

Supplemental Digital Content is available in the text
Keywords: differentially expressed genes, osteoarthritis, protein-protein interaction network, regulatory network, rheumatoid arthritis, underlying pathogenesis
Abstract
Rheumatoid arthritis (RA) and osteoarthritis (OA) comprise the most common forms of arthritis. The aim of this study was to identify differentially expressed genes (DEGs) and associated biological processes between RA and OA using a bioinformatics approach to elucidate their potential pathogenesis.
The gene expression profiles of the GSE55457 datasets, originally produced through use of the high-throughput Affymetrix Human Genome U133A Array, were downloaded from the Gene Expression Omnibus (GEO) database. The GSE55457 dataset contains information from 33 samples, including 10 normal control (NC) samples, 13 RA samples, and 10 OA samples. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes pathway (KEGG) enrichment analyses were performed to identify functional categories and associated molecular and biochemical pathways, respectively, for the identified DEGs, and a protein-protein interaction (PPI) network of the DEGs was constructed using Cytoscape software.
GO and KEGG results suggested that several biological pathways (ie, “immune response,” “inflammation,” and “osteoclast differentiation”) are commonly involved in the development of both RA and OA, whereas several other pathways (eg, “MAPK signaling pathway,” and “ECM-receptor interaction”) presented significant differences between these disorders.
This study provides further insights into the underlying pathogenesis of RA and OA, which may facilitate the diagnosis and treatment of these diseases.
1. Introduction
Rheumatiod arthritis (RA) is a complex, chronic multi-systemic autoimmune disease that mainly affects the flexibile joints.[1] Approximately 0.5% to 1% of the adult population is affected with RA worldwide 5 to 50 per 100,000 new cases reported annually.[2] Although disease etiology remains unknown, the progressive, chronically inflammatory, and destructive joint characteristics of RA are perpetuated by an invasive inflammatory tissue also known as pannus tissue.[3] In addition, various inflammatory cells in the synovial membrane (including monocytes/macrophages, T and B cells, osteoclasts, dendritic cells, endothelial cells, and synovial fibroblasts) contribute to the development and progression of RA.[4] Similarly, osteoarthritis (OA) constitutes another main type of common and chronic disease that affects the joints. OA mainly affects individuals older than with a reported disease incidence as high as 80%.[1] OA is also a progressive disorder that is characterized by degradation of the cartilage and bone, along with osteophyte formation, narrowing of the joint space and subchondral sclerosis.[5] Although RA and OA share similar symptoms, OA differs from RA in that these diseases exhibit different pathological manifestations for the synovial. The characteristics of RA are synovial cell hypertrophy and hyperplasia along with infiltration of lymphocytes and inflammatory cells, whereas OA has a fewer leukocytic infiltrates.[6,7] Despite significant advances toward an understanding of the pathophysiology of RA and OA, early diagnosis, therapeutic intervention, and underlying pathogenesis remain a challenging.[8] Therefore, elucidating the unique characteristics belonging to RA or OA is paramount in developing therapies to improve patient outcome.
Recently, numerous research strategies have explored the molecular characteristics of RA and OA. Among these research strategies, high-throughput microarray methodologies have received extensive attention and engendered substantial progress in field such as medical oncology with marked clinical applications ranging from molecular diagnosis to molecular classification, patient stratification to prognosis prediction, and new drug targets discovery to response prediction.[9–11] In addition, multiple gene expression profiling studies on OA and RA have been performed using microarray technology and several key genes and diagnostic biomarkers have been identified for these diseases, including the profiling of hundreds of differentially expressed genes (DEGs) involved in different pathways, biological processes, or molecular functions.[12,13] However, owing to the underlying shortcomings of microarray technology, such as small sample size, measurement error, and information not sufficient, the integrative analysis of multiple factors that contribute to the development of RA and OA has proven to be a major challenge, and underlying pathogenesis of RA and OA thus remaining poorly understood.
Therefore, in the present study, we downloaded the original data (GSE55457), provided by Dirk Woetzel, from the publically available Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo/) to identify DEGs and the associated biological processes between OA and RA using comprehensive bioinformatics analyses. The DEGs were subjected to functional enrichment and pathway analyses; moreover, a protein-protein interaction (PPI) network was constructed to screen for key gene nodes. Analysis of the biological functions and pathways shared between and unique to these disorders may shed light on further insights regarding OA and RA development at the molecular level and pave the way toward understanding potential disease pathogenesis mechanisms to facilitate diagnosis, prognosis, and the identification of drug targets.
2. Methods
2.1. Data resources
Gene expression profile dataset GSE55457[14] was downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). The data was produced using an Affymetrix Human Genome U133A Array (Agilent Technologies, Santa Clara, CA). The GSE55457 dataset contained data from 33 samples, including 10 NC, 13 RA, and 10 OA samples.
2.2. Data acquisition and preprocessing
The original datasets including soft-formatted family files, serial Matrix files, and Robust Multi-array Averaging (RMA) tar files, were downloaded for further analysis. The analysis was carried out using GeneSpring software (version 11.5, Agilent). Arrays from the Affymetrix platform were preprocessed by robust multi-array. We used a classical t test to identify DEGs with P < .05 and fold change ≥2 as being statistically significant. Ethical approval was not necessary in our study because the expression profiles were downloaded from the public database and no new experiments in patients or animals were performed.
2.3. Gene ontology (GO) and pathway enrichment analysis of DEGs
The GO analysis[15] database (http://geneontology.org/), a set of a large number of gene annotation terms, can be used to annotate genes and gene products and identify characteristic biological attributes for high-throughput genome or transcriptome data.[15,16] The Kyoto Encyclopedia of Genes and Genomes (KEGG)[17] knowledge database (http://www.kegg.jp/) is a knowledge base for the systematic analysis of gene functions, linking genomic information with higher-order functional information.[18] The Database for Annotation, Visualization, and Integrated Discovery (DAVID) (http://david.ncifcrf.gov/)[19] constitutes an essential online tool for data synthesis that serves as a foundation for successful high-throughput gene functional analyses.[20] In this study, the identified DEGs were subjected to GO enrichment and KEGG pathway analysis using DAVID. P < .05 was used as the cutoff criterion for the functional enrichment analysis.
2.4. Integration of the PPI network and analysis of key genes therein
The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING: http://string-db.org/) is online biological database and website designed to evaluate PPI information.[21] Proteins associated with DEGs (species: Homo) were selected based on information in the STRING database (PPI score >0.7), and then PPI networks were constructed using Cytoscape software (http://cytoscape.org/).[22] In this study, degree centrality, which constitutes a fundamental parameter in network theory, was adopted to evaluate the nodes in a network. Degree centrality is defined as the number of adjacent links; that is, the number of interactions that connect one protein to its neighbors. The degree centrality method was calculated using Cytoscape plugin CytoHubba.
2.5. Module analysis
The plug-in Molecular Complex Detection (MCODE)[23] is used to detect densely connected regions in PPI networks. In this study, we selected significant modules from the constructed PPI network using MCODE. The criteria settings were as follows: node score cutoff = 0.2, K-Core = 2, and degree cutoff = 2. The MCODE score was then calculated. In addition, the functional and pathway enrichment analyses of DEGs in each module were performed using DAVID, with a P < .05 used as the cutoff criterion for the functional enrichment analysis.
3. Results
3.1. Identification of DEGs
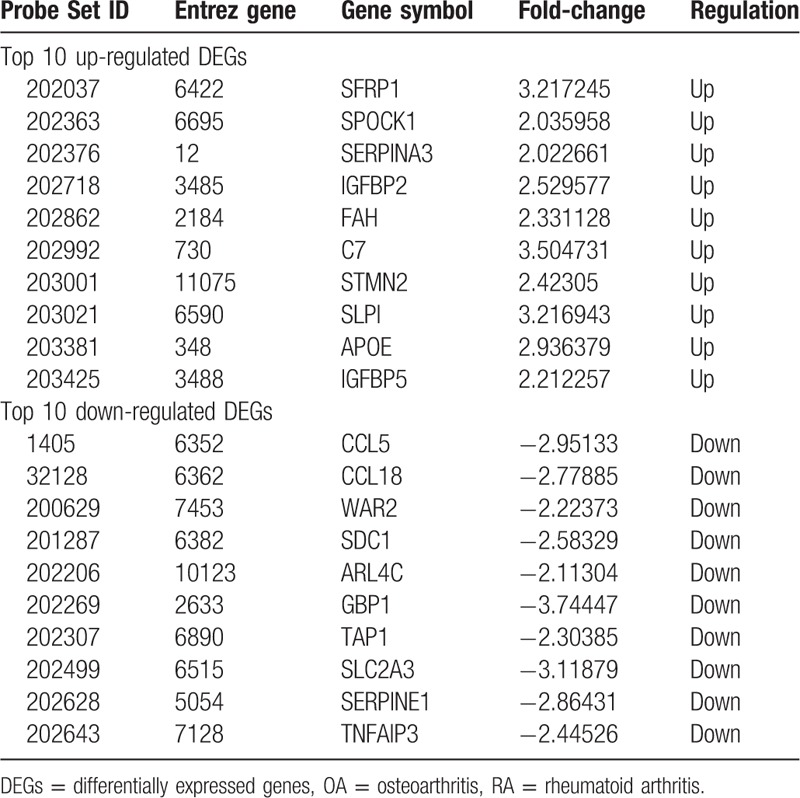
A total of 313 genes were identified to be differentially expressed between RA and NC samples with the threshold of P < .05 and a minimal 2-fold change of expression. Among these DEGs, 101 were up-regulated and 212 down-regulated in RA compared with NC samples (Supplementary list 1). The top 10 up- and down-regulated genes for RA and NC are list in Table 1. Similarly, 208 DEGs were identified to be differentially expressed between OA and NC samples, including 150 up-regulated and 58 down-regulated genes (Supplementary list 2). In addition, a total of 160 DEGs between RA and OA were identified, including 82 up-regulated and 78 down-regulated genes (Supplementary list 3). The top 10 DEGs for OA vs. NC and RA vs. OA samples are listed in Table 2 and Table 3, respectively.
Table 1.
The top 10 up- and down-regulated DEGs in RA and NC.
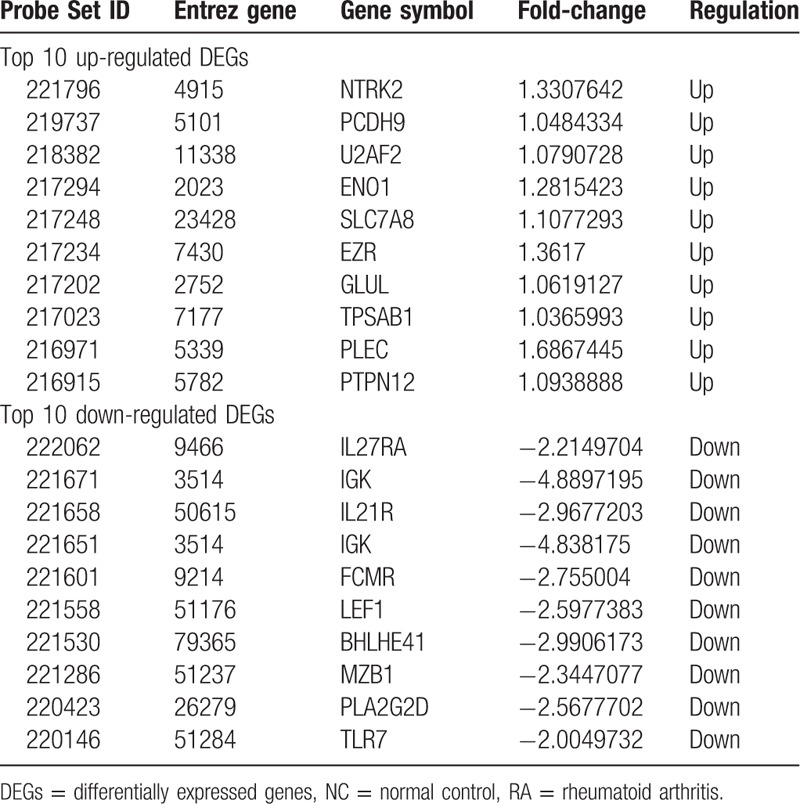
Table 2.
The top 10 up- and down-regulated DEGs in OA and NC.
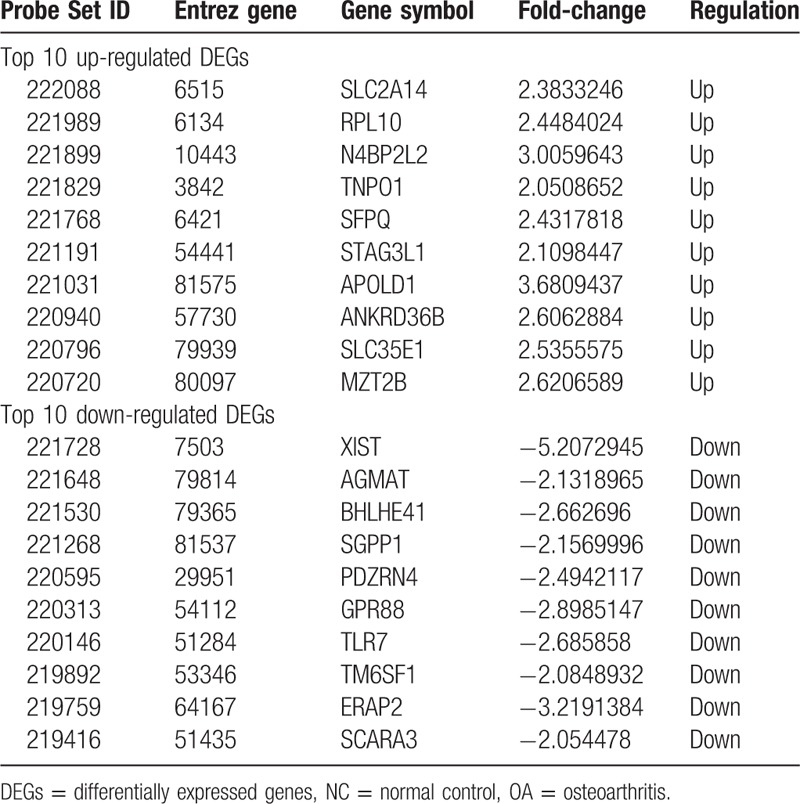
Table 3.
The top 10 up- and down-regulated DEGs in RA and OA.
3.2. Enrichment analysis of DEGs
To gain insights into the biological roles of the DEGs from RA and OA versus NC samples, we performed GO categories enrichment analysis. In this study, all DEGs were uploaded to the online software DAVID in order to identify overrepresented GO categories. GO term enrichment analysis results varied according to GO classification and expression change of DEGs. With the criterion of P < .05, “immune response” (P = 1.7E- 20), “response to drug” (P = 2.3E- 6), and “inflammatory response” (P = 3.6E- 11) exhibited highly significant enrichment within the GO biological process category. For the cellular component category, DEGs were significantly enriched in “external side of plasma membrane” (P = 5.3E-12), “nucleoplasm” (P = 3.9E- 5), and “extracellular space” (P = 1.4E-11). In addition, the molecular function category contained DEGs significantly enriched in “antigen binding” (P = 2.9E-9), “poly(A) RNA binding” (P = 3.0E-9), and “heparin binding” (P = 5.6E-9). KEGG pathway enrichment analysis was then used to understand the signaling pathway enrichment of DEGs. With the criterion of P < .05, the top enriched biological pathways associated with RA and OA included “cytokine-cytokine receptor interaction”, “mitogen-activated protein kinase (MAPK) signaling pathway,” “chemokine signaling pathway,” “human T-lymphotropic virus 1 infection,” and “extracellular matrix (ECM)-receptor interaction” (Figs. 1–3).
Figure 1.

The biological process, cellular component ,and molecular function terms, and KEGG pathways enriched by the DEGs between RA and NC. DEGs = differentially expressed genes, KEGG = Kyoto Encyclopedia of Genes and Genomes, NC = normal control, RA = rheumatoid arthritis.
Figure 3.
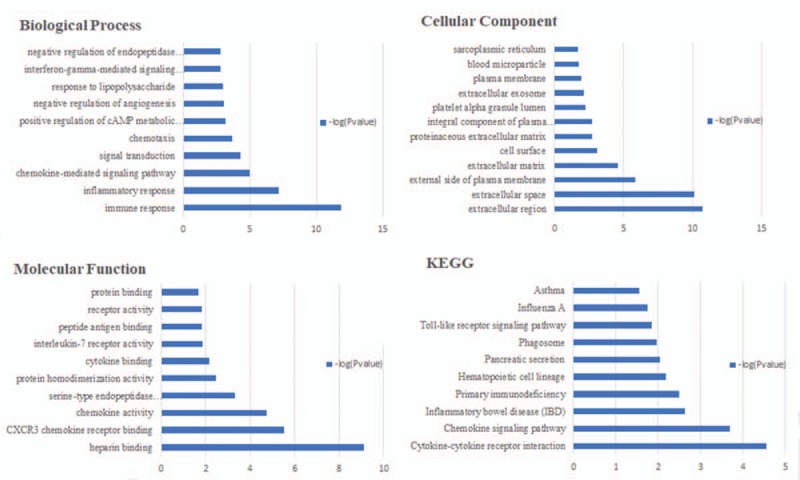
The biological process, cellular component and molecular function terms, and KEGG pathways enriched by the DEGs between RA and OA. DEGs = differentially expressed genes, KEGG = Kyoto Encyclopedia of Genes and Genomes, OA = osteoarthritis, RA = rheumatoid arthritis.
Figure 2.
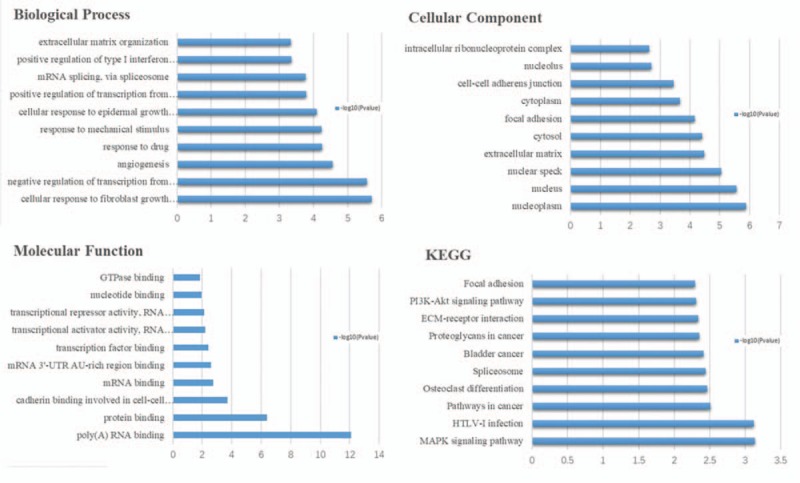
The biological process, cellular component and molecular function terms, and KEGG pathways enriched by the DEGs between OA and NC. DEGs = differentially expressed genes, KEGG = Kyoto Encyclopedia of Genes and Genomes, NC = normal control, OA = osteoarthritis.
3.3. PPI network analysis of DEGs
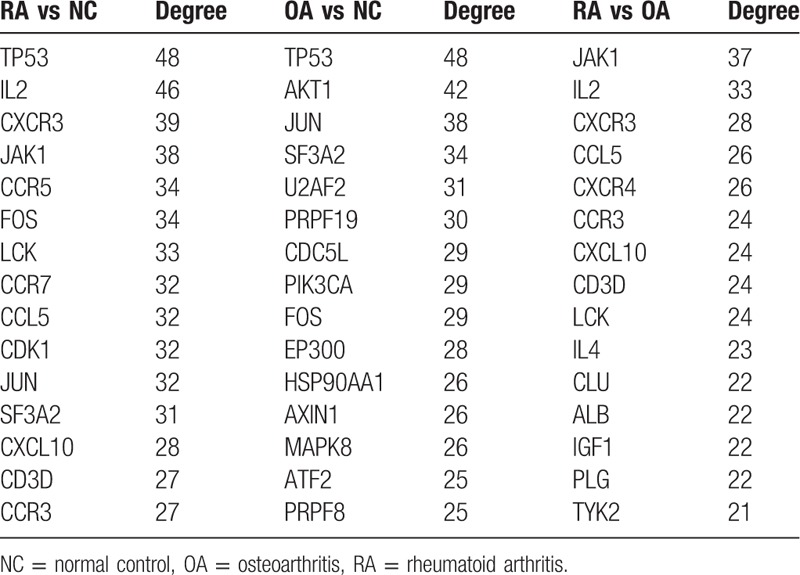
To further explore the relationships between DEGs at the protein level, the PPI networks were constructed based on the interactions of DEGs. With the predefined criterion of combined score > 0.7, a total of 3605 interactions and 797 nodes were screened to establish the PPI network between RA and NC samples (Supplementary Fig. 1). In this network, the top 15 key genes with highest degree scores are shown in Table 3. Similarly, the PPI networks regarding OA versus NC and RA versus OA samples were constructed including 696 nodes, 2991 edges and 466 nodes, 1923 edges, respectively (Supplementary Figs. 2–3). Among these nodes, the top 15 hub genes with the highest degree scores are shown in Table 4.
Table 4.
The top 15 hub genes with highest degree scores.
3.4. Module analysis
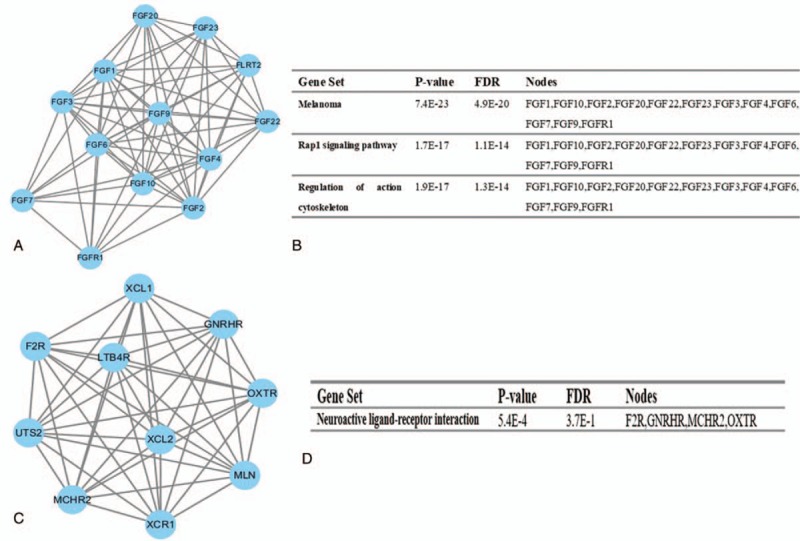
The top 2 significant modules of RA versus NC, OA versus NC, as well as RA versus OA samples were selected, respectively, and functional annotation of the genes involved in the modules was analyzed (Figs. 4–6). KEGG enrichment analysis showed that the genes of RA versus NC samples in modules 1 to 2 and OA versus NC samples were mainly associated with “melanoma,” “neuroactive ligand-receptor interaction,” “Rap1 signaling pathway,” “Wnt singaling pathway,” and “MAPK signaling pathway.” Furthermore, the DEGs of RA versus OA were significantly clustered in “chemokine signaling pathway” and “Jak-STAT signaling pathway.”
Figure 4.
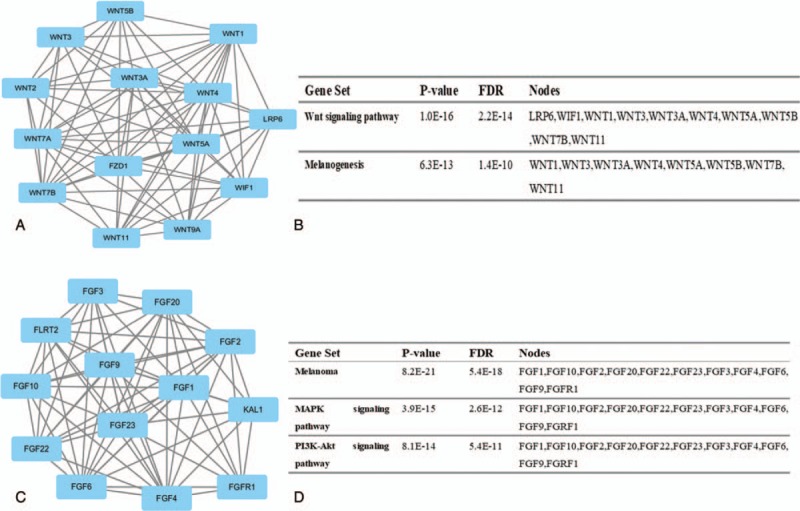
Top 2 modules from the protein-protein interaction network between RA and NC: (A) module 1, (B) the KEGG enriched pathways of module 1, (C) module 2, (D) the KEGG enriched pathway of module. KEGG = Kyoto Encyclopedia of Genes and Genomes, NC = normal control, RA = rheumatoid arthritis.
Figure 6.

Top 2 modules from the protein-protein interaction network between RA and OA: (A) module 1, (B) the KEGG enriched pathways of module 1, (C) module 3, (D) the KEGG enriched pathway of module. KEGG = Kyoto Encyclopedia of Genes and Genomes, OA = osteoarthritis, RA = rheumatoid arthritis.
Figure 5.
Top 2 modules from the protein-protein interaction network between OA and NC: (A) module 1, (B) the KEGG enriched pathways of module 1, (C) module 2, (D) the KEGG enriched pathway of module 2. KEGG = Kyoto Encyclopedia of Genes and Genomes, NC = normal control, OA = osteoarthritis.
4. Discussion
RA and OA, the most common forms of arthritis, constitute chronic painful conditions that affect an individual's quality of life.[24] However, the potential causes of RA and OA remain uncertain. Understanding the underlying pathogenesis of RA and OA is of critical importance for diagnosis, prognosis, and identifying drug targets. As high-throughput sequencing and microarray technology can provide information regarding the expression levels of thousands of genes in the human genome simultaneously, this methodology has been widely used to predict the potential diagnostic and therapeutic targets for OA and RA. In the present study, we extracted the data from GSE55457, which includes 10 NC samples, 13 RA samples, and 10 OA samples. We identified 101 up-regulated and 212 down-regulated DEGs between RA and NC samples using bioinformatics analysis. As cumulative evidence has shown that co-expressed genes normally represent those with similar expression profiles that also frequently participate in similar biological processes,[25] to better understand the interactions of the identified DEGs, we further performed GO and KEGG functional annotation and pathway enrichment analyses. These analyses suggested that the identified DEGs were mainly involved in “immune response,” “chemokine-mediated signaling pathway,” “B cell receptor signaling pathway,” “integral component of plasma membrane,” “antigen binding,” “cytokine-cytokine receptor interaction,” “osteoclast differentiation,” and “tumor necrosis factor (TNF) signaling pathway.”
In addition, a total of 208 DEGs including 150 up- and 58 down-regulated DEGs were identified between OA and NC samples. The majority of DEGs were enriched in a limited number of functional categories, such as “negative regulation of transcription from RNA polymerase promoter,” “nucleoplasm, focal adhesion,” “osteoclast differentiation,” “EMC-receptor interaction,” “MAPK signaling pathway,” and “phosphatidylinositol 3-kinase-protein kinase B (PI3K-Akt) signaling pathway.” Moreover, 160 DEGs were identified between RA and OA samples, including 82 up- and 78 down-regulated DEGs. In this case, significantly enriched functions included “negative regulation of endopeptidase,” “sarcoplasmic reticulum,” “protein binding,” and “blood microparticle.” Overall, the top enriched biological pathways for the both RA and OA included “chemokine signaling pathway,” “osteoclast differentiation,” “MAPK signaling pathway,” and “focal adhesion.” Furthermore, by constructing PPI networks, we identified some key genes that provide new insights for RA and OA diagnosis, prognosis, and drug target identification.
Chemokines comprises a family of small cytokines or proteins secreted by immune cells that regulate and control cells of the immune system during the processes of immune response.[1] Accumulating evidence suggested that chemokines can become involved at RA development and progression.[26] In the present study, DEGs involved in the chemokine signaling pathway were identified to be altered in both RA and OA, including the gene clusters chemokine (C-C motif) ligand 13 (CCL13), CCL18, CCL5, chemokine (C-C motif) receptor 2 (CCR2), CCR5, chemokine (C-X-C motif) ligand 10 (CXCL10), CXCL11, CXCL13, CXCL9, interleukin 2-inducible T-cell kinase, chemokine (C motif) ligand 1, neutrophil cytosolic factor 1, protein kinase, cAMP-dependent, catalytic, beta, and signal transducer, and activator of transcription 1. Specifically, these chemokine-related DEGs were down-regulated in RA samples. Therefore, increased expression of these genes in RA might not have a potential role for inflammation activation in the pathogenesis of RA.
Osteoclasts derived from multinucleated giant cells are responsible for remodeling and resorption of bone[27,28] comprise specialized monocyte/macrophage family members that differentiate from hematopoietic precursors that appear to stem from a common monocyte precursor. The process of osteoclast differentiation from precursor cells involves the expression of proteins that together identify cells of the osteoclast lineage, including cell surface receptors (calcitonin receptors, c-fms), lysosomal enzymes (TRAP), cathepsin K, intracellular signaling molecules (c-src), and transcription factors (PU.1). Numerous studies have shown that osteoclast differentiation plays an important role in RA bone destruction.[29,30] In the present study, several DEGs of RA were enriched in osteoclast differentiation, such as FBJ murine osteosarcoma viral oncogene homolog, transcription factor family JUN (JUN), Jun B proto-oncogene, interleukin 1 receptor type 1 (IL1R1), suppressor of cytokine signaling 3, and transforming growth factor, beta receptor 2 (TGFBR2). As these were up-regulated in the RA samples, increased expression of these genes in RA might constitute a potential therapeutic target of RA.
The MAPK family includes a serious of serine/threonine protein kinases that are involved in regulating numerous cell functions, such as differentiation, proliferation, apoptosis, and inflammatory responses.[31] In comparison, the involvement of MAPK in the regulation of the inflammatory response and the development and progression of RA has been widely identified.[32] MAPK pathway inhibition can be regarded as an anti-inflammatory therapy, thus suggesting these DEGs as potential targets of anti-inflammatory therapy for OA and RA.[33] In the present study, DEGs that were found to be associated with the MAPK signaling pathway included JUN, dual specificity phosphatase 1 (DUSP1), DUSP5, growth arrest and DNA-damage-inducible 45 beta, IL1R1, nuclear factor of kappa light polypeptide gene enhancer in B-cells 2, nuclear receptor subfamily 4, group A, member 1, TGFBR2, and myelocytomatosis oncogene, which were up-regulated in the OA samples. Therefore, the increased expression of these genes in OA might indicate potential mechanisms of inflammation activation in OA pathogenesis.
An increasing number of studies suggested that members of focal adhesion kinase (FAK) associated with cell adhesion, cell proliferation, cell migration, and osteoclast differentiation.[34] In this study, we identified several DEGs of OA samples as being enriched in the focal adhesion pathway, including JUN, cartilage oligomeric matrix protein, catenin (cadherin-associated protein), beta 1, collagen type IV, alpha 2, collagen type VI alpha 1 placental growth factor, and vascular endothelial growth factor A, which were up-regulated in the OA samples. Therefore, the increased expression of these genes in OA might also constitute a therapeutic and diagnostic target.
The subsequent construction of the PPI network using the respective DEGs identified 15 DEGs as potential key genes involved in OA and RA. Tumor protein 53 (TP53) was identified as one of the key genes exhibiting the highest degree of differential expression in both RA and OA samples. TP53, also known as the tumor suppressor gene p53, induces cell growth arrest or apoptosis depending on the physiological circumstances and cell type. Tak et al[35] reported that the development of mutations in TP53 and other key regulatory genes could help convert inflammation into chronic disease in RA and other inflammatory disorders. In addition, Zhu et al[36] reported that the expression of the TP53 gene is correlated with disease grades of OA. In turn, IL2, the second key gene of RA, is crucial to regulation of the immune response, which can stimulate B-cells, monocytes, lymphokine-activated killer cells, natural killer cells, and glioma cells. Notably, recent studies have proposed that IL2 is associated with the development and progression of RA.[37] The protein encoded by Akt murine thymoma viral oncogene homolog 1 (AKT1), the second key gene of OA, is activated by phosphorylation at Thr308 and Ser473. Activated AKT1 promotes cell growth, survival pathways, and inactivates various components of apoptotic cascade.[38] Accumulating evidence has demonstrated that AKT1 appears to be constitutively activated with increases in both native and phosphorylated forms in OA tissues.[39]
Furthermore, module analysis of the PPI network revealed that the development of RA was associated with the PI3K-Akt signaling pathway. This pathway is crucial to many aspects of cell growth and survival in both physiological as well as in pathological conditions.[40] It has been demonstrated that the PI3K-Akt signaling pathway can be activated by several cytokines such as TNF-α in RA synoviocytes.[41] Moreover, module analysis of the PPI network of OA suggested that the development and progression of OA were related to the Wnt signaling pathway. Recent studies have suggested that the Wnt signaling pathway is involved in cartilage distortion and subchondral bone changes.[42] Moreover, the increased expression of Wnt-induced signaling protein 1 has been found in both experimental and human OA.[43]
5. Conclusions
In conclusion, TP53, IL2, and AKT1may be key genes for RA and OA. Furthermore, the results of the present study suggested that several biological pathways (ie, “the immune response,” “inflammation,” and “osteoclast differentiation”) are commonly involved in the development of both RA and OA. In comparison, several other pathways (ie, “MAPK signaling pathway,” “ECM-receptor interaction”) presented significant differences between RA and OA. This study thus provides further insights into the underlying pathogenesis of OA and RA, which may facilitate the diagnosis and treatment of these diseases.
Author contributions
Conceptualization: Naiqiang Zhu, Jinxin Liu.
Methodology: Naiqiang Zhu.
Visualization: Jinxin Liu.
Writing – original draft: Naiqiang Zhu, Jinxin Liu.
Writing – review & editing: Jingyi Hou, Guiyun Ma, Yuanhao Wu, Geng Li, Bin Chen, Youxin Song.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Footnotes
Abbreviations: AKT1 = Akt murine thymoma viral oncogene homolog 1, CCL = chemokine (C-C motif) ligand, CCR = chemokine (C-C motif) receptor, CXCL = chemokine (C-X-C motif) ligand, DAVID = Database for Annotation, Visualization and Integrated Discovery, DEG = differentially expressed genes, DUSP = dual specificity phosphatase, ECM = extracellular matrix, FAK = focal adhesion kinase, GEO = Gene Expression Omnibus, GO = gene ontology, IL1R1 = interleukin 1 receptor type 1, KEGG = Kyoto Encyclopedia of Genes and Genomes, MAPK = mitogen-activated protein kinase, MCODE = Molecular Complex Detection, NC = normal control, OA = osteoarthritis, PI3K-Akt = phosphatidylinositol 3-kinase-protein kinase B, PPI = protein-protein interaction, RA = rheumatoid arthritis, STRING = Search Tool for the Retrieval of Interacting Genes/Proteins, TGFBR2 = transforming growth factor, beta receptor 2, TNF = tumor necrosis factor, TP53 = tumor protein 53.
This study was supported by China Postdoctoral Science Foundation (2017M610815); Heibei Natural Science Foundation (H2017406031); Young Talents Project of Hebei Education Department (BJ201602); National Natural Science Foundation of China (81641136, 81703659, 81503506) and Chengde City Science and Technology Research and Development Projects (20151048). Science and Technology Program of Tianjin (No. 15ZXLCSY00020).
The authors declare no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- [1].Li ZC, Xiao J, Peng JL, et al. Functional annotation of rheumatoid arthritis and osteoarthritis associated genes by integrative genome-wide gene expression profiling analysis. PLoS One 2014;9:e85784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet 2010;376:1094–108. [DOI] [PubMed] [Google Scholar]
- [3].de Lange-Brokaar BJ, Ioan-Facsinay A, van Osch GJ, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthritis Cartilage 2012;20:1484–99. [DOI] [PubMed] [Google Scholar]
- [4].Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford) 2012;51(Suppl 5):v3–11. [DOI] [PubMed] [Google Scholar]
- [5].JCh R, Garnero P. Biological markers in osteoarthritis. Bone 2012;51:265–77. [DOI] [PubMed] [Google Scholar]
- [6].Baecklund E, Iliadou A, Askling J, et al. Association of chronic inflammation, not its treatment, with increased lymphoma risk in rheumatoid arthritis. Arthritis Rheum 2006;54:692–701. [DOI] [PubMed] [Google Scholar]
- [7].Franklin J, Lunt M, Bunn D, et al. Incidence of lymphoma in a large primary care derived cohort of cases of inflammatory polyarthritis. Ann Rheum Dis 2006;65:617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].da MLM, dos SNLL, de Carvalho JF. Autoantibodies and other serological markers in rheumatoid arthritis: predictors of disease activity. Clin Rheumatol 2009;28:1127–34. [DOI] [PubMed] [Google Scholar]
- [9].Kulasingam V, Diamandis EP. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat Clin Pract Oncol 2008;5:588–99. [DOI] [PubMed] [Google Scholar]
- [10].Nannini M, Pantaleo MA, Maleddu A, et al. Gene expression profiling in colorectal cancer using microarray technologies: results and perspectives. Cancer Treat Rev 2009;35:201–9. [DOI] [PubMed] [Google Scholar]
- [11].Bustin SA, Dorudi S. Gene expression profiling for molecular staging and prognosis prediction in colorectal cancer. Expert Rev Mol Diagn 2004;4:599–607. [DOI] [PubMed] [Google Scholar]
- [12].Heruth DP, Gibson M, Grigoryev DN, et al. RNA-seq analysis of synovial fibroblasts brings new insights into rheumatoid arthritis. Cell Biosci 2012;2:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang R, Fang H, Chen Y, et al. Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS One 2012;7:e32356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Woetzel D, Huber R, Kupfer P, et al. Identification of rheumatoid arthritis and osteoarthritis patients by transcriptome-based rule set generation. Arthritis Res Ther 2014;16:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 2000;25:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].The Gene Ontology (GO) project in 2006. Nucleic Acids Res 2006;34:D322–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Altermann E, Klaenhammer TR. PathwayVoyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics 2005;6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huang DW, Sherman BT, Tan Q, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 2007;35:W169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dennis G, Sherman BT, Hosack DA, et al. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 2003;4:3. [PubMed] [Google Scholar]
- [21].Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003;13:2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 2003;4:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Murphy G, Nagase H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: destruction or repair. Nat Clin Pract Rheumatol 2008;4:128–35. [DOI] [PubMed] [Google Scholar]
- [25].Liang B, Li C, Zhao J. Identification of key pathways and genes in colorectal cancer using bioinformatics analysis. Med Oncol 2016;33:111. [DOI] [PubMed] [Google Scholar]
- [26].Molendijk M, Hazes JM, Lubberts E. From patients with arthralgia, pre-RA and recently diagnosed RA: what is the current status of understanding RA pathogenesis. RMD Open 2018;4:e000256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Suda T, Takahashi N, Martin TJ. Modulation of osteoclast differentiation. Endocr Rev 1992;13:66–80. [DOI] [PubMed] [Google Scholar]
- [28].Väänänen HK, Laitala-Leinonen T. Osteoclast lineage and function. Arch Biochem Biophys 2008;473:132–8. [DOI] [PubMed] [Google Scholar]
- [29].Gravallese EM, Manning C, Tsay A, et al. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum 2000;43:250–8. [DOI] [PubMed] [Google Scholar]
- [30].Shigeyama Y, Pap T, Kunzler P, et al. Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum 2000;43:2523–30. [DOI] [PubMed] [Google Scholar]
- [31].Jazirehi AR, Wenn PB, Damavand M. Therapeutic implications of targeting the PI3Kinase/AKT/mTOR signaling module in melanoma therapy. Am J Cancer Res 2012;2:178–91. [PMC free article] [PubMed] [Google Scholar]
- [32].Schett G, Zwerina J, Firestein G. The p38 mitogen-activated protein kinase (MAPK) pathway in rheumatoid arthritis. Ann Rheum Dis 2008;67:909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Saklatvala J. The p38 MAP kinase pathway as a therapeutic target in inflammatory disease. Curr Opin Pharmacol 2004;4:372–7. [DOI] [PubMed] [Google Scholar]
- [34].Shahrara S, Castro-Rueda HP, Haines GK, et al. Differential expression of the FAK family kinases in rheumatoid arthritis and osteoarthritis synovial tissues. Arthritis Res Ther 2007;9:R112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Tak PP, Zvaifler NJ, Green DR, et al. Rheumatoid arthritis and p53: how oxidative stress might alter the course of inflammatory diseases. Immunol Today 2000;21:78–82. [DOI] [PubMed] [Google Scholar]
- [36].Zhu X, Yang S, Lin W, et al. Roles of cell cyle regulators cyclin D1, CDK4, and p53 in knee osteoarthritis. Genet Test Mol Biomarkers 2016;20:529–34. [DOI] [PubMed] [Google Scholar]
- [37].Kurreeman FA, Daha NA, Chang M, et al. Association of IL2RA and IL2RB with rheumatoid arthritis: a replication study in a Dutch population. Ann Rheum Dis 2009;68:1789–90. [DOI] [PubMed] [Google Scholar]
- [38].Zhou W, Fu XQ, Liu J, et al. RNAi knockdown of the Akt1 gene increases the chemosensitivity of gastric cancer cells to cisplatin both in vitro and in vivo. Regul Pept 2012;176:13–21. [DOI] [PubMed] [Google Scholar]
- [39].Sieghart D, Liszt M, Wanivenhaus A, et al. Hydrogen sulphide decreases IL-1β-induced activation of fibroblast-like synoviocytes from patients with osteoarthritis. J Cell Mol Med 2015;19:187–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chang L, Graham PH, Ni J, et al. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit Rev Oncol Hematol 2015;96:507–17. [DOI] [PubMed] [Google Scholar]
- [41].Zhang HG, Wang Y, Xie JF, et al. Regulation of tumor necrosis factor alpha-mediated apoptosis of rheumatoid arthritis synovial fibroblasts by the protein kinase Akt. Arthritis Rheum 2001;44:1555–67. [DOI] [PubMed] [Google Scholar]
- [42].Velasco J, Zarrabeitia MT, Prieto JR, et al. Wnt pathway genes in osteoporosis and osteoarthritis: differential expression and genetic association study. Osteoporos Int 2010;21:109–18. [DOI] [PubMed] [Google Scholar]
- [43].Blom AB, Brockbank SM, van Lent PL, et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: prominent role of Wnt-induced signaling protein 1. Arthritis Rheum 2009;60:501–12. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.