

Abstract
Background:
Mulvihill–Smith syndrome is a rare sporadic condition that was first recognized in 1975. A total of 11 cases have been described in the literature. The aim of this study was to describe the orofacial signs and dental anomalies, their frequency, and the relationship between Mulvihill–Smith syndrome and other progeroid syndromes via a review of the literature.
Methods:
A systematic PubMed search was performed to retrieve articles published between 1975 and the present day that described patients affected by Mulvihill–Smith syndrome. The search identified 14 articles, and data on 11 patients were extracted from the selected articles.
Results:
A total of 7 patients (63.6%) affected by Mulvihill–Smith syndrome were described as having a typical “bird” face. Dental abnormalities, including irregular shape, enamel defects, hypodontia, and taurodontism, were described in 6 patients (54.5%). All patients (100%) had multiple pigmented nevi on the face and a lack or thinning of subcutaneous tissue around the neck and face. Three patients with Mulvihill–Smith syndrome exhibited early onset of tumors of the gastrointestinal tract, including the tongue.
Conclusion:
Mulvihill–Smith syndrome is a clinically complex disease that may be caused by a single gene mutation. Numerous different tissues of the body are affected. This analysis of the orofacial signs may help clinicians to diagnose this rare pathology.
Keywords: dental abnormalities, management progerodi syndrome, Mulvihill-Smith syndrome, oro-facial signs, progerian pathology, rare disease
1. Introduction
Aging and death are unavoidable events in the life cycle of an organism. In recent decades, researchers have given considerable attention to the physiological events and underlying biological processes that are involved in aging.[1] Aging is a consequence of nuclear metabolic defects that promote the accumulation of mutations and chromosomal anomalies and lead to macromolecule and DNA damage.[1,2] Due to the features of premature aging, the rare progeroid syndromes have attracted much attention.[3]
In 1975, Mulvihill and Smith reported a case entitled “Another disorder with prenatal shortness of stature and premature aging,” which described a 17-year-old male with a mild intellectual disability, microcephaly, very short stature, hypodontia, numerous pigmented nevi and freckles, chronic infections, and insulin dependent diabetes mellitus.[4] Since the recognition of this syndrome, 11 patients have been reported.[4–14] Mulvihill–Smith syndrome is a rare genetic disorder [Online Mendelian Inheritance in Man (OMIM) number: %176690] that is characterized by a progeria-like phenotype.[12] Its inheritance mode is likely to be autosomal recessive, as the syndrome affects females and males and those born to consanguineous parents[9,14]; however, the causative gene has not yet been identified.[5] The first cases reported by Mulvihill and Smith,[4] Elliott,[6] and Baraitser et al[8] described isolated male patients with no parental consanguinity; however, in 1993, Ohashi et al[9] reported the case of a 30-year-old Japanese woman who was immunodeficient and whose parents were first cousins. They suggested autosomal recessive inheritance of the pathology in this patient and indicated that the female patient described in 1979 by Wong et al[7] might have been affected by the same syndrome.[9] In 1994, Bartsch et al[10] reported another male patient with Mulvihill–Smith syndrome and immunodeficiency, while de Silva et al[11] described the seventh case of Mulvihill–Smith progeria-like syndrome in a 5-year-old boy in 1997. The mutation responsible for this syndrome was not identified due to its rarity and the diagnosis was therefore purely clinical.[12] A total of 30 progeroid syndromes are currently known, including Ehlers–Danlos syndrome (OMIM number: #130070), progeroid syndrome neonatal (OMIM number: %264090), Werner syndrome (OMIM number: #277700), Cockayne syndrome A (OMIM number: #216400), Nestor–Guillermo progeria syndrome (OMIM number: #614008), and Hutchinson–Gilford progeria syndrome (OMIM number: #176670). The progeroid syndromes are characterized by signs of premature aging, including alopecia, decreased subcutaneous fat, skin pigmentation, dry and wrinkled skin, hypogonadism, and clinical and laboratory findings that indicate a decline in cellular and humoral immunocompetence.[9]
The first mention of progeria in the medical literature was reported in the case of a 3-year-old boy in 1886 by Hutchinson,[2] who suggested that this disorder was a type of ectodermal dysplasia. Hutchinson[2] described a second patient in 1895; however, this case was explained in more detail by Gilford who had taken care of the patient for 17 years.[1–3] Gilford first used the term progeria for this disease: “pro” meaning before and “geras” meaning old age in Ancient Greek.[15] Progeroid syndromes have a very low incidence rate of one per 8 million live births.[16] Like other accelerated aging diseases, they are caused by damaged DNA repair.[17] Approximately 100 cases have been recognized in the literature.[18] Patients affected by this disorder normally live for 13 years; however, many have been described to live into their late teens and early 20s but very few reach their 40s.[19,20] The causes of death in individuals with this lethal premature aging disease are stroke or myocardial infarction.[21]
The signs and symptoms reported in the 11 cases of Mulvihill–Smith syndrome include short stature, microcephaly, pigmented nevi, loud and raucous voice, increased facial fat, hypertelorism, alopecia, brachydactyly, genital anomalies, visual change, hypospadias, diabetes, recurrent infections, deafness, tumor development (e.g., gastric, tongue, pancreas, and melanoma), intellectual disability, sleep disorders, hepatic dysfunction, and orofacial and dental abnormalities. Children with progeroid disorders are at risk of serious oral complications and require an evaluation by an oral specialist at the time of diagnosis. They are also at risk of serious ophthalmic complications due to ocular surface disease.[1] Some reported ophthalmological findings in patients affected by Mulvihill–Smith syndrome include myopia, amblyopia, astigmatism, dry eye disease, meibomian gland dysfunction, allergic conjunctivitis, band keratopathy, keratoconus, endothelial dystrophy, cataracts, and retinal abnormalities.[22]
This article aims to review the findings concerning the clinical features of Mulvihill–Smith syndrome, particularly the orofacial and dental abnormalities, their frequency, and the relationship between Mulvihill–Smith syndrome and other progeroid syndromes, to define which clinical features permit the distinction between Mulvihill–Smith syndrome and other better known progeroid syndromes.
2. Patients and methods
A systematic PubMed search was performed to retrieve articles published between 1975 and the current day that described patients affected by Mulvihill–Smith syndrome. The search strategy included the combination of 3 keywords: Mulvihill–Smith, Mulvihill–Smith syndrome, and progeroid disorder. This was completed with a manual search of the reference lists of the selected studies. Only articles that provided a clinical description of the case were included.
As this is a literature review, it was granted exemption in writing by the institutional review board of the Catholic University of Sacred Heart, Rome. The Declaration of Helsinki guidelines were followed in this investigation.
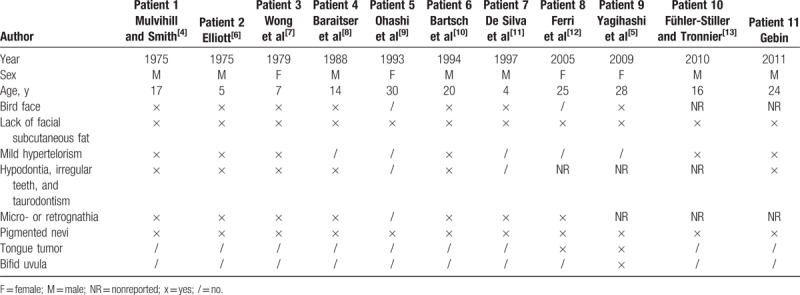
The search identified 43 articles, 14 of which described Mulvihill–Smith cases. Three articles did not describe a new case, while 11 articles described unique cases. The first case was described in 1975[4] and the last in 2011.[14] Seven of the patients were male and 4 were female, and the median age of the subjects was 17 years (range: 4–30). The demographic features of the study population are reported in Table 1.
Table 1.
Orofacial signs in the 11 patients.
3. Results
Seven of the 11 patients (63.6%) affected by Mulvihill–Smith syndrome were described as having a typical “pinched” or “bird” face that was characterized by hypoplasia of the lower half of the face (micro- or retrognathia). The lack or thinning of subcutaneous tissue around the neck and face was reported in all (100%) patients. Mild hypertelorism was present in 6 patients (54.5%), while all patients (100%) had multiple pigmented nevi on the face. Dental abnormalities such as irregular shape, enamel defects, hypodontia, and taurodontism were described in 6 patients (54.5%). A bifid uvula was reported in 1 patient (9.1%).
The third reported case of Mulvihill–Smith syndrome was described in 1979 by Wong et al.[7] The 14-year-old female patient had a triangular face with central cutaneous atrophy, reduced lower facial height, and micrognathia. Intraoral and radiographic examinations revealed oligodontia, incomplete dental root development, protruding maxillary central incisors, periodontitis, and reduced alveolar bone height. No development of the adenoids and tonsils was observed.[7]
Ohashi et al[9] reported that their patient's teeth first erupted at 7 months and developed in normal alignment but were mostly lost as a consequence of severe caries.
Orofacial and dental situations in the sixth patient were carefully described by Bartsch et al.[10] The patient had malpositioned, hypoplastic deciduous, and permanent teeth with enamel defects; a narrow and highly arched palate; malocclusion (overbite); and micrognathia. In addition, the second premolars and lower third molars were absent (hypodontia), the upper lateral incisors were cone-shaped, the molars had enlarged coronal pulp and reduced roots (hypotaurodontism), and the lower first molars and upper third molars were tilted forwards.[10]
The eighth patient, described by Ferri et al in 2005,[12] underwent surgical removal of the left part of her tongue because of the occurrence of a squamous cell carcinoma when she was 22 years old. Another female patient with Mulvihill–Smith syndrome underwent surgical removal of a tongue tumor at age 27 years. In this case, the histopathologic diagnosis was an ulcer of the tongue with chronic inflammation; however, no evidence of malignancy was disclosed.[5] Agenesis of the second premolars and the right maxillary central incisors and poor oral health condition were reported in the eleventh case of Mulvihill–Smith syndrome in the literature.[14]
4. Discussion
Mulvihill–Smith syndrome is a clinically complex disease that may be caused by a single gene mutation. The disorder affects numerous different tissues of the body.[14] In their subject with Mulvihill–Smith syndrome, Ferri et al[12] reported an absence of chromosomal abnormalities and that the karyotype was normal. Therefore, they suggested that a new mutation could be the genetic cause of this syndrome. Since the first report of a 17-year-old boy with prenatal short stature, a pinched face, premature aging, multiple pigmented nevi, intellectual disability, photosensitivity, thin skin, sparse hair, and oligodontia was reported in 1975, 10 other patients with Mulvihill–Smith syndrome have been identified.[4–14]Table 1 provides a summary of the orofacial signs described in the 11 reported cases in the literature. The presence of rhinitis, allergic conjunctivitis, and recurrent infections associated with low levels of immunoglobulins and T- and B-cells in almost all cases strengthens the idea that immunodeficiency is one of the characteristics of the syndrome.[10,23] There is currently no known gene responsible for Mulvihill–Smith syndrome; however, considering that the genetic causes of many other progeroid syndromes have been discovered, it is pertinent to determine whether any of the previously reported patients may actually suffer from a different progeroid syndrome with a somewhat atypical presentation. Previous authors diagnosed their patients with Mulvihill–Smith syndrome based on phenotypical (e.g., facial features and pigmented nevi) and pathological aspects.[9–11] For example, Ohashi et al[9] reported that their patient presented with a possible manifestation of Werner syndrome (another premature aging disease) due to the short stature and hypogonadism; however, they excluded this diagnosis, as patients with Werner syndrome are usually of normal intelligence and do not show signs of immunodeficiency.
4.1. Dental abnormalities
Wong et al[7] suggested that oligodontia, alveolar and dental development deficiency, and periodontitis further contributed to reduced facial height in patients with Mulvihill–Smith syndrome. De Silva et al[11] underlined the association between taurodontism, which results from delayed development of Hertwig– Bruhn epithelial division between the dental roots, and autosomal dominant inheritance of Klinefelter syndrome and suggested the trait be used as a diagnostic feature in future cases. Gebin observed that a high-pitched voice in 8 of the cases may be a reflection of the abnormal face structure.[14]
4.2. Tumors of the gastrointestinal tract
Three patients with Mulvihill–Smith syndrome were diagnosed with early-onset tumors of the gastrointestinal tract: a 23-year-old patient had a signet ring cell carcinoma of the stomach,[10] a 20-year-old patient had a squamous cell carcinoma of the tongue,[12] and a 17-year-old patient had a solid pseudopapillary cystic tumor of the pancreas.[5] Yagihashi et al[5] suggested that early-onset tumors could represent an important adult Mulvihill–Smith syndrome phenotype that requires attention and that patients with Mulvihill–Smith syndrome may be susceptible to specific types of gastrointestinal tract tumors. Although histological examination by Yagihashi et al[5] did not reveal tumor cells in the abnormal tongue mass, Ferri et al[12] reported a possible increased risk of Mulvihill–Smith syndrome in patients developing squamous cell carcinoma of the tongue or oral mucosa.
4.3. Pigmented nevi
Multiple pigmented nevi is a typical feature of Mulvihill–Smith syndrome; however, it also occurs in a large number of genetic immunodeficiency syndromes such as N syndrome (OMIM number: 310465), Fanconi pancytopenia (OMIM number: #22765), and Maraschio–Peretti type chromosomal instability. Heyne et al[24] described junctional-type nevus cell nevi after leukemia therapy in a monozygotic twin; in this case, the possible mutagenic effects of cytostatic therapy on somatic cells in a state of induced immunoincompetence were considered.[10] Thus, Bartsch et al[7] suggested that immunodeficiency and nevi are possibly related to Mulvihill–Smith syndrome.
4.4. Orofacial signs in other progeroid syndromes
Other progeroid syndromes have many facial characteristics in common with Mulvihill–Smith syndrome, including loss of subcutaneous fat; a convex nasal ridge; a narrow, compressed, and deformed point of the nose; a small chin; prominent outer ears that lack lobules; and anatomical eye deformation. Almost all children have a highly pitched voice and normally speak quite well, and a mild conductive loss was reported in the majority of European patients.[2,21,25,26]
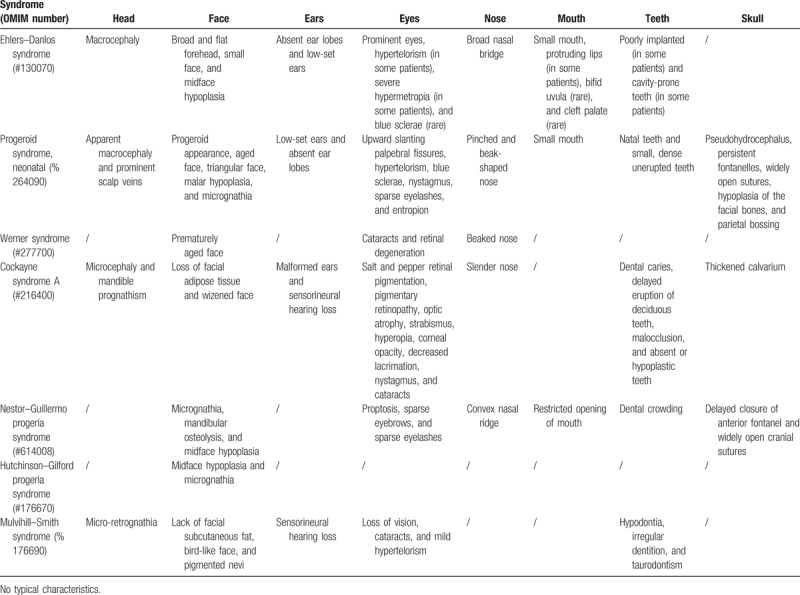
The loss of subcutaneous fat generally starts at 6 months of age but becomes more visible at 3 or 4 years of age. This loss of subcutaneous fat first becomes visible in the limbs and thorax, then the face and buccal, and it finally disappears in the pubic area; almost all children with progeroid syndromes have a prominent belly due to intraabdominal fat. Prominent appearance of the eyes, thin skin, and easily visible blood vessels are caused by the vanishing intraorbital and subcutaneous fat; skin appears wrinkled in the mouth area. Elevated collagen type IV and elastin production are recognized as the cause of the skin irregularity.[2,26,27] Caries and periodontal pathology affect patients suffering from Mulvihill–Smith syndrome and other progeroid syndromes. Of the oro-dental abnormalities, hypodontia and taurodontism were most commonly described in Mulvihill–Smith syndrome. Conversely, patients suffering from other progeroid syndromes reported ankyloglossia, an ogival palate, vertical chewing where rotatory chewing is supposed to develop, delayed tooth eruption, irregular teeth showing elevated decay due to maxilla dental crowding, limited mandible size, and hard dental care caused by a minute oral aperture. Poor compliance is also seen in patients who are normally jovial, agile, and with normal psychosocial growth.[1] The typical orofacial signs in other progeroid syndromes and Mulvihill–Smith syndrome are reported in Table 2.
Table 2.
Typical orofacial signs in some progeroid syndromes.
4.5. Ophthalmological findings
The reported ophthalmological findings in the 11 patients with Mulvihill–Smith syndrome include astigmatism, myopia, endothelial dystrophy, keratoconus, cataracts, amblyopia, and allergic conjunctivitis. Irahim et al analyzed ocular complications in a Japanese patient and described new ophthalmic features, including band keratopathy, meibomian glands dysfunction, posterior subcapsular cataracts, and dry eye disease. The authors also proposed oxidative stress disorder due to low manganese superoxide dismutase levels as an explanation for the aging manifestation of Mulvihill–Smith syndrome.[28] Tyagi et al[22] described retinal features in a 25-year-old man with Mulvihill–Smith syndrome. The authors stated that Mulvihill–Smith syndrome showed phenotypic similarities with other progeroid syndromes such as Cockayne syndrome, which exhibits pigmentary retinopathy and characteristic electrodiagnostic changes; however, the functional electroretinography changes and the absence of fundus pigmentation suggest a different underlying pathophysiology. Other described retinal features included schisis, diffuse thickening, and folding of the retinal layers in both eyes. The authors reported that structural changes in the retina were progressive with loss of foveal contours and wrinkling of the inner retinal layers.[22]
4.6. Lipid disorders
Bartsch et al[7] and Wong et al[10] described hypercholesterolemia in patients suffering from Mulvihill–Smith syndrome. It is well known that lipid metabolism disorders are common in progeroid syndromes such as Hutchinson–Gilford progeria; however, further analysis is required to establish whether hypercholesterolemia represents a typical manifestation of Mulvihill–Smith syndrome.
4.7. Anesthetic difficulties
A few papers have discussed the anesthetic management in patients with progeroid disorders. Stevic et al[29] underlined that the anesthetic management and positioning of patients with Mulvihill–Smith syndrome may be difficult due to bone deformities, arthritic joint changes, and atherosclerosis. The small mouth opening of these patients also makes it difficult to establish an airway. [29]
4.8. Urogenital features
The urogenital features described include hypospadias, cryptorchidism, under descended testicles, cryptorchidism, and amenorrhea.[9]
4.9. Immunological findings
De Silva and Bartsch described lymphopenia with reduced T- and B-cell numbers. The in vitro functional immune deficits described by De Silva et al[11] appeared more limited and, differently from the results by Bartsch et al,[10] the pattern of immunoglobulin (Ig) abnormalities was restricted to low IgG2 and IgG4 subclasses.
4.10. Fibroblast abnormalities
Mulvihill–Smith syndrome has similarities, such as onset in the first decade, with other progeroid disorders [i.e., Hutchison–Gilford, progeroid syndrome neonatal, Rothmund–Thompson (OMIM number #268400), and Cockayne Syndrome A]. DNA repair defects were demonstrated in Cockayne syndrome and involved excision repair or the post-ultraviolet (post-UV) damage replication pathway.[11] In 1975, Mulvihill and Smith[4] demonstrated UV fibroblast sensitivity in their patient, while the patient in the study by Ohashi et al[9] was reported to have X-ray fibroblast sensitivity when compared with a healthy patient. The patient described by De Silva et al[11] showed demonstrable evidence of UV or X-ray fibroblast sensitivity; therefore, the authors stated that these findings were not constant in Mulvihill–Smith syndrome and did not provide evidence that DNA excision repair pathway defects were involved in the pathogenesis of this disorder.[11] Slow growth of cultured fibroblasts from patients with Mulvihill–Smith syndrome was documented by Mulvihill and Smith in 1975, Ohashi et al[9] in 1993, and De Silva et al[11] in 1997. The distinctive differences in the fibroblasts from the patient in the study by De Silva et al[11] compared with those by Mulvihill and Smith and Ohashi et al[9] suggest that further studies are needed in patients with this and other progeria-like syndromes to identify if fibroblast abnormalities are a consistent finding.
4.11. Sleep disorders
Sleep disorders were described in 3 cases by Yagihashi et al,[5] Ferri et al,[12] and Lugaresi and Provini.[30] Clinical and neurophysiological studies suggested that a particular sleep disruption condition, which was in some ways similar to the sleep alterations generally found in fibrillary chorea and fatal familial insomnia by Morvan et al,[31] may be associated with Mulvihill–Smith syndrome.[12,32–34] A mutation in the prion protein gene (PRNP gene) causes the substitution of asparagine for aspartic acid at codon 178, which in conjunction with methionine at polymorphic codon 129, causes fatal familial insomnia.[35] The patient reported by Ferri et al[12] had a cognitive status characterized by progeria and early-onset dementia and showed clinical traits that may have been due to similar genetic alterations. More specifically, the patient's sleep disruption, called agrypnia excitata, was similar to that reported in people suffering from fatal familial insomnia. For this reason, Ferri et al[12] analyzed the PRNP gene; however, no mutations were observed. Therefore, the clinical picture of their patient could not be related to mutations in this gene and the genetic basis of Mulvihill–Smith syndrome remains unknown.[12] Agrypnia excitata has some peculiar polysomnographic findings, including the complete loss of slow-wave sleep, the absence of sleep spindles and K-complexes, and abnormal rapid eye movement sleep with lack of muscle atonia.[5] Yagihashi et al[5] reported distinctive sleep pattern features in their Japanese patient, including severe insomnia with the marked disappearance of sleep spindles and K complexes, loss of slow-wave sleep, and persisting muscle tone. As the same sleep pattern abnormality was described by Ferri et al,[12] Yagihashi et al[5] proposed that agrypnia excitata could be a feature of Mulvihill–Smith syndrome.
4.12. Cognitive deterioration
Yagihashi et al[5] underlined that cognitive deterioration, in addition to developmental delay, was an underappreciated feature in patients with Mulvihill–Smith syndrome. Their patient's cognitive function started to decline when they were 26 years old, and at 28 years old they showed distinct cognitive impairments resembling dementia, such as memory disorder, executive dysfunction, and intellectual deficits. In 2005, Ferri et al[12] also described the progressive decline in cognitive function in a patient with Mulvihill–Smith syndrome aged 25 years. Yagihashi et al[5] suggested that cognitive deterioration, severe insomnia accompanied by agrypnia excitata, and the early onset of tumors could represent an emerging phenotype in adults with Mulvihill–Smith syndrome.
5. Conclusion
The orofacial and dental features reported in Mulvihill–Smith syndrome highlight the importance of special and early dental care by a general dentist, orthodontist, oral surgeon, and maxillofacial surgeon to reduce oral pathologies and lead to a higher quality of life. All interventions should be strictly goal-oriented, and the dental specialist must consider that dental treatment is difficult, as these patients have a small oral aperture and present potential anesthesia risks and other medical disorders.[36] Intensive prophylaxis and goal-oriented interceptive orthodontic and periodontal treatment produces better oral health in patients with this progeria-like syndrome.
This literature review analyzing the medical and dental features in patients with Mulvihill–Smith syndrome may help clinicians to diagnose this rare pathology. Dental abnormalities (i.e., hypodontia and taurodontism) and pigmented nevi on the face could help clinicians to distinguish between Mulvihill–Smith syndrome and other progeroid disorders.
Author contributions
Conceptualization: Roberta Condò.
Data curation: Pier Carmine Passarelli, Loredana Cerroni, Manuele Mancini.
Formal analysis: Guido Pasquantonio.
Funding acquisition: Pier Carmine Passarelli.
Investigation: Pier Carmine Passarelli, Roberta Condò.
Methodology: Pier Carmine Passarelli.
Project administration: Pier Carmine Passarelli.
Supervision: Guido Pasquantonio.
Validation: Guido Pasquantonio, Loredana Cerroni, Manuele Mancini.
Writing – original draft: Pier Carmine Passarelli.
Writing – review & editing: Paolo Francesco Manicone, Antonio D’Addona.
Footnotes
Abbreviations: Ig = immunoglobulin, OMIM = Online Mendelian Inheritance in Man, PRNP gene = Prion protein gene, UV = ultraviolet.
The authors declare that there is no conflict of interest regarding the publication of this paper.
References
- [1].Ahmed MS, Ikram S, Bibi N, et al. Hutchinson-Gilford progeria syndrome: a premature aging disease. Mol Neurobiol 2018;55:4417–27. [DOI] [PubMed] [Google Scholar]
- [2].Hutchinson J. Congenital absence of hair and mammary glands with atrophic condition of the skin and its appendages, in a boy whose mother had been almost wholly bald from alopecia areata from the age of six. Med Chir Trans 1886;69:473–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gilford H. On a condition of mixed premature and immature development. Med Chir Trans 1897;80:17–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mulvihill JJ, Smith DW. Another disorder with prenatal shortness of stature and premature aging. Birth Defects Orig Artic Ser 1975;11:368–70. [PubMed] [Google Scholar]
- [5].Yagihashi T, Kato M, Izumi K, et al. Case report: adult phenotype of Mulvihill-Smith syndrome. Am J Med Genet A 2009;149A:496–500. [DOI] [PubMed] [Google Scholar]
- [6].Elliott DE. Undiagnosed syndrome of psychomotor retardation, low birthweight dwarfism, skeletal, dental, dermal and genital anomalies. Birth Defects 1975;11:364–7. [PubMed] [Google Scholar]
- [7].Wong W, Cohen MM, Miller M, et al. Case report for syndrome identification. Cleft Palate J 1979;16:286–90. [PubMed] [Google Scholar]
- [8].Baraitser M, Insley J, Winter R. A recognizable short stature syndrome with premature ageing and pigmented naevi. J Med Genet 1988;25:53–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ohashi H, Tsukahara M, Murano I, et al. Premature aging and immunodeficiency: Mulvihill-Smith syndrome. Am J Med Genet 1993;45:597–600. [DOI] [PubMed] [Google Scholar]
- [10].Bartsch O, Tympner KD, Schwinger E, et al. Mulvihill-Smith syndrome: case report and review. J Med Genet 1994;31:707–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].De Silva DC, Wheatley DN, Herriot R, et al. Mulvihill-Smith progeria-like syndrome: a further report with delineation of phenotype, immunologic deficits, and novel observation of fibroblast abnormalities. Am J Med Genet 1997;69:56–64. [DOI] [PubMed] [Google Scholar]
- [12].Ferri R, Lanuzza B, Cosentino FI, et al. Agrypnia excitata in a patient with progeroid short stature and pigmented nevi (Mulvihill-Smith syndrome). J Sleep Res 2005;14:463–70. [DOI] [PubMed] [Google Scholar]
- [13].Fühler-Stiller M, Tronnier M. Suspicious pigmented tumor in Mulvihill-Smith syndrome. J Dtsch Dermatol Ges 2011;9:308–11. [DOI] [PubMed] [Google Scholar]
- [14].Breinis P, Alves FG, Alves CA, et al. The eleventh reported case of Mulvihill-Smith syndrome in the literature. BMC Neurol 2014;14:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gilford H. Progeria: a form of senilism. Practitioner 1904;73:188–217. [Google Scholar]
- [16].Coppedè F. The epidemiology of premature aging and associated comorbidities. Clin Interv Aging 2013;8:1023–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bennett GCJ, Ebrahim S. The Essentials of Health Care in Old Age. Oxford University Press; 1995; 2:3-10. [Google Scholar]
- [18].Sternberg S. Gene found for rapid aging disease in children. USA Today 2003;12–3. [Google Scholar]
- [19].Cambridge University Press, Steve RE, Miller VS. Neurocutaneous disorders. 2004;36:150. [Google Scholar]
- [20].Rakha P, Gupta A, Dhingra G, et al. Hutchinson-Gilford progeria syndrome: a review. Der Pharmacia Sinica 2011;2:110–7. [Google Scholar]
- [21].Merideth MA, Gordon LB, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 2010;358:592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tyagi P, Juma Z, Reddy AR. Retinal features in Mulvihill-Smith syndrome. Ophthalmic Genet 2017;38:183–6. [DOI] [PubMed] [Google Scholar]
- [23].Bartsch O, Ludwig D, Schwinger E, et al. Severe complications and gastric carcinoma in Mulvihill-Smith syndrome. J Med Genet 1999;36:175. [PMC free article] [PubMed] [Google Scholar]
- [24].Heyne K, Hof M, Hansen HG. Pigmented naevi after therapy of leukaemia (ALL) in a monozygotic twin. Eur J Pediatr 1984;142:70. [DOI] [PubMed] [Google Scholar]
- [25].Gorlin RO, Sedano HO. Progeria Hutchinson-Gilford syndrome. Mod Med 1968;46:62. [Google Scholar]
- [26].Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet 2006;140:2603–24. [DOI] [PubMed] [Google Scholar]
- [27].Rosenbloom AL, DeBusk FL. Progeria of Hutchinson-Gilford: a caricature of aging. Am Heart J 1971;82:287–9. [DOI] [PubMed] [Google Scholar]
- [28].Irahim OM, Takefumi Y, Dogru M, et al. Ocular complications in Mulvihill-Smith syndrome. Eye (Lond) 2010;24:1123–4. [DOI] [PubMed] [Google Scholar]
- [29].Stevic M, Simic D, Milojevic I. Anesthesia in a child with Mulvihill-Smith syndrome. J Anesth 2014;28:313. [DOI] [PubMed] [Google Scholar]
- [30].Lugaresi E, Provini F. Agrypnia excitata: clinical features and pathophysiological implications. Sleep Med Rev 2001;5:313–22. [DOI] [PubMed] [Google Scholar]
- [31].Morvan A. About the fibrillary chorea. Gazette Hebdomadaire Med Chir 1890;27:173–200. [Google Scholar]
- [32].Lugaresi E, Medori R, Baruzzi A, et al. Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 1986;315:997–1003. [DOI] [PubMed] [Google Scholar]
- [33].Montagna P, Lugaresi E. Agrypnia excitata: a generalized overactivity syndrome and a useful concept in the neurophysiopathology of sleep. Clin Neurophysiol 2002;113:552–60. [DOI] [PubMed] [Google Scholar]
- [34].Josephs KA, Silber MH, Fealey RD, et al. Neurophysiologic studies in Morvan syndrome. J Clin Neurophysiol 2004;21:440–5. [DOI] [PubMed] [Google Scholar]
- [35].Gambetti P, Parchi P, Petersen RB, et al. Fatal familial insomnia and familial Creutzfeldt-Jakob disease: clinical, pathological and molecular features. Brain Pathol 1995;5:43–51. [DOI] [PubMed] [Google Scholar]
- [36].Reichert C, Gölz L, Götz W, et al. Dental and craniofacial characteristics in a patient with Hutchinson-Gilford progeria syndrome. J Orofac Orthop 2014;75:251–63. [DOI] [PubMed] [Google Scholar]