

Abstract
Polymerase delta interacting protein 2 (Poldip2) is a multi-functional protein with numerous roles in the vasculature, including the regulation of cell apoptosis and migration as well as extracellular matrix deposition; however, its role in VSMC proliferation and neointimal formation is unknown. In this study, we investigated the role of Poldip2 in intraluminal wire-injury induced neointima formation and proliferation of vascular smooth muscle cells in vitro and in vivo. Poldip2 expression was observed in the intima and media of human atherosclerotic arteries, where it colocalized with proliferating cell nuclear antigen (PCNA). Wire injury of femoral arteries of Poldip2+/+ mice induced robust neointimal formation after two weeks, which was impaired in Poldip2+/− mice. PCNA expression was significantly reduced and expression of the cell cycle inhibitor p21 was significantly increased in wire-injured arteries of Poldip2+/− animals compared to wild type controls. No difference was observed in apoptosis. Down-regulation of Poldip2 in rat aortic smooth muscle cells significantly reduced serum-induced proliferation and PCNA expression, but upregulated p21 expression. Downregulation of p21 using siRNA reversed the inhibition of proliferation induced by knock down of Poldip2. These results indicate that Poldip2 plays a critical role in the proliferation of VSMCs.
Introduction
Atherosclerosis is a major contributing factor to coronary heart disease and peripheral arterial disease (PAD), which together account for nearly $200B in health care costs.1 It has been clearly established that vascular smooth muscle cell (VSMC) migration and proliferation play major roles in the progression of this disease. Clinical symptoms become noticeable when blood flow is occluded and are often treated with corrective measures such as angioplasty or stent placement. While drug-eluting stents, drug eluting balloons and biodegradable scaffolds have greatly improved the therapeutic benefits of these interventions, they are far less effective for PAD, suggesting a need for improved therapies.2
VSMCs are plastic; that is, they can de-differentiate from the contractile phenotype characteristic of adult vessels to a more synthetic phenotype that can migrate and proliferate.3 Various treatments have been used to control VSMC proliferation ranging from controlled release of drugs from implanted stents to systemic administration of the drugs.4 Pharmacological agents that target the cell cycle have been shown to be a beneficial treatment option for restenosis,4 but have limited benefit because they also prevent endothelial coverage of the stent.
Proliferation is a complex process and various protein-protein interactions have been reported that ensure accurate cell division. While cyclins and cyclin-dependent kinase (CDK) complexes positively regulate cell cycle progression, cyclin complexes with CDK inhibitors (CDKIs) such as p21 are negative regulators of the cell cycle.5 One relatively unstudied protein that has the potential to impact the cell cycle is polymerase delta interacting protein-2 (Poldip2), which was originally identified as an interacting protein of DNA polymerase δ and proliferating cell nuclear antigen (PCNA),6 and has been implicated in mitotic spindle formation, NADPH oxidase 4 (Nox4) activation, DNA repair, and cellular adhesion 7–10. Our recent data suggest that homozygous deletion of Poldip2 is lethal, indicating a critical role in development.11 Poldip2+/− animals have reduced vascular H2O2 levels and excess and disordered extracellular matrix in their large vessels. Moreover, Poldip2 depletion delays cell cycle progression in cultured mouse embryonic fibroblasts and impairs proliferation of cultured endothelial cells,11, 12 suggesting that it may play a similar role in proliferative cells in the adult. However, there have been no studies on the role of Poldip2 on proliferation in vivo, and the mechanisms by which it influences growth are incompletely understood.
In this study, we tested the hypothesis that Poldip2 regulates VSMC growth in vivo and, using in vitro systems, investigated the mechanism by which it modifies proliferation. We showed that Poldip2 is present in human atherosclerotic lesions and used heterozygous Poldip2 mice subjected to wire injury of their femoral artery to investigate the role of Poldip2 in neointima formation, which has a large proliferative component. To elucidate the mechanism by which Poldip2 affects cell proliferation, we tested the effect of Poldip2 downregulation on two cell cycle regulators: PCNA and p21. Our results provide the first evidence that Poldip2 plays a role in the vascular response to injury.
Materials and Methods
Reagents and antibodies
All reagents used in this study were purchased from standard suppliers. Poldip2 goat antibody was custom made by GenScript Corporation (Piscataway, NJ) against the peptide sequence NPAGHGSKEVKGKTC as reported previously.10 When available, commercial antibodies were used: PCNA (Abcam, Cambridge, MA), p21 (EMD Millipore, Billerica, MA and Abcam), and β-actin (Abcam). Secondary antibodies were from Jackson ImmunoResearch (West Grove, PA), Bio-Rad (Hercules, CA) or ThermoFisher Scientific.
Human Coronary Arteries
Human coronary arteries were harvested from patients with end-stage heart failure undergoing heart transplantation at Emory University Hospital as described previously.13 All procedures were approved by the Emory Institutional Review Board. Briefly, hearts were harvested during surgery, placed in ice-cold normal saline, and transferred immediately to ice-cold Krebs buffer. Left anterior descending, left circumflex and right coronaries were excised, fixed in 10% buffered formaldehyde up to three days, followed by 70% ethanol. Tissues were embedded in paraffin before sectioning and processing for morphology and PCNA and Poldip2 co-staining. The severity of the atherosclerosis was ranked based on histological classification as described previously14 and sections of stage II, IV and VI were used in this study. Stage II arteries exhibit established neointima with high smooth muscle cell density but without a lipid core; stage IV arteries have an established lipid core but no thrombosis; and stage VI arteries have a disrupted lesion surface with hematoma, hemorrhage or thrombosis.14 To quantify the colocalization of Poldip2 and PCNA staining, a Pearson coefficient was calculated using Image-Pro Plus 6.2 software (Media Cybernetics, Rockville, MD) from images taken using a Zeiss LSM 510 META confocal microscope with 20X, 40X or 63X lenses.
Poldip2 Gene Trap Mice
Mice with a gene trap insertion in the first intron of Poldip2 (chromosome 11, NCBI Gene ID: 67811) were produced at the Texas A&M Institute for Genomic Medicine (College Station, TX) as previously described.15 Mice were produced directly in C57BL/6 background. Because homozygous deletion of Poldip2 is lethal,11, 15 Poldip2+/− mice were used for this study.
Mouse Femoral Artery Injury Model
Transluminal mechanical injury of the right femoral artery was induced by introducing a guide wire as previously reported.16, 17 In brief, the right femoral artery was exposed by blunt dissection and was looped proximally and distally with 6–0 silk suture for temporary control of blood flow during the procedure. Arteriotomy was performed distal to the epigastric branch between the rectus femoris and vastus medialis muscles. A straight spring wire of 0.38 mm in diameter (Cook, Bloomington, IN), was carefully inserted into the femoral artery through the epigastric artery toward the iliac artery and left in place for 1 minute to denude and dilate the artery. After removing the wire, the epigastric artery was secured by silk suture at the proximal portion. Blood flow in the femoral artery was restored by releasing the proximal and distal femoral sutures. After the procedure mice were maintained on a standard chow diet. Animals were euthanized at weeks 1, 2 and 3 after surgery and arteries were pressure-perfused at 100 mm Hg with 0.9% sodium chloride, followed by pressure-fixation with 10% formalin. Femoral arteries were then carefully excised and embedded in paraffin. Sections (5 μm) were double-labeled with Poldip2 and PCNA antibodies. For nuclear labeling of p21, following deparaffinization, antigen retrieval was performed in citrate buffer, pH 6.0, at 120°C for 10 min, followed by 1% Triton-X 100 for 15 min. All sections were stained with 4’,6-diamidino-2-phenylindole and mounted in Shandon Immu-Mount (both from Thermo Scientific). Images were taken using a Zeiss Axioskop 2 microscope with an Axiocam CCD camera and processed using AxioVision 4.8 software. For additional immunohistochemistry and DHE staining studies, arteries were collected immediately after saline perfusion, embedded in OCT solution and stored at −80°C. To assess the early apoptotic response to injury, arteries were obtained 2 hours after injury induction, as described previously16, 17. All procedures were approved by the Emory University Institutional Animal Care and Use Committee.
Detection of Superoxide
Intracellular superoxide (O2.-) production was evaluated by measuring the conversion of dihydroethidium (DHE) to 2-hydroxyethidium using high-performance liquid chromatography (DHE-HPLC) as described previously.18 To investigate localization of ROS production, in-situ evaluation of ROS was performed by DHE staining as previously reported 13.
Cell Culture
Rat aortic VSMCs were isolated from thoracic aorta by enzymatic digestion as described previously.19 Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% calf serum, 100 U/mL penicillin and 100 μg/mL streptomycin. After confirming VSMC identity by immunostaining for smooth muscle α-actin and calponin, cells were passaged by using trypsin. Passages between 7 and 12 were used for experiments.
siRNA
For transfection with siRNA, VSMCs were trypsinized and plated at 40–50% confluence in six well plates or on 100-mm cell culture dishes. After 4–6 h, cells were washed with serum-free Opti-MEM, and incubated with siRNA and Oligofectamine. Cells were incubated in OPTI-MEM for an additional 1 – 2 days as described previously.10, 15 A stealth siRNA against human, mouse and rat Poldip2 (siPoldip2; sense: 5’GCCCACAUAUAUCUCAGAGAUCUCA3’, antisense: 5’UGAGAUCUCUGAGAUAUAUGUGGGC3’) and the stealth control siRNA (siControl) with corresponding GC content were purchased from Invitrogen. A rat siRNA against p21 (sip21; sense: 5’CUCCAAACUUAGUUAUUUA[dTdT]3’, antisense: 5’UAAAUAACUAAGUUUGGAG[dGdC]-3’) and the AllStars negative control siRNA were purchased from Qiagen. Cells were transfected with a final siRNA concentration of 15 nmol/L.
Cell Proliferation
VSMC proliferation was assessed by measuring the cell number as described previously with some modifications.17 Briefly, VSMCs were seeded at 200,000 cells/100 mm dish and treated with siPoldip2 alone or in combination with sip21. After 72 hours of siRNA treatment, cells were serum deprived overnight and then stimulated with 10% calf serum. Cell number was counted using a Scepter Cell Counter (Millipore) at days 1–5. Proliferation rate was further confirmed using the Click-iT® EdU Cell Proliferation Assay kit (Life science) according to the manufacturer’s instructions.
Immunoblotting
Protein expression was measured as described previously.15, 20 Briefly, to measure the expression levels of Poldip2, p21 and PCNA in vessels, femoral arteries were harvested at week 2 after injury. Two to four arteries were pooled per sample. Vessels were homogenized in Hunter’s buffer (25 mmol/L HEPES, 150 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EGTA, 10 mmol/L Na-pyrophosphate, 10 mmol/L NaF, 0.1 mmol/L Na-orthovanadate, 1% Na deoxycholate, 1% Triton X 100, 0.1% SDS, 10% glycerol, and protease inhibitors) on ice and centrifuged at 10,000 x g for 5 min and supernatants were utilized for western blot analysis.
To measure the expression patterns of Poldip2, p21 and PCNA in cultured VSMCs, cells were washed twice with phosphate-buffered saline at the end of treatment period and lysed in Triton X-100 buffer (50 mmol/L HEPES, 5 mmol /L EDTA, 50 mmol/L NaCl, 1% Triton X-100) containing protease inhibitors (10 μg/mL aprotinin, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL leupeptin), and phosphatase inhibitors. Lysates were sonicated and cleared at 10,000 x g for 5 minutes. Protein concentration in the supernatant was determined using Bradford reagent (Bio-rad). Proteins were separated using SDS-PAGE, transferred to nitrocellulose membranes, blocked, and incubated with the indicated primary antibodies. Proteins were detected using enhanced chemiluminescence (ECL, GE), and band intensity was quantified by densitometry using ImageJ 1.43 software.
Histological Analysis of Neointima
Sections of 7 μm thicknesses were prepared from paraffin-embedded arteries. Standard Hematoxylin and Eosin (H&E) staining was used for morphometric analysis of mouse samples and for determining the severity of atherosclerosis in human samples. The intima:media ratio in arteries taken from wire-injured Poldip2+/− mice was calculated as described previously16, 17 on 5–7 sections from each artery (40 X) using image J software (Ver. 1.43). Apoptosis was measured using a commercially available kit (In Situ Cell Death Detecting Kit - TMR red (Roche)) according to the manufacturer’s instructions.
Immunofluorescence
PCNA and Poldip2 co-staining was carried out on 7 μm sections of paraffin-embedded human atherosclerotic arteries. Antigen retrieval was performed in citrate buffer (pH 6.0) before incubating with Poldip2 antibody (1: 250 in 2% BSA, in-house goat polyclonal antibody) and PCNA antibody (1:100 in 2% BSA, rabbit polyclonal antibody; Abcam) followed by incubation with biotinylated anti-goat secondary antibody (Vector Labs, Burlingame, CA) and then with Streptavadin QDot 655 (1:200 in 2% BSA; Invitrogen). After Poldip2 staining was completed, biotin was blocked using the Biotin Blocking kit (Vector Labs) as described in the manufacturer’s instructions. Sections were then incubated with biotinylated rabbit anti-goat secondary antibody (Vector Labs, Burlingame, CA) and then incubated with Streptavadin QDot 605 (1:200 in 2 % BSA; Invitrogen).
PCNA expression in wire-injured femoral arteries was assessed using a rabbit polyclonal antibody as described previously.21 Briefly, formalin-fixed, paraffin embedded tissue sections were deparaffinized and then rehydrated in a series of xylene, ethanol and PBS. Following blocking, sections were incubated with a rabbit polyclonal anti-PCNA primary antibody (1:100 in 3% BSA, Abcam) at 4°C overnight. Alexa Fluor 568 donkey anti-rabbit antibody (Vector Labs, 1:200 in 3% BSA) was used as secondary antibody for 1 hour. The tissue sections were then counterstained with DAPI (1:1000 in PBS) for 10 min. Slides were mounted with Shandon Immu-Mount (Thermo Scientific). For negative controls in each experiment, incubation without the primary antibody (PCNA) was performed. Images were acquired with an Axioskop microscope and Axiocam CCD camera (Carl Zeiss, Inc, Göttingen, Germany).
Statistical Analysis
Results are expressed as mean ± SEM from at least 3 independent experiments. Statistical significance was assessed using either t-test or analysis of variance, followed by Tukey’s multiple comparison post hoc test (GraphPad Prism software, version 7). A value of P < 0.05 was considered significant.
Results
Poldip2 and PCNA co-localize in human atherosclerotic arteries
It has been reported that Poldip2 is associated with Polymerase δ and PCNA.6 To begin to investigate the potential role of Poldip2 in atherosclerosis, we first investigated its expression and potential co-localization with PCNA in human atherosclerotic arteries. Using H&E staining of human coronary arteries, we selected arteries with progressively greater disease (stages II, IV and VI as shown in Figure 1a-d, e-h and i-l). Interestingly, in stage II arteries, Poldip2 (Figure 1b 1d) was highly expressed in endothelium and in the underlying proliferating VSMC, where it colocalized with PCNA. Poldip2 and PCNA expression decreased in the advanced stages of disease, but still showed similar colocalization (Figure 1, f-h and j-l).
Figure 1.
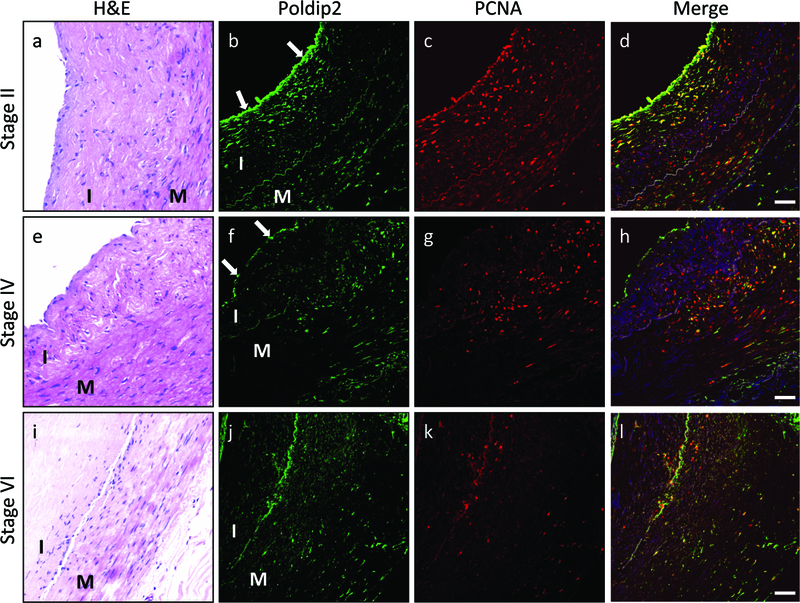
Poldip2 and PCNA are co-expressed in early stages of human atherosclerosis. Sections of human coronary arteries were classified into 3 atherosclerotic stages using H&E staining, as described in Methods: stage II (a-d), stage IV (e-h) and stage VI (i-l). Immunofluorescence was used to detect Poldip2 (green) and PCNA (red). Poldip2 is more highly expressed in the endothelial layer of stage II arteries (indicated by arrows) than in neointima and media, but is apparent throughout the vessel wall in stage IV and VI arteries. PCNA is highly expressed in the medial and intimal layers of stage II and IV arteries, but is limited in stage VI arteries. Pearson coefficients, indicating colocalization of Poldip2 and PCNA were high in all analyzed images: stage II: 0.66±0.02 (n=28); stage IV: 0.68±0.02 (n=22); stage VI: 0.62±0.04 (n=22), suggesting that Poldip2 is upregulated in proliferating cells. Arterial layers are indicated by letters: I (intima), M (media). Nuclei were counterstained in blue with DAPI. Images are representative of sections from at least 4 patients for each stage of atherosclerosis. Scale bars: 50μm.
Role of Poldip2 in neointimal formation in vivo
To study the importance of Poldip2 in lesion formation in vivo, we investigated its role in experimentally induced neointimal formation using Poldip2+/+ and Poldip2+/− mice subjected to wire injury of their femoral arteries. As shown, in Figure 2A, femoral arteries from Poldip2+/− mice developed a smaller neointima than arteries from Poldip2+/+ mice. The intima:media ratio was reduced in Poldip2+/− animals compared to that in control animals at 2 weeks and began to recover by 3 weeks post injury (Figure 2B). At week 1, a similar trend was already present but did not reach statistical significance, presumably because of small sample size. No significant difference was found in medial thickness between Poldip2+/+ and Poldip2+/− animals (data not shown). To determine whether altered apoptosis contributes to the difference in neointimal formation, we measured apoptosis 2 hours after injury. As shown in Figure 2C & D, we found no difference in the number of apoptotic cells between the two genotypes.
Figure 2.
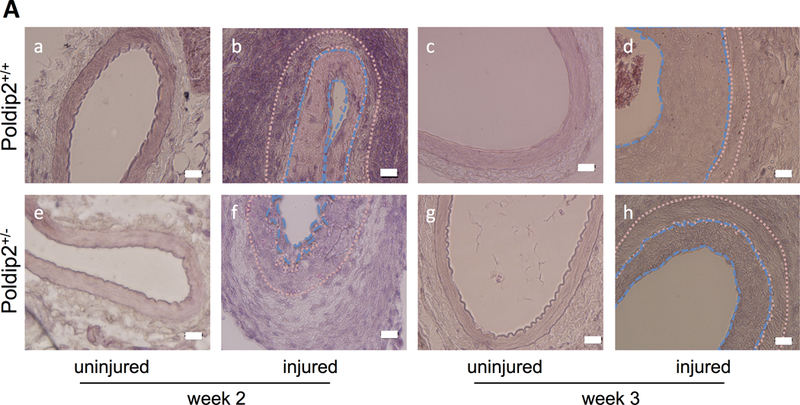
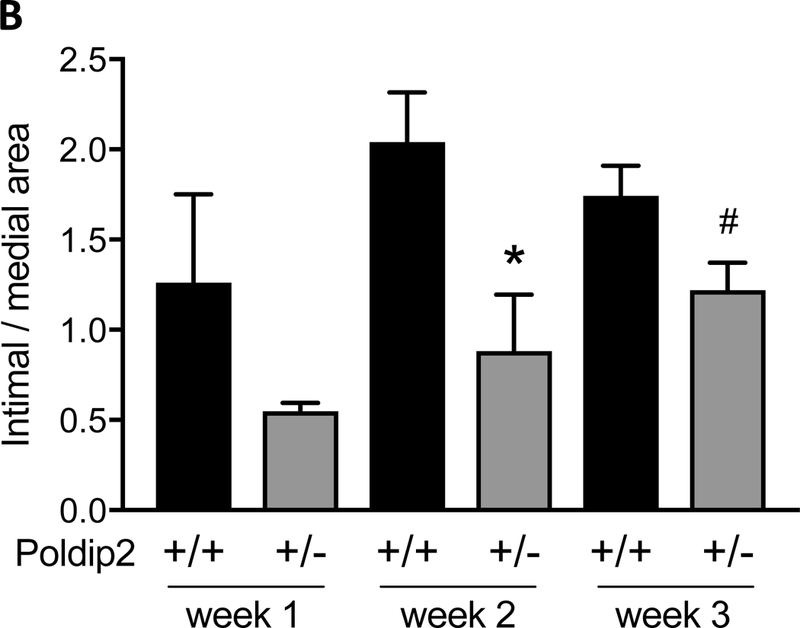

Poldip2 knockdown reduces neointima formation after vascular injury. Intraluminal injury was induced by insertion of a guide wire into the right femoral artery in mice. Both injured and uninjured vessels were harvested at various time points after surgery for histological studies. (A) Representative H&E images of cross-sections from Poldip2+/+ and Poldip2+/− femoral arteries harvested 2 or 3 weeks after surgery. Tracing in ImageJ software was used to measure intimal and medial areas. The endothelium and internal elastic lamina were marked with blue dashed lines, while the internal and external elastic laminae were marked with pink dotted lines in selected images. The scale bars are 20 μm. (B) Intimal and medial areas were measured in duplicate cross-sections from each vessel. Bars represent average ± SEM of intimal/medial area ratios from 3–15 animals. Neointimal area was reduced in Poldip2+/− mice vs. wild-type at week 2: *P<0.03, and remained almost statistically significant by week 3: #P<0.06 (C) Femoral arteries were harvested 2 hours after injury from wild-type (a) and Poldip2+/− mice (b). Apoptotic TUNEL-positive cells were stained in red and nuclei were counterstained with DAPI. Negative controls (c) were produced in sections from uninjured arteries. (D) Quantification of images similar to those in panel C. Apoptotic TUNEL-positive cells were counted in duplicate femoral artery sections and expressed as percent of total cell number assessed by DAPI staining. Values are expressed as mean ± SEM. H&E and TUNEL images are representative of sections from at least 3 animals in each genotype.
Superoxide levels in blood vessels is regulated by Poldip2
We have previously shown that Poldip2 can regulate NADPH oxidases and that NADPH oxidase-derived reactive oxygen species are involved in neointimal formation.10, 22 To determine if manipulation of Poldip2 altered O2.- production after injury, we used dihydroethidium (DHE) high-performance liquid chromatography (HPLC) and in-situ O2.- staining by DHE to measure O2. As shown in Figure 3A, O2.- levels increased by intraluminal wire injury were abrogated by Poldip2 knockdown. Interestingly, O2.- levels in uninjured arteries were also reduced in Poldip2+/−, compared to wild type animals, which is in consistent with previous observations in these mice.15 In-situ staining with DHE corroborated these results. In the uninjured arteries, O2.- was mainly concentrated in the medial layer, but wire-injury clearly increased O2.- production throughout the vessel wall, especially in the adventitia and in the neointima, where proliferating VSMCs are found (Figure 3B). DHE staining was reduced in Poldip2+/− mice, both basally and after injury.
Figure 3 :
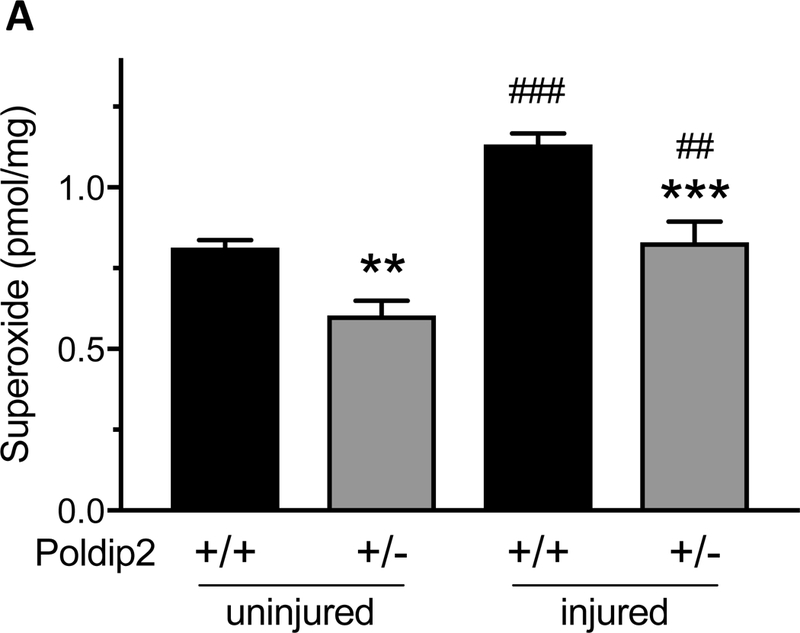

Poldip2 knockdown reduces superoxide production in femoral arteries. Intraluminal injury was induced by insertion of a guide wire into the right femoral artery in mice. Injured and uninjured arteries were harvested after 3 weeks. (A.) Superoxide was quantified in femoral arteries by measuring the conversion of dihydroethidium (DHE) to 2-hydroxyethidium using high-performance liquid chromatography (DHE-HPLC) and normalizing to protein. Superoxide was increased by vascular injury (##P<0.01, ###P<0.001 vs. uninjured) and reduced by Poldip2 knockdown (**P<0.01, ***P<0.001 vs. wild-type). Bars represent average ± SEM of results from 6–8 animals. (B.) Superoxide was detected in femoral artery sections by staining with DHE (red fluorescence). Green autofluorescence was used to identify vessel structure. All images were acquired with identical settings and are representative of results from at least 3 animals. Superoxide was increased by injury and reduced by Poldip2 knockdown throughout the vessels. Nuclei were counterstained in blue with DAPI.
Role of Poldip2 in cell proliferation
Because there was no change in apoptosis in the face of an increase in neointima (Figure 2) and because Poldip2 has been previously shown to regulate cell cycle proteins,11 we compared proliferation in injured arteries from Poldip2+/+ and Poldip2+/− mice using PCNA immunofluorescence. Intense PCNA staining was apparent at week 2 in Poldip2+/+ mice and was noticeably decreased in Poldip2+/− animals (Figure 4A), while the reverse was true for p21 (Figure 4B). Western blot analysis revealed that PCNA expression is decreased in wire-injured arteries from Poldip2+/− mice, compared to wild type (Figure 5, 29 ± 16% decrease, n = 4). In contrast, expression of the cell cycle inhibitory protein p21, which we have previously shown to be regulated by reactive oxygen species,23 is augmented after injury in Poldip2+/− animals compared to wild type (Figure 5, 48 ± 32% increase, n = 3). These observations suggest that Poldip2 may regulate p21 as well as PCNA in arteries.
Figure 4:

Expression of PCNA and p21 after intraluminal injury of the femoral artery. Right femoral arteries were harvested from mice two weeks after induction of intraluminal injury by insertion of a guide wire. PCNA (A) and p21 (B) were detected by immunofluorescence (red) as described in the methods section and nuclei were counter stained with DAPI (blue). Autofluorescence is shown in green. While the numbers of cells positive for PCNA were decreased, those expressing p21 were increased in arteries from Poldip2+/− vs. wild type mice. Images are representative of sections from at least 3 animals of each genotype. Scale bars are 50 μm.
Figure 5.
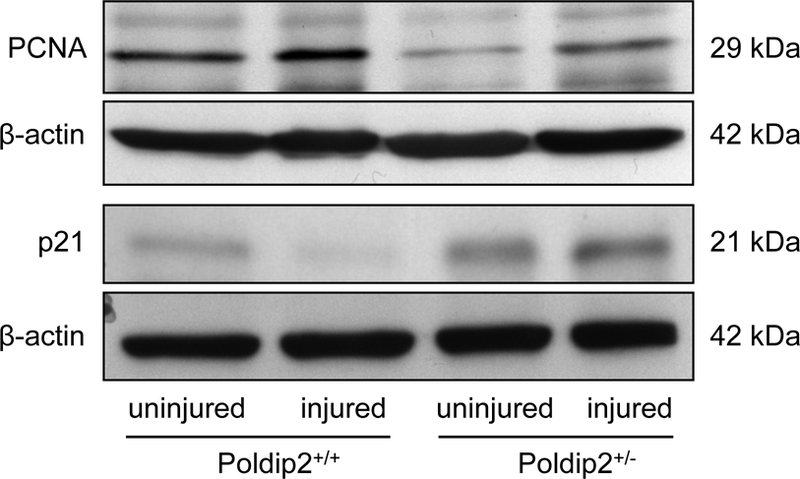
Poldip2 knockdown normalizes cell cycle markers following vascular injury. Intraluminal injury was induced by insertion of a guide wire into the right femoral artery in mice. Arteries were harvested 2 weeks after surgery and pooled samples from 2–4 animals were processed for protein analysis using Western blotting. In injured vessels, PCNA was reduced (by 29±16%, n=5, P<0.05) and p21 was increased (by 74±27%, n=3, P<0.05) in arteries from Poldip2+/− vs. wild type mice. Thus, Poldip2 knockdown appears to normalize cell cycle markers affected by neointimal proliferation. Equal loading between lanes was verified using β-actin. Data from injured arteries are representative from 3–5 independent experiments. Two lanes of uninjured arteries from a single experiment are also included for comparison.
Because Poldip2 is expressed in multiple cell types in the vessel wall, we used a culture system and siRNA-mediated knockdown to specifically examine the role of Poldip2 in VSMC proliferation. Rat aortic VSMCs were treated with siPoldip2 or siControl, serum-deprived overnight and stimulated with 10% calf serum. Cells were counted at days 1–5. As shown in Figure 6A, Poldip2 knockdown significantly decreased VSMC proliferation. These results were confirmed using an EdU incorporation assay, which showed that DNA synthesis is reduced after 1 day of serum stimulation in siPoldip2, compared to siControl-treated VSMC (Figure 6B).
Figure 6 :
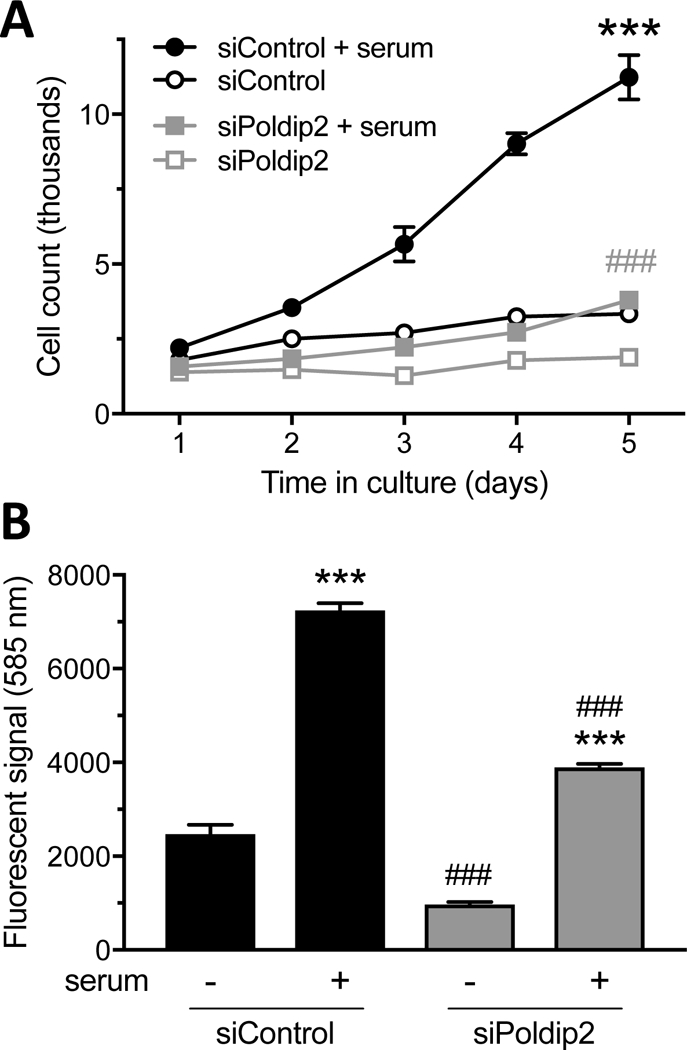
Poldip2 knockdown decreases proliferation in cultured VSMC. VSMC were transfected with control (siControl) or Poldip2 (siPoldip2) siRNA. (A) Transfected cells were counted at the time of plating (day 0, not shown) and maintained in serum-free medium or stimulated with 10% calf serum (days 1–5). Data points represent average number of cells in each group ± SEM from 6 independent experiments. In some conditions, the error bar is smaller than the symbol. While control cells multiplied during that time (***P<0.001 vs. no serum at days 3–5), Poldip2 knockdown abrogated serum-induced proliferation (###P<0.001 vs. siControl + serum at days 3–5). (B) Transfected cells were incubated with the nucleoside analog Edu (10 μM) for 1 day in the presence or absence of 10% calf serum. Edu incorporation was measured by fluorescence. Stimulation with serum increased Edu incorporation in both control and Poldip2 knockdown cells (***P<0.001 vs. no serum). Poldip2 knockdown abrogated Edu incorporation in the absence or presence of serum (###P<0.001 vs. siControl). Bars represent the mean ± SEM from 3 independent experiments.
To determine if the reduction in PCNA and increase in p21 observed in vivo was also true in isolated VSMCs, we measured protein expression of these targets at day 0, 2 and 4 after siRNA treatment followed by serum stimulation. The effectiveness of siRNA was measured by Poldip2 western blot (Figure 7A). As expected, PCNA expression is significantly increased at day 2 and 4 in siControl-treated cells, while p21 is largely unchanged. The increase in PCNA was significantly inhibited in siPoldip2 treated cells, while p21 expression was robustly elevated (Figure 7B-D). To investigate the role of the elevated p21 in the reduction of proliferation engendered by Poldip2 knockdown, we co-treated VSMCs with siPoldip2 and sip21. Efficacy of knockdown was confirmed by western analysis (Figure 8A). As shown in Figure 8B., sip21 co-treatment almost completely reversed the siPoldip2-mediated reduction in serum-induced proliferation. Taken together, these results support the notion that Poldip2 regulates VSMC proliferation by modulating the levels of p21, which likely accounts, at least in part, for the impaired neointimal formation in Poldip2+/− mice subjected to wire injury.
Figure 7 :
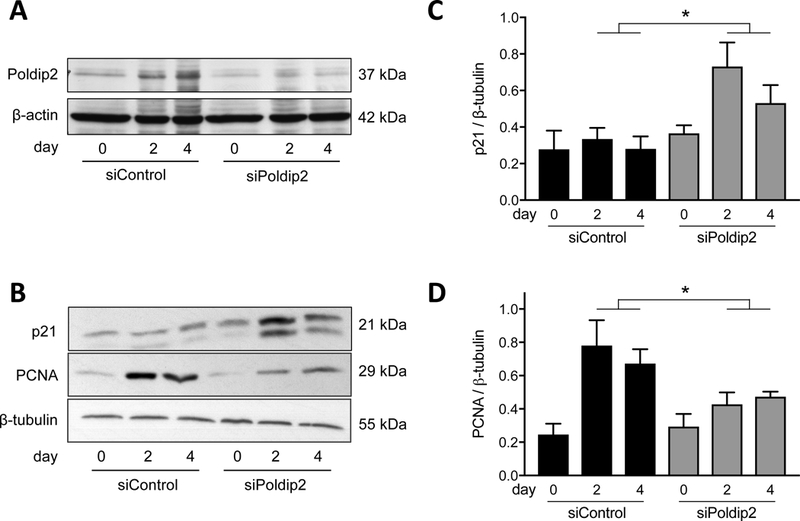
Poldip2 knockdown affects cell cycle markers in cultured VSMC. VSMC were transfected with control (siControl) or Poldip2 (siPoldip2) siRNA and processed for Western blotting after the indicated number of days in culture in the presence of 10% calf serum. Membranes were probed for Poldip2 (A), p21 or PCNA (B) using corresponding primary antibodies. Equal loading was verified using β-actin (A) or β-tubulin (B) antibodies. (C and D) Quantification of results from panel B. At days 2 and 4, p21 and PCNA were significantly upregulated and downregulated, respectively, by siPoldip2 (*P<0.05 vs. siControl). Bars represent the average ± SEM of data from 5 (C) and 7 (D) independent experiments. The effectiveness of siPoldip2 treatment from one experiment is shown (A).
Figure 8:

Knockdown of p21 rescues the inhibition of cell proliferation induced by siPoldip2. VSMC were transfected with a pair of control (siControl), Poldip2 (siPoldip2) or p21 (sip21) siRNAs, as indicated. (A) Transfected cells were grown for 2 days in the presence of 10% calf serum before harvesting for Western blotting. Membranes were probed with p21, Poldip2 or β-actin primary antibodies to verify the effectiveness of siRNA treatments. These blots are representative of 3–4 independent experiments. (B) Transfected cells were counted after 2 days in the presence of 10% calf serum. Knockdown of Poldip2 reduced proliferation (**P<0.01 vs. siControl) and sip21 rescued this inhibition (#P<0.05 vs. siPoldip2 alone). Bars represent the average ± SEM of cell counts from 3 independent experiments.
Discussion
In this study, we identify Poldip2 as an important regulator of VSMC proliferation in vitro and in vivo during neointima formation. Poldip2 colocalizes with PCNA in human atherosclerotic arteries and PCNA expression is reduced in injured arteries from Poldip2+/− mice and in VSMC treated with siPoldip2. Consistent with previous work in mouse embryonic fibroblasts, the reduction in VSMC proliferation when Poldip2 is depleted is accompanied by upregulation of the cell cycle inhibitor, p21, and importantly, knockdown of p21 returns proliferation to control levels.
Poldip2 is a multifunctional protein that has no homology to other known mammalian proteins, consisting of an N-terminal mitochondrial targeting sequence and two main functional domains: a DUF525 domain and a YccV-like domain.24 Although Poldip2 is transcribed from a single gene, its mitochondrial targeting sequence results in cleavage of the 42-kDa protein to a 37-kDa form. Poldip2 binds to a variety of proteins, including p22phox,25 polymerase-δ and PCNA,6, 9 and carcinoembryonic antigen-related cell adhesion molecule-1 (CEACAM-1).8. With regard to proliferation, it has been shown to play a role in mitotic spindle formation,7 angiogenesis,12 and expression of Cdk1, CyclinA2 and p21.11 In this study, we show that not only does Poldip2 colocalize with PCNA, but also that upregulation of p21 by depletion of Poldip2 is sufficient to inhibit VSMC proliferation. This is the first study to show a role for Poldip2 control of growth in this cell type.
One potential mechanism by which Poldip2 may regulate neointimal formation is via regulation of hydrogen peroxide production from Nox4. Our original work showed that Poldip2 increases Nox4 activity10 and ROS generation. In the present study we measured the production of superoxide, the primary product of NADPH oxidases, which is then converted to the important signaling mediator hydrogen peroxide. A role for ROS has been identified in cell cycle progression. For example, ROS regulate the G0 to G1 transition of the cell cycle by activating the signaling pathways that promote cyclin D.26, 27 Nox4 itself has been implicated in proliferation of endothelial 28, pulmonary vascular smooth muscle cells 29, and VSMC 30. In Nox4 knockdown cells, TGF-β-induced Rb phosphorylation and proliferation are inhibited.31 Similarly, Salmeen et al.32 showed that knockdown of Nox4 causes an increase in p53 and p21 levels. However, because Poldip2 knockout animals die at birth,15 but Nox4 knockout mice survive,33 it is likely that Poldip2 has additional roles in proliferation and neointimal formation.
The ability of Poldip2 depletion to inhibit injury-induced neointima formation is clear at 2 weeks (Figure 2A & B). This reduction in neointimal formation was not associated with a change in apoptosis (Figure 2C & D), but was accompanied by a decrease in PCNA and an increase in p21 expression (Figure 4). In fact, decreased PCNA and increased p21 were also evident in uninjured Poldip2+/− femoral arteries (Figure 5). It is noteworthy that gene transfer of p27 or p21 into balloon injured vessels significantly reduces VSMC proliferation and neointimal formation.34, 35 In addition, it has been previously shown that adenoviral or chemical mediated p21 overexpression can decrease neointima formation 34, 36 and deficiency of p21 leads to accelerated atherogenesis in apoE KO mice.37 These observations strongly support the conclusion that the increase in p21 expression in Poldip2+/− arteries is in part responsible for the reduction in neointimal formation.
The mechanism by which downregulation of Poldip2 modulates PCNA and p21 expression is unclear. It has been shown that p21 interacts with PCNA and hence blocks cell cycle progression.38, 39 It has also been suggested that p21 prevents or displaces binding of polymerase δ to PCNA at the G1/S phase transition.40 Lu et al. 41 showed that PCNA directly interacts with the N-terminus of the p50 subunit of polymerase δ and that this binding is inhibited by p21, suggesting that p21 competes for the same binding site on PCNA. It is thus possible that Poldip2 is required for stabilization of PCNA binding to polymerase δ and subsequent DNA synthesis. Our results support this possibility as siPoldip2-treated cells failed to proliferate in response to serum. Bendjennat et al.42 showed that p21 downregulation is required for efficient PCNA dependent-DNA synthesis after UV irradiation, implying that p21 accumulation might be sufficient to inhibit PCNA-dependent DNA repair. This possibility is also consistent with our data. In fact, it has been shown that binding of p21 to PCNA prevents proteasomal degradation of p21, which could leads to increased p21 expression.43 In addition to interaction with PCNA, p21 can bind to CDK2/cyclinE,44 which could control replication of DNA. Interaction with the CDK/cyclin complex increases proteasome-mediated degradation of p21, whereas binding to PCNA protects p21 from proteasome-mediated degradation.43 It has also been suggested that PCNA interaction regulates p21 expression levels and stability in vivo.45 We speculate that Poldip2 knockdown affects the PCNA and Polymerase δ interaction, allowing p21 to interact with PCNA, which ultimately leads to decreased cell cycle progression. This effect presumably also takes place in human arteries with stage VI atherosclerosis when expression of Poldip2 and PCNA appears to be reduced and may thus contribute to plaque stabilization.
Physiologically, Poldip2 has a role in development, because homozygous deletion of Poldip2 is perinatal lethal.11 Adult heterozygous animals have stiffer arteries, increased extracellular matrix accumulation and impaired collateral formation.12, 46 The role of Poldip2 in matrix regulation is potentially relevant here as well. Heterozygous deletion of Poldip2 results in a reduction of both MMP-2 and MMP-9 activity during hindlimb ischemia12 and following stroke.47 This suggests another mechanism that might contribute to the reduction in neointimal formation we see following wire injury of Poldip2+/− mice. Reduced MMP activity impairs breakdown of matrix, limiting VSMC migration into the intimal area. In human atherosclerotic arteries, colocalization of Poldip2 and PCNA is mainly found in the neointima of stage II arteries, and in the neointima and shoulders of the plaques in stage IV arteries. Poldip2 and PCNA expression in stage VI arteries is minimal, which could be because samples were collected at terminal stages of the disease. It is interesting to speculate that the reduced expression of Poldip2 in advanced atherosclerosis might contribute to a reduction in VSMC proliferation in late-stage plaques but an increase in extracellular matrix. Examination of a potential role for MMPs in this phenotype requires further study.
In summary, our data suggest that Poldip2 is an important regulator of VSMC proliferation and neointimal formation, likely through multiple mechanisms that involve PCNA, ROS and p21. Targeted disruption of the interaction of Poldip2 with specific binding partners may represent a novel therapeutic strategy to control unwanted proliferation in atherosclerosis or restenosis.
Supplementary Material
Acknowledgments
This work was supported by NIH grants HL38206 and HL095070.
Footnotes
Conflicts of interest
The authors have no conflicts of interest to report.
References
- 1.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017;135:e146–e603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van den Berg JC. Drug-eluting balloons for treatment of SFA and popliteal disease - A review of current status. Eur J Radiol 2017;91:106–115. [DOI] [PubMed] [Google Scholar]
- 3.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 2012;95:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoffmann R, Mintz GS. Coronary in-stent restenosis - predictors, treatment and prevention. Eur Heart J 2000;21:1739–1749. [DOI] [PubMed] [Google Scholar]
- 5.Ekholm SV, Reed SI. Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol 2000;12:676–684. [DOI] [PubMed] [Google Scholar]
- 6.Liu L, Rodriguez-Belmonte EM, Mazloum N, et al. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J Biol Chem 2003;278:10041–10047. [DOI] [PubMed] [Google Scholar]
- 7.Klaile E, Kukalev A, Obrink B, et al. PDIP38 is a novel mitotic spindle-associated protein that affects spindle organization and chromosome segregation. Cell Cycle 2008;7:3180–3186. [DOI] [PubMed] [Google Scholar]
- 8.Klaile E, Muller MM, Kannicht C, et al. The cell adhesion receptor carcinoembryonic antigen-related cell adhesion molecule 1 regulates nucleocytoplasmic trafficking of DNA polymerase delta-interacting protein 38. J Biol Chem 2007;282:26629–26640. [DOI] [PubMed] [Google Scholar]
- 9.Tissier A, Janel-Bintz R, Coulon S, et al. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Poleta. DNA Repair 2010;9:922–928. [DOI] [PubMed] [Google Scholar]
- 10.Lyle AN, Deshpande NN, Taniyama Y, et al. Poldip2, a Novel Regulator of Nox4 and Cytoskeletal Integrity in Vascular Smooth Muscle Cells. Circ Res 2009:CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown DI, Lassegue B, Lee M, et al. Poldip2 knockout results in perinatal lethality, reduced cellular growth and increased autophagy of mouse embryonic fibroblasts. PLos One 2014;9:e96657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amanso AM, Lassegue B, Joseph G, et al. Polymerase delta-interacting protein 2 promotes postischemic neovascularization of the mouse hindlimb. Arterioscler Thromb Vasc Biol 2014;34:1548–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorescu D, Weiss D, Lassegue B, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002;105:1429–1435. [DOI] [PubMed] [Google Scholar]
- 14.Stary HC, Chandler AB, Dinsmore RE, et al. A Definition of Advanced Types of Atherosclerotic Lesions and a Histological Classification of Atherosclerosis : A Report From the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb Vasc Biol 1995;15:1512–1531. [DOI] [PubMed] [Google Scholar]
- 15.Sutliff RL, Hilenski LL, Amanso AM, et al. Polymerase Delta Interacting Protein 2 Sustains Vascular Structure and Function. Arteriosclerosis, Thrombosis, and Vascular Biology 2013;33:2154–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sata M, Maejima Y, Adachi F, et al. A mouse model of vascular injury that induces rapid onset of medial cell apoptosis followed by reproducible neointimal hyperplasia. J Mol Cell Cardiol 2000;32:2097–2104. [DOI] [PubMed] [Google Scholar]
- 17.Lee MY, San Martin A, Mehta PK, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol 2009;29:480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension 2007;49:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griendling KK, Taubman MB, Akers M, et al. Characterization of phosphatidylinositol-specific phospholipase C from cultured vascular smooth muscle cells. J Biol Chem 1991;266:15498–15504. [PubMed] [Google Scholar]
- 20.Ushio-Fukai M, Alexander RW, Akers M, et al. p38 Mitogen-activated Protein Kinase Is a Critical Component of the Redox-sensitive Signaling Pathways Activated by Angiotensin II: Role in Vascular Smooth Muscle Cell Hypertrophy Journal of Biological Chemistry 1998;273:15022–15029. [DOI] [PubMed] [Google Scholar]
- 21.Liu S, Xu SW, Blumbach K, et al. Expression of integrin beta1 by fibroblasts is required for tissue repair in vivo. J Cell Sci 2010;123:3674–3682. [DOI] [PubMed] [Google Scholar]
- 22.Szocs K, Lassegue B, Sorescu D, et al. Upregulation of Nox-based NAD(P)H oxidases in restenosis after carotid injury. Arterioscler Thromb Vasc Biol 2002;22:21–27. [DOI] [PubMed] [Google Scholar]
- 23.Deshpande NN, Sorescu D, Seshiah P, et al. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid Redox Signal 2002;4:845–854. [DOI] [PubMed] [Google Scholar]
- 24.Hernandes MS, Lassegue B, Griendling KK. Polymerase delta-interacting Protein 2: A Multifunctional Protein. J Cardiovasc Pharmacol 2017;69:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyle AN, Deshpande NN, Taniyama Y, et al. Poldip2, a Novel Regulator of Nox4 and Cytoskeletal Integrity in Vascular Smooth Muscle Cells. Circ Res 2009;105:249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burch PM, Heintz NH. Redox regulation of cell-cycle re-entry: cyclin D1 as a primary target for the mitogenic effects of reactive oxygen and nitrogen species. Antioxid Redox Signal 2005;7:741–751. [DOI] [PubMed] [Google Scholar]
- 27.Burhans WC, Heintz NH. The cell cycle is a redox cycle: Linking phase-specific targets to cell fate. Free Radical Biology and Medicine 2009;47:1282–1293. [DOI] [PubMed] [Google Scholar]
- 28.Datla SR, Peshavariya H, Dusting GJ, et al. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler Thromb Vasc Biol 2007;27:2319–2324. [DOI] [PubMed] [Google Scholar]
- 29.Green DE, Murphy TC, Kang B-Y, et al. The Nox4 Inhibitor GKT137831 Attenuates Hypoxia-Induced Pulmonary Vascular Cell Proliferation. American Journal of Respiratory Cell and Molecular Biology 2012;47:718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng D, Lv DD, Fang J. Insulin-like growth factor-I induces reactive oxygen species production and cell migration through Nox4 and Rac1 in vascular smooth muscle cells. Cardiovasc Res 2008;80:299–308. [DOI] [PubMed] [Google Scholar]
- 31.Sturrock A, Huecksteadt TP, Norman K, et al. Nox4 mediates TGF-beta1-induced retinoblastoma protein phosphorylation, proliferation, and hypertrophy in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007;292:L1543–1555. [DOI] [PubMed] [Google Scholar]
- 32.Salmeen A, Park BO, Meyer T. The NADPH oxidases NOX4 and DUOX2 regulate cell cycle entry via a p53-dependent pathway. Oncogene 2010;29:4473–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kleinschnitz C, Grund H, Wingler K, et al. Post-Stroke Inhibition of Induced NADPH Oxidase Type 4 Prevents Oxidative Stress and Neurodegeneration. PLoS Biology 2010;8:2109–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang MW, Barr E, Lu MM, et al. Adenovirus-mediated over-expression of the cyclin/cyclin-dependent kinase inhibitor, p21 inhibits vascular smooth muscle cell proliferation and neointima formation in the rat carotid artery model of balloon angioplasty. Journal of Clinical Investigation 1995;96:2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen D, Krasinski K, Sylvester A, et al. Downregulation of cyclin-dependent kinase 2 activity and cyclin A promoter activity in vascular smooth muscle cells by p27(KIP1), an inhibitor of neointima formation in the rat carotid artery. J Clin Invest 1997;99:2334–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granada JF, Ensenat D, Keswani AN, et al. Single Perivascular Delivery of Mitomycin C Stimulates p21 Expression and Inhibits Neointima Formation in Rat Arteries. Arteriosclerosis, Thrombosis, and Vascular Biology 2005;25:2343–2348. [DOI] [PubMed] [Google Scholar]
- 37.Akyürek LM, Boehm M, Olive M, et al. Deficiency of cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1 accelerates atherogenesis in apolipoprotein E-deficient mice. Biochemical and Biophysical Research Communications 2010;396:359–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cayrol C, Knibiehler M, Ducommun B. p21 binding to PCNA causes G1 and G2 cell cycle arrest in p53-deficient cells. Oncogene 1998;16:311–320. [DOI] [PubMed] [Google Scholar]
- 39.Ando T, Kawabe T, Ohara H, et al. Involvement of the Interaction between p21 and Proliferating Cell Nuclear Antigen for the Maintenance of G2/M Arrest after DNA Damage. Journal of Biological Chemistry 2001;276:42971–42977. [DOI] [PubMed] [Google Scholar]
- 40.Cazzalini O, Perucca P, Riva F, et al. p21CDKN1A does not interfere with loading of PCNA at DNA replication sites, but inhibits subsequent binding of DNA polymerase delta at the G1/S phase transition. Cell Cycle 2003;2:596–603. [PubMed] [Google Scholar]
- 41.Lu X, Tan C-K, Zhou J- Q, et al. Direct Interaction of Proliferating Cell Nuclear Antigen with the Small Subunit of DNA Polymerase δ. Journal of Biological Chemistry 2002;277:24340–24345. [DOI] [PubMed] [Google Scholar]
- 42.Bendjennat M, Boulaire J, Jascur T, et al. UV Irradiation Triggers Ubiquitin-Dependent Degradation of p21WAF1 to Promote DNA Repair. Cell 2003;114:599–610. [DOI] [PubMed] [Google Scholar]
- 43.Cayrol C, Ducommun B. Interaction with cyclin-dependent kinases and PCNA modulates proteasome-dependent degradation of p21. Oncogene 1998;17:2437–2444. [DOI] [PubMed] [Google Scholar]
- 44.Luo Y, Hurwitz J, Massague J. Cell-cycle inhibition by independent CDK and PCNA binding domains in p21Cip1. Nature 1995;375:159–161. [DOI] [PubMed] [Google Scholar]
- 45.Chang MW, Barr E, Lu MM, et al. Adenovirus-mediated over-expression of the cyclin/cyclin-dependent kinase inhibitor, p21 inhibits vascular smooth muscle cell proliferation and neointima formation in the rat carotid artery model of balloon angioplasty. The Journal of Clinical Investigation 1995;96:2260–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sutliff RL, Hilenski LL, Amanso AM, et al. Polymerase delta interacting protein 2 sustains vascular structure and function. Arterioscler Thromb Vasc Biol 2013;33:2154–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hernandes MS, Lassegue B, Hilenski LL, et al. Polymerase delta-interacting protein 2 deficiency protects against blood-brain barrier permeability in the ischemic brain. J Neuroinflammation 2018;15:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.