

We report the cryo-EM structure of rod photoreceptor phosphodiesterase 6.
Abstract
Cyclic nucleotide phosphodiesterases (PDEs) work in conjunction with adenylate/guanylate cyclases to regulate the key second messengers of G protein–coupled receptor signaling. Previous attempts to determine the full-length structure of PDE family members at high-resolution have been hindered by structural flexibility, especially in their linker regions and N- and C-terminal ends. Therefore, most structure-activity relationship studies have so far focused on truncated and conserved catalytic domains rather than the regulatory domains that allosterically govern the activity of most PDEs. Here, we used single-particle cryo–electron microscopy to determine the structure of the full-length PDE6αβ2γ complex. The final density map resolved at 3.4 Å reveals several previously unseen structural features, including a coiled N-terminal domain and the interface of PDE6γ subunits with the PDE6αβ heterodimer. Comparison of the PDE6αβ2γ complex with the closed state of PDE2A sheds light on the conformational changes associated with the allosteric activation of type I PDEs.
INTRODUCTION
The phosphodiesterase (PDE) family displays a conserved catalytic phosphohydrolase domain, whose activity is controlled by diverse domain structures and regulatory mechanisms (1, 2) (figs. S1 and S2). Because of their association with various pathologies and distinct cellular and subcellular distributions, PDEs are targets of several widely used drugs and remain a major target for drug development (3). Most structure-activity relationship studies have so far focused on their conserved catalytic domains that share 25 to 52% sequence homology among type I PDE family members. As a result, most PDE inhibitors display a substantial degree of cross-reactivity within the PDE family (4, 5) and other related enzymes (6). In particular, inhibitors of PDE5, including sildenafil and vardenafil, are widely used for the treatment of erectile dysfunction and pulmonary hypertension (7). However, PDE5 inhibitors have been associated with several ocular side effects, including blurred vision, changes in color vision, transient alterations in the electroretinogram, conjunctival hyperemia, ocular pain, photophobia, and, in extreme cases, damage to the optic nerve (8). These secondary effects are mediated by the binding of PDE5 inhibitors to a closely related phosphohydrolase, PDE6 (9), with about 10% of the inhibitory effect as compared to PDE5 (10). PDE6 is an essential component of visual phototransduction that catalyzes the hydrolysis of guanosine 3′,5′-monophosphate (cGMP) to GMP in response to light (9, 11). The reduction in cGMP concentration results in the closure of Na+ and Ca2+ ion channels in the photoreceptor plasma membrane, leading to their hyperpolarization. Because of its catalytic efficiency operating close to the diffusion limit of cGMP and a ~100-fold activation by transducin (12), PDE6 is crucial for the visual phototransduction. Rod photoreceptor PDE6 is an atypical phosphohydrolase that consists of a heterodimeric catalytic core composed of PDE6α and PDE6β subunits (PDE6αβ heterodimer) and two inhibitory PDE6γ subunits (11). All other members of type I PDE harbor a homodimeric catalytic core, including PDE6 from cone photoreceptors that is composed of two α′ subunits (13). All PDE6 catalytic subunits contain two regulatory N-terminal GAF domains (GAF-A and GAF-B) and a C-terminal catalytic domain (14). GAF domains and the tightly bound inhibitory PDE6γ subunits regulate the activity of catalytic domain allosterically (15). Binding of cGMP to the GAF-A domain of PDE6 increases affinity for the PDE6γ subunit that inhibits the catalytic activity of the PDE6αβ heterodimer when bound. Reciprocally, the binding of PDE6γ to the PDE6αβ heterodimer catalytic subunits enhances the affinity of cGMP for noncatalytic sites, which, in turn, increases the affinity of the catalytic domains for PDE6γ (15–17). During phototransduction, activated transducin [Gαt-GTP (guanosine 5′-triphosphate)] reduces the inhibitory constraint of the PDE6γ C-terminal region on the catalytic site of PDE6 (16), resulting in a reduced binding affinity of cGMP to one of the two GAF-A domains and the release of cGMP. Gαt-GTP has been previously shown to bind amino acid residues 24 to 45 (18–21), 54 to 55 (22), and 55 to 62 (22) and the C terminus of PDE6γ (23). Among these interaction sites, the glycine-rich region of PDE6γ (residues 55 to 62) has been implicated in facilitating the interaction of Gαt-GTP with the N-terminal polycationic region of PDE6γ (18–22).
The absence of a detailed understanding of the allosteric modulation of PDE isoenzymes has limited the commercial success of PDE inhibitors due to cross-reactivity within the PDE family. Several high-resolution structures of individual domains of PDE isoenzymes, including PDE1, PDE2, PDE3, PDE4, PDE5, PDE6, PDE7, PDE8, PDE9, and PDE10, are available (fig. S1). A crystal structure of near full-length PDE2A that includes both GAF and catalytic domains was reported previously (24). In addition, several low-resolution cryo–electron microscopy (cryo-EM) structures of PDE6 that describe the overall architecture of the isoenzyme have been reported (14, 25, 26). Despite the wealth of knowledge that these partial structures have provided, the structural relationships between the regulatory domains and the catalytic domain of PDE family members remain elusive.
RESULTS
To gain structural insights into this allosteric mechanism, we purified native bovine PDE6 from rod outer segments (ROS) and used cryo-EM to determine the structure of the PDE6αβ2γ complex. The structure of the PDE6αβ2γ complex resolved at 3.4-Å resolution shows a linear organization of the three domains (GAF-A, GAF-B, and the catalytic phosphohydrolase domain, respectively) connected by long α helices in both PDE6α and PDE6β subunits (Fig. 1). The overall structure of PDE6 shows a trilobed architecture with each lobe corresponding to the GAF-A, GAF-B, and phosphohydrolase catalytic domains of the PDE6αβ heterodimer (Figs. 1 and 2, A and D). The PDE6αβ heterodimer attains a pseudo-twofold symmetry where the three domains of PDE6α and PDE6β are organized in a head-to-head arrangement with a dimension of 154 Å by 115 Å by 74 Å. The heterodimeric interface of the PDE6αβ heterodimer (~5036 Å2) extends over the entire length of the molecule. Major interfaces lay in the N-terminal pony-tail helical structure (Pt motif, ~895 Å2) and extended linker helices that connect GAF-A to GAF-B (LH1; ~510 Å2) and GAF-B to the catalytic domain (LH2; ~685 Å2) of PDE6α and PDE6β. Local resolution analysis of the PDE6αβ heterodimer shows a resolution better than 3.4 Å in the core and 5 to 7 Å in peripheral regions (Fig. 1A, bottom). The Pt motif is among the most flexible regions within the PDE6αβ2γ complex with a local resolution of 7 to 8 Å (Fig. 1A, bottom), and therefore has uncertainty in the de novo model. The final model is composed of amino acid residues 10 to 822 of PDE6α and 11 to 822 of PDE6β (Fig. 1C). The amino acid residues 2 to 9 and 823 to 856 of PDE6α as well as 2 to 10 and 823 to 850 of PDE6β could not be modeled because of their flexibility. This flexibility is exemplified by the two-dimensional (2D) class averages that show blurring of the flexible C-terminal ends of both PDE6α and PDE6β (fig. S3D, arrows). These long C-terminal helices extend in opposite directions and serve as sites for lipid modifications that anchor PDE6αβ2γ in the ROS disk membranes. Although the distal C-terminal helices of the PDE6αβ heterodimer were averaged out during the final 3D cryo-EM reconstruction, this is the first instance where 2D snapshots of these long C-terminal helices have been visualized.
Fig. 1. The cryo-EM structure of PDE6αβ2γ ternary complex at 3.4-Å resolution.
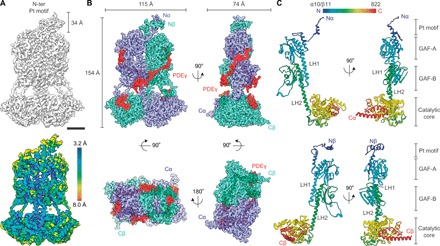
(A) Overall cryo-EM 3D reconstruction of the PDE6αβ2γ complex displaying the 34-Å-long N-terminal pony-tail domain (Pt motif) (top) and the local resolution estimation map (bottom). Resolution keys are labeled from 3.2 (blue) to 8.0 Å (red). (B) Four orthogonal views of the cryo-EM structure of PDE6αβ2γ complex displaying PDE6α (purple), PDE6β (green cyan), and two molecules of PDE6γ (red) subunits. (C) Structures of PDE6α (top, 10 to 822) and PDE6β (bottom, 11 to 822) in two orthogonal views shown in cartoon representation. Scale bar, 30 Å.
Fig. 2. Structural features of the PDE6αβ2γ complex.
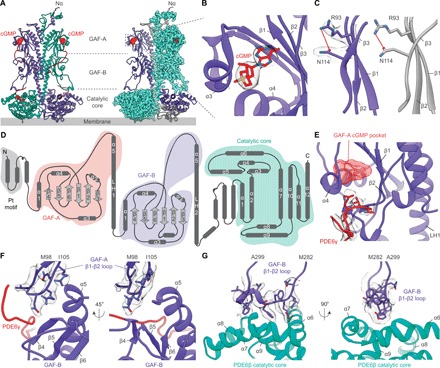
(A) Structure of the PDE6αβ heterodimer showing the domain distribution within PDE6α (purple) and PDE6β (green cyan) subunits where regulatory GAF domains are trailed by the phosphohydrolase catalytic domain in a trilobed organization. One molecule of PDE6γ (red ribbon) wraps around PDE6α and PDE6β individually for a tighter regulation of PDE6 activity. One molecule of cGMP (red spheres) is bound to each of the GAF-A domains of the PDE6αβ heterodimer. (B) Zoomed-in view of the GAF-A cGMP-binding pocket showing the orientation of the cGMP molecule (red sticks) with respect to the surrounding secondary structures. The electron density corresponding to the cGMP molecule was calculated after the final refinement and is displayed as gray mesh. (C) The cGMP-bound GAF-A domains of both PDE6αβ2γ complex (left) and PDE6C (gray, right) feature a salt bridge between the partial negative charge of Asn114 carbonyl oxygen and the guanidine group of Arg93. Amino acid residue Asn114 provides important cGMP-specific protein-ligand hydrogen bonds that stabilize cGMP binding. (D) Topology diagram showing the secondary structure elements of the Pt motif (gray), GAF-A (red), GAF-B (purple), and catalytic domain (green). (E) Zoomed-in view of the cGMP-binding pocket (red mesh) of PDE6α GAF-A domain. The N terminus of PDE6γ (red cartoon) forms a lid over the buried cGMP molecule and thereby prevents its release from the GAF-A domain until a threshold concentration gradient of cGMP has been reached in the cell. The electron density of the N terminus of PDE6γ after the final refinement is displayed as gray mesh. (F) Interaction interface between the GAF-A β1-β2 loop and the GAF-B domain of PDE6α. PDE6γ (red) staples the GAF-A β1-β2 loop interaction to the GAF-B domain and likely senses changes associated with cGMP binding in the GAF-A domain. The refined electron density around the GAF-A β1-β2 loop is displayed as gray mesh. (G) The interface between GAF-B β1-β2 loop of PDE6α and the catalytic domain of PDE6β features a short two-turn helical structure that forms hydrophobic interactions with α6, α7, α8, and α9 helices of the catalytic domain. The refined electron density corresponding to the key amino acid residues is displayed as gray mesh.
The N-terminal region of the PDE6αβ heterodimer displays an asymmetric double-stranded Pt motif that extends to more than 34 Å in length (Fig. 1). The Pt motif likely provides structural stability to the PDE6αβ heterodimer by providing an interaction interface of ~895 Å2 between PDE6α and PDE6β subunits. The asymmetry of the Pt motif can be attributed to the low sequence identity between the N-terminal regions of the two subunits of PDE6αβ heterodimer, with 27% identity in the first 70 amino acids as compared to 77.5% identity in the rest of the sequence (fig. S2). The GAF-A domain of the PDE6αβ heterodimer shows a high structural similarity to previously published GAF-A domains of PDE2A (24), PDE5 (27), and PDE6C (28) with root mean square deviations (RMSDs) of 1.34, 1.17, and 1.12 Å, respectively. The conserved structural features of the GAF-A domain include an antiparallel β-sheet cluster with strand order 3-2-1-6-5-4 (Fig. 2D and fig. S4A). This β-sheet cluster is sandwiched by α1, α2, and α5 helices that lay toward the heterodimer interface as well as α3 and α4 helices on the distal side (Fig. 2A and fig. S4A). In comparison to previously determined structures of GAF-A domains, a substantial structural change is observed in the 14–amino acid–long loop that connects the β1 with the β2 strand (β1-β2 loop) of the GAF-A six-stranded β-sheet cluster. The GAF-A β1-β2 loop directly interacts with the β4 strand and the β6-α5 loop of the GAF-B regulatory domain, suggesting a potential allosteric signal transduction route from the GAF-A to GAF-B domain upon cGMP binding (Fig. 2F).
The refined cryo-EM map shows one molecule of cGMP bound to the GAF-A domain of both PDE6α and PDE6β (Figs. 2, A and B, and 3A). The 3′,5′-cyclic phosphate group of cGMP points toward the α3 and α4 helices, whereas its guanine ring makes several well-defined contacts with the β1 and β2 strands (Fig. 2B and fig. S4C). The buried cGMP molecule makes several electrostatic contacts with the side chains of Ser95, Asn114, Val166, and Thr174 in addition to hydrophobic contacts with Phe113, Phe134, Val140, Tyr172, and M193 that determine its orientation in the cGMP binding pocket (Fig. 2B and fig. S4C). As observed in the crystal structure of cGMP-bound PDE6C GAF-A domain (28), the GAF-A domain of PDE6αβ2γ features Asn114 locked into position by a 3.0-Å salt bridge between the partial negative charge of its carbonyl oxygen and the guanidine group of Arg93 (Fig. 2C). The N-terminal polycationic region of PDE6γ binds close to the GAF-A cGMP-binding site, suggesting its potential role in preventing the release of bound cGMP unless a certain threshold of cGMP gradient has been established in the cell and thereby lowering the dissociation constant (KD) of cGMP for the GAF-A domain (Figs. 2E and 3A). In addition, PDE6γ staples the GAF-A β1-β2 loop interaction site to GAF-B domain and likely senses changes upon cGMP binding (Figs. 2, A and F, and 3A). One molecule of PDE6γ interacts exclusively with GAF-A and the catalytic domain of the same subunit in the PDE6αβ heterodimer, suggestive of a tighter allosteric regulation (Figs. 1B, 2A, and 3A). PDE6γ subunits display a resolution range of 3.2 to 9 Å with the highest resolution achieved in the N- and C-terminal ends due to their direct interaction with GAF-A and catalytic domains, respectively (Figs. 2, A, E, and F, and 3, A and B). Amino acid residues 30 to 70 of PDE6γ were omitted from the final de novo model of the PDE6αβ2γ complex because of poor cryo-EM density near that region (Fig. 3, A and B). This could be explained by the flexible nature of the polypeptide segment and the lack of direct interface contacts with the PDE6αβ heterodimer. Overall, these findings suggest a direct communication between the cGMP-bound GAF-A and GAF-B regulatory domains in PDE6αβ2γ complex through the β1-β2 loop of GAF-A.
Fig. 3. The PDE6γ subunit stabilizes the open-state conformation of PDE6αβ heterodimer.
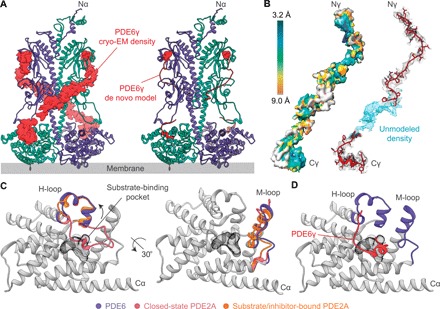
(A) Cryo-EM density (left, red surface) and partial de novo model (right, red ribbon) of PDE6γ around the PDE6αβ heterodimer (PDE6α, purple; PDE6β, green cyan). One molecule of PDE6γ wraps around PDE6α and PDE6β individually for a tighter regulation of PDE6 activity. (B) Local resolution estimation map (left) and partial de novo model (right) of PDE6γ showing a good correlation between the 3D map and the refined PDE6γ model. The density of PDE6γ after the final refinement is displayed as gray mesh. (C) Comparison between the catalytic domains of PDE6α (purple), closed-state PDE2A (pink, PDBID: 3IBJ), and inhibitor-bound PDE2A (orange, PDB ID: 4D08) displaying different orientations of the H-loop (606 to 629, left) and M-loop (745 to 767, right) regions. The catalytic domain of closed-state PDE2A shows the H-loop folded into the catalytic site close to the substrate-binding pocket (black mesh). In contrast, the catalytic domain of PDE6α features an open H-loop similar to the inhibitor-bound PDE2A crystal structure. The M-loop region of PDE6α (purple) shows a similar conformation as the inhibitor-bound PDE2A structure (orange). Notably, residues 840 to 850 of the M-loop of the closed-state PDE2A crystal structure (pink) are disordered. (D) The C terminus of PDE6γ (red cartoon) binds near the substrate-binding pocket of the PDE6 catalytic domain. PDE6γ binding to the catalytic domain of PDE6 mimics a substrate/inhibitor-bound form where the H-loop displays an open conformation and the substrate-binding pocket is occluded by the C terminus of PDE6γ. Structurally conserved regions of the catalytic domain (RMSD ≤ 0.2 Å between PDE6α and PDE2A) are shown in gray.
The GAF-A C-terminal α5 helix (206 to 232) is linked to the N-terminal α1 helix of GAF-B (254 to 280) by LH1 (Figs. 1C and 2D). Although the GAF-B domain maintains a familiar topology to those of previously published GAF-B domain structures of PDE2A (RMSD = 1.120 Å between 59 pruned atom pairs) (24) and PDE5 (RMSD = 1.06 Å between 56 pruned atom pairs) (27), substantial structural differences were observed in the orientation of LH1 (~20° tilt), α2 (~10° tilt), and LH2 (~30° tilt). A major structural difference was found in the 34–amino acid–long β1-β2 loop (280 to 313) that connects β1 with β2 in the six-stranded β-sheet cluster of the GAF-B domain (Fig. 2D and fig. S4B). The GAF-B β1-β2 loop of one subunit of the PDE6αβ heterodimer directly interacts with the catalytic domain of the other subunit (Fig. 2G). This cross-talk is mediated by the intertwined architecture of the PDE6αβ heterodimer, where LH2 helices cross over the pseudo-twofold axis such that the catalytic domain of one subunit lies directly below the GAF-B domain of the other subunit of the heterodimer (Figs. 1, B and C, and 2A). The interacting region of the GAF-B β1-β2 loop adopts a short two-turn helical structure that forms hydrophobic interactions with α6, α7, α8, and α9 helices of the catalytic domain (Fig. 2G). The direct interaction suggests a potential allosteric signal transduction route from the cGMP-bound GAF-A to the catalytic domain via GAF-B. This feature makes the PDE6αβ2γ complex the only PDE isoenzyme showing the second direct association between the GAF-B domain and the catalytic phophohydrolase domain in addition to the LH2 helix that links the two domains together. This finding suggests a role of the GAF-B β1-β2 loop in the direct regulation of PDE6 catalytic domains transducing the allosteric signal from cGMP-bound GAF-A domains to the catalytic domains.
The GAF-B C-terminal α5 helix (414 to 441) is linked to the N-terminal α1 helix of the catalytic domain (558 to 577) through the LH2 helix and a helix-loop-helix region comprising a four-helix bundle (Figs. 1C and 2, A and D). The all–α-helical structure of the catalytic domain in PDE6 (PDE6α, 558 to 799; PDE6β, 556 to 796) is structurally conserved with previously published crystal structures of the catalytic domains of several PDE isoenzymes (24, 29). However, a substantial conformational change was seen in the H-loop region (606 to 629) that flanks the substrate-binding site (Fig. 3C). The H-loop is a characteristic loop that changes its conformation upon ligand binding. During PDE2A activation, the H-loop swings out and attains an open-loop conformation that allows the binding of a substrate or an inhibitor to the PDE catalytic domain (Fig. 3C) (24). Whereas during PDE2A inactivation, the H-loop occludes the substrate-binding pocket, which results in deactivation of the isoenzyme (Fig. 3C) (24). Unlike the structure of closed-state PDE2A, the H-loop in the PDE6αβ2γ complex features an open conformation that is reminiscent of the substrate/inhibitor-bound state of PDE2A and PDE5 catalytic domains (Fig. 3, C and D, and fig. S5). This open H-loop conformation is stabilized by the C terminus of PDE6γ that binds near the substrate-binding site and occludes substrate binding (Fig. 3D).
In contrast to the closed-state structure of PDE2A homodimer, the M-loop region (745 to 767) that connects α14 and α15 of the catalytic domain does not participate in heterodimer formation in the PDE6αβ2γ complex (Fig. 4A). In light of the direct interaction of GAF β1-β2 loops connecting the regulatory cGMP-bound GAF-A to the catalytic domain through the GAF-B domain of the PDE62αβγ complex and the comparison with the closed state of PDE2A (Fig. 4A) (24), a model for allosteric activation of PDE isoenzymes can be deduced to reveal the representative conformational changes (Fig. 4B). These conformational changes include a downward movement of the GAF-A β1-β2 loop, a 10° inward twist of LH1, an 80° outward twist of the GAF-B β1-β2 loop, and an 80° rotation of the catalytic domains (Fig. 4A). However, the rearrangement of catalytic domains is unlikely to occur during the allosteric activation of PDE6 because of its association with PDE6γ. In an attempt to visualize some of these structural changes, we performed cryo-EM on the PDE6αβ2γ complex in the presence of fivefold molar excess of sildenafil. High-resolution 2D class averages of PDE6αβ2γ complex treated with sildenafil displays a reduced density of the GAF-B β1-β2 loop associated with its increased flexibility (Fig. 4C; compare panels marked with asterisks). The observed structural changes are consistent with partial occupancy of sildenafil in the catalytic site of PDE6αβ2γ complex due to a competition with the inhibitory PDE6γ subunit for the same binding site (30). Notably, the catalytic domains did not show notable structural changes likely because of the partially bound PDE6γ that stabilizes the PDE6αβ2γ complex in its open state. Overall, these results further support the role of GAF β1-β2 loops in the allosteric regulation of PDE catalytic activity.
Fig. 4. The β1-β2 loop of regulatory GAF domains as potential allosteric triggers to PDE allosteric regulation.
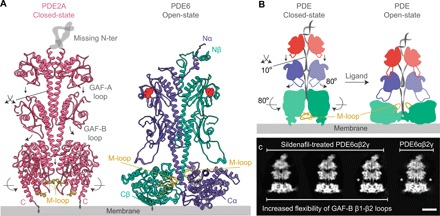
(A) Comparison between overall structures of the closed-state PDE2A homodimer and the PDE6αβ heterodimer. M-loop regions of the closed-state PDE2A and the PDE6αβ heterodimer are shown in gold. (B) Schematic representation showing the extent of potential conformational changes that occur during PDE activation. These conformational changes include a downward movement of the GAF-A β1-β2 loop, a 10° inward twist of the LH1 coiled coil, an 80° outward twist of the GAF-B β1-β2 loop, and an 80° rotation of the catalytic domains. M-loop regions are shown in gold. (C) A comparison between the 2D class averages of the untreated PDE6αβ2γ complex and PDE6αβ2γ treated with sildenafil recapitulates some of the key structural differences illustrated in (B). The face views of PDE6αβ2γ treated with sildenafil feature changes in the interaction profile of the GAF-B β1-β2 loop with the catalytic domain of the PDE6αβ heterodimeric core. The GAF-B β1-β2 loops are denoted by asterisks. Scale bar, 60 Å.
DISCUSSION
This study provides structural insights into the allosteric regulation of PDE6 catalytic activity by both N-terminal GAF domains and the PDE6γ subunit. A comparison of the PDE6αβ2γ complex structure with the closed-state structure of PDE2A shows reorganization of the GAF β1-β2 loop regions that form potential allosteric signal transduction routes from GAF-A to the catalytic domain through the GAF-B domain upon cGMP binding (Fig. 4). These allosteric signals are then translated into the outward movement of the H-loop region that allows substrate binding and hydrolysis (Figs. 3 and 4). The N-terminal regulatory domains are crucial for tightly regulating the cyclic nucleotide signaling by PDE isoenzymes. The type I PDE family features a variety of N-terminal domains including calcium/calmodulin-binding domains, protein kinase–regulated upstream conserved regions, and GAF domains. However, the allosteric regulation of the majority of PDEs is governed through N-terminal GAF domains (fig. S1). Notably, PDE2A activation is mediated by binding of cGMP to the GAF-B domain, whereas cGMP-induced activation of PDE5 is mediated by cGMP binding to the GAF-A domain. On the other hand, the activation of PDE6 is more complex and involves the binding of Gαt-GTP, which releases the inhibitory constraint of PDE6γ on the catalytic site of PDE6 (16) and reduces the binding affinity of cGMP to one of the two GAF-A domains. Therefore, the proposed model displaying the representative structural changes associated with PDE allosteric regulation (Fig. 4B) is likely limited to the closely related structural homologs of PDE6.
The structure of PDE6αβ2γ complex displays several features that are in agreement with previously published chemical cross-linking and mass spectrometric data (31). These features include a tandem organization of GAF domains, parallel organization of the two catalytic subunits, and presence of juxtaposed α-helical segments in the PDE6αβ heterodimer (31). PDE6 is the only type I PDE family member that has inhibitory subunits bound to the catalytic dimer (32). Previous cross-linking studies have indicated the ability of PDE6γ to bind the catalytic domains of the PDE6αβ heterodimer, whereby its C-terminal residues block the substrate-binding pocket (30). PDE6γ binding has also been proposed to modulate cGMP binding at the regulatory GAF-A domain (33). This unusual interdomain allosteric control for a 9.7-kDa PDE6γ polypeptide has been debated often because of the lack of structural evidence. This study provides the direct structural confirmation that PDE6γ adopts an extended and mostly unstructured conformation to enable its interaction with both the GAF-A cGMP-binding pocket and the catalytic domain of the PDE6αβ heterodimer. In addition, the interaction of PDE6γ C terminus with the catalytic subunits stabilizes the open-state conformation of the PDE6αβ2γ complex.
Overall, the high-resolution structure of the PDE6αβ2γ complex and its comparison with the closed-state PDE2A structure provide a solid structural framework for rational design of candidate molecules that are selective to specific PDE isoenzymes to alleviate side effects induced by conventional therapeutic approaches.
MATERIALS AND METHODS
Purification of PDE6
All experimental procedures were carried out in a darkroom under dim red light (>670 nm). Bovine ROS were prepared as described elsewhere (34). ROS were diluted in isotonic buffer containing 20 mM Hepes (pH 7.5), 5 mM MgCl2, 1 mM dithiothreitol (DTT), and 100 mM NaCl. The suspension was then centrifuged at 30,000g at 4°C for 25 min to remove soluble and some membrane-associated proteins (35, 36). The pellet was resuspended in hypotonic buffer containing 5 mM Hepes (pH 7.5), 0.1 mM EDTA, and 1 mM DTT and gently homogenized three times by manually passing the suspension through a glass-to-glass homogenizer. The homogenate was centrifuged at 40,000g for 30 min at 4°C. Supernatants from the two hypotonic washes were pooled and centrifuged multiple times at 40,000g for 30 min at 4°C to completely remove any residual ROS pellet. The clear supernatant was dialyzed against buffer containing 10 mM Hepes (pH 7.5), 6 mM MgCl2, and 1 mM DTT for 3 hours at 4°C. The hypotonic solution was supplemented with ROS membranes (25 μM rhodopsin) and 250 μM GTPγS (Sigma-Aldrich), followed by light illumination for 30 min with a 150-W fiber light (NCL-150, Volpi, USA) delivered through a 480- to 520-nm band-pass filter (Chroma Technology Corporation, USA). The resuspension was then centrifuged multiple times at 40,000g for 30 min at 4°C to completely remove any residual ROS pellet. The supernatant was loaded onto a C10/10 column (GE Healthcare) with 6 ml of propyl-agarose resin pre-equilibrated with 10 mM Hepes (pH 7.5), 2 mM MgCl2, and 1 mM DTT. Next, the column was washed with 30 resin volumes of the equilibration buffer followed by 2 resin volumes of buffer containing 10 mM Hepes (pH 7.5), 2 mM MgCl2, 1 mM DTT, and 50 mM NaCl. Bound proteins were eluted with 30 ml of equilibration buffer containing 0.4 M NaCl. The eluate was then dialyzed against buffer containing 10 mM Hepes (pH 7.5), 6 mM MgCl2, 1 mM EDTA, and 1 mM DTT.
The dialyzed eluate was loaded onto a C10/20 column (GE Healthcare) with 15 ml of Blue Sepharose CL-6B resin (Sigma-Aldrich) pre-equilibrated with 10 mM Hepes (pH 7.5), 6 mM MgCl2, 1 mM EDTA, and 1 mM DTT. The flow-through was supplemented with a nanobody that specifically binds to Gβ1γ1 (37) to accomplish its removal from the sample (fig. S3). After 30 min of incubation, Ni2+–nitrilotriacetic acid resin pre-equilibrated with 10 mM Hepes (pH 7.5), 6 mM MgCl2, 1 mM EDTA, and 1 mM DTT was added. Following 30 min of incubation, the resin bound with Gβ1γ1 was removed by passing the resuspension through a Pierce disposable column (Thermo Fisher Scientific). The flow-through containing Gαt and PDE6 obtained from the immobilized-Ni2+ affinity chromatography was then concentrated and loaded onto a Superdex 200 10/300 GL column equilibrated with buffer containing 10 mM Hepes (pH 7.5), 2 mM MgCl2, 1 mM DTT, and 100 mM NaCl (fig. S3, A and B). Fractions containing PDE6 were combined, concentrated to about 0.7 mg ml−1, and used for cryo-EM analyses. The functional characterization of PDE6 has been described previously (26, 38).
Cryo-EM specimen preparation, data acquisition, and movie processing
Three microliters of the purified PDE6αβ2γ or PDE6αβ2γ with 5 M excess of sildenafil at a concentration of 0.7 mg ml−1 were applied to a Quantifoil R2/2 400 mesh grid (Electron Microscopy Sciences) without a prior glow discharge. The grids were plunge-frozen in liquid ethane with a FEI Vitrobot Mark IV (Thermo Fisher Scientific) under these conditions: temperature, 4°C; humidity, 100%; blotting time, 2 s; and blotting force set to −10. Frozen grids were imaged in a FEI Titan Krios (300 kV, Thermo Fisher Scientific) equipped with a Gatan Quantum-LS energy filter (20-eV zero-loss filtering) connected to a Gatan K2 Summit detector operating in super-resolution counting mode. Super-resolution movies of 50 frames were acquired at a magnification of ×47,259 in the nanoprobe mode using the SerialEM software (39). A total dose of 80 e− Å−2 and a pixel size of 0.529 Å for the super-resolution pixels were used during data collection (fig. S3C). The acquired movies were processed during the imaging session with the Focus program (40), which included (i) gain reference application and binning 2× by the clip and resample_mp.exe programs from the IMOD (41) and Frealign (42) packages, respectively; (ii) motion correction and dose weighting by MotionCor2 (43); and (iii) contrast transfer function (CTF) estimation by CTFFIND4 (44). A total of 3134 aligned movies were used for further single-particle processing. Images displaying a resolution lower than 7 Å during CTF correction or average drifts higher than 2 Å per frame were excluded from the analysis.
Image processing
Particles in micrographs were selected with the EMAN2 boxer (45) and Gautomatch (www.mrc-lmb.cam.ac.uk/kzhang/Gautomatch/) programs. During the initial processing, 2176 particles were automatically selected from 100 preselected images with reference-free Gautomatch localization. The selected particles were inspected with the e2boxer.py script and 2D classified in Relion 2.1 (46). A few high-resolution 2D classes were low-pass–filtered to 30 Å and used as templates for Gautomatch particle selection on all aligned images. This larger dataset of particles was 2D classified in Relion 2.1, and 30 high-resolution class averages covering a broader range of views were selected as templates for another round of Gautomatch particle selection. The final particle selection resulted in a total of 197,171 particles, which were reprocessed with Relion 2.1. After seven rounds of 2D classification, a 2D class average representing the face-view of PDE6 (fig. S3D) was used to create a rotationally symmetric starting model for 3D processing. The Fourier transform of the selected projection was low-pass–filtered to 60 Å and rotated around the long axis of the class average. The created 3D Fourier volume was back-transformed to obtain the initial real-space 3D model.
The 2D class averages comprising 85,929 particles were subjected to three rounds of 3D classification with the 60-Å rotationally symmetric model as a reference (fig. S6A). The resulting five 3D classes were used to generate the initial map of PDE6 at a resolution of 8.2 Å, which was subjected to the e2project.py script of EMAN2 to compute 30 reprojections covering views from all directions to serve as templates for the final round of Gautomatch particle selection. The final dataset comprising 199,658 particles was processed by Relion 2.1. Five rounds of 2D classification reduced the dataset to 82,558 particles, which were subjected to three rounds of 3D classification (with seven, five, and five 3D classes, respectively) with the rotationally symmetric initial 3D model as a reference (fig. S6A). Two identical 3D classes displaying a resolution of 8.3 Å and comprising a total of 43,597 particles were combined into a single dataset. The combined dataset was then refined against one of the two 8.3-Å 3D classes, which was low-pass–filtered to 35 Å. Upon masking and modulation transfer function correction, the reconstructions reached a resolution of 4.1 Å, as measured by Fourier shell correlation (FSC) of a refinement in which two halves of the dataset were refined separately and combined only when building the final map (fig. S6A). The same dataset was then reprocessed with the cisTEM program (47) that uses per-particle CTF refinement and B-factor particle weighting. The final 3D reconstruction reached a resolution of 3.4 Å at an FSC of 0.143 (fig. S6B) (48). Local resolution distribution of the final map was determined by ResMap (49). The data acquisition and processing of PDE6αβ2γ treated with sildenafil was done under identical conditions as the untreated PDE6αβ2γ holoenzyme. In total, 4780 images were acquired, from which 223,656 particles were isolated and subjected to 2D classification.
Model building and refinement
The cryo-EM map was put into an artificial crystal lattice to calculate its structure factor using the phenix.map_to_structure_factors script in the PHENIX program (50). The 3D density map was sharpened by applying a negative B-factor of −148 Å2 as determined with the phenix.auto_sharpen script (50) and by manual intervention. De novo modeling of PDE6α, PDE6β, and PDE6γ subunits was performed in Coot 0.8.8 (51) using secondary structure predictions calculated by PSIPRED (52). The densities of bulky side chains were used as references during the backbone tracing. A partial PDE6 model (PDE6α, 10 to 822; PDE6β, 11 to 822; and PDE6γ 1 to 30 and 70 to 87) was used for rigid body fitting into the 3D density map with Chimera (53). The model was then refined by rigid body refinement of individual chains in the PHENIX program (50), where the amplitudes and phases of the structural factors were used as pseudo-experimental diffraction data for model refinement. Initial models were improved by multiple rounds of PHENIX real-space refinement (50) and REFMAC version 5.8 (54) refinement against the overall map at a resolution of 3.4 Å. Each round of refinement was followed by manual model building and adjustments with Coot 0.8.8 (51). Simultaneous optimization of the stereochemical and molecular clashes was performed during the refinements, resulting in a final model to map a cross-correlation coefficient of 0.79 (fig. S6C).
The stereochemical quality of the final PDE6αβ2γ complex model was assessed with the Molprobity server (55). The final PDE6αβ2γ model was cross-validated with previously available crystal structures of GAF-A (27, 28), GAF-B (27, 56), and catalytic (29, 30) domains of PDE family members. Details of the cryo-EM data collection and structural refinement statistics are provided in table S1. Protein coordinates and the EM map were deposited in the Protein Data Bank (PDB) (PDB accession number: 6MZB and Electron Microscopy Data Bank ID: EMD-9297). The raw image data have been deposited in the Electron Microscopy Public Image Archive (EMPIAR) database (www.ebi.ac.uk/pdbe/emdb/empiar/; access number: EMPIAR-10228). All structural and density representations were generated using either Chimera (53) or Pymol (www.pymol.org).
Supplementary Material
Acknowledgments
We thank A. Sears, R. Zimmerman, and other members of the Palczewski laboratory for their helpful comments regarding this manuscript and J. Thorne-Wallis from C-CINA for assistance with the preparation of cryo-EM grids. K.P. is the Leopold Chair of Ophthalmology. Funding: This research was supported in part by grants from the National Institutes of Health (NIH) (R24EY024864 and R01EY027283 to K.P.) and Research to Prevent Blindness (RPB) to The Department of Ophthalmology at UCI, the Canadian Institute for Advanced Research (CIFAR), Alcon Research Institute (ARI), and the Swiss Commission for Technology, and Innovation (CTI) grant 18272.1 PFLS-LS (H.S.). Author contributions: S.G. and K.P. designed the research and wrote the paper. S.G. performed the protein purifications and optimizations. S.G. prepared PDE6 samples for cryo-EM specimen preparation. S.G., L.K., and H.S. performed the cryo-EM work. L.K. did the cryo-EM data processing and contributed to the written paper. S.G. carried out de novo model building and refinement and analyzed the structure of the PDE6αβ2γ complex. A.E. made intellectual contributions to the study and contributed to the written paper. All authors reviewed and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data supporting the findings of this study are available within this paper. Additional data supporting the findings of this manuscript are available from the corresponding authors upon reasonable request. The cryo-EM map of PDE6αβ2γ complex has been deposited in the Electron Microscopy Data Bank under accession code EMD-9297. The modeled structure of the PDE6αβ2γ complex has been deposited at the PDB under accession code 6MZB. The cryo-EM movie files have been deposited at the EMPIAR under accession code EMPIAR-10228.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav4322/DC1
Fig. S1. Domain organization in the type I phosphodiesterase superfamily.
Fig. S2. Domain organization and primary sequence comparison between PDE6α and PDE6β subunits.
Fig. S3. Biochemical characterization and cryo-EM of the PD6αβ2γ complex.
Fig. S4. Structural features of regulatory GAF domains of the PDE6αβ2γ complex.
Fig. S5. Comparison between the H-loop orientations of PDE6 and PDE5.
Fig. S6. Cryo-EM reconstruction and data fitting of the PDE6αβ2γ complex.
Table S1. Cryo-EM data collection, refinement, and validation statistics for the PDE6αβ2γ complex.
REFERENCES AND NOTES
- 1.Conti M., Beavo J., Biochemistry and physiology of cyclic nucleotide phosphodiesterases: Essential components in cyclic nucleotide signaling. Annu. Rev. Biochem. 76, 481–511 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Halpin D. M. G., ABCD of the phosphodiesterase family: Interaction and differential activity in COPD. Int. J. Chron. Obstruct. Pulmon. Dis. 3, 543–561 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.DeNinno M. P., Future directions in phosphodiesterase drug discovery. Bioorg. Med. Chem. Lett. 22, 6794–6800 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Zhang X., Feng Q., Cote R. H., Efficacy and selectivity of phosphodiesterase-targeted drugs in inhibiting photoreceptor phosphodiesterase (PDE6) in retinal photoreceptors. Invest. Ophthalmol. Vis. Sci. 46, 3060–3066 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bischoff E., Potency, selectivity, and consequences of nonselectivity of PDE inhibition. Int. J. Impot. Res. 16 (Suppl. 1), S11–S14 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Boess F. G., Hendrix M., van der Staay F. J., Erb C., Schreiber R., van Staveren W., de Vente J., Prickaerts J., Blokland A., Koenig G., Inhibition of phosphodiesterase 2 increases neuronal cGMP, synaptic plasticity and memory performance. Neuropharmacology 47, 1081–1092 (2004). [DOI] [PubMed] [Google Scholar]
- 7.Maurice D. H., Ke H., Ahmad F., Wang Y., Chung J., Manganiello V. C., Advances in targeting cyclic nucleotide phosphodiesterases. Nat. Rev. Drug Discov. 13, 290–314 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moschos M. M., Nitoda E., Pathophysiology of visual disorders induced by phosphodiesterase inhibitors in the treatment of erectile dysfunction. Drug Des. Devel. Ther. 8, 3407–3413 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beavo J. A., Cyclic nucleotide phosphodiesterases: Functional implications of multiple isoforms. Physiol. Rev. 75, 725–748 (1995). [DOI] [PubMed] [Google Scholar]
- 10.Ballard S. A., Gingell C. J., Tang K., Turner L. A., Price M. E., Naylor A. M., Effects of sildenafil on the relaxation of human corpus cavernosum tissue in vitro and on the activities of cyclic nucleotide phosphodiesterase isozymes. J. Urol. 159, 2164–2171 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Baehr W., Devlin M. J., Applebury M. L., Isolation and characterization of cGMP phosphodiesterase from bovine rod outer segments. J. Biol. Chem. 254, 11669–11677 (1979). [PubMed] [Google Scholar]
- 12.Lamb T. D., Heck M., Kraft T. W., Implications of dimeric activation of PDE6 for rod phototransduction. Open Biol. 8, 180076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolandaivelu S., Chang B., Ramamurthy V., Rod phosphodiesterase-6 (PDE6) catalytic subunits restore cone function in a mouse model lacking cone PDE6 catalytic subunit. J. Biol. Chem. 286, 33252–33259 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z., He F., Constantine R., Baker M. L., Baehr W., Schmid M. F., Wensel T. G., Agosto M. A., Domain organization and conformational plasticity of the G protein effector, PDE6. J. Biol. Chem. 290, 17131–17132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mou H., Cote R. H., The catalytic and GAF domains of the rod cGMP phosphodiesterase (PDE6) heterodimer are regulated by distinct regions of its inhibitory γ subunit. J. Biol. Chem. 276, 27527–27534 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Norton A. W., D’Amours M. R., Grazio H. J., Hebert T. L., Cote R. H., Mechanism of transducin activation of frog rod photoreceptor phosphodiesterase. Allosteric interactiona between the inhibitory γ subunit and the noncatalytic cGMP-binding sites. J. Biol. Chem. 275, 38611–38619 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki M., Li N., Bondarenko V. A., Yamazaki R. K., Baehr W., Yamazaki A., Binding of cGMP to GAF domains in amphibian rod photoreceptor cGMP phosphodiesterase (PDE). Identification of GAF domains in PDE αβ subunits and distinct domains in the PDE γ subunit involved in stimulation of cGMP binding to GAF domains. J. Biol. Chem. 277, 40675–40686 (2002). [DOI] [PubMed] [Google Scholar]
- 18.Morrison D. F., Cunnick J. M., Oppert B., Takemoto D. J., Interaction of the γ-subunit of retinal rod outer segment phosphodiesterase with transducin. Use of synthetic peptides as functional probes. J. Biol. Chem. 264, 11671–11681 (1989). [PubMed] [Google Scholar]
- 19.Artemyev N. O., Rarick H. M., Mills J. S., Skiba N. P., Hamm H. E., Sites of interaction between rod G-protein α-subunit and cGMP-phosphodiesterase γ-subunit. Implications for the phosphodiesterase activation mechanism. J. Biol. Chem. 267, 25067–25072 (1992). [PubMed] [Google Scholar]
- 20.Artemyev N. O., Mills J. S., Thornburg K. R., Knapp D. R., Schey K. L., Hamm H. E., A site on transducin α-subunit of interaction with the polycationic region of cGMP phosphodiesterase inhibitory subunit. J. Biol. Chem. 268, 23611–23615 (1993). [PubMed] [Google Scholar]
- 21.Artemyev N. O., Binding of transducin to light-activated rhodopsin prevents transducin interaction with the rod cGMP phosphodiesterase γ-subunit. Biochemistry 36, 4188–4193 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Zhang X. J., Gao X. Z., Yao W., Cote R. H., Functional mapping of interacting regions of the photoreceptor phosphodiesterase (PDE6) γ-Subunit with PDE6 catalytic dimer, transducin, and regulator of G-protein signaling9-1 (RGS9-1). J. Biol. Chem. 287, 26312–26320 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slep K. C., Kercher M. A., He W., Cowan C. W., Wensel T. G., Sigler P. B., Structural determinants for regulation of phosphodiesterase by a G protein at 2.0 Å. Nature 409, 1071–1077 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Pandit J., Forman M. D., Fennell K. F., Dillman K. S., Menniti F. S., Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. U.S.A. 106, 18225–18230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qureshi B. M., Behrmann E., Schöneberg J., Loerke J., Bürger J., Mielke T., Giesebrecht J., Noé F., Lamb T. D., Hofmann K. P., Spahn C. M. T., Heck M., It takes two transducins to activate the cGMP-phosphodiesterase 6 in retinal rods. Open Biol. 8, 180075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goc A., Chami M., Lodowski D. T., Bosshart P., Moiseenkova-Bell V., Baehr W., Engel A., Palczewski K., Structural characterization of the rod cGMP phosphodiesterase 6. J. Mol. Biol. 401, 363–373 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H., Robinson H., Ke H., Conformation changes, N-terminal involvement, and cGMP signal relay in the phosphodiesterase-5 GAF domain. J. Biol. Chem. 285, 38149–38156 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez S. E., Heikaus C. C., Klevit R. E., Beavo J. A., The structure of the GAF A domain from phosphodiesterase 6C reveals determinants of cGMP binding, a conserved binding surface, and a large cGMP-dependent conformational change. J. Biol. Chem. 283, 25913–25919 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang K. Y., Card G. L., Suzuki Y., Artis D. R., Fong D., Gillette S., Hsieh D., Neiman J., West B. L., Zhang C., Milburn M. V., Kim S. H., Schlessinger J., Bollag G., A glutamine switch mechanism for nucleotide selectivity by phosphodiesterases. Mol. Cell 15, 279–286 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Barren B., Gakhar L., Muradov H., Boyd K. K., Ramaswamy S., Artemyev N. O., Structural basis of phosphodiesterase 6 inhibition by the C-terminal region of the γ-subunit. EMBO J. 28, 3613–3622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng-Elmore X., Gao X.-Z., Pellarin R., Schneidman-Duhovny D., Zhang X.-J., Kozacka K. A., Tang Y., Sali A., Chalkley R. J., Cote R. H., Chu F., Molecular architecture of photoreceptor phosphodiesterase elucidated by chemical cross-linking and integrative modeling. J. Mol. Biol. 426, 3713–3728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deterre P., Bigay J., Forquet F., Robert M., Chabre M., cGMP phosphodiesterase of retinal rods is regulated by two inhibitory subunits. Proc. Natl. Acad. Sci. U.S.A. 85, 2424–2428 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arshavsky V. Y., Dumke C. L., Bownds M. D., Noncatalytic cGMP-binding sites of amphibian rod cGMP phosphodiesterase control interaction with its inhibitory γ-subunits. A putative regulatory mechanism of the rod photoresponse. J. Biol. Chem. 267, 24501–24507 (1992). [PubMed] [Google Scholar]
- 34.Papermaster D. S., Preparation of retinal rod outer segments. Methods Enzymol. 81, 48–52 (1982). [DOI] [PubMed] [Google Scholar]
- 35.Gulati S., Jastrzebska B., Banerjee S., Placeres Á. L., Miszta P., Gao S., Gunderson K., Tochtrop G. P., Filipek S., Katayama K., Kiser P. D., Mogi M., Stewart P. L., Palczewski K., Photocyclic behavior of rhodopsin induced by an atypical isomerization mechanism. Proc. Natl. Acad. Sci. U.S.A. 114, E2608–E2615 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baker B. Y., Gulati S., Shi W., Wang B., Stewart P. L., Palczewski K., Crystallization of proteins from crude bovine rod outer segments. Methods Enzymol. 557, 439–458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gulati S., Jin H., Masuho I., Orban T., Cai Y., Pardon E., Martemyanov K. A., Kiser P. D., Stewart P. L., Ford C. P., Steyaert J., Palczewski K., Targeting G protein-coupled receptor signaling at the G protein level with a selective nanobody inhibitor. Nat. Commun. 9, 1996 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker B. Y., Palczewski K., Detergents stabilize the conformation of phosphodiesterase 6. Biochemistry 50, 9520–9531 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Biyani N., Righetto R. D., McLeod R., Caujolle-Bert D., Castano-Diez D., Goldie K. N., Stahlberg H., Focus: The interface between data collection and data processing in cryo-EM. J. Struct. Biol. 198, 124–133 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Kremer J. R., Mastronarde D. N., McIntosh J. R., Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 116, 71–76 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Grigorieff N., FREALIGN: High-resolution refinement of single particle structures. J. Struct. Biol. 157, 117–125 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Zheng S. Q., Palovcak E., Armache J.-P., Verba K. A., Cheng Y., Agard D. A., MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rohou A., Grigorieff N., CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J., EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grant T., Rohou A., Grigorieff N., cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scheres S. H. W., Chen S., Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Buchan D. W., Minneci F., Nugent T. C., Bryson K., Jones D. T., Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Collaborative Computational Project, Number 4 , The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994). [DOI] [PubMed] [Google Scholar]
- 55.Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. III, Snoeyink J., Richardson J. S., Richardson D. C., MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Russwurm M., Schlicker C., Weyand M., Koesling D., Steegborn C., Crystal structure of the GAF-B domain from human phosphodiesterase 5. Proteins 79, 1682–1687 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/2/eaav4322/DC1
Fig. S1. Domain organization in the type I phosphodiesterase superfamily.
Fig. S2. Domain organization and primary sequence comparison between PDE6α and PDE6β subunits.
Fig. S3. Biochemical characterization and cryo-EM of the PD6αβ2γ complex.
Fig. S4. Structural features of regulatory GAF domains of the PDE6αβ2γ complex.
Fig. S5. Comparison between the H-loop orientations of PDE6 and PDE5.
Fig. S6. Cryo-EM reconstruction and data fitting of the PDE6αβ2γ complex.
Table S1. Cryo-EM data collection, refinement, and validation statistics for the PDE6αβ2γ complex.