (AML12) and human (HepG2) liver cell lines. The HepG2 cell line was used to confirm that the fatty acid supplementation and other treatments performed in the study elicit a similar response in murine and human cells, since the latter is in some cases not appropriately modeled by the former. Targeted LC-MS/MS experiments confirmed significant uptake of both palmitic and oleic acids in both cell types, although at least a fraction these pools remained non-esterified as FFAs.
While elevation of serum FFAs is an indicator of the metabolic syndrome (Zhang et al., 2014), hepatic lipid accumulation more specifically indicates NAFLD. Targeted analysis for major FFAs confirmed that palmitic, stearic, and myristic acids were significantly elevated in livers of HFD-fed mice. The most abundant unsaturated fatty acid measured in our study is oleate, although its level in the liver was not significantly altered by the HFD. Palmitic acid can act as an inflammatory signaling molecule, whereas oleic acid is a quantitatively important substrate for esterification and promotes lipid accumulation through modulation of SREBP-1c activity (Ricchi et al., 2009). We supplemented the culture medium of hepatocytes with both fatty acids to induce lipid loading and present an inflammatory stimulus.
Two common denominators of NAFLD are lipid accumulation and elevation of pro-inflammatory factors in the liver. Lipid accumulation occurs primarily in hepatocytes, whereas proinflammatory cytokines are produced by resident and/or infiltrating macrophages (Stojsavljević et al., 2014). We found that FFA exposure along with LPS synergistically promotes TNF-α, IL-1β, and MCP-1 production and secretion by macrophages. We selected TNF-α as a representative cytokine to mimic the effects of local macrophage activation on hepatocytes. In both AML12 and HepG2 cells, TNF-α significantly increased the levels of intracellular FFAs. Similar observations were reported in vivo by Endo et al. (2007), who found that induction of TNF-α expression through LPS injection in mice significantly enhances the expression of SREBP-1c and Fasn and elevates liver steatosis. We show that the effects of TNF-α are nearly abolished by treating the cells with I3A. Treatment with I3A also attenuated the effects of TNF-α on bile acid metabolism. Clinical studies (Aranha et al., 2008; Chow et al., 2017) have suggested that an alteration in both serum and liver bile acid compositions could be a biomarker for NAFLD; in particular, CDCA was found to be higher in NASH patients compared to healthy subjects. In our two-hit in vitro model, FFA and cytokine treatments independently and together increased the ratio of CDCA to CA, which was partially reversed by I3A treatment, again suggesting a normalization of lipid metabolism.
While the mechanism whereby I3A counters the effects of TNF-α on lipid metabolism remains to be fully elucidated, our results, together with previously reported findings, point to activation of the AhR. We, and others (Hubbard et al., 2015; Jin et al., 2014; Sridharan et al., 2014), have shown that I3A is an agonist for the AhR in several cell types, including hepatocytes (Figure 7D). The present study shows that I3A also modulates the expression of two AhR regulated genes, SREBP-1c and Fasn, in a manner consistent with ligand activation of AhR. Rats administered the AhR agonist TCDD exhibited decreased fatty acid synthesis in the liver (Lakshman et al., 1988). Liver-specific knockout (KO) of AhR exacerbated the effects of a HFD on liver steatosis, inflammation, and lipotoxicity (Wada et al., 2016). The same study also found increased de novo lipogenesis in AhR KO mice. Conversely, TNF-α appears to interfere with transcriptional regulation by the AhR (Gharavi and El-Kadi, 2005). However, the mechanism remains to be elucidated, and conflicting findings have been reported in other cell types (e.g., MCF7 cells) (Drozdzik et al., 2014).
The protective role of the AhR in NAFLD likely depends on the degree of activation and the cellular context. Gorski et al. (1988) reported that TCDD increased fatty acid synthesis in rat livers when administered at a very high dose (10-fold higher than Lakshman et al. [1988]). We observed similar dose dependence in both murine and human hepatocytes. At the highest dose (500 μM), we found that I3A by itself increased the levels of both palmitic and oleic acids compared to vehicle control, an effect we did not observe at lower doses (data not shown). On the other hand, I3A consistently lowered intracellular palmitic and oleic acids when presented together with FFAs, TNF-α, or both. One possible explanation for these different findings is that negative regulation of lipogenesis by the AhR depends on whether the nuclear receptor is activated in a naive state or in a heightened state of inflammation when expression of AhR is suppressed.
Of the two ‘‘hits’’ applied in the present study, I3A more strongly attenuated the effects of TNF-α. Hierarchical clustering (Figure S3A) and PLS-DA of untargeted LC-MS/MS data indicate that the global metabolite profiles of I3A and TNF-α-treated cells (Figure S3B) more closely resemble the vehicle control compared to I3A- and FFA-treated cells (Figure S3C). Interestingly, treatment with I3A alone led to a metabolite profile that is distinct from the vehicle control, suggesting that this metabolite has broad effects on cellular metabolism. While our results indicate that these effects are at least partially mediated by the AhR, the involvement of other regulatory pathways cannot be ruled out. Furthermore, the stronger attenuation of TNF-α effects could reflect the timing of I3A treatment, which preceded TNF-α exposure and followed FFA supplementation.
In conclusion, we have shown that the gut microbiota-dependent metabolite I3A can directly modulate inflammatory responses of hepatocytes and macrophages. Our results suggest that I3A could modulate liver inflammatory responses in at least two ways. Acting on the macrophages, I3A could attenuate the release of pro-inflammatory cytokines that induce the liver to synthesize FFAs, which in turn stimulate the macrophages. Acting on the hepatocytes, I3A could attenuate the cytokine-mediated upregulation in lipogenesis. These actions of I3A on the hepatocytes are AhR-dependent, as inhibition of AhR by a specific antagonist suppressed the effects of I3A. The AhR is likely but one of several host cellular receptors mediating host-microbiota cross-talk. In this regard, the approach presented in this paper could serve as a useful template to characterize other microbiota-dependent ligands and their effects on specific host cell types and pathways. Finally, further studies are warranted in animal models and human subjects to determine whether I3A or other microbiota metabolites can effectively intervene in the pathogenesis of NAFLD.
EXPERIMENTAL PROCEDURES
Reagents
RAW 264.7, AML12, and HepG2 cells were purchased from ATCC (Manassas, MA, USA). DMEM, alpha minimal essential medium (αMEM), RPMI-1640, penicillin/streptomycin, PBS, TRIzol, RNase-free water, DNase-free water, LPS (from Salmonella minnesota), and fetal bovine serum (FBS) for culture of HepG2 and AML12 cells were purchased from Invitrogen (Carlsbad, CA, USA). FBS for culture of RAW 264.7 was purchased from Atlanta Biologicals (Flowery Branch, GA). Insulin-transferrin-selenium (ITS) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). The AhR inhibitor CH-223191 was developed in the Sherr lab at Boston University. H1G1.1c3 reporter cells were obtained from the Denison lab at UC Davis and utilized in the Sherr lab. Unless otherwise noted, all other reagents were purchased from Sigma Aldrich (St. Louis, MO, USA).
Comparison of GF and Conventionally Raised Mice
To screen for intestinal metabolites that depend on the microbiota, we performed untargeted and targeted analysis of cecal contents and fecal material collected from conventionally raised (CONV-R) and GF female C57BL/6N mice. The mice were purchased from Taconic (Albany, NY, USA) at 6 weeks of age and immediately sacrificed by euthanasia upon receipt at Texas A&M Health Science Center. Cecal contents and fecal material were collected as described previously (Sridharan et al., 2014) and stored at −80°C until further analysis.
Sample Collection from Mice Fed a Low- or High-Fat Diet
Two groups of male C57BL/6J mice 14 weeks of age were purchased from Jackson labs. One group (n = 5) was initially raised on a LFD consisting of 10% calories from fat (D12450B, Research Diets, New Brunswick, NJ) until 6 weeks of age, after which the mice were fed a HFD consisting of 60% calories from fat (D12492, Research Diets), whereas the other group (n = 5) continued on a LFD. Mice from both groups had access to the respective diets during transit and prior to sacrifice (within 16 hr of receipt). Mice were sacrificed by euthanasia, and blood was collected via heart puncture and centrifuged at 4,000 × g for 30 min to obtain serum. The liver was quickly excised, rinsed with 10× volume of ice-cold PBS, flash frozen in liquid nitrogen, and weighed. Cecal contents were gently squeezed out of the excised cecum into cold tubes and flash frozen. Serum, liver, and cecal content samples were stored at −80° C until further analysis. All animals were handled in accordance with the Texas A&M University Health Sciences Center Institutional Animal Care and Use Committee guidelines under an approved animal use protocol (2015–0346).
HepG2 and AML12 Hepatocyte Culture
HepG2 cells were seeded into 12-well plates at a density of ~1.25 × 105 cells/cm2 and cultured in a humidified incubator at 37°C and 5% CO2. The culture medium was low-glucose DMEM supplemented with 10% (v/v) heat-inactivated FBS, penicillin (200 U/mL), and streptomycin (200 μg/mL). The medium was changed every 24 hr for 3 days after seeding. AML12 cells were seeded into 12-well plates at a density of ~1.25 × 105 cells/cm2 and cultured in a humidified incubator at 37° C and 5% CO2. The culture medium was a 1:1 mixture of DMEM and Ham’s F12 medium supplemented with 5 μg/mL insulin, 5 μg/mL transferrin, 5 ng/mL selenium, 40 ng/mL dexamethasone, 10% (v/v) heat-inactivated FBS, 1.2 g/L sodium bicarbonate, penicillin (200 U/mL), and streptomycin (200 μg/mL). The medium was changed every 24 hr until the culture reached ~80% confluence. For a ‘‘one-hit’’ inflammation model, the cells were treated for 24 hr with dimethylsulfoxide (DMSO) or 10, 100, or 500 μM I3A or TA and then treated with either DMSO or 25 ng/mL TNF-α for another 24 hr. For a ‘‘two-hit’’ steatosis and inflammation model, the cells were grown for 48 hr in medium supplemented with 0.5 mM palmitic acid and 0.5 mM oleic acid, then treated for 24 hr with DMSO or 500 μM I3A, and finally treated for 24 hr with vehicle or 25 ng/mL TNF-α. The AhR inhibitor CH-223191 was prepared as a stock solution of 20 mM in DMSO. AML12 cultures were dosed with either 1 μL of inhibitor solution or 0.1% DMSO 10 min prior to I3A treatment and again 10 min prior to TNF-α treatment.
Murine Macrophage Culture
Raw 264.7 murine macrophages were seeded into 24-well plates at a density of 4 × 105 cells/ml and cultured in a humidified incubator at 37°C and 5% CO2. The culture medium was DMEM supplemented with 10% heat-inactivated FBS, penicillin (200 U/mL), and streptomycin (200 μg/mL). Cells were pretreated with different concentrations of I3A or TA for 4 hr, followed by addition of 300 μσM palmitate complexed with BSA. After 18 hr, 10 ng/mL LPS was added to the culture medium. The cells were then incubated for an additional 6 hr. BMDMs were isolated from the femur of wild-type C57/BL6 mice as previously described (Ying et al., 2013). The isolated progenitor cells were resuspended in RPMI-1640 medium supplemented with 10 ng/mL macrophage colony-stimulating factor (M-CSF) (Invitrogen), seeded in polystyrene dishes, and incubated for 3 days at 37° C and 5% CO2 in a humidified incubator. The culture medium was replenished on day 3, and the cells were incubated for an additional 4 days. At the end of the 7-day culture period, > 95% of the cells were positive for macrophages markers F4/80 and CD11b.
Metabolite Extraction
Metabolites were extracted from tissue samples using a solvent-based method. Liver tissue samples were weighed and placed into a pre-cooled garnet bead beating tube followed by adding 500 μL ice cold methanol and 250 μL ice-cold chloroform. After homogenization (6,500 rpm for 30 s), the sample tube was centrifuged under refrigeration at 700 × g for 10 min. The supernatant was then transferred to a new tube through a 70-μm cell strainer; 1 mL ice-cold water was added; and the sample centrifuged at 3,700 × g for 10 min. The upper and lower phases were each separately transferred into new tubes while taking care not to disturb the interface. The liver tissue pellet was extracted again with ice-cold methanol (500 μL). The polar phases from both extractions were combined and concentrated using a centrifugal evaporator (Vacufuge, Eppendorf, Hauppauge, NY, USA). Metabolites from cecal content samples were extracted as described above without the homogenization step. Serum metabolites were extracted as previously described (Wikoff et al., 2009). Extracted samples were concentrated using the Vacufuge and stored at −80°C until analysis. Prior to metabolite analysis, the samples were reconstituted in 100 μL methanol/water (50% v/v).
Untargeted Metabolomics
The extracted samples were analyzed for global metabolite profiles using information-dependent acquisition (IDA) experiments (Tables S1 and S5) performed on a triple-quadrupole time-of-flight (TOF) instrument (AB Sciex 5600+). Raw data were processed in MarkerView (v. 1.2, AB Sciex) to determine the ion peaks. The peaks were aligned based on accurate mass (m/z) and retention time (RT) (30 ppm and 2.5 min tolerance, respectively) and then filtered based on intensity (100 cps threshold) to eliminate low-quality peaks. An additional filter was applied to retain only monoisotopic ions. The remaining ions were organized into a feature table, with each feature specified by m/z and RT. In the case a precursor ion detected by the TOF survey scan triggered an MS/MS scan, the corresponding MS/MS spectrum was extracted from the product ion scan data and added to the feature table. Each feature was searched against spectral libraries in METLIN (Smith et al., 2005) and HMDB (Wishart et al., 2007) and analyzed using in silico fragmentation tools MetFrag (Wolf et al., 2010) and CFM-ID (Allen et al., 2014). In some cases, the database search or in silico analysis returned more than one matching compound for a given feature. In these cases, the highest-ranked match was selected as the search result. The search results from all four sources were used together to assign a putative identity (ID) to the features. An ID was categorized as a confident assignment, if the search results for the feature were consistent across at least one experimental database and one other data source.
Targeted Analysis of Bile Acids and Fatty Acids
The extracted samples were analyzed for specific metabolites (I3A, TA, bile acids, and fatty acids) using product ion experiments performed on the Triple-TOF instrument (Tables S1, S2, S3, and S4). Metabolite-specific parameters for product ion scans were determined from runs using high-purity chemical standards.
Metabolite Quantitation
For each metabolite with a confirmed or putative identity, the corresponding peak in the ion chromatogram was integrated using MultiQuant (v.2.1, AB SCIEX) to determine the area under the curve (AUC). For comparisons of metabolite levels between samples, fold changes were calculated based on the AUC values normalized to the corresponding tissue weight (liver and cecum samples) or DNA content (in vitro cell culture samples). Cellular DNA content was measured using Hoechst dye.
RNA Extraction and qRT-PCR
Cells were washed with PBS and stored at −80° C until RNA extraction. Total RNA was extracted from RAW 264.7 cells using the EZNA Total RNA Kit (Omega Bio-Tek, Norcross, GA, USA). RT-PCR analysis was carried out using the qScript One-Step PCR Kit (Quanta Biosciences, Gaithersburg, MD, USA) on a LightCycler 96 System (Roche, Indianapolis, IN, USA). For hepatocytes and AML12 cells, RNA was extracted using TRIzol (Invitrogen). Total RNA was reverse-transcribed using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). Quantification of RNA was performed using Brilliant II SYBR Green qPCR Master Mix (Agilent Technologies) on a Stratagene Mx3000P qPCR System (Agilent Technologies). Fold change values were calculated using the 2−∆∆Ct method, with β-actin and GAPDH as the housekeeping genes for macrophages and hepatocytes, respectively. The primer sequences are listed in Table S6.
Proteomics
Proteins were extracted from cell lysates, purified, digested, and analyzed using LC-MS/MS experiments as described previously (Manteiga and Lee, 2017). An IDA scan was used to generate an ion library in ProteinPilot (v.5.1, AB Sciex) of all proteins and their corresponding peptides in the sample, and a data-independent acquisition (DIA) scan was used to obtain high-quality MS/MS data for quantification.
Cytokine Quantification
Raw 264.7 cells were treated with I3A or TA for 4 hr, palmitate for 18 hr, and followed by LPS for another 24 hr. Culture supernatants were centrifuged at 5,000 × g for 15 min and secreted cytokines were quantified using a multiplex kit (Mouse Inflammation Panel, BioLegend, San Diego, CA, USA) using the manufacturer’s suggested protocol.
Macrophage Migration Assay
BMDM migration was investigated in transwell cell culture chambers with polycarbonate membranes (8-μm pore size, Corning Costar, Corning, NY, USA). Cells were added to the upper chamber, and either vehicle or different concentrations of I3A or TA was added to both the upper and the lower chambers. After 4 hr, 20 ng/mL MCP-1 (Invitrogen) was added to the bottom chamber. After 24 hr, cells remaining on the upper side of the membrane were scraped off with a cotton swab. The migrated cells in the bottom chamber were fixed with methanol for 15 min, stained with 0.1% crystal violet for 30 min, and counted under a microscope (Axiovert 200M, Zeiss). Three replicate wells were analyzed in each experiment, with cells counted in 15 randomly chosen fields of view per well.
AhR Ligand Activation Assay
A previously described AhR reporter assay (Nagy et al., 2002) using murine H1G1.1c3 hepatoma cells was adapted to test ligand activation of the AhR by I3A and TA. These experiments were performed at the Sherr lab. Indoxyl sulfate (IS) and 6-formylindolo(3,2-b)carbazole (FICZ) were used as positive controls. Fluorescence readings were recorded at 24 and 48 hr using a plate reader (SpectraFluor Plus, Tecan, Ma¨ nnedorf, Switzerland). Relative fluorescence as a measure of induced AhR activity was calculated by subtracting the average background fluorescence of untreated cells from the experimental values.
CARS Microscopy
Lipid accumulation in hepatocytes grown in fatty-acid-supplemented medium was visualized using coherent anti-stokes Raman scattering (CARS) microscopy. Images were recorded every 24 hr by tuning the Stokes (1,064 nm) and pump (817 nm) beams to excite the Raman peak at 2,845 cm−1 (663 nm) corresponding to the CH2 stretch in lipid molecules. The microscopy system was configured to detect both forward- and epi-CARS signals. All images were averaged over 64 frames. Overlays of forward-(green) and epi-CARS (red) signals were generated using raw images (LAS X Software v.3.0.2, Leica Microsystems).
Statistical Analysis
Metabolite level comparisons from in vivo data were performed using the Wilcoxon rank-sum test. Statistical significance of in vitro data was calculated using one-way ANOVA for single treatment comparisons and two-way ANOVA for multiple treatment comparisons. A p value of less than 0.05 was considered statistically significant. All heatmaps were generated using pareto-scaled data. The similarity of the metabolite profiles was assessed based on group centroid distances using PLS-DA. Ellipses in PLS-DA plots indicate 95% confidence regions for each treatment group.
Supplementary Material
Highlights.
A high-fat diet depletes the microbiota metabolites indole-3-acetate and tryptamine
Indole-3-acetate and tryptamine attenuate indicators of inflammation in macrophages
Indole-3-acetate attenuates cytokine-mediated lipogenesis in hepatocytes
ACKNOWLEDGMENTS
This work was supported by grants from the NSF (12645502, 1337760, and 0821381), the National Institute of Medical Sciences (GM106251) (to K.L. and A.J.) and the Nesbitt Chair (to A.J.). We thank the Geourgakoudi lab for training and access to the CARS microscope (NSF 1531683). We thank Alexandra Ramirez-Cardenas from the Sherr lab at Boston University for AhR reporter assays.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Allen F, Pon A, Wilson M, Greiner R, and Wishart D (2014). CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res 42, W94–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranha MM, Cortez-Pinto H, Costa A, da Silva IB, Camilo ME, de Moura MC, and Rodrigues CM (2008). Bile acid levels are increased in the liver of patients with steatohepatitis. Eur. J. Gastroenterol. Hepatol 20, 519–525. [DOI] [PubMed] [Google Scholar]
- Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min HK, Mirshahi F, et al. (2016). A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol 65, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeck C, Wehr A, Karlmark KR, Heymann F, Vucur M, Gassler N, Huss S, Klussmann S, Eulberg D, Luedde T, et al. (2012). Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61, 416–426. [DOI] [PubMed] [Google Scholar]
- Bansal T, Alaniz RC, Wood TK, and Jayaraman A (2010). The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 107, 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti E, Pinzani M, and Tsochatzis EA (2016). The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 65, 1038–1048. [DOI] [PubMed] [Google Scholar]
- Chow MD, Lee YH, and Guo GL (2017). The role of bile acids in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mol. Aspects Med 56, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day CP (2011). Non-alcoholic fatty liver disease: a massive problem. Clin. Med. (Lond.) 11, 176–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowman JK, Tomlinson JW, and Newsome PN (2010). Pathogenesis of non-alcoholic fatty liver disease. QJM 103, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdzik A, Dziedziejko V, and Kurzawski M (2014). IL-1 and TNF-α regulation of aryl hydrocarbon receptor (AhR) expression in HSY human salivary cells. Arch. Oral Biol 59, 434–439. [DOI] [PubMed] [Google Scholar]
- Endo M, Masaki T, Seike M, and Yoshimatsu H (2007). TNF-alpha induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c). Exp. Biol. Med. (Maywood) 232, 614–621. [PubMed] [Google Scholar]
- Eslam M, Valenti L, and Romeo S (2018). Genetics and epigenetics of NAFLD and NASH: clinical impact. J. Hepatol 68, 268–279. [DOI] [PubMed] [Google Scholar]
- García-Ruiz I, Solís-Muñoz P, Ferna´ndez-Moreira D, Muñoz-Yagüe T, and Solı´s-Herruzo JA (2015). In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis. Model. Mech 8, 183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharavi N, and El-Kadi AO (2005). Down-regulation of aryl hydrocarbon receptor-regulated genes by tumor necrosis factor-alpha and lipopolysaccharide in murine hepatoma Hepa 1c1c7 cells. J. Pharm. Sci 94, 493–506. [DOI] [PubMed] [Google Scholar]
- Ghazalpour A, Cespedes I, Bennett BJ, and Allayee H (2016). Expanding role of gut microbiota in lipid metabolism. Curr. Opin. Lipidol 27, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JR, Weber LW, and Rozman K (1988). Tissue-specific alterations of de novo fatty acid synthesis in 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-treated rats. Arch. Toxicol 62, 146–151. [DOI] [PubMed] [Google Scholar]
- Haukeland JW, Dama˚s JK, Konopski Z, Løberg EM, Haaland T, Goverud I, Torjesen PA, Birkeland K, Bjøro K, and Aukrust P (2006). Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol 44, 1167–1174. [DOI] [PubMed] [Google Scholar]
- Hubbard TD, Murray IA, Bisson WH, Lahoti TS, Gowda K, Amin SG, Patterson AD, and Perdew GH (2015). Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep 5, 12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin UH, Lee SO, Sridharan G, Lee K, Davidson LA, Jayaraman A, Chapkin RS, Alaniz R, and Safe S (2014). Microbiome-derived tryptophan metabolites and their aryl hydrocarbon receptor-dependent agonist and antagonist activities. Mol. Pharmacol 85, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KA, Gu W, Lee IA, Joh EH, and Kim DH (2012). High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS ONE 7, e47713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshman MR, Campbell BS, Chirtel SJ, and Ekarohita N (1988). Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on de novo fatty acid and cholesterol synthesis in the rat. Lipids 23, 904–906. [DOI] [PubMed] [Google Scholar]
- Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, et al. (2013). Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 62, 1787–1794. [DOI] [PubMed] [Google Scholar]
- Lee JH, Wada T, Febbraio M, He J, Matsubara T, Lee MJ, Gonzalez FJ, and Xie W (2010). A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 139, 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, and Sanyal AJ (2013). The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol 10, 686–690. [DOI] [PubMed] [Google Scholar]
- Malhi H, Bronk SF, Werneburg NW, and Gores GJ (2006). Free fatty acids induce JNK-dependent hepatocyte lipoapoptosis. J. Biol. Chem 281, 12093–12101. [DOI] [PubMed] [Google Scholar]
- Manteiga S, and Lee K (2017). Monoethylhexyl phthalate elicits an inflammatory response in adipocytes characterized by alterations in lipid and cytokine pathways. Environ. Health Perspect 125, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Masciana` R, Forgione A, Gabrieli ML, Perotti G, et al. (2009). Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 49, 1877–1887. [DOI] [PubMed] [Google Scholar]
- Murphy EA, Velazquez KT, and Herbert KM (2015). Influence of high-fat diet on gut microbiota: a driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 18, 515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy SR, Sanborn JR, Hammock BD, and Denison MS (2002). Development of a green fluorescent protein-based cell bioassay for the rapid and inexpensive detection and characterization of ah receptor agonists. Toxicol. Sci 65, 200–210. [DOI] [PubMed] [Google Scholar]
- Pagadala M, Kasumov T, McCullough AJ, Zein NN, and Kirwan JP (2012). Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol. Metab 23, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang J, Xi C, Huang X, Cui J, Gong H, and Zhang T (2016). Effects of excess energy intake on glucose and lipid metabolism in C57BL/6 mice. PLoS ONE 11, e0146675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelclova´ D, Fenclova´ Z, Preiss J, Procha´zka B, Spa´cil J, Dubska´ Z, Okrouhlı´k B, Luka´s E, and Urban P (2002). Lipid metabolism and neuropsy- chological follow-up study of workers exposed to 2,3,7,8-tetrachlordibenzo-p-dioxin. Int. Arch. Occup. Environ. Health 75 (Suppl), S60–S66. [DOI] [PubMed] [Google Scholar]
- Reid DT, and Eksteen B (2015). Murine models provide insight to the development of non-alcoholic fatty liver disease. Nutr. Res. Rev 28, 133–142. [DOI] [PubMed] [Google Scholar]
- Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, Fantoni LI, Marra F, Bertolotti M, Banni S, et al. (2009). Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol 24, 830–840. [DOI] [PubMed] [Google Scholar]
- Schilling JD, Machkovech HM, He L, Sidhu R, Fujiwara H, Weber K, Ory DS, and Schaffer JE (2013). Palmitate and lipopolysaccharide trigger synergistic ceramide production in primary macrophages. J. Biol. Chem 288, 2923–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schippa S, and Conte MP (2014). Dysbiotic events in gut microbiota: impact on human health. Nutrients 6, 5786–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz EA, Zhang WY, Karnik SK, Borwege S, Anand VR, Laine PS, Su Y, and Reaven PD (2010). Nutrient modification of the innate immune response: a novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler. Thromb. Vasc. Biol 30, 802–808. [DOI] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, and Flier JS (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest 116, 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, O’Maille G, Want EJ, Qin C, Trauger SA, Brandon TR, Custodio DE, Abagyan R, and Siuzdak G (2005). METLIN: a metabolite mass spectral database. Ther. Drug Monit 27, 747–751. [DOI] [PubMed] [Google Scholar]
- Sridharan GV, Choi K, Klemashevich C, Wu C, Prabakaran D, Pan LB, Steinmeyer S, Mueller C, Yousofshahi M, Alaniz RC, et al. (2014). Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat. Commun 5, 5492. [DOI] [PubMed] [Google Scholar]
- Stojsavljevic S, Gomerc-ic, Palc-ic M, Virovic, Jukic L, Smirc-ic, Duvnjak L, and Duvnjak M (2014). Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol 20, 18070–18091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, and Zimmermann HW (2014). Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol 60, 1090–1096. [DOI] [PubMed] [Google Scholar]
- Tanos R, Murray IA, Smith PB, Patterson A, and Perdew GH (2012). Role of the Ah receptor in homeostatic control of fatty acid synthesis in the liver. Toxicol. Sci 129, 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernocchi P, Del Chierico F, and Putignani L (2016). Gut microbiota profiling: metabolomics based approach to unravel compounds affecting human health. Front. Microbiol 7, 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Sunaga H, Miyata K, Shirasaki H, Uchiyama Y, and Shimba S (2016). Aryl hydrocarbon receptor plays protective roles against high fat diet (HFD)-induced hepatic steatosis and the subsequent lipotoxicity via direct transcriptional regulation of Socs3 gene expression. J. Biol. Chem 291, 7004–7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, and Ting JP (2011). Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol 12, 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, and Siuzdak G (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. USA 106, 3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BB, Van Benschoten AH, Cimermancic P, Donia MS, Zimmermann M, Taketani M, Ishihara A, Kashyap PC, Fraser JS, and Fischbach MA (2014). Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 16, 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Tzur D, Knox C, Eisner R, Guo AC, Young N, Cheng D, Jewell K, Arndt D, Sawhney S, et al. (2007). HMDB: the Human Metabolome Database. Nucleic Acids Res 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf S, Schmidt S, Mu€ller-Hannemann M, and Neumann S (2010). In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinformatics 11, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Cheruku PS, Bazer FW, Safe SH, and Zhou BY (2013). Investigation of macrophage polarization using bone marrow derived macrophages. J. Vis. Exp 76 10.3791/50323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhao Y, Xu C, Hong Y, Lu H, Wu J, and Chen Y (2014). Association between serum free fatty acid levels and nonalcoholic fatty liver disease: a cross-sectional study. Sci. Rep 4, 5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Degroot DE, Hayashi A, He G, and Denison MS (2010). CH223191 is a ligand-selective antagonist of the Ah (Dioxin) receptor. Toxicol. Sci 117, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, and Gill SR (2013). Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology 57, 601–609. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.