

Abstract
In vitro, microRNA-126 (miR-126) inhibits SLK cell proliferation, inhibits the cell cycle, induces cell apoptosis, and reduces cell invasiveness. Double luciferase assays have shown that phosphatidylinositol-3 kinase (PI3K) is the miR-126 target in SLK cells. We aimed to investigate the influence of miR-126 on the phosphate and tension homology deleted on chromosome ten (PTEN)/PI3K/protein kinase B (AKT) pathway members in SLK cells and to determine the expression of these pathway members in Kaposi's sarcoma (KS). The mimic and inhibitor of miR-126 were transfected into SLK cells and PTEN and AKT1 expression was assayed in SLK cells by real-time quantitative PCR and western blotting. PTEN, AKT1, phosphorylated (P)-PTEN, and phosphorylated (P)-AKT expression in KS and paraneoplastic skin were assayed by immunohistochemistry. AKT1 expression was downregulated in SLK cells that overexpressed miR-126, while there was no significant difference in PTEN expression between SLK cells overexpressing miR-126 and those in which its expression was knocked down. PTEN and AKT1 were expressed in KS and paraneoplastic skin but P-AKT was not. Interestingly, P-PTEN was not expressed in paraneoplastic skin but it was expressed in 90% of KS biopsies (P < .05). P-PTEN expression was also significantly higher in visceral than in cutaneous KS (P = .01) and was higher in indoor than in outdoor workers (P = .018). In vitro, miR-126 negatively regulated AKT1 expression but no regulation of PTEN expression was evident. Results indicated that in KS, PTEN is activated and may therefore be a potential therapeutic target for KS. In addition, these results also indicate that sunlight may not be the cause of KS.
Keywords: AKT1, Kaposi's sarcoma, miR-126, PTEN, PTEN/PI3K/AKT, regulation
1. Introduction
Kaposi's sarcoma (KS) is a heterogeneous angioproliferative neoplasm that requires HHV-8 infection and presents in 4 recognized epidemiological forms. Pathogenesis of all types of this disease is not clear, and there is no standard treatment.[1] Previous studies have reported the involvement of micro RNAs (miRNAs) in the etiology, diagnosis, prognosis, and treatment of many cancers.[2,3] miR-126 expression is upregulated in KS, and miR-126 has been shown to inhibit cell proliferation, arrest cell cycle progression, induce cell apoptosis, and inhibit cell invasiveness of KS SLK cells.[4–6] The results of bioinformatics analysis and luciferase reporter assays revealed that phosphatidylinositol-3 kinase (PI3K) was a direct target of miR-126 and that aberrant expression of genes in the phosphate and tension homology deleted on chromosome ten (PTEN)/PI3K/ protein kinase B (AKT) signaling pathway was involved in tumorigenesis and regulation of gene expression, the cell cycle, cell proliferation, differentiation, proliferation, angiogenesis, and metastasis in tumor cells.[6–9] PTEN is a tumor suppressor gene for phosphatases with lipid and protein substrates[10] and its dysfunction can activate the PI3K/AKT signaling pathway, leading to malignant cell proliferation and cancer.[11] AKT is a proto-oncogene, and a downstream effector of the PTEN/PI3K/AKT signaling pathway. P-AKT, the active form of AKT, is strongly expressed in various malignancies.[12,13] PI3K, a second messenger system located on the plasma membrane. To promote cell proliferation, PI3K activates AKT by transferring a phosphate from PTEN.[14]
Here, we investigated the regulation of miR-126 in SLK cells on the PTEN/PI3K/AKT signaling pathway in vitro, and the expression of its members in KS tissue.
2. Material and methods
2.1. SLK cells
The human KS-derived SLK cell line was purchased from the National Institutes of Health AIDS Reagent Program (Bethesda, Maryland) and cultured in Dulbecco's Modified Eagle Medium (DMEM) with 4.5 g/L glucose (Gibco, Grand Island, NY) and 10% fetal bovine serum (Gibco, Grand Island, NY). Cells were cultured in a humidified atmosphere of 95% air and 5% CO2 at 37°C.
2.2. Transfection of SLK cells by miR-126 mimic and inhibitor
2.2.1. Determination of the optimal transfection conditions
HiPerFect Transfection Reagent (Qiagen, Hilden, Germany) and AllStars Hs Cell Death Control siRNA (Qiagen) were mixed according to the manufacturer's instructions. The mixture was then added to 96-well plates containing SLK cell cultures and an MTT Cell Proliferation Cytotoxicity Assay Kit (Sang Biotech, Shanghai, China) was used to measure the cell viability at 6, 24, 48, and 72 hours, respectively, by measuring the absorbance at 450 nm using a spectrophotometer (NanoDrop, NY). Six wells were measured for each time point and all experiment were done in triplicate. Cell viability (%) = (As – Ab)/(Ac – Ab) × 100%, with data expressed as the mean (Ab: blank control group; Ac: negative control group; As: experimental group).[6]
According to the above optimal transfection conditions, the miR-126 mimic (miR-126m, cat. no. MSY0000445), miR-126 inhibitor (miR-126i, cat. no. MIN0000445), AllStars Negative Control siRNA (miR-126NC, cat. no.1027272), and miScript Inhibitor Negative Control miR-126 (miR-126iNC, cat no.1027271) were purchased from Qiagen (Qiagen). Cells were transfected with HiPerFect Transfection Reagent (Qiagen) following the manufacturer's instructions. SLK cells were used as blank control to exclude the effect of small nucleotide fragments on SLK cells.
2.2.2. Quantitative real-time reverse transcription PCR (qRT-PCR)
After 48 hours transfection, total RNA was extracted from cells using an RNeasy Mini Kit (Qiagen). The cDNA was reverse transcribed with an miRcute miRNA cDNA Kit (Tiangen, Beijing, China) and a PrimeScript First strand cDNA Synthesis Kit (TaKaRa, Dalian, China) following the manufacturer's instructions. The RNA concentration was determined by fluorometric quantitation (Invitrogen, NY), and RNA integrity was assayed by gel electrophoresis. miR-126, PTEN, and AKT1 expressions were assayed by qRT-PCR using SYBR Green (Tiangen). Real-time PCR experiments were performed in a ThermoFisher PCR system (Thermo Fisher Scientific, NY), under the following conditions: 1 cycle of 95°C for 2 minutes, 40 cycles of 95°C for 15 seconds, and 60°C for 10 seconds. The primers used were miR-126 sense (5′-3′, 100354638, Sang Biotech, Shanghai, China), U6 (Hs_RNU6 miScript Primer Assay, MS00033740, Tiangen), PTEN (F, 9407365113; R, 9407365114, Sang Biotech), AKT1 (F, 9407365109; R, 9407365110, Sang Biotech), and β-actin (assay kit QT00095431, Qiagen). Each sample was assayed in triplicate and the values (expressed as mean ± standard deviation (SD)) were calculated using the 2–ΔΔCt method.
2.2.3. Western blotting assay (WB)
After transfection for 48 hours, the cells were collected in cold PBS and splitted in radioimmunoprecipitation assay buffer (RIPA) buffer with protease inhibitors (e.g., 1 mmol/L phenylmethylsulfonyl fluoride, 2 mmol/L NaVO4, 50 mmol/L NaF, 1 μg/mL aprotinin, and 10 μg/mL leupeptin) at 4°C for 30 minutes. Protein concentration was determined with a Pierce (bicinchoninic acid) BCA assay kit (Thermo Fisher Scientific) following the manufacturer's instructions. Equal amounts of protein were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose membranes. Membranes were blocked with 5% goat serum for 2 hours at room temperature and washed 3 times with tris-buffered saline and Tween 20 (TBST). The membrane was incubated overnight at 4°C with primary PTEN, AKT1, or β-actin antibodies (diluted 1:200; Abcam, London, UK), washed 3 times with TBST and incubated with HRP-conjugated secondary antibody (diluted 1: 10,000; Abcam, London, UK). Signals were detected using enhanced chemiluminescence reagents (ECL) (BosterBio, Wuhan, Hubei, China). Image J software was used to calculate the optical density of the target proteins relative to β-actin protein levels. Each sample was assayed in triplicate.
2.2.4. Participants
Participants gave written consent before inclusion. The ethics committee of the People's Hospital of Xinjiang Uygur Autonomous Region (China) approved the study protocol (no. 16-87-KY). Paraffin blocks of tumor tissue obtained from 40 KS patients treated between 2010 and 2016 were selected from the hospital dermatology laboratory. The patients included 29 Uygur, 3 Han, 7 Hakesa, and 1 Siberian Chinese. Thirty-three were males and 7 were females with ages that ranged from 26 to 85 (mean 57.30 ± 17.56) years; 24 were at the patch/plaque stage and 16 were at the nodular stage when treated. Ten cases were human immunodeficiency virus (HIV) seropositive and 35 cases were human herpesvirus 8 (HHV8) seropositive. The HHV8 status of 2 patients was unknown. Chemotherapy, radiation, laser, immunization, cryotherapy, and other treatments had not been performed in the 3 months before specimen preparation. Paraffin blocks of paraneoplastic skin were used as controls.
2.2.5. Immunohistochemistry (IHC)
Forty formalin-fixed, paraffin-embedded KS and 40 normal adjacent biopsy tissue blocks were evaluated by 2 independent pathologists who were blinded to the experimental groups. PTEN, P-PTEN, AKT1, and P-AKT1 expression were assayed in sections by IHC. After fixation, the slides were deparaffinized by placing them in xylene followed by absolute, 95%, and 70% ethanol and finally water for 2 minutes each. Antigen retrieval was performed in a pressure-cooker with pH 9.0 Tris/EDTA buffer, followed by a 20 minutes cool-down. Then the slides were treated with 3% H2O2 in methanol for 15 minutes to inhibit endogenous peroxidase. After washing, the slides were blocked with 10% normal goat serum in a humid chamber for 30 minutes at room temperature, and incubated with PTEN, P-PTEN, AKT1, and P-AKT primary antibodies (1:300 dilution; Abcam, London, England) overnight at 4°C. The slides were counterstained with hematoxylin, dehydrated, and mounted. Positive control slides were provided by the reagent company (Abcam), and an isotype antibody instead of the primary antibody was used as a negative control. Staining was evaluated by 2 pathologists who were blinded to the experimental conditions.
The percentage of positive cells in 10 randomly selected high-power visual fields (400×) was calculated. Brownish to yellow staining was considered as a positive reaction. Staining intensity was scored as 0 (colorless), 1 (light yellow), 2 (light brown), and 3 (dark brownish yellow). Percent positivity was scored as 0 (no positive cells), 1 (< 25% positive cells), 2 (25%–50% positive cells, 3 (51%–75% positive cells), and 4 (> 75% positive cells). An integral IHC score was calculated by adding the intensity and percentage scores; total scores of 0–1 were recorded as –, scores of 1–3 as +, scores of 4–8 as ++, and scores of 9–12 as +++.[15]
2.2.6. Statistical analysis
Repeated measure analysis of variance (ANOVA) was used to compare the expression between genes (and proteins), with data expressed as mean ± SD. The results of staining and clinical parameters were compared by Chi-square or Fisher's exact tests. All analyses were performed with SPSS, version 20.0 software (IBM Corp. Armonk, NY). A 2-tailed P < .05 was considered to be statistically significant.
3. Results
3.1. Transfection efficiency
3.1.1. Transfection efficiency
SLK cells were transfected with the AllStars Hs Cell Death Control siRNA to determine the optimal conditions. The cell survival percentage was the lowest, and the transfection efficiency was the highest with 1 μL of transfection reagent and a transfection time of 48 hours (Fig. 1A). SLK cells were then transfected with either miR-126m, miR-126i, miR-126NC, or miR-126iNC according to the above conditions and SLK cells were treated as blank controls. miR-126 expression in each group was assayed by RT-PCR. The expression in the miR-126NC, miR126iNC, and blank control cell groups did not differ significantly, but was higher in the miR126i than in miR126m cell groups (Fig. 1B and C). The results confirmed successful transfection of miR-126m, miR-126i, miR-126NC, and miR-126iNC according to above transfection conditions.
Figure 1.
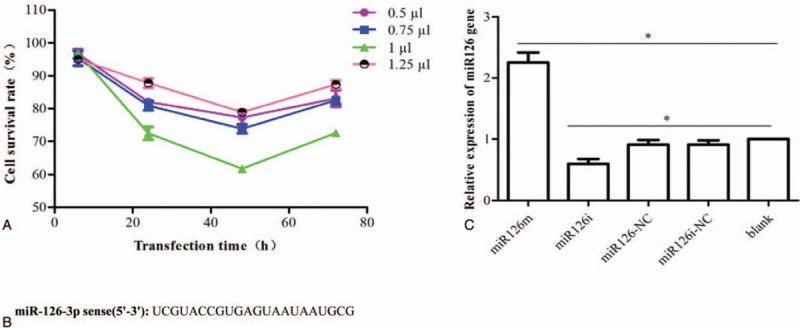
Validation of transfection efficiency. (A) The optimal transfection conditions were determined in miR-126NC cells. (B, C) The expression of miR-126 in SLK cells transfected with miR-126m, miR-126i, miR-126NC, and miR-126iNC, as well as in blank control cells for 48 hours as assessed by qRT-PCR. miR-126m = miR-126 mimic, miR-126NC = AllStars Negative Control siRNA.
3.1.2. Regulatory effects of miR-126 on AKT1 and PTEN expression
AKT1 and PTEN expression in SLK cells transfected with either miR-126m, miR-126i, miR-126NC, or miR-126iNC, as well as in blank control cells was assayed by both RT-PCR and western blot. AKT1 mRNA and protein expression were significantly higher in miR-126i-transfected cells than in cells transfected with miR-126m (Fig. 2). These results suggest that miR-126 overexpression was associated with reduced expression of both AKT1 mRNA and protein. PTEN mRNA and protein expression in miR-126m-transfected, miR-126i-transfected, miR-126NC, miR-126iNC, and blank control cells did not differ significantly (Fig. 3). These results suggest that PTEN expression was not affected by miR-126.
Figure 2.
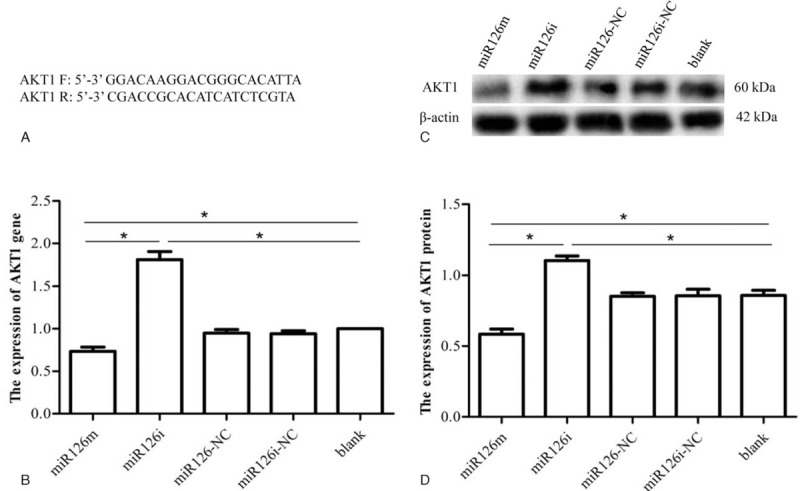
Regulatory effects of miR126 on AKT1 expression. (A) AKT1 primer sequence. (B) The expression of AKT1 mRNA in SLK cells after transfection with either the miR126 mimic, inhibitor, negative control, and inhibitor negative control, as well as in blank control cells at 48 hours, as assessed by qRT-PCR. (C, D) Western blot assays of AKT1 protein expression in SLK cells transfected with either the miR-126 mimic, inhibitor, negative control, or inhibitor negative control, as well as in blank control cells. ∗P < .05. AKT = protein kinase B, miR-126m = miR-126 mimic.
Figure 3.
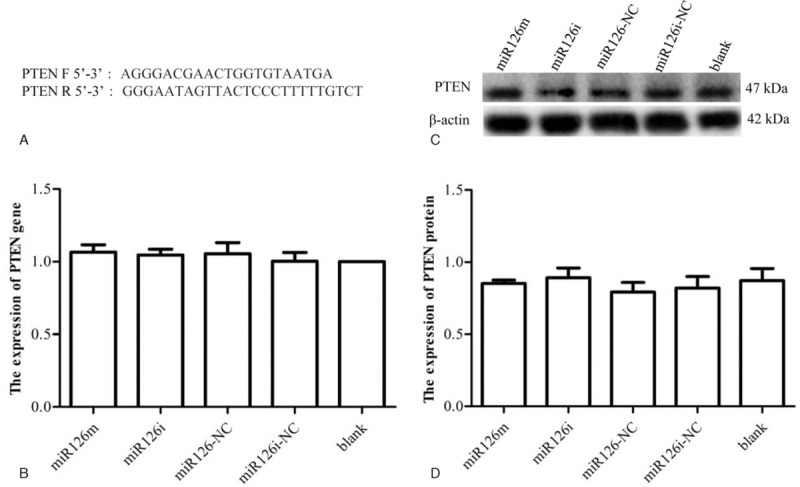
Regulatory effects of miR126 on PTEN expression. (A) PTEN primer sequence. (B) PTEN mRNA expression in SLK cells after transfection with either the miR-126 mimic, inhibitor, negative control, or inhibitor negative control, as well as in blank control at 48 hours. (C, D) Western blot assays of PTEN protein expression in SLK cells transfected with either the miR-126 mimic, inhibitor, negative control, or inhibitor negative control, as well as in blank control cells. ∗P < .05. PTEN = phosphate and tension homology deleted on chromosome ten.
3.1.3. PTEN, P-PTEN, AKT, and P-AKT expression and clinicopathological characteristics
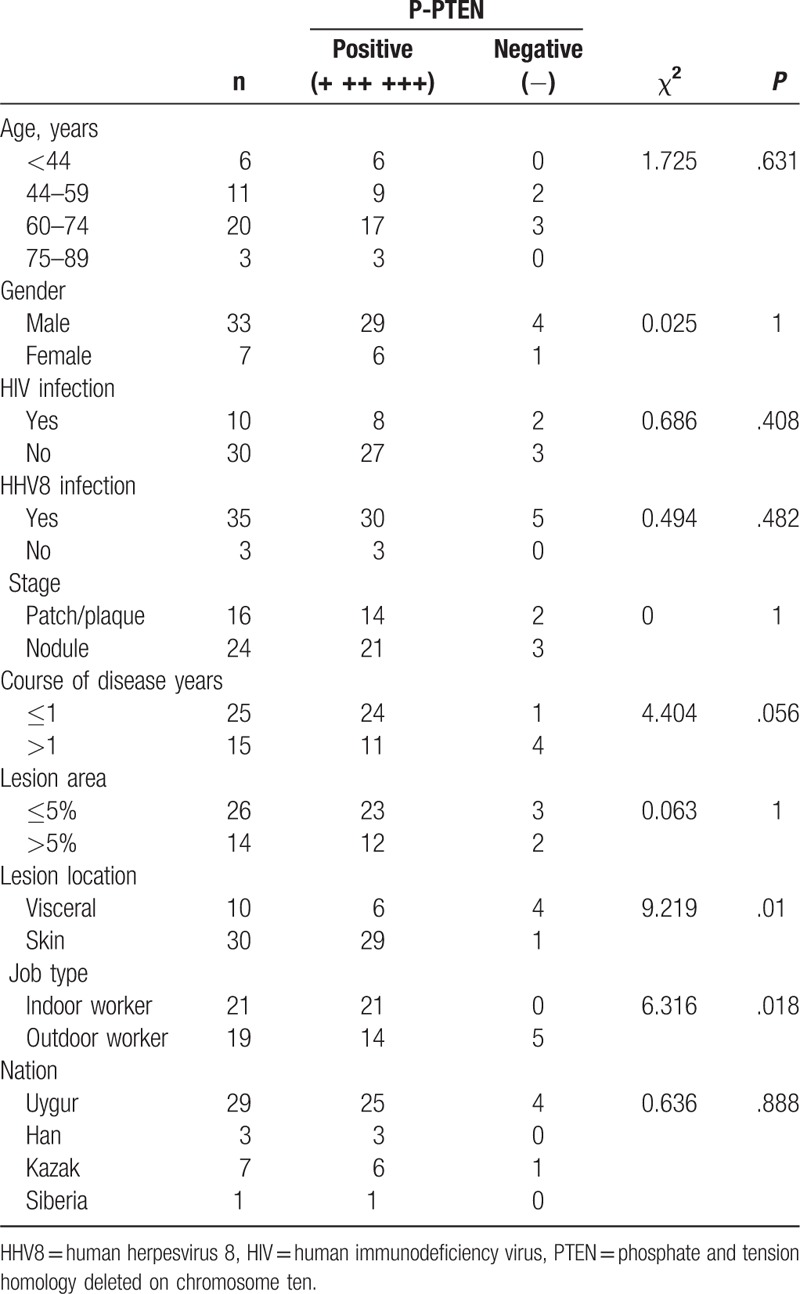
The expression of PTEN, P-PTEN, AKT, and P-AKT in KS tissue was assayed by IHC. As expected, PTEN and AKT were positive in both KS and paraneoplastic skin tissues, primarily in the cytoplasm of tumor cells (Fig. 4 C, D, G, H), which was similar to previous reports.[16,17] P-AKT was negative in KS tissue (Fig. 4I and J) and in paraneoplastic skin tissue. P-PTEN was positive in 36 of 40 KS biopsies, was negative in 4 (Fig. 4E and F), and was negative in all normal samples. The differential expression of P-PTEN in KS and paraneoplastic skin tissues was statistically significant (P < .001; Table 1). These results suggest P-PTEN is highly expressed in KS. The association of P-PTEN expression and the clinical manifestations of KS were found to be significant. There were also significant differences in P-PTEN expression in KS skin and visceral KS samples (P = .01) and in indoor and outdoor workers (P = .018; Table 2).
Figure 4.
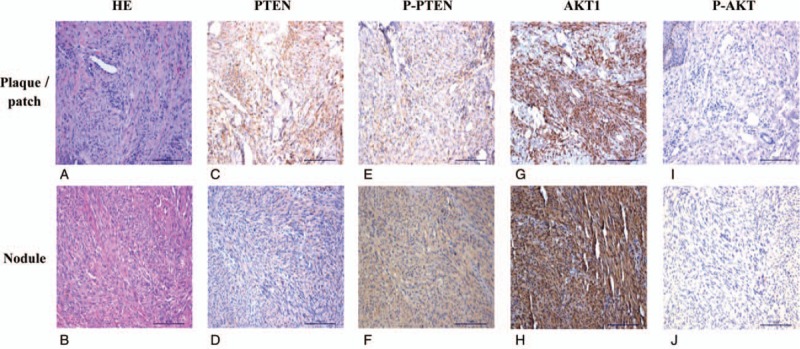
PTEN, P-PTEN, AKT, and P-AKT expression in patch/plaque phase and nodular phase KS (×20 magnification). (A, B) Hematoxylin and eosin staining of plaque/patch and nodular phase KS. (C, D) PTEN is expressed in the cytoplasm of spindle cells and vascular endothelial cells at both plaque/patch and nodular phase KS. (E, F) P-PTEN is expressed in the cytoplasm of spindle cells and vascular endothelial cells at both plaque/patch and nodular phase KS. (G, H) AKT is expressed in the cytoplasm of spindle cells and vascular endothelial cells at both plaque/patch and nodular phase KS. (I, J) P-AKT expression is not seen in the cytoplasm of spindle cells and vascular endothelial cells at both plaque/patch and nodular phase KS. AKT = protein kinase B, KS = Kaposi's sarcoma, PTEN = phosphate and tension homology deleted on chromosome ten.
Table 1.
Expression of PTEN, P-PTEN, AKT1 and P-AKT1 in the tissues of Kaposi's sarcoma and its paraneoplastic skin.
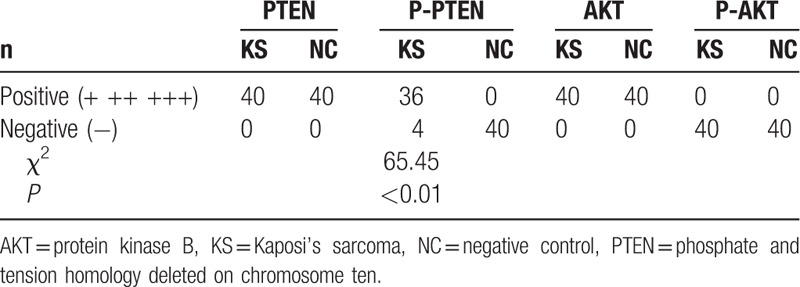
Table 2.
The relationship between the expression of P-PTEN and clinical features.
4. Discussion
KS is a skin tumor with a high recurrence rate, and there is no effective treatment. The mechanism and regulation of growth in KS have not been extensively studied. Future investigations will help to provide a molecular basis for targeted treatment of KS. miRNAs participate in tumor development by degrading or inhibiting the translation of target mRNAs. miR-126 expression is downregulated in endometrial carcinoma and non-small cell lung cancer,[18–20] and its downregulation has been associated with shortened survival.[21,22] We hypothesized that low miR-126 expression promoted malignant proliferation and metastasis of tumor cells, but the mechanism of growth regulation was not clear. Previous studies have shown that the expression of miR-126 in KS was significantly upregulated, and that its target was PI3K.[4–6] The PTEN/PI3K/AKT signaling pathway regulates cell growth in breast, lung, and gastric cancer.[23–27] The PTEN gene integrates the activity of multiple upstream and downstream signaling pathways, and inactivation and mutations in this gene have been associated with endometrial cancer, hepatocellular carcinoma, and breast cancer.[28–30] Recent studies have found that transfection of human prostate cancer cells with wild-type PTEN reduced tumor angiogenesis; furthermore, AKT inhibitors are currently in clinical trials as anticancer treatments.[31–33] PTEN and AKT were chosen as research targets because they are key regulators of tumor cell growth. AKT1 was chosen because of previous studies confirming its extensive tissue distribution and regulation of cell growth and apoptosis.[34–35]
In this study, the SLK cell line was transfected in vitro with miR-126 mimic and inhibitor as a cytological method to confirm the regulation of PTEN/PI3K/AKT pathway components. AKT1 expression was significantly lower in the miR-126 mimic group than in the miR-126 inhibitor group at both gene and protein levels. There were no significant differences in the expression of PTEN mRNA or protein in SLK cells transfected with the miR-126 mimic, miR-126 inhibitor, or miR-126 negative control group, or in the blank control cells, which indicated that miR-126 did not regulate PTEN expression in SLK cells in vitro.
PTEN and AKT were expressed in both KS and paraneoplastic skin tissues, indicating that they were not downregulated in KS. P-AKT was not expressed in KS tissues or paraneoplastic skin, which indicated that AKT was inactive in KS. We speculate that P-AKT, a downstream effector in the PTEN/PI3K/AKT signaling pathway, had no effect on the growth of KS cells, indicating that KS growth in tumors was regulated by other growth factors. The difference in expression of PTEN in KS and paraneoplastic skin tissues was statistically significant and the expression of P-PTEN (the active state of PTEN) in KS tissue was higher than that in paraneoplastic skin, indicating that PTEN is active in KS. As PTEN is the upstream gene of AKT, it might be involved in the inactivation of AKT. P-AKT is overexpressed in various malignancies,[36–38] but was not expressed in KS tissues, suggesting an absence of malignant hyperplasia or a regulation of malignant hyperplasia by other factors; Therefore, we propose that AKT is not a suitable treatment target in KS.
Both PTEN and P-PTEN were expressed in KS tissues, indicating partial activation. P-PTEN expression differed with the location of the lesion and the patient's job type, being higher in skin than in visceral tumors, and being more prevalent in tumors of indoor workers than in those of outdoor workers, which indicated that sun exposure was not a factor in the growth of KS. We therefore propose that PTEN and its activated form, P-PTEN, participate in the growth and regulation of KS. Combined with previous studies,[6] results from the current study reveal that PTEN and miR-126 have similar roles in regulating the growth of KS cells, and in theory, PTEN may be a potential therapeutic target in KS.
The KS tissues used in this study are all archive specimens, of which RNA has been degraded and difficult to extract. But the expression of miR126 in KS has been detected with gene chip technology and published in previous articles by our research team.[4] The interaction between miR126 and PTEN, P-PTEN, AKT and P-AKT in vivo will be studied in future experiments.
5. Conclusions
In vitro, miR-126 negatively regulated AKT1 expression, but did not influence the expression of PTEN. In KS tissue, P-PTEN was highly expressed but P-AKT expression was not observed. We speculate that AKT1 activation was negatively regulated by PTEN and that PTEN/PI3K/AKT signaling was not the only influence on the growth of KS. The lack of P-AKT expression in KS suggested the absence of malignant proliferation or that malignancy was regulated by other growth factors. Sunlight did not affect the growth of KS. PTEN may be a therapeutic target for KS, while AKT inhibitors may not be suitable for the treatment of KS. The regulation of miR-126 on AKT and PTEN in vivo needs further study.
Acknowledgements
We thank the Clinical Laboratory Center of the Dermatological Department of Xinjiang Uygur Autonomous Region People's Hospital who graciously provided experimental platform.
Author contributions
Conceptualization: Gaihui Lu, Xiongming Pu.
Data curation: Gaihui Lu.
Formal analysis: Gaihui Lu, Yuan Ding, Peng Wang, Caoying Wu.
Funding acquisition: Xiujuan Wu.
Investigation: Gaihui Lu.
Methodology: Gaihui Lu, Zongfeng Zhao, Caoying Wu.
Resources: Gaihui Lu.
Software: Gaihui Lu, Zongfeng Zhao.
Supervision: Xiaojing Kang, Xiongming Pu.
Writing – original draft: Gaihui Lu.
Writing – review & editing: Gaihui Lu.
Footnotes
Abbreviations: AKT = protein kinase B, HHV8 = human herpesvirus 8, HIV = human immunodeficiency virus, KS = Kaposi's sarcoma, miR-126 = microRNA-126, miR-126i = miR-126 inhibitor, miR-126iNC = miScript Inhibitor Negative Control miR-126, miR-126m = miR-126 mimic, miR-126NC = AllStars Negative Control siRNA, PI3K = phosphatidylinositol-3 kinase, PTEN = phosphate and tension homology deleted on chromosome ten, SD = standard deviation.
GL and XW, contributed equally to this work and should be considered as the first co-authors.
Grant support: This work was supported by grants from the Natural Science Foundation of China Xinjiang Uygur Autonomous Region (No. 81660454).
Conflicts of interest: The authors declared no potential conflict of interest with respect to the authorship and/or publication of this article.
References
- [1].Bolognia JL, Jorizzo JL, Rapini RP. Dermatology 2015;1961–4. Translated into Chinese by Xuejun Zhu. [Google Scholar]
- [2].Zhang B, Pan X, Cobb GP, et al. microRNAs as oncogenes and tumor suppressors. Dev Biol 2007;302:1–2. [DOI] [PubMed] [Google Scholar]
- [3].Landgraf P, Rusu M, Sheridan R, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell1 2007;29:1401–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wu XJ, Pu XM, Zhao ZF, et al. The expression profiles of microRNAs in Kaposi's sarcoma. Tumour Biol 2015;36:437–46. [DOI] [PubMed] [Google Scholar]
- [5].Ene AMC, Borze I, Guled M. MicroRNA expression profiles in Kaposi's sarcoma. Pathol Oncol Res 2014;20:153–9. [DOI] [PubMed] [Google Scholar]
- [6].Wu XJ, Zhao ZF, Kang XJ, et al. MicroRNA-126-3p suppresses cell proliferation by targeting PIK3R2 in Kaposi's sarcoma cells. Oncotarget 2016;7:36614–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Haga S, Ozaki M, Inoue H, et al. The survival pathwaysphosphatidylinositol-3 kinase (PI3-K)/phosphoinositide-dependent protein kinase 1 (PDK1)/Akt modulate liver regeneration through hepatocytesize rather than proliferation. Hepatology 2009;49:204–14. [DOI] [PubMed] [Google Scholar]
- [8].Patsoukis N, Brown J, Petkova V, et al. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal 2012;5:ra46doi: 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Limon JJ, Fruman DA. Akt and mTOR in B cell activation and differentiation. Front Immunol 2012;3:228–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumor suppressor gene MMAC1 at chromosome q23.3 that is mutated in multiple advanced cancers. Nat Genet 1997;15:356–62. [DOI] [PubMed] [Google Scholar]
- [11].Vivanco I, Sawyers CL. The phosphatidylinositol 3 kinase Akt pathway in human cancer. Nat Rev Cancer 2002;2:489–501. [DOI] [PubMed] [Google Scholar]
- [12].Bellacosa A, Testa JR, Staal SP, et al. A retroviral oncogene, Akt, encoding a serine-threonine kinase containing an SH2-like region. Science 1991;254:274–7. [DOI] [PubMed] [Google Scholar]
- [13].Matheny RW, Adamo ML. Current perspectives on Akt Akt-ivation and Akt-ions. Exp Biol Med (Maywood) 2009;234:1264–70. [DOI] [PubMed] [Google Scholar]
- [14].Foster K, Wang Y, Zhou D, et al. Dependence on PI3K/Akt signaling for malignant rhabdoid tumor cell survival. Cancer Chemother Pharmacol 2009;63:783–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ma Y, Yang J, Wang R, et al. Aurora-A affects radiosenstivity in cervical squamous cell carcinoma and predicts poor prognosis. Oncotarget 2017;8:31509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kremer CL, Klein RR, Mendelson J, et al. Expression of mTOR signaling pathway markers in prostate cancer progression. Prostate 2006;66:1203–12. [DOI] [PubMed] [Google Scholar]
- [17].Bhattacharyya SS, Paul S, Mandal SK, et al. A synthetic coumarin (4-methyl-7 hydroxy coumarin) has anti-cancer potentials against DMBA-induced skin cancer in mice. Eur J Pharmacol 2009;614:128–36. [DOI] [PubMed] [Google Scholar]
- [18].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009;136:215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jayaraman M, Radhakrishnan R, Mathews CA, et al. Identification of novel diagnostic and prognostic miRNA signatures in endometrial cancer. Genes Cancer 2017;8:566–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang K, Chen M, Wu W. Analysis of microRNA (miRNA) expression profiles reveals 11 key biomarkers associated with non-small cell lung cancer. World J Surg Oncol 2017;15:175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fiala O, Pitule P, Hosek P, et al. The association of miR-126-3p, miR-126-5p and miR-664-3p expression profiles with outcomes of patients with metastatic colorectal cancer treated with bevacizumab. Tumour Biol 2017;39: 1010428317709283. [DOI] [PubMed] [Google Scholar]
- [22].Li J, Ping JL, Ma B, et al. Deregulation of miR-126-3p in basal-like breast cancers stroma and its clinical significance. Pathol Res Pract 2017;213:922–8. [DOI] [PubMed] [Google Scholar]
- [23].Xue M, Ji X, Xue C. Caspase-dependent and caspase-independent induction of apoptosis in breast cancer by fucoidan via the PI3K/AKT/GSK3( pathway in vivo and in vitro. Biomed Pharmacother 2017;94:898–908. [DOI] [PubMed] [Google Scholar]
- [24].Rädler PD, Wehde BL, Wagner KU. Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol Cell Endocrinol 2017;82:31–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhong JT, Yu J, Wang HJ, et al. Effects of endoplasmic reticulum stress on the autophagy, apoptosis, and chemotherapy resistance of human breast cancer cells by regulating the PI3K/AKT/mTOR signaling pathway. Tumour Bio 2017;39: 1010428317697562. [DOI] [PubMed] [Google Scholar]
- [26].Wang T, Cai Z, Hong G, et al. MicroRNA-21 increases cell viability and suppresses cellular apoptosis in non-small cell lung cancer by regulating the PI3K/Akt signaling pathway. Mol Med Rep 2017;16:6506–11. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [27].Geng W, Zhang HY. Research on the mechanism of HP mediated PI3K/AKT/GSK3 ( pathways in gastric cancer. Eur Rev Med Pharmacol Sci 2017;21:33–7. [PubMed] [Google Scholar]
- [28].Bian X, Gao J, Luo F, et al. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene 2017;37:341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang B, Xu J, Wang H, et al. Effect and mechanism of sophoridine to suppress hepatocellular carcinoma in vitro and vivo. Biomed Pharmacother 2017;95:324–30. [DOI] [PubMed] [Google Scholar]
- [30].Golmohammadi R, Rakhshani MH, Moslem AR, et al. Prognostic role of PTEN gene expression in breast cancer patients from North-East Iran. Asian Pac J Cancer Prev 2016;17:4527–31. [PubMed] [Google Scholar]
- [31].Anai S, Goodison S, Shiverick K, et al. Combination of PTEN gene therapy and radiation inhibits the growth of human prostate cancer xenografts. Human Gene Therapy 2006;17:975–84. [DOI] [PubMed] [Google Scholar]
- [32].Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nature Rev Cancer 2009;9:550–62. [DOI] [PubMed] [Google Scholar]
- [33].Marone R, Cmiljanovic V, Giese B, et al. Targeting phosphoinositide 3-kinase—moving towards therapy. Biochim Biophys Acta 2008;1784:159–85. [DOI] [PubMed] [Google Scholar]
- [34].Yang ZZ, Tschopp O, Baudry A, et al. Physiological functions of protein kinase B/Akt. Biochem Soc Trans 2004;32(pt 2):350–4. [DOI] [PubMed] [Google Scholar]
- [35].Chang Z, Zhang Q, Feng Q, et al. Deletion of Akt1 causes heart defects and abnormal cardiomyocyte proliferation. Dev Biol 2010;347:384–91. [DOI] [PubMed] [Google Scholar]
- [36].Liu L, Gao H, Wang H, et al. Catalpol promotes cellular apoptosis in human HCT116 colorectal cancer cells via microRNA-200 and the downregulation of PI3K-Akt signaling pathway. Oncol Lett 2017;14:3741–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Shan ZZ, Chen PN, Wang F, et al. Expression of P-EGFR and P-Akt protein in esophageal squamous cell carcinoma and its prognosis. Oncol Lett 2017;14:2859–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Gao HY, Han CX. The role of PTEN up-regulation in suppressing glomerular mesangial cells proliferation and nephritis pathogenesis. Eur Rev Med Pharmacol Sci 2017;21:3634–41. [PubMed] [Google Scholar]