

Abstract
Decay of transcribed mRNA is a key determinant of steady state mRNA levels in cells. Global analysis of mRNA decay in cultured cells has revealed amazing heterogeneity in rates of decay under normal growth conditions, with calculated half-lives ranging from several minutes to many days. The factors that are responsible for this wide range of decay rates are largely unknown, although our knowledge of trans-acting RNA binding proteins and non-coding RNAs that can control decay rates is increasing. Many methods have been used to try to determine mRNA decay rates under various experimental conditions in cultured cells, and transcription inhibitors like actinomycin D have probably the longest history of any technique for this purpose. Despite this long history of use, the actinomycin D method has been criticized as prone to artifacts, and as ineffective for some promoters. With appropriate guidelines and controls, however, it can be a versatile, effective technique for measuring endogenous mRNA decay in cultured mammalian and insect cells, as well as the decay of exogenously-expressed transcripts. It can be used readily on a genome-wide level, and is remarkably cost-effective. In this short review, we will discuss our utilization of this approach in these cells; we hope that these methods will allow more investigators to apply this useful technique to study mRNA decay under the appropriate conditions.
Keywords: transcription shut-off, mRNA turnover, post-transcriptional gene expression, RNA-binding proteins, Drosophila cells
1. Introduction
Inhibition of gene transcription using actinomycin D (ActD) is a widely used, classical technique in biochemistry that, when applied appropriately can be a powerful tool for measuring messenger RNA (mRNA) decay rates. However, use of the drug has developed a somewhat checkered reputation and has been supplanted for some applications by newer techniques. We believe that part of the reason for its mixed reputation is its improper use, and incomplete appreciation of its advantages and limitations. In this review, we will describe in detail the use of this inhibitor in studies from our laboratories using cultured cells from Drosophila melanogaster and vertebrates. Our hope is that other investigators will be able to use these methodological details in successful applications of this technique. We refer the reader to several recent reviews that have discussed specific aspects of this technique, along with a discussion of alternative transcriptional inhibitors [1–6]. Our discussion will be limited to Drosophila and vertebrate tissue culture cells, since, to our knowledge, ActD has not proven to be useful as a transcriptional inhibitor in commonly used laboratory yeast species, nor is it widely used in Dictyostelium discoideum.
2. General considerations: Actinomycin D
ActD is an antibiotic and antineoplastic compound derived from Streptomyces parvullus that is comprised of phenoxazine connected to two cyclic peptides. It is used clinically in the treatment of childhood tumors, as well as choriocarcinoma in women. Its mechanism of action is complex, but its major mechanism is thought to involve direct binding to DNA, mainly through intercalating between guanine-cytosine pairs. The net result is inhibition of RNA polymerases and decreased transcription [7, 8]. This global inhibition of transcription is presumably one of the major mechanisms behind its effective use as an antitumor compound, ideally resulting in preferential death of tumor cells. Its ability to rapidly shut off transcription in cultured cells makes it an extremely useful experimental tool for studying RNA stability within a short timeframe, taking into consideration the ultimate cytotoxicity of the compound after longer exposures [9].
We will focus here on the use of ActD to rapidly shut off transcription in cultured metazoan cells. Although certain types of cultured cells are resistant to the actions of ActD [10], presumably at the level of transport and cellular uptake, most commonly used cultured cells seem to be susceptible. Use of ActD to shut off mRNA transcription is widely used to study the effects of different cellular perturbations on the decay rates of endogenous mRNAs. Such perturbations include permanent genetic modifications of the cells, such as the use of cells derived from mice deficient in one or more trans-acting mRNA regulatory factors, or cells in which mRNA sequences have been modified to include or delete cis-regulatory elements. Alternatively, mRNA stability can be assessed with more transient modifications, such as knockdown or overexpression of mRNA turnover-modifying proteins or RNAs; effects of external agents, such as hormones, environmental agents, or enzyme inhibitors; and comparisons of mRNA decay in the same cell type over time and/or differentiation status, or between different cell types. ActD is also widely used to study the decay rates of exogenously expressed mRNAs, with certain caveats (discussed below) about the susceptibility of commonly used vector promoters to ActD. Given the proper use of conditions and controls, there is vast potential for this technique to provide valuable information on mRNA decay rates in response to diverse genetic modifications and/or external stimuli.
Before describing the specific experimental procedures used in our laboratories, we will describe some general cautions to consider before embarking on these types of experiments. First, ActD is toxic (see package insert from Sigma-Aldrich for product A1410 for specific toxicities and precautions), and appropriate care should be exercised when handling. It is also very light sensitive and hygroscopic, and should be stored in the dark in high concentration stock solutions and diluted only prior to use, if possible; while not ideal, frozen ActD aliquots can be stored in amber tubes (to block light) at −20°C for up to one month. The manufacturer recommends solubilizing it in acetonitrile, acetone or dimethyl sulfoxide (DMSO), but we have found that it is also quite soluble and stable in water, as long as dissolution takes place in the cold and dark and sufficient time is allowed (see below). PubChem states that 1 gram dissolves in about 8 mL of ethanol and 25 mL of water at 10° C (https://pubchem.ncbi.nlm.nih.gov/compound/actinomycin_D#section=Solubility).
Another important general consideration is that commonly used cultured cells exhibit differing sensitivities to the cytotoxic effects of ActD [9]. For example, [11] found that HeLa cells were extremely sensitive to rapid ActD-induced cell death, even at concentrations that are widely used today in cultured cells (1-10 μg/ml). This potential differential sensitivity of different cells types suggests that a time course of cytotoxicity at several concentrations of ActD should be performed in each new cell type proposed for study, and that time courses of mRNA decay should take this eventual cell toxicity into account. In general, we recommend the shortest possible time courses of ActD treatment, so as to avoid secondary effects of cytotoxicity. In practice, we try to limit time courses in most cells to four hours, and try to use shorter times if possible. The extremely rapid cellular uptake and onset of action of ActD [3] make this type of short time course feasible.
However, a practical consequence of this recommendation in favor of short time courses is that many cellular mRNAs will be too stable for this technique to be useful in determining their decay rates. For example, in a recent study of human diploid fibroblasts grown in normal, serum-containing medium at about 80% confluence, we found using ActD (5 μg/ml) and Affymetrix microarrays that only 4,992 of 54,613 original probe sets (9.1%) decayed by > 25% by 4 hours; this would equate to mRNA half-lives of < 8 hours [12]. This means that in practice it would be difficult to determine differences in stability caused by, for example, genetic manipulations, of more than 90% of the fibroblast transcripts. We have noticed in the literature examples of attempts to use ActD to determine mRNA stability differences in very stable mRNAs, i.e., with estimated half-lives of > 8 hours, and we do not believe that this is an appropriate technique to use for these kinds of stable transcripts. An exception is when a given mRNA or group of mRNAs is going to be used as control, for example, as denominators in NanoString experiments; in those cases, it is also useful to establish by another technique, for example, northern blotting, that a given transcript is stable in response to ActD. Taking examples from our own recent experiments, we have found that Actb and Gapdh m RNAs were extremely stable in mouse trophoblast stem cells [13], and the human equivalents were extremely stable in human diploid fibroblasts [12].
Conversely, we have found that it is important to identify very unstable mRNAs in a given experimental system. These should be used as additional internal controls to ensure that the ActD is working properly and rapidly. For example, in mouse trophoblast stem cells, we used Fos and Myd116 mRNAs as examples of rapidly degrading mRNAs [13]. In human diploid fibroblasts, we found that the following mRNAs were the most rapidly degraded following ActD treatment, and we used these for internal controls: DUSP1, CYR61, SGK1 and DUSP6 [12]. In mouse bone marrow-derived macrophages (BMDM), we used Dusp2 and Socs3 mRNAs as reasonably well-expressed and very unstable mRNAs [14]. In that last BMDM experiment, we found that Dusp2 mRNA was already decreased by 50% 10 min after adding ActD, and by nearly 100% within 30 min; this is very reassuring evidence that the ActD is acting immediately and effectively.
Another general practical concern is that transcript stability can change in response to cellular stimulation, growth medium, cellular confluence, genetic factors, and probably many other factors. We found recently that decay patterns exhibited by a given transcript, measured after ActD treatment, were different at different times after LPS stimulation of primary mouse BMDM [14]. We have also found that certain transcripts that are quite stable under steady state growth conditions in cultured cells are very unstable after short-term stimulation with, for example, LPS in macrophages.
Finally, an important consideration concerns the use of ActD in cellular transfection experiments, particularly when the stability of transiently expressed mRNA is measured. On one hand, some prokaryotic promoters are not only resistant to ActD, but are actually activated by it [1]. We have found that the CMV promoters commonly used in mammalian transfection experiments are resistant to ActD, at least at the concentrations we normally use, so that use of ActD to determine turnover of transcripts in transfection experiments often requires the use of eukaryotic promoters determined to be sensitive to the inhibitor. In mammalian cell transfection experiments, we have relied on the use of a promoter that normally drives expression of the protein MARCKS-L1; this promoter is sensitive to ActD, and its expression in mammalian cells is normally quite high [15]. In Drosophila cells, we routinely use the constitutive actin 5c promoter, which similarly yields high expression and is sensitive to transcription shut off with ActD. Nonetheless, a disturbing feature of these types of experiments is that turnover rates of a given transcript after transfection and expression in standard tissue culture cells are often quite different from those seen with the endogenous transcript in other cell types. Part of this difference may stem from the high level of expression often used in these types of transfection experiments, compared to endogenous levels of expression, but there are undoubtedly many other factors at play.
2.1. General considerations: Measuring mRNA concentrations.
The ultimate success of an ActD experiment is dependent on the methods used to quantitate cellular RNA. In general, for genome-wide analysis of mRNA levels after treatment of cells with ActD, microarrays or RNA-Seq are used. These are useful techniques for initial genome-wide surveys; examples of their recent use for this purpose from our laboratories include [12, 16] for microarrays, and [13] for RNA-Seq. Detailed descriptions of these techniques are beyond this scope of this review, but the reader is referred to some recent general reviews [17–19]. One disadvantage is that both techniques are relatively expensive and time consuming; however, they are essentially the only techniques that can offer the major advantage of genome-wide scale. For confirmation and lower-throughput analyses, we and many other investigators have relied on northern blotting, real-time RT-PCR, and solution hybridization techniques like NanoString. Each of these methods has advantages and disadvantages, and some of these are described in recent reviews (reviewed in [20–25]). For example, northern blotting is probably the “gold standard” for its ability to determine mRNA concentrations accurately, to not require amplification, and to also display decay intermediates that often provide valuable information (see below). However, it is obviously very low throughput, relies on phosphorimager quantitation, and often is dependent on the total cellular RNA concentration in the sample for normalization. This works quite well for ActD decay experiments, however, since ribosomal RNA contributes to much of the measured cellular RNA concentration, resulting in less upward “drift” of stable transcripts than is seen, for example, with RNA-Seq. Expression data can also be normalized by quantitating the expression of one or more stable control transcripts in the same sample, often insisted upon by reviewers.
Real-time RT-PCR relies on amplification and is often used with a single denominator transcript, both of which can introduce inaccuracies, but is rapid and relatively inexpensive [23, 24]. Numerous authors have weighed in on the best approaches to conducting and analyzing these types of experiments, and some of these considerations are discussed in a recent review [26]. Finally, we have recently used “digital mRNA profiling”, involving the NanoString reagents and methods [25, 27, 28], to quantitate mRNA levels after ActD. This method uses nCounter technology in solution to hybridize each transcript with sequence-specific, bar-coded capture probes and reporter probes, and then reads out the probe-RNA complexes with a fluorescent detector. A major advantage of this technique is that no amplification is involved. Other advantages include rapid turnaround, the relatively high throughput, and digital output. Disadvantages include the need to develop of a custom “codeset” for most specific applications, the need for a cell-specific set of unchanging transcripts that can be used as the denominators in the final concentration calculations, and the expense relative to real-time RT-PCR and northern blotting.
2.2. General considerations: Expressing and comparing results.
A topic subject to much debate concerns how best to express the results of ActD experiments and compare them statistically. Our approaches to this are described below, but it is fair to say that no single approach has emerged as ideal. Many papers attempt to calculate mRNA half-lives, but in our experience cellular mRNAs rarely decay with classical first-order kinetics, which is perhaps unsurprising for what is presumably a multi-step process. In contrast to experiments involving shutoff of a single promoter in an otherwise normal cell, the use of ActD complicates matters because the inhibitor is also blocking transcription and synthesis of transcripts encoding mRNA decay-promoting and inhibiting factors. Attention to several factors can make the results of such experiments more convincing, including: comparing decay curves between otherwise identical cells after changing a single variable, like the genetic origin of the cells; using adequate numbers of biological replicates, in addition to technical replicates; using rapidly degrading and stable positive and negative control transcripts as internal controls; and normalizing the starting value to 100%, if experimental conditions warrant. Examples of our specific approaches are shown in the sections that follow.
3. ActD in mammalian cell cultures
To illustrate the usefulness of ActD incubation in evaluating mRNA stability in mammalian cell cultures, we will use as examples our standard techniques in mouse embryo fibroblasts (MEF) and mouse bone marrow derived macrophages (BMDM). Since we are often trying to determine whether a specific genetic perturbation has resulted in changes in mRNA decay patterns of target transcripts for members of the tristetraprolin (TTP) family of mRNA binding and destabilizing proteins [29], we will refer to examples from our work in that area.
3.1. Primary BMDM
We normally culture the primary BMDM in 10-cm plates, to provide sufficient RNA for many types of analysis. Depending on experimental needs, we sometimes grow them in 6-cm or 6-well plates. Briefly, bone marrow was obtained from mouse femurs, and BMDM were cultured in 10-cm plates in macrophage growth medium (RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), 30% L929 cell culture supernatant, 100 U/ml penicillin, 100 μg/ml streptomycin and 6 mM L-glutamine). When the cells reached 70-80% confluency, the growth medium was replaced with RPMI1640 medium, supplemented with 1% FBS and the antibiotics, and incubated for 16 to 24 h.
3.2. Primary MEF
Primary MEF were prepared from E14.5 embryos, as previously described [14, 30], and cultured in DMEM medium supplemented with 10% FBS, 2 mM L-glutamine and antibiotics in 10-cm plates. We generally use MEF at relatively early passages (3-8). The cells (at 80 to 90% confluence) were prepared for experiments by incubating in DMEM medium supplemented with 0.5% FBS and antibiotics for 16 to 24 h.
3.3. Reagents
3.3.1. LPS (Sigma, catalogue number L6529):
Reconstituted to 1 mg/ml in DEPC treated water, store at −20°C for up to 6 months.
3.3.2. Recombinant mouse IL-1β (R&D systems):
Reconstituted to 10 ng/μl in PBS with 0.2% bovine serum albumin, store in aliquots at −80°C for up to 6 months.
3.3.3. Actinomycin D (ActD) (Sigma catalogue number A4262):
Carefully open the vial (pay attention to the rubber stopper, for a large amount of the reddish-orange powder often stays on the rubber stopper), add 5 ml DEPC treated water into the vial that contains 10 mg of ActD, carefully put back the stopper, seal tightly with Parafilm along the seam between the rubber stopper and the vial, wrap the vial in aluminum foil, and store the vial upside down at 4°C for 4-7 days. At this point the ActD is entirely in solution (2 mg/ml) and can be kept at 4°C for up to 1 year. We view the extended incubation period as one of the most important steps, since no solvents other than water are used; however, short-term attempts to dissolve ActD in water are unlikely to work, and will result in variable concentrations of the active compound.
3.4. Cell treatment
3.4.1. Agonist stimulation of mRNA accumulation.
Many experiments with ActD are conducted in cells under normal culture conditions, sometimes pre-defined with, for example, restrictions on the extent of cell confluence. An example of an experiment like this is in [12], in which we added ActD to human diploid fibroblasts under normal growth conditions that had achieved a certain percentage of confluence. In that experiment, we screened ActD time courses by microarray, and then focused on specific transcripts that were assayed by real-time RT-PCR. In a second such experiment, we added ActD to cultured mouse trophoblast stem cells without stimulation, screening with RNA-Seq before confirming the quantitation of selected transcripts by NanoString [13]. In both cases, control and experiment cells differed by one critical element, e.g., arsenic treatment in the former study, and a genetic knockout in the latter. In these and similar cases, two-way comparisons of cells of control and treatment groups treated in parallel often yield clear cut differences in decay rates between experimental groups.
Another common experimental manipulation that we have used extensively is based on the fact that one of our favorite genes, Zfp36, encoding the protein tristetraprolin or TTP, is highly inducible in cultured cells, and we are often interested in comparing the effect of highly induced TTP in cells with the effect of knocking out TTP in parallel. Before attempting to determine the decay profile of a transcript after its expression is induced by some sort of agonist in cell culture (e.g., by LPS in macrophages), it is preferable first to perform one or more time-course experiments to characterize the mRNA levels after different times of accumulation (see, for example, [14]). In our experiments, in which we are often investigating the effect of an RNA binding and destabilizing protein, TTP, on mRNA decay, the main purposes of these time-course experiments are to identify the profiles of potential or known mRNA targets of TTP, including their times of peak expression, and to characterize the peak times of TTP protein expression. In practice, since many transcripts are usually evaluated at one time, and TTP protein may peak at a different time, it is sometimes necessary to pick an “average” time for ActD treatment that is close to the maxima for several transcripts of interest as well as the “perpetrator” protein. If resources permit, or the experimental plan dictates it, the stimulation time course can be stopped with ActD at several different times after the initial stimulation, as we have recently done with LPS-stimulated BMDM [14].
3.4.2. Time points after ActD.
After the pre-defined treatment length, we then treat the cells with ActD and isolate total cellular RNA at intervals, generally to compare the effects of the genetic modification on mRNA decay rates. To illustrate details of one of our standard protocols, we will use the example of treating the MEFs with IL-1β for 30 min, followed by a time course after ActD treatment. This experiment required 23, 10 cm plates of MEF of each genotype, prepared as described above. We used 4 plates each for untreated cells (without IL-1β or ActD), and 4 plates each for cells treated for 30 min with IL-1β (without ActD); we then needed 15 plates pretreated for 30 min with IL-1β, 3 of which were used at each of the following ActD incubation times: 10, 20, 30, 60, and 120 min.
3.4.3. Protocol
For reproducible results, it is important to incubate the cells in IL-1β and ActD for the exact intended times, since at least some transcripts of interest can decay very rapidly, and small changes in timing can greatly affect reproducibility. To minimize the chance of making mistakes, separate the plates of cells into groups corresponding to each time point (e.g., IL-1β only, IL-1β + actD 10 min, and so on) before the actual experiment. It is very helpful to use repeater pipets (e.g., Eppendorf Repeater pipette, and pipette tips of different sizes) to apply reagents to the cells in culture. In general, for cells to be incubated with ActD after agonist stimulation, there is no need to change the culture medium before adding ActD, i.e., it is fine to leave the agonist in the medium. This minimizes variability that would undoubtedly be introduced by a complete change of medium.
On the day before the experiment, we apply our usual serum-deprivation protocol to induce cell quiescence. To do this, rinse the cell monolayers with 5 ml of DMEM, and incubate each 10 cm plate of cells in 10 ml of 0.5% FBS/DMEM with antibiotics for the desired length of time (usually overnight). To start the cellular stimulation, add IL-1β (final concentration in culture: 10 ng/ml) to one group of plates, and wait for 50 sec to before adding IL-1β to the next group. Beginning with the IL-1β + ActD 120 min group, quickly add 10 μl of 10 ng/μl IL-1β to each of these six plates (3 plates of each genotype) using a repeater pipet, and start the 30 min timer. Tilt the plates side to side carefully to mix in the agonist, and return these plates to the incubator. Then, apply the same procedure for the IL-1β + ActD 60 min group, the IL-1β + ActD 30 min group, the IL-1β + ActD 20 min group, and finally the IL-1β + ActD 10 min group. After returning the IL-1β + ActD 10 min group to the incubator, wait for 3 min to lapse, then add IL-1β to the 8 plates (4 plates of each genotype) of the IL-1β only group, start the 30 min timer and return these plates to the incubator.
For the ActD incubation (final concentration in culture: 5 μg/ml), it is important to keep the ActD in the dark as much as possible; we actually dim the lights in the work room while working with ActD, given its light sensitivity. It is also toxic, so wear gloves. At the end of the 30 min IL-1β treatment period for the IL-1β + ActD 120 min group, quickly add 25 μl of 2 mg/ml ActD to each of these six plates using a repeater pipet, starting the 120 min timer. Tilt the plates carefully side to side to mix in the reagent, and return these plates to the incubator. Take the IL-1β + ActD 60 min group out of the incubator, wait until the end of the incubation with the IL-1β (in about 30 to 40 sec), quickly add ActD to this group, and start the 60 min timer. Apply the same procedure for the IL-1β + ActD 30 min group, the IL-1β + ActD 20 min group, and finally the IL-1β + ActD 10 min group. After returning the IL-1β + ActD 10 min group to the incubator, there should be about 2 min remaining before the 30 min incubation will end for the IL-1β only group. Make sure that there will be ice trays and ice-cold PBS available for stopping the reactions.
3.4.4. Isolation of total cellular RNA.
At the end of each group’s final incubation, quickly pour off the culture medium from each plate into a waste vessel, and place the plates on a tray of ice. Pour about 10 ml of ice-cold PBS into each plate to stop the reaction. Discard the cold PBS and try to aspirate all of the PBS off the plates. Add 0.5 ml of Lysis Buffer (containing 1% (v/v) ρ-mercaptoethanol) to each plate (Illustra RNAspin mini Isolation Kit, GE Healthcare), and make sure the Lysis Buffer covers all the cells by tipping the plates carefully side to side.
After all cells from a given group are stopped and lysed, transfer the lysate from each plate to one RNAspin Mini filter unit, centrifuge, and collect the filtrate. We generally pool filtrates from three or four plates of identical genotype and treatment conditions. At this point we either proceed to RNA extraction and DNase treatment, following the manufacturer’s protocol, or store the filtrates at −80°C for up to a few months until RNA can be extracted. We generally use two RNAspin Mini columns to extract RNA from filtrates from pooled lysates from three or four plates of cells.
3.5. mRNA quantitation.
The focus of this methods review is on the optimal use of the ActD technique in the determination of mRNA decay in cultured cells. Obviously, a key element in these experiments is the final quantitation of mRNA levels. We have used several different techniques for this, including microarray, RNA-Seq, NanoString, real-time RT-PCR, and northern blotting, and a detailed discussion of all of them is beyond the scope of this review. All of them have inherent advantages and disadvantages, as noted above. In general, we prefer to use screening techniques such as microarrays and RNA-Seq for genome-wide studies, and then use the other techniques to focus on smaller numbers of transcripts for verification and quantitation.
Some of these techniques have disadvantages when applied to the specific situation of measuring mRNA levels after transcription shutoff. For example, when using RNA-Seq or microarrays to survey transcriptome-wide mRNA levels, it should be kept in mind that large numbers of transcripts begin to decay immediately upon addition of ActD, so the denominator, often total reads in the sample, will be decreasing in absolute terms. This means that the commonly applied normalization techniques will often show a gradual increase with time in the levels of stable mRNAs. It is possible to try to normalize the values of interest to the values from a group of housekeeping transcripts, but in our hands this has not helped the problem of denominator drift very much. In general, we feel that it is preferable to compare two different experimental conditions in the same experiment, e.g., parallel experiments in WT vs. mutant cells, so the differences between the decay curves will persist, even in the setting of a drifting baseline.
An example of such an experiment is shown in Fig. 1A, which shows data from an RNA-Seq experiment of MEF stimulated with Il1β for 30 minutes, and then treated with ActD, with samples removed at intervals thereafter. In Fig. 1A, each data point represents the mean of three biological replicates. Samples from WT MEF are indicated by black triangles and a solid black line, and from TTP zinc finger point mutant knock-in (KI) MEF as open red triangles and a red dashed line. These cells express a mutated form of TTP that does not bind mRNA and is therefore inactive. The fraction of mRNA remaining was calculated relative to initial levels at the time of ActD addition (set to 1). A detailed description of the experiment is contained in [14]. The two left-most panels show two very labile mRNAs that we often use as internal controls for ActD activity in this cell type, Dusp1 and Nfkbia. In both cases, the mRNA had essentially disappeared by 1 h, with the curves from the two genotypes being essentially superimposable. This extremely rapid initial decay, with about 50% disappearance by 10 min in the case of Dusp1 mRNA, is very encouraging evidence that the ActD is acting rapidly and effectively. At the other extreme, in the two rightmost panels, are two abundant mRNAs that are often stable in our experiments, depending on the cell type: Gapdh and Rpl14 mRNAs. Again, the WT and KI curves are superimposable, but data from these stable mRNAs demonstrate the type of baseline drift that is common in RNA-Seq experiments.
Fig. 1. mRNA decay in MEF.
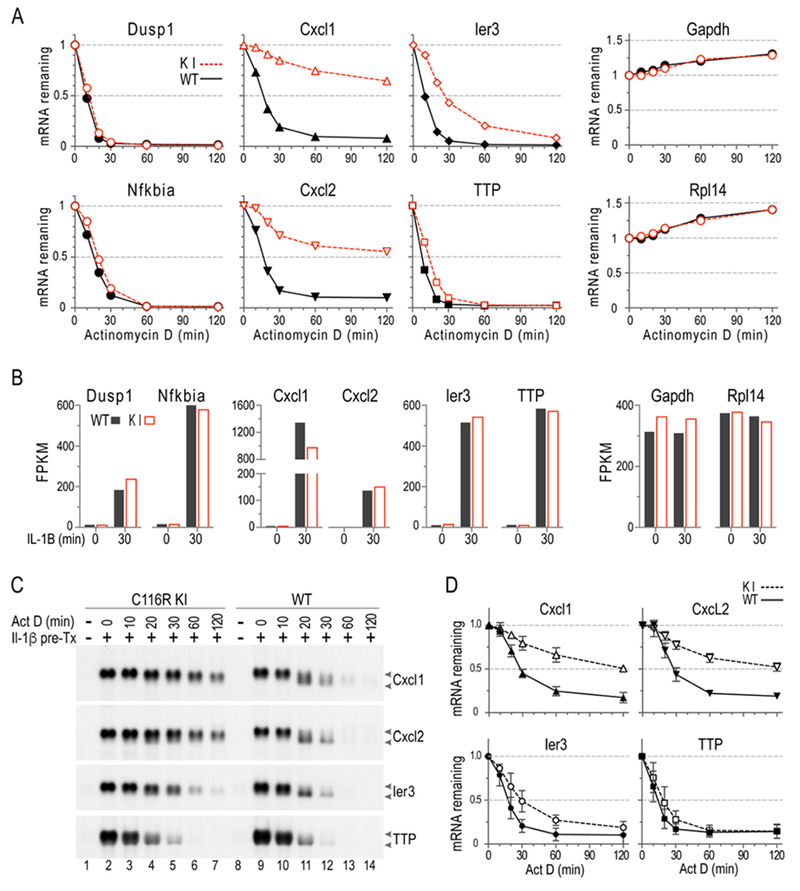
MEF were pretreated with IL-1α (10 ng/ml) for 30 min, followed by addition of ActD; samples were then harvested at the indicated times. Northern blots were hybridized to Cxcl1, Cxcl2, Ier3, or TTP cDNA probes. (A) Decay curves are shown of RNAseq data, using a subset of the same RNA samples used in B. Each point represents the average of three biological replicates, in all cases shown as a fraction of the initial sample. (B) Shows northern blots from a representative experiment. The mRNA migration positions are indicated by arrows, with the upper arrow pointed to the fully polyadenylated species, and the lower arrow pointing to the deadenylated species. (C) Decay curves are shown, with means plotted as fractions of the original mRNA levels (mean ± SD). These data are from northern blots of the type shown in B, and the data represent probe-bound mRNA volumes generated from three independent experiments like the one shown in (B), using 3 pairs of WT and C116R littermates. (A) and (B) are adopted from [14], with permission.
Nonetheless, the data from the four internal panels in Fig. 1A, showing three TTP target mRNAs, demonstrate the ability of the ActD technique to distinguish between the decay curves of cells of two different genotypes. In all four cases, it is clear that the transcripts from the TTP KI cells decayed more slowly than in the WT cells, and these differences are easy to see (and usually statistically significant), despite the baseline drift in the stable mRNAs.
When these kinds of differences have been identified by an initial screen, such as RNA-Seq or microarray, they can be confirmed and quantitated by northern blotting, real-time RT-PCR, or NanoString. We have used all three methods, but in the section below, we will focus on the use of northern blotting in the verification of a small number of transcripts, because of some of its unique advantages in the identification of transcriptional and decay intermediates, among others. Northern blotting suffers from the same potential problem of “denominator decay” described above for RNA-Seq and microarray experiments, but it is generally not as severe, since the denominator for northern blotting is generally the total RNA concentration in the sample, not, for example, polyA+ selected mRNA. Other disadvantages include low throughput, the requirement for relatively large amounts of RNA, somewhat lower sensitivity than other techniques, and the use of radioactivity. In the example described below, we will describe the results of northern blotting to confirm, and add additional information to, the results from genome-wide screening techniques; we will also show results from northern blotting compared to NanoString as an example of two different confirmatory assays.
3.6. Northern blotting
If RNA quantities are sufficient, northern blotting is often advantageous because it does not require RNA amplification, and it can often uncover additional information not provided by other quantitation techniques. Because the electrophoretic process separates RNA by size in a denaturing gel, northern blots can not only detect the size and quantity of the mature mRNA, but they can also provide important information about, for example, mature mRNA vs. pre-mRNA, and fully polyadenylated vs. deadenylated mRNA intermediates that can accumulate during a decay experiment (for example, see [31]).
Fig. 1B and C demonstrate some of the advantages of northern blotting in this kind of experiment. The same RNA samples used for the RNA-Seq experiments shown in Fig. 1A were used in Figs. 1B and 1C. The examples shown in Fig. 1B demonstrate the accumulation of mature Cxcl1, Cxcl2, Ier3 and TTP mRNA in response to 30 min of IL-1β stimulation in MEF (Fig. 1B, compare lane 2 to lane 1 in C116R mutant cells, and lane 9 to lane 8 in WT cells). As noted above, the C116R mutation is a knock-in mutation in mouse TTP that prevents RNA binding, and the protein is therefore non-functional. In subsequent lanes of the time courses, northern blotting reveals the appearance of lower molecular weight intermediates of Cxcl1, Cxcl2, and Ier3 mRNAs in the WT cells, but not in the mutant cells, during the ActD treatment time-course (Fig. 1B, compare lanes 11-14 to lanes 3-7). These intermediate species probably represent semi-stable deadenylated mRNA species, as previously shown for GM-CSF mRNA [31]; these intermediates, while evident on northern blotting, would probably not have been detected by any other technique. The presence of these intermediates allowed us to conclude that the absence of functional TTP was preventing deadenylation of the target mRNAs. As seen in the later lanes of these blots, there is also clearly a slowing in the overall decay in the C116R mutant cells for all three of these fibroblast TTP target transcripts, Cxcl1, Cxcl2 and Ier3 mRNAs. If appropriate, decay curves generated in this way can be used to obtain the time at which the mRNA decreased to 50% of its original value (decay t50) during the ActD time-course, as analyzed by interpolation (GraphPad Prism 7) using non-linear regression [14].
Several identical experiments were quantitated using probe-bound mRNA volumes (ImageQuant, Molecular Dynamics) from a phosphorimager, and the results are plotted in Fig. 1C. These results confirmed the RNA-Seq results, and also allowed for the statistical determination that TTP mRNA decay was significantly slowed in the mutant cells, although the magnitude of the difference was obviously not very large [14].
Northern blotting can also reveal other details that might be missed by other techniques. For example, although Cxcl1 and Cxcl2 mRNAs appeared as single mRNA species after IL-1β stimulation in MEF (Fig. 1A, lanes 2 and 9) that persisted throughout the 24 h incubation (not shown), the Cxcl1 and Cxcl2 mRNA were each expressed as two different molecular weight species when the cells were stimulated with TNF [14]. In both WT and C116R mutant cells, this two-species appearance of Cxcl1 and Cxcl2 mRNA was seen as early as 15 min of TNF stimulation, and continued for 24 h (not shown). It is not clear what these lower molecular weight mRNA species represent and why they appeared when the cells were stimulated by TNF but not by IL-1β. It should be noted that in both the Cxcl1 (NM_008176.3) and Cxcl2 (NM_009140.2) mRNA, there are additional upstream potential polyadenylation signals, AUUAAA, that could produce shorter forms of these two mRNAs of approximately the sizes we observed after TNF stimulation.
In these ActD mRNA decay experiments, the normal molecular weight mRNA species of Cxcl1 or Cxcl2 decayed in a genotype-dependent manner (i.e., WT vs C116R mutant), and this pattern was the same as seen in the IL-1β stimulated cells (Fig. 1B). Interestingly, the lower molecular weight species of Cxcl1 and Cxcl2 mRNA decayed in a similar fashion in both WT and mutant cells, raising the possibility that at least some of the TTP family member target sequences in these transcripts that are present between the two polyadenylation signals are differentially affecting the decay of the longer but not the shorter versions of the mRNAs [14].
These types of observations, of possible agonist-specific alternative use of polyadenylation signals, and genotype-dependent differential decay of different size transcripts, would probably not have been observed with other standard mRNA quantitation techniques, such as real-time RT-PCR or NanoString.
Fig. 2 demonstrates that ActD can be used at different times after cell stimulation, depending on the time course of induction of an inducible gene. In this case, BMDM were stimulated with LPS for 1, 3 and 6 h, and ActD was added at the end of each incubation, and decay curves calculated, by both northern blotting (Fig. 2A) and NanoString (Fig. 2B). Fig. 2 is focused on the decay of Tnf mRNA, which exhibits an LPS time course that allows transcription to be stopped at these intervals [14]. In this case, it is clear that the differences in mRNA decay patterns between the two cell genotypes are present at all three times after LPS stimulation. The same samples were used in a NanoString experiment (Fig. 2B), which also used Dusp2 mRNA as a very rapidly degrading internal control. Both northern blotting and NanoString assays gave essentially the same results in this case, demonstrating that the mutant form of TTP was relatively ineffective in promoting the decay of the Tnf mRNA at all three times after its initial induction.
Fig. 2. mRNA decay in BMDM.
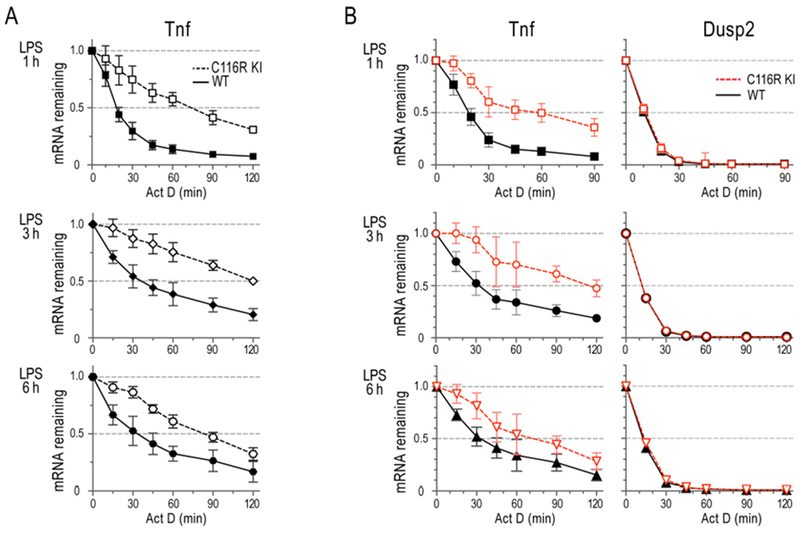
BMDM were pretreated with LPS (1 μg/ml) for 1 h, 3h, or 6h, as indicated to the left of the graphs, followed by incubation with ActD for the times indicated. Data are shown as fractions of the original mRNA levels (mean ± SD). (A) TNF decay curves shown are from data of northern blotting probe-bound mRNA volumes generated from five independent experiments, using five pairs of WT and C116R BMDM. (B) Decay curves shown are from NanoString data, using four sets of RNA samples similar to those used in (A). Adopted from [14], with permission.
Although the technique of northern blotting is not the focus of this review, we have included below some of the technical details that we normally use in this type of experiment, when phosphorimager quantitation of results is important. For probe labeling, we routinely use cDNA as templates, and α-[32P]-dCTP (3,000 Ci/mmol) random-primed (Prime-a-gene labeling system, Promega) body-labeled probes, to hybridize to northern blots [14, 32], although random prime incorporation of biotin-dNTP or a fluorophore-dNTP has gained popularity recently [33].
It is important to use an excess amount of labeled probe (5-8 × 106 cpm/ml of hybridization solution, 5 ml for a 10 × 10 cm membrane) to hybridize with northern blots, to complete hybridization. We also use constant conditions for prehybridization (in 50% formamide, 5X SSC (1X SSC is 150 mM NaCI, 15 mM sodium citrate (pH 7) , 50 mM sodium phosphate (pH 6.5)), 1X Denhardt’s solution (1X Denhardt’s solution is 0.02% BSA, 0.02% Ficoll 400, 0.02% polyvinylpyrrolidone), 0.5% SDS, and 0.15 mg/ml salmon sperm DNA, at 42 °C for at least 1h) and probe hybridization (to every 4 ml of pre-hybridization solution, add 1 ml of 50% Dextran sulphate, at 42°C overnight). After rinsing 3 times at room temperature with 1 X SSC and 0.1% SDS, we wash the blots 2 times in 0.1 X SSC and 0.1% SDS at 60°C. These conditions are suitable for most probe-hybridized blots, but some probes produce high backgrounds on the membrane and require higher temperatures (70°C or 75°C) for the final two washes.
For moderately abundant mRNAs, for example, TTP and TNF mRNAs from LPS-stimulated BMDM, or TTP and Ier3 mRNAs from IL-1β stimulated MEF, the optimal exposure time for a probe-hybridized northern blot to an X-ray film is usually 2-4 h at −80°C. The exposure time for Cxcl1 and Cxcl2 mRNAs from IL-1β-stimulated fibroblasts is usually 8 h to overnight. When we expose a northern blot to a phorsphorimager storage screen, the length of time required is about a third to a half of that required for an X-ray film.
Although a northern blot membrane can be stripped and hybridized with other probes for 4-5 times, the method is clearly low throughput, and not suitable for quantitating more than a handful of mRNAs at one time. The amount of total cellular RNA required per lane is also large (3 to 10 μg), further limiting the number of assays that can be performed in any given experiment. Northern blots are also less useful for quantitating the levels or decay patterns of low abundance mRNAs. Finally, the use of 32P-labeled probes requires additional considerations pertaining to the use of radioactive materials. Nonetheless, for obtaining certain types of information about mRNA species, levels and decay rates, northern blotting is unparalleled in the types of information that can be obtained.
4. Considerations for ActD use in Drosophila cells.
Described here is a method for using ActD in d.mel-2 cells, but these guidelines should be applicable for most, if not all types of cultured Drosophila cells. Drosophila Schneider-2 (S2) cells [34] (ATCC CRL-1963) and their derivatives (such as the d.mel-2 cells used in the method described here) are widely used to study gene regulation, including post-transcriptional control, in insects [35–38]. They are amenable to transfection and simple to maintain, as they can be grown at room temperature in the absence of serum. They are also not fully adherent, thus circumventing the requirement for trypsinization. S2 cells can also be efficiently transfected, making it possible to study regulators of interest through transient overexpression. For transfection of reporters and/or effectors, we use the FuGene HD (Promega) transfection reagent (described below).
In addition to the advantages in growth conditions, S2 cells are also a user-friendly, cost-effective system in which to knock down expression of specific genes and in which to perform RNAi screens. This advantage is conferred by the ability of S2 cells take in double-stranded RNAs (dsRNAs) via receptor-mediated endocytosis [39]. Thus, for RNAi, one can design one’s own short (|300-600 bp) double-stranded RNAs (dsRNA), which can be transcribed in vitro. These dsRNAs are then delivered to cells through the “bathing” method (DRSC/TRiP Functional Genomics Resources), without the need of transfection, further reducing the cost and labor of RNAi experiments. For dsRNA design principles and minimizing off-target effects, as well as suggested RNAi protocols and optimization, we refer the reader to the Drosophila RNAi Screening Center (https://fgr.hms.harvard.edu/). It should be noted that RNAi will likely require initial optimization experiments to determine conditions for the best knockdown efficiency; this time period will depend on the abundance and stability of the target protein.
Considerations for controls in insect cells are similar to those described above for mammalian cells. For internal control normalization, we use the stable, ActD-resistant 18S ribosomal RNA; other stable non-coding RNAs can be used, such as the 7SL RNA of the Signal Recognition Particle. One may also consider using an mRNA which would not be affected by the experimental conditions as an additional comparison, as described above. For measuring relative changes mRNA half-life using overexpressed effectors, we compare WT with an RNA-binding defective mutant. For RNAi experiments, knockdown of a regulatory factor is compared with a Non-targeting control (NTC), such as a dsRNA corresponding to the e. coli lacZ gene.
As there are many similarities between the ActD utilization in mammalian and insect cells, many of the considerations described in the above sections also apply to transcription shutoff in Drosophila cells; following RNA isolation, downstream applications and data analysis can be identical to that described mammalian cells. In this section, we will describe a basic protocol, highlighting key differences in using this method between the two cell types, such as culturing conditions and use of drug. Note that methods may need to be adapted slightly for adherent versus suspension cells.
4.1. Protocol for insect cells
4.1.1. Growth and maintenance
For general maintenance, d.mel-2 cells (ATCC CRL-1963) are grown in Sf-900 III serum-free media (SFM) (Gibco). While antibiotics are not necessary, we typically include 100 μg/ml Penicillin and 100 μg/ml Streptomycin for regular passaging. Note that these antibiotics should be omitted for transfections using Fugene HD. Prior to setting up transcription shut-off experiments, cells are grown for 1-2 days to a confluency of 5-20 million cells/ml. For transcription shut off, dilute 15.8 million d.mel-2 cells into a new T-75 flask to a total volume of 15.8 ml (1×106 cells/ml) in Sf-900 III SFM. Overexpression via transient transfection and/or RNAi via double-stranded RNAs can be performed at this time if desired, as described below.
4.1.2. Plasmid transfection
For transfection of plasmids, we use FuGene HD (Promega) transfection reagent, following the manufacturer’s guidelines. Per the manufacturer, the optimal ratio of reagent volume to total weight of DNA will need to be optimized. For d.mel-2 cells, we use a 4:1 ratio of volume of reagent (in pi) to total weight of DNA (in μg). Amounts of plasmid DNA effectors and reporters are determined empirically and can be scaled proportionally to the surface area of the growth vessel. For transient overexpression of Pumilio in a T-75 flask, we use 23.7 μg of total effector and 158 ng of Nanoluciferase (Nluc) reporter (this amount was scaled up from 6-well plate proportionally by surface area by a factor of 7.9).
4.1.3. RNAi
For RNAi, we transcribe 300-600 bp dsRNAs from DNA templates bearing T7 promoters on both strands using the HiScribe T7 High Yield RNA Synthesis Kit (NEB). DsRNAs are then treated with DNase (RQ1, Promega) and purified using the Clean and Concentrator kit (Zymo). Cells are then incubated with 12 μg of dsRNA per million cells (190 μg per T-75 flask), by simple addition of the dsRNA stock straight to the cell dilution and mixing well.
4.1.4. Transcription shut off
Incubate cells at 25°C for 3 days (note that RNAi may require longer incubation period depending on stability of target protein). Prepare a working solution of 39.5 μg/ml ActD in Sf-900 III SFM (diluted from a 1 mg/ml stock solution in DMSO), preparing enough for 2 ml media per T-75 flask. We prepare a stock solution of ActD in DMSO (in light safe amber tubes), and then dilute it into culture medium immediately before use. Before adding ActD, remove 2 ml of the cell suspension from each flask for the first time point. Then add 2 ml of the ActD working solution to each flask, for a final concentration of 5 μg/ml. For each time point, we remove 2.5 ml of cells (from the same flask) for RNA isolation, with 3.6 ml taken at the final time point; note that this volume can be adjusted for the needs of the specific experiment (for example, larger volumes of cells can be taken at later time points to compensate for cell death, or lower volume to take more time points). To collect cells for each time point, shake flask for approximately 5-10 seconds to dislodge cells, and carefully collect desired volume of culture. Then, pellet cells (1000 ×g for 3 minutes) and proceed to RNA extraction. We generally isolate RNA using the Maxwell RSC SimplyRNA cells or tissue kit (Promega), adhering to the manufacturer’s guidelines. DNase treatment is included with the Simply RNA Cells system and described by the manufacturer; however, an additional DNase step may be performed after RNA isolation, if desired. RNA can then be used for quantitation by one or more of the methods described above for mammalian cells, and the experimenter can proceed to downstream applications, such as RT-qPCR and/or northern blotting.
4.1.5. RNA detection and analysis
As detailed above, we routinely use northern blotting to analyze and quantitate RNA for visualization and size determination, which we will briefly describe. For mRNA detection, we generate antisense 32P-labeled riboprobes using in vitro transcription incorporating α-[32P] rUTP (800 Ci/mmol) using T7 polymerase. We generate transcription templates by PCR, using oligonucleotides with a promoter for T7 RNA polymerase appended to the reverse strand. For in vitro transcription, we use the MAXIscript T7 transcription kit (Thermo-Fisher). We prehybridize the blot, incubating at 68°C for 45 minutes prior to addition of probe. We then purify riboprobes via size exclusion over a G25 sephadex column, and then add to the blot in hybridization buffer. We incubate the blot overnight at 68°C, and then wash twice with 2× SSC and 0.1% SDS and twice with 0.1× SSC and 0.1%SDS (15 minutes per wash).
For 18S rRNA detection (as a control not affected by ActD), we radiolabel a DNA oligo probe using γ-[32P] rATP (6000 Ci/mmol) via Polynucleotide Kinase (NEB). We prehybridize the blot with ULTRAhyb-Oligo hybridization buffer for 45 minutes at 42°C. We purify the probe using size exclusion with G25 sephadex, and add it to the blot in hybridization buffer, incubating overnight at 42°C. We then wash the blots twice with 2× SSC and 0.5% SDS for 30 minutes for each wash.
Northern blot signals are quantitated using ImageQuant, and non-linear regression is performed using GraphPad Prism software, fitting to first order exponential decay if possible, as described for mammalian cells. For appropriate statistical analyses, at least three independent replicates should be analyzed, ideally between independent experiments. In most cases, it is appropriate to determine and report confidence intervals for half-life measurements.
4.2. Drosophila cells for transient transfection of mRNA regulators.
Here, we present example experiments pertaining to mRNA decay in d.mel-2 cells; however, it should be noted that the ActD transcription shut off approach has been utilized to investigate several different effectors in mRNA decay in various types of Drosophila cells [35–38]. In order to study regulation by RBPs and miRNAs, a cell-based reporter assay can be performed wherein the effector of interest is transiently overexpressed, and enhancements in decay rates are measured over control. Drosophila Pumilio (Pum) is a sequence-specific RBP that regulates mRNA expression through recognition of cis elements, termed Pum Response Elements (PREs) within the 3’UTR of target transcripts [40–44]. Similar to how TTP is used as an RNA regulator in the above example, here we use Pum to stimulate decay of a Nanoluciferase (Nluc) reporter mRNA bearing PREs (Figure 3). As a negative control, we use an RNA-binding defective Pum, with mutations in the R7 repeat module (mut R7). Effector and reporter plasmids were introduced via transient transfection of d.mel-2 cells with FuGene HD. Nluc signal was detected via northern blot using a 32P-labeled riboprobe, and normalized to 18S rRNA as a stable internal/loading control unaffected by ActD. Under these conditions, Pum accelerates degradation of the reporter, corresponding to a 2.3-fold decrease in half-life.
Fig. 3. Pumilio enhances decay of a reporter mRNA bearing Pum Response Elements (PREs) in d.mel-2 (S2) insect cells.
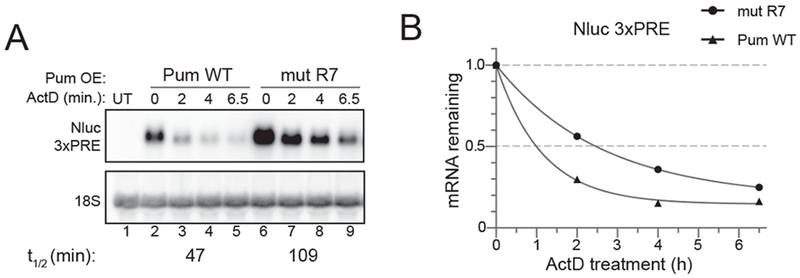
A) Northern blot of a transcription shut-off experiment. Plasmids bearing either wild type (WT) or RNA binding defective (mut R7) Pum were expressed in d.mel-2 cells, along with the Nanoluc (Nluc) 3xPRE reporter. Nluc mRNA was detected with a probe to the open reading frame of Nluc. mRNA from untransfected (UT) cells served as a negative control to demonstrate specificity of the probe. B) Decay curves from transcription shut-off experiments,generated by Graphpad Prism. The fraction of mRNA remaining was plotted versus time (hours).
4.3. Drosophila cells for RNAi of mRNA decay factors.
Transcription shut off with ActD can also be used to measure the contribution of RNA degradation enzymes to the decay of transcripts. For instance, removal of the poly(A) tail, or deadenylation, is often the rate-limiting step of mRNA decay. Trans-acting factors, such as the microRNA-induced Silencing Complex (miRISC) and RBPs like and TTP and Pumilio proteins can engage with the deadenylase machinery to cause degradation of target mRNAs [29, 38, 45–51]. In Drosophila S2 cells, the Pop2 deadenylase is the primary catalytic component of the Ccr4-Not deadenylase complex [52]. In the given example, we used RNAi via double-stranded RNA (dsRNA) to deplete Pop2 in d.mel-2 cells (Figure 4). Depletion of Pop2 stabilizes cycB mRNA (detected via northern blotting with a 32P-labeled riboprobe), corresponding to greater than a 5.9-fold decrease in its half-life, reflecting its critical role in modulating mRNA stability.
Figure 4: Depletion of the Pop2 deadenylase slows decay of cyclin B mRNA in d.mel-2 (S2) insect cells.
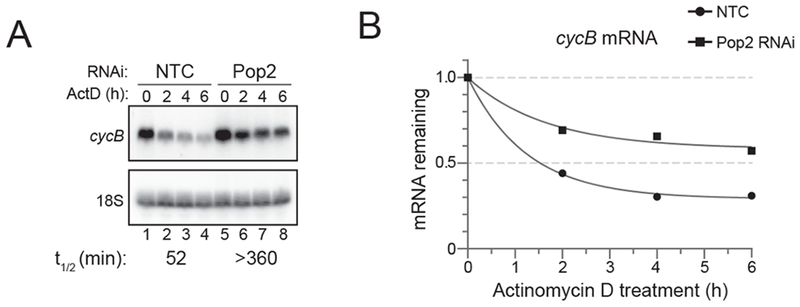
A) Northern blot of transcription shut-off experiment to measure endogenous cyclin b mRNA decay in d.mel-2 cells treated with dsRNA to either a non-targeting control (NTC) or the Pop2 deadenylase. B) Decay curves from a transcription shut-off experiment, generated by Graphpad Prism. The fraction of mRNA remaining was plotted versus time (hours).
5. Conclusions
Transcriptional shut-off using ActD is a venerable and widely used method that is useful for determining the patterns of transcript decay in cultured metazoan cells. It can be used on a genome-wide basis when coupled with global RNA analysis techniques like RNA-Seq and microarray, and it also can be used to interrogate the stability of smaller numbers of mRNAs using lower throughput methods of RNA quantitation. When appropriate precautions are taken, it is a convenient, inexpensive, flexible and powerful technique that can be used in many experimental situations, particularly when comparing the effects of an experimental manipulation to the effects seen in control cells.
This review discusses a facile, versatile method for analyzing mRNA decay in cultured cells.
Actinomycin D mediated transcriptional shut-off coupled with quantitative mRNA detection methods can be used to measure decay of many mRNAs in most types of cultured metazoan cells.
This method has been successfully applied to many mRNAs and their trans-acting regulators.
This chapter provides practical information and examples to enable new users to implement the approach successfully.
6. Acknowledgments
We thank Drs. Michael Fessler and Donald Cook for constructive comments on the manuscript. This work was supported in part by the Intramural Research Program of the National Institute of Environmental Health Sciences, NIH (WSL and PJB). It was also supported by grant R01GM105707 from the National Institute of General Medical Sciences, National Institutes of Health (ACG). René M. Arvola was supported by graduate research fellowship DGE 1256260 from the National Science Foundation and the University of Michigan Genetics Training Program NRSA 5T32GM007544.
Abbreviations
- ActD
actinomycin D
- TTP
tristetraprolin
- DMSO
dimethyl sulfoxide
- BMDM
bone marrow-derived macrophages
- LPS
lipopolysaccharide
- MEF
mouse embryo fibroblasts
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- WT
wild type
- KI
knock-in
- NTC
non-targeting control
- SFM
serum-free medium
- Nluc
Nanoluciferase
- Pum
pumilio
- MiRISC
microRNA-induced Silencing Complex
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- [1].Casse C, Giannoni F, Nguyen VT, Dubois MF, Bensaude O, The transcriptional inhibitors, actinomycin D and alpha-amanitin, activate the HIV-1 promoter and favor phosphorylation of the RNA polymerase II C-terminal domain, J Biol Chem 274(23) (1999) 16097–106. [DOI] [PubMed] [Google Scholar]
- [2].Ayupe AC, Reis EM, Evaluating the Stability of mRNAs and Noncoding RNAs, Methods Mol Biol 1468(2017)139–53. [DOI] [PubMed] [Google Scholar]
- [3].Bensaude O, Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity?, Transcription 2(3) (2011) 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Raghavan A, Ogilvie RL, Reilly C, Abelson ML, Raghavan S, Vasdewani J, Krathwohl M, Bohjanen PR, Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes, Nucleic Acids Res 30(24) (2002) 5529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sharova LV, Sharov AA, Nedorezov T, Piao Y, Shaik N, Ko MS, Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells, DNA Res 16(1) (2009) 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE Jr., Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes, Genome Res 13(8) (2003) 1863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Perry RP, Kelley DE, Inhibition of RNA synthesis by actinomycin D: characteristic dose-response of different RNA species, J Cell Physiol 76(2) (1970) 127–39. [DOI] [PubMed] [Google Scholar]
- [8].Sobell HM, Actinomycin and DNA transcription, Proc Natl Acad Sci U S A 82(16) (1985) 5328–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Valeriote F, Vietti T, Tolen S, Kinetics of the lethal effect of actinomycin D on normal and leukemic cells, Cancer Res 33(11) (1973) 2658–61. [PubMed] [Google Scholar]
- [10].Papahadjopoulos D, Poste G, Vail WJ, Biedler JL, Use of lipid vesicles as carriers to introduce actinomycin D into resistant tumor cells, Cancer Res 36(9 pt.1) (1976) 2988–94. [PubMed] [Google Scholar]
- [11].Sawicki SG, Godman GC, On the differential cytotoxicity of actinomycin D, J Cell Biol 50(3) (1971) 746–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Qiu LQ, Abey S, Harris S, Shah R, Gerrish KE, Blackshear PJ, Global analysis of posttranscriptional gene expression in response to sodium arsenite, Environ Health Perspect 123(4) (2015) 324–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stumpo DJ, Trempus CS, Tucker CJ, Huang W, Li L, Kluckman K, Bortner DM, Blackshear PJ, Deficiency of the placenta- and yolk sac-specific tristetraprolin family member ZFP36L3 identifies likely mRNA targets and an unexpected link to placental iron metabolism, Development 143(8) (2016) 1424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lai WS, Stumpo DJ, Qiu L, Faccio R, Blackshear PJ, A Knock-In Tristetraprolin (TTP) Zinc Finger Point Mutation in Mice: Comparison with Complete TTP Deficiency, Mol Cell Biol 38(4) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Choi YJ, Lai WS, Fedic R, Stumpo DJ, Huang W, Li L, Perera L, Brewer BY, Wilson GM, Mason JM, Blackshear PJ, The Drosophila Tis11 protein and its effects on mRNA expression in flies, J Biol Chem 289(51) (2014) 35042–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ, Novel mRNA targets for tristetraprolin (TTP) identified by global analysis of stabilized transcripts in TTP-deficient fibroblasts, Mol Cell Biol 26(24) (2006) 9196–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Consortium SM-I, A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium, Nat Biotechnol 32(9) (2014) 903–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang C, Gong B, Bushel PR, Thierry-Mieg J, Thierry-Mieg D, Xu J, Fang H, Hong H, Shen J, Su Z, Meehan J, Li X, Yang L, Li H, Labaj PP, Kreil DP, Megherbi D, Gaj S, Caiment F, van Delft J, Kleinjans J, Scherer A, Devanarayan V, Wang J, Yang Y, Qian HR, Lancashire LJ, Bessarabova M, Nikolsky Y, Furlanello C, Chierici M, Albanese D, Jurman G, Riccadonna S, Filosi M, Visintainer R, Zhang KK, Li J, Hsieh JH, Svoboda DL, Fuscoe JC, Deng Y, Shi L, Paules RS, Auerbach SS, Tong W, The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance, Nat Biotechnol 32(9) (2014) 926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Grassmann F, Conduct and Quality Control of Differential Gene Expression Analysis Using High-Throughput Transcriptome Sequencing (RNASeq), Methods Mol Biol 1834 (2019) 29–43. [DOI] [PubMed] [Google Scholar]
- [20].Segundo-Val IS, Sanz-Lozano CS, Introduction to the Gene Expression Analysis, Methods Mol Biol 1434(2016)29–43. [DOI] [PubMed] [Google Scholar]
- [21].Josefsen K, Lee YC, Validation of RNAi by real time PCR, Methods Mol Biol 703 (2011) 205–17. [DOI] [PubMed] [Google Scholar]
- [22].Josefsen K, Nielsen H, Northern blotting analysis, Methods Mol Biol 703 (2011) 87–105. [DOI] [PubMed] [Google Scholar]
- [23].Navarro E, Serrano-Heras G, Castano MJ, Solera J, Real-time PCR detection chemistry, Clin Chim Acta 439 (2015) 231–50. [DOI] [PubMed] [Google Scholar]
- [24].Wong ML, Medrano JF, Real-time PCR for mRNA quantitation, Biotechniques 39(1) (2005) 75–85. [DOI] [PubMed] [Google Scholar]
- [25].Tsang HF, Xue VW, Koh SP, Chiu YM, Ng LP, Wong SC, NanoString, a novel digital color-coded barcode technology: current and future applications in molecular diagnostics, Expert Rev Mol Diagn 17(1) (2017) 95–103. [DOI] [PubMed] [Google Scholar]
- [26].Bustin SA, Benes V, Garson J, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley G, Wittwer CT, Schjerling P, Day PJ, Abreu M, Aguado B, Beaulieu JF, Beckers A, Bogaert S, Browne JA, Carrasco-Ramiro F, Ceelen L, Ciborowski K, Cornillie P, Coulon S, Cuypers A, De Brouwer S, De Ceuninck L, De Craene J, De Naeyer H, De Spiegelaere W, Deckers K, Dheedene A, Durinck K, Ferreira-Teixeira M, Fieuw A, Gallup JM, Gonzalo-Flores S, Goossens K, Heindryckx F, Herring E, Hoenicka H, Icardi L, Jaggi R, Javad F, Karampelias M, Kibenge F, Kibenge M, Kumps C, Lambertz I, Lammens T, Markey A, Messiaen P, Mets E, Morais S, Mudarra-Rubio A, Nakiwala J, Nelis H, Olsvik PA, Perez-Novo C, Plusquin M, Remans T, Rihani A, Rodrigues-Santos P, Rondou P, Sanders R, Schmidt-Bleek K, Skovgaard K, Smeets K, Tabera L, Toegel S, Van Acker T, Van den Broeck W, Van der Meulen J, Van Gele M, Van Peer G, Van Poucke M, Van Roy N, Vergult S, Wauman J, Tshuikina-Wiklander M, Willems E, Zaccara S, Zeka F, Vandesompele J, The need for transparency and good practices in the qPCR literature, Nat Methods 10(11) (2013) 1063–7. [DOI] [PubMed] [Google Scholar]
- [27].Fortina P, Surrey S, Digital mRNA profiling, Nat Biotechnol 26(3) (2008) 293–4. [DOI] [PubMed] [Google Scholar]
- [28].Veldman-Jones MH, Brant R, Rooney C, Geh C, Emery H, Harbron CG, Wappett M, Sharpe A, Dymond M, Barrett JC, Harrington EA, Marshall G, Evaluating Robustness and Sensitivity of the NanoString Technologies nCounter Platform to Enable Multiplexed Gene Expression Analysis of Clinical Samples, Cancer Res 75(13) (2015) 2587–93. [DOI] [PubMed] [Google Scholar]
- [29].Wells ML, Perera L, Blackshear PJ, An Ancient Family of RNA-Binding Proteins: Still Important!, Trends Biochem Sci 42(4) (2017) 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Qiu LQ, Lai WS, Bradbury A, Zeldin DC, Blackshear PJ, Tristetraprolin (TTP) coordinately regulates primary and secondary cellular responses to proinflammatory stimuli, J Leukoc Biol 97(4) (2015) 723–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carballo E, Lai WS, Blackshear PJ, Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability, Blood 95(6) (2000) 1891–9. [PubMed] [Google Scholar]
- [32].Lai WS, Stumpo DJ, Blackshear PJ, Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein, J Biol Chem 265(27) (1990) 16556–63. [PubMed] [Google Scholar]
- [33].Rombouts K, Braeckmans K, Remaut K, Fluorescent Labeling of Plasmid DNA and mRNA: Gains and Losses of Current Labeling Strategies, Bioconjug Chem 27(2) (2016) 280–97. [DOI] [PubMed] [Google Scholar]
- [34].Schneider I, Cell lines derived from late embryonic stages of Drosophila melanogaster, J Embryol Exp Morphol 27(2) (1972) 353–65. [PubMed] [Google Scholar]
- [35].Haas G, Braun JE, Igreja C, Tritschler F, Nishihara T, Izaurralde E, HPat provides a link between deadenylation and decapping in metazoa, J Cell Biol 189(2) (2010) 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Raisch T, Bhandari D, Sabath K, Helms S, Valkov E, Weichenrieder O, Izaurralde E, Distinct modes of recruitment of the CCR4-NOT complex by Drosophila and vertebrate Nanos, EMBO J 35(9) (2016) 974–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sgromo A, Raisch T, Bawankar P, Bhandari D, Chen Y, Kuzuoglu-Ozturk D, Weichenrieder O, Izaurralde E, A CAF40-binding motif facilitates recruitment of the CCR4-NOT complex to mRNAs targeted by Drosophila Roquin, Nat Commun 8 (2017) 14307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Weidmann CA, Raynard NA, Blewett NH, Van Etten J, Goldstrohm AC, The RNA binding domain of Pumilio antagonizes poly-adenosine binding protein and accelerates deadenylation, RNA 20(8) (2014) 1298–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ulvila J, Parikka M, Kleino A, Sormunen R, Ezekowitz RA, Kocks C, Ramet M, Double-stranded RNA is internalized by scavenger receptor-mediated endocytosis in Drosophila S2 cells, J Biol Chem 281(20) (2006) 14370–5. [DOI] [PubMed] [Google Scholar]
- [40].Arvola RM, Weidmann CA, Hall T.M. Tanaka, Goldstrohm AC, Combinatorial control of messenger RNAs by Pumilio, Nanos and Brain Tumor Proteins, RNA Biol 14(11) (2017) 1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gerber AP, Luschnig S, Krasnow MA, Brown PO, Herschlag D, Genome-wide identification of mRNAs associated with the translational regulator PUMILIO in Drosophila melanogaster, Proc Natl Acad Sci U S A 103(12) (2006) 4487–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Murata Y, Wharton RP, Binding of pumilio to maternal hunchback mRNA is required for posterior patterning in Drosophila embryos, Cell 80(5) (1995) 747–56. [DOI] [PubMed] [Google Scholar]
- [43].Wharton RP, Sonoda J, Lee T, Patterson M, Murata Y, The Pumilio RNA-binding domain is also a translational regulator, Mol Cell 1(6) (1998) 863–72. [DOI] [PubMed] [Google Scholar]
- [44].Zamore PD, Williamson JR, Lehmann R, The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins, RNA 3(12) (1997) 1421–33. [PMC free article] [PubMed] [Google Scholar]
- [45].Bulbrook D, Brazier H, Mahajan P, Kliszczak M, Fedorov O, Marchese FP, Aubareda A, Chalk R, Picaud S, Strain-Damerell C, Filippakopoulos P, Gileadi O, Clark AR, Yue WW, Burgess-Brown NA, Dean JLE, Tryptophan-Mediated Interactions between Tristetraprolin and the CNOT9 Subunit Are Required for CCR4-NOT Deadenylase Complex Recruitment, J Mol Biol 430(5) (2018) 722–736. [DOI] [PubMed] [Google Scholar]
- [46].Fabian MR, Frank F, Rouya C, Siddiqui N, Lai WS, Karetnikov A, Blackshear PJ, Nagar B, Sonenberg N, Structural basis for the recruitment of the human CCR4-NOT deadenylase complex by tristetraprolin, Nat Struct Mol Biol 20(6) (2013) 735–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Goldstrohm AC, Seay DJ, Hook BA, Wickens M, PUF protein-mediated deadenylation is catalyzed by Ccr4p, J Biol Chem 282(1) (2007) 109–14. [DOI] [PubMed] [Google Scholar]
- [48].Jonas S, Izaurralde E, Towards a molecular understanding of microRNA-mediated gene silencing, Nat Rev Genet 16(7) (2015) 421–33. [DOI] [PubMed] [Google Scholar]
- [49].Lykke-Andersen J, Wagner E, Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1, Genes Dev 19(3) (2005) 351–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sandler H, Kreth J, Timmers HT, Stoecklin G, Notl mediates recruitment of the deadenylase Cafl to mRNAs targeted for degradation by tristetraprolin, Nucleic Acids Res 39(10) (2011) 4373–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Van Etten J, Schagat TL, Hrit J, Weidmann CA, Brumbaugh J, Coon JJ, Goldstrohm AC, Human Pumilio proteins recruit multiple deadenylases to efficiently repress messenger RNAs, J Biol Chem 287(43) (2012) 36370–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Temme C, Zhang L, Kremmer E, Ihling C, Chartier A, Sinz A, Simonelig M, Wahle E, Subunits of the Drosophila CCR4-NOT complex and their roles in mRNA deadenylation, RNA 16(7) (2010) 1356–70. [DOI] [PMC free article] [PubMed] [Google Scholar]