

Abstract
RNA-sequencing is a popular next-generation sequencing technique for assaying genome-wide gene expression profiles. Nonetheless, it is susceptible to biases that are introduced by sample handling prior gene expression measurements. Two of the most common methods for preserving samples in both field-based and laboratory conditions are submersion in RNAlater and flash freezing in liquid nitrogen. Flash freezing in liquid nitrogen can be impractical, particularly for field collections. RNAlater is a solution for stabilizing tissue for longer-term storage as it rapidly permeates tissue to protect cellular RNA. In this study, we assessed genome-wide expression patterns in 30 day old fry collected from the same brood at the same time point that were flash-frozen in liquid nitrogen and stored at −80°C or submerged and stored in RNAlater at room temperature, simulating conditions of fieldwork. We show that sample storage is a significant factor influencing observed differential gene expression. In particular, genes with elevated GC content exhibit higher observed expression levels in liquid nitrogen flash-freezing relative to RNAlater-storage. Further, genes with higher expression in RNAlater relative to liquid nitrogen experience disproportionate enrichment for functional categories, many of which are involved in RNA processing. This suggests that RNAlater may elicit a physiological response that has the potential to bias biological interpretations of expression studies. The biases introduced to observed gene expression arising from mimicking many field-based studies are substantial and should not be ignored.
Keywords: Liquid nitrogen, RNAlater, gene expression, gene length, GC proportion, technical variation
Introduction
High throughput sequencing technologies, such as RNA-sequencing methods, have revolutionized the quantification of genome-wide expression patterns across a broad range of fields in biological sciences (López-Maury et al. 2008; Wang et al. 2009). However, storage and RNA extraction methods prior to RNA-seq library preparation exert substantial impacts on biological studies, and often account for the majority of variation in a dataset if conditions and protocols are not identical across all samples (Todd et al. 2016). With the rise of RNAlater (Ambion, Invitrogen) as a popular storage method in field-based studies (De Smet et al. 2017; Wille et al. 2018), it is important to quantify if there are systematic biases in gene expression when samples are preserved in RNAlater versus flash-frozen in liquid nitrogen. In our literature review, however, we could find few direct comparisons of RNAseq data obtained from the most common field-preservation method RNAlater and the “gold standard” of flash freezing samples in liquid nitrogen (Alvarez et al. 2015; Wolf 2013) (but see(Cheviron et al. 2011; Choi et al. 2016)). Further, few studies examine whether a systematic bias due to gene characteristics exists for samples preserved in RNAlater (Bray et al. 2010).
Currently, two of the most common methods for RNA preservation and storage are flash freezing in liquid nitrogen and preservation in aqueous sulfate salt solutions, such as commercially available RNAlater. Flash freezing, usually through the use of immersing the sample in dry ice or liquid nitrogen, is the most preferred means of stabilizing tissue samples for downstream analysis (Wolf 2013). While preferred, it can often be difficult to access and transport dry ice or liquid nitrogen, particularly in field conditions (Mutter et al. 2004). Hence, in the past decade, it has become common practice, especially in field environments, to store RNAseq-destined samples in RNAlater, which minimizes the need to readily process samples or chill the tissue. RNAlater can rapidly permeate tissue to stabilize and protect RNA (Chowdary et al. 2006; Florell et al. 2001). Likewise, RNAlater-immersed samples can be stored safely at room temperature for a week and longer when stored at colder temperatures. Though, common practice in field conditions is to store samples in RNAlater for much longer than a week (Camacho-Sanchez et al. 2013; Gorokhova 2005). While the exact ingredients of commercial RNAlater are proprietary, the Material Safety Data Sheet lists inorganic salt as the major component and the homemade versions contain ammonium sulfate, sodium citrate, ethylenediaminetetraacetic acid (EDTA), and adjustment of pH using sulfuric acid.
In this study, we quantified the effects of storage condition on gene expression and examined differentially expressed genes for specific characteristics to assay for systematic bias. Individual, Mexican tetra fry (Astyanax mexicanus), were collected from the same brood and stored immediately in liquid nitrogen (N = 6) or RNAlater (N = 5). We specifically asked (1) Does storage condition affect patterns of differential gene expression and if so, (2) Are these effects on gene expression non-random, such that genes with certain features are differentially affected by storage condition? We found that a majority of the variation in gene expression was explained by storage condition. Likewise, we found that genes with higher GC content exhibited higher expression values in liquid nitrogen than RNAlater. Based on these findings, we conclude that RNAlater-storage at room temperature for extended periods of time may potentially bias biological conclusions of RNAseq experiments.
Methods
Sample Collection
Samples for the transcriptome analyses were collected from a surface population of Astyanax mexicanus (total of 8 parents) that had been reared in the Keene laboratory at Florida Atlantic University for multiple generations. Parental fish were derived from wild-caught Río Choy stocks originally collected by William Jeffery. To minimize variation outside of storage methods, all individuals were collected from the same clutch (fertilized on 2016-12-08). Fish were raised in standard conditions, and three days prior to experiment, fish were transferred into dishes with 12-21 fish per dish in a 14:10 light-dark cycle. These fish were a part of a larger experiment, so fish were kept in total darkness for 24 hours prior to sampling, and sampled at 16:00h (10pm). Five individuals were sampled with forceps and stored in RNAlater and six individuals were flash frozen in liquid nitrogen and stored at −80°C. Fry at 30 days post fertilization (dpf) were < 5mm long, transparent, and highly permeable. To mimic field conditions, RNAlater individuals were stored at room temperature for 17 days (Camacho-Sanchez et al. 2013; Kono et al. 2016). Procedures for all experiments performed were approved by the Institutional Animal Care and Use Committee at Florida Atlantic University (Protocol #A15-32).
RNA extraction, library preparation and sequencing
For RNA isolation, all individuals were processed within a week of each other (between 2017-01-19 and 2017-01-24), and RNAlater stored individuals were processed 17 days after initial storage (2017-01-24) (Table S1) with the same researcher performing all extractions. Whole organisms (< 30 mg of tissue) were homogenized using Fisherbrand pellet pestles and cordless motor (Fisher Scientific) in the lysate buffer RLT plus. Total RNA was extracted using the Qiagen RNAeasy Plus Mini Kit (Qiagen) and quantified using NanoDrop Spectrophotometer (Thermo Fisher Scientific), Ribogreen assay (Thermo Fisher Scientific), and Bioanalyzer RNA 6000 Nano assay (Agilent) to obtain RNA integrity numbers (RIN). All cDNA libraries were constructed at the University of Minnesota Genomics Center on the same day in the same batch. In brief, a total of 400 ng of RNA was used to isolated mRNA via oligo-dT purification. dsDNA was constructed from the mRNA by random-primed reverse transcription and second-strand cDNA synthesis. Strand-specific cDNA libraries were then constructed using TruSeq Nano Stranded RNA kit (Illumina), following manufacturer protocol. Library quality was assessed using Agilent DNA 1000 assay on a Bioanalyzer. To minimize batch effects, barcoded libraries were then pooled and sequenced across multiple lanes of an Illumina HiSeq 2500 to produce 125-bp paired-end reads at University of Minnesota Genomics Center (Table S1). All sequence data were deposited in the short read archive (Study Accession ID: RNAlater: SRX3446133, SRX3446136, SRX3446135, SRX3446155, SRX3446156; liquid nitrogen: SRS2736519, SRS2736520, SRS2736523, SRS2736524, SRS2736525,SRS2736526).
RNAseq quality check
The raw RNA-seq reads were quality checked using Fastqc (Andrews 2014) and trimmed to removed adapters using the program Trimmomatic version 0.33; (Bolger et al. 2014). Trimmed reads were mapped to the Astyanax mexicanus reference genome (version 1.0.2; GenBank Accession Number: GCA_000372685.1; (McGaugh et al. 2014)). Mapping was conducted using the splice-aware mapper STAR (Dobin et al. 2013), because it yielded the higher alignment percentage and quality compared to a similar mapping program (HISAT2, results not shown (Kim et al. 2015)). We used Stringtie (version 1.3.3d; (Pertea et al. 2016; Pertea et al. 2015)) to quantify number of reads mapped to each gene in the reference annotation set of the A. mexicanus genome, and used the python script provided with Stringtie (prepDE.py) to generate a gene counts matrix (Pertea et al. 2016). R (Team 2014) was used to compare RIN between liquid nitrogen and RNAlater treatments using a nonparametric Kruskal-Wallis test.
Variation in gene expression
To visualize changes in observed gene expression, we performed principal components analysis on a gene counts matrix. Genes with less than 100 counts across all samples were removed from the matrix because genes with low counts bias the differential expression tests (Love et al. 2014). The resulting counts were decomposed into a reduced dimensionality data set with the prcomp() function in R (Team 2014). To understand the extent storage method affected the ability to detect inter-individual variation, we calculated the coefficient of variation in gene expression for each gene under both storage conditions.
To identify genes that showed the largest difference in observed gene expression between storage conditions, we performed a differential expression analysis between samples flash frozen in liquid nitrogen (N = 6) and samples stored in RNAlater (N = 5) using DESeq2 (Love et al. 2014). DESeq2 normalizes expression counts for each sample and then fits a negative binomial model for counts for each gene. Samples with the same storage condition were treated as replicates, (i.e., the variation due to storage was assumed to be greater than variation among biological samples). This was confirmed in the PCA plot (Figure 1), where PC1 linearly separated samples based on their treatments. P-values for differential expression were adjusted based on the Benjamini-Hochberg algorithm, using a default false discovery rate of at most 0.05 (Love et al. 2014). Genes were labeled as differentially expressed if the Benjamini-Hochberg adjusted P-value was less than 0.05. Log2(RNAlater/liquid nitrogen) values were calculated with DESeq2 and exported for further analysis.
Figure 1:
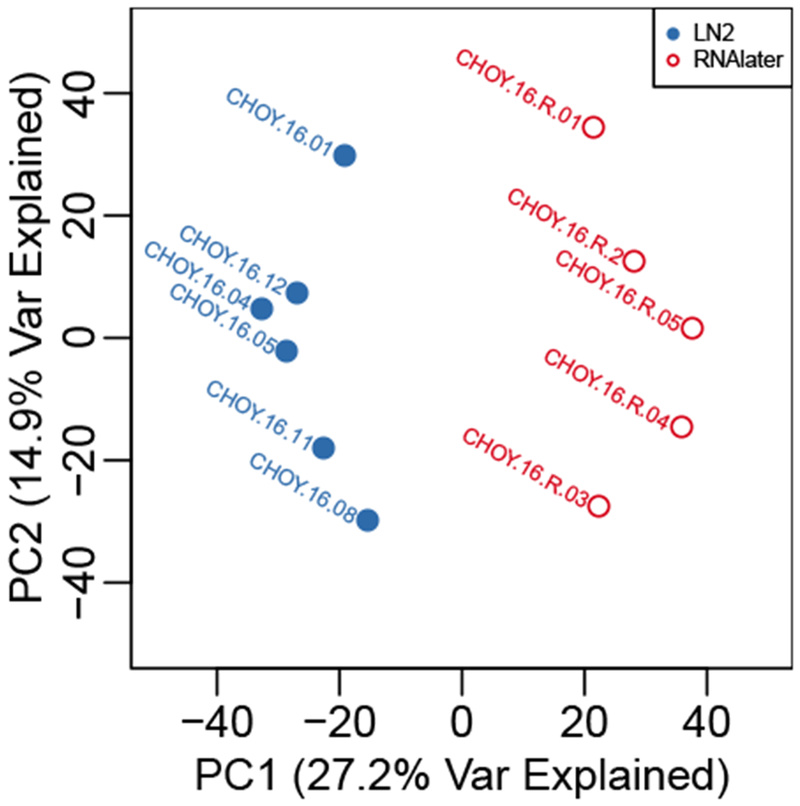
Principal components analysis plot showing PC1 and PC2 for each sample. RNAlater samples (red, open circles) are linearly separated from liquid nitrogen samples (blue, closed circles) by PC1.
Linear model to determine factors influencing differential expression
To identify the factors that contribute to the variability in gene expression between preservation methods, we fit a linear model of observed gene expression of all genes as a function of various genomic characteristics. We tested the contributions of mean expression level, annotated coding gene length, exon number, GC content, presence or absence of simple sequence repeats, and presence or absence of a homopolymer tract to differences in observed gene expression between preservation methods. We used the log2(RNAlater/liquid nitrogen) values from DESeq2 as the measure of change in observed gene expression and the mean of normalized counts across all samples as the mean expression level. The annotated gene length was calculated as the length of the coding region of the longest transcript from each gene. A simple sequence repeat was defined as two or more nucleotides repeated at least three times in tandem, and a homopolymer tract was defined as a single nucleotide repeated at least six times in tandem in the reference genome. Repeat presence or absence was based only on the reference genome sequence and were not scored to be polymorphic in the sample. Reference data was downloaded from Ensembl BioMart (Durinck et al. 2005; Durinck et al. 2009) and custom Python scripts were used to extract exon number and calculate coding length and GC content. Presence/absence of a simple sequence repeat, and presence/absence of a homopolymer repeat were scored with a custom Python script. All scripts used for analysis are available on our GitHub repository. Notably, the reference genome is a Pachón cavefish, and it is conceivable that some homopolymers and sequence repeats may not be identical in the surface fish.
We performed model selection on a series of linear models using likelihood ratio tests of nested models. The “full model” was as follows:
where Y is log2(RNAlater/liquid nitrogen) of expression between treatments, M is the the normalized mean expression value across all samples, G is GC content, L is coding gene length, E is the total number of exons in the gene, S is simple sequence repeats (SSR) presence/absence, and H is homopolymer presence/absence. GC content, coding length of the gene, and exon number were treated as continuous variables, and SSR presence and homopolymer presence were treated as categorical variables. Model selection proceeded by testing the contributions of the interaction terms to the variance explained and removing them if not significant. We tested the terms with the lowest non-significant t-values in the regression and removed them if they did not significantly improve model fit.
Annotation of differentially expressed genes
Because we expected most of the variation was going to be explained by a technical variable (i.e., preservation and storage), we did not expect biologically meaningful functional annotations. However, we conducted annotation analyses using differentially expressed genes at the 0.05 false discovery rate. Zebrafish (Danio rerio) genes that were one-to-one orthologs with Astyanax were used for a gene ontology (GO) term enrichment analysis. PANTHER analysis (Mi et al. 2016) (http://pantherdb.org/tools/compareToRefList.jsp) was run using only 1:1 orthologs between zebrafish and Asytanax with database current as of 2018-04-30. Within the PANTHER suite, we used PANTHER v13.1 overrepresentation tests (i.e., Fisher’s exact tests with FDR multiple test correction) with the Reactome v58, PANTHER proteins, GoSLIM, GO, and PANTHER Pathways. The target list was the zebrafish genes that were orthologous to differentially expressed Astyanax genes, and the background list was all zebrafish genes genome-wide. We confirmed these results by performing GO term enrichment with the GOrilla webserver (Eden et al. 2009) (http://cbl-gorilla.cs.technion.ac.il/), with a database current as of 2018-10-06. The target list was the zebrafish genes that were orthologous to differentially expressed Astyanax genes, and the background list was all 1-to1 orthologs between zebrafish and cavefish in our expression dataset.
Script Availability
Scripts to perform all data QC and processing are available at https://github.com/TomJKono/CaveFish_RNAlater
Results
Mapping statistics and annotation
RNA sequencing from whole, 30-days post fertilization individuals yielded a total of 108,874,500 reads for individuals stored in liquid nitrogen (mean = 18,145,750 ± stdev 1,938,410 per individual; N = 6) and 82,448,455 reads for individuals stored in RNAlater (mean = 16,489,691 ± stdev 1,890,519 per individual; N = 5) (Table 1). While all RIN scores from the extracted total RNA passed the threshold (> 7) (Table S1), to proceed into library preparation, RIN scores were significantly different between RNAlater and liquid nitrogen treatments (Kruskal-Wallis chi-squared = 7.6744, df = 1, p-value = 0.0056; RNAlater mean RIN = 8.60, liquid nitrogen mean RIN = 9.83).
Table 1:
Reported are the number of reads (after adapter trimming) used as input for the mapping software (STAR), number of reads that uniquely mapped to the reference genome, and the percent of reads that mapped to the reference genome. “Liquid N2” stands for liquid nitrogen.
| Sample Name | Treatment | Input reads | Uniquely mapped reads | % Mapped |
|---|---|---|---|---|
| CHOY-16-01 | Liquid N2 | 20,162,412 | 18,125,738 | 89.90% |
| CHOY-16-04 | Liquid N2 | 15,760,631 | 13,812,190 | 87.64% |
| CHOY-16-05 | Liquid N2 | 18,025,208 | 16,015,383 | 88.85% |
| CHOY-16-08 | Liquid N2 | 16,368,007 | 14,584,314 | 89.10% |
| CHOY-16-11 | Liquid N2 | 17,997,036 | 15,126,300 | 89.61% |
| CHOY-16-12 | Liquid N2 | 20,561,206 | 18,221,558 | 88.62% |
| CHOY-16-R-01 | RNAlater | 17,984,846 | 15,643,479 | 86.98% |
| CHOY-16-R-03 | RNAlater | 17,064,911 | 14,913,653 | 87.39% |
| CHOY-16-R-04 | RNAlater | 13,585,649 | 11,809,525 | 86.93% |
| CHOY-16-R-05 | RNAlater | 15,692,250 | 13,716,160 | 87.41% |
| CHOY-16-R-2 | RNAlater | 18,120,799 | 15,851,038 | 87.47% |
Total yield of reads and number of uniquely mapping reads were not significantly different between treatments (t = 1.4301; P = 0.1875). On average, samples mapped 88.17% of the reads to the Astyanax mexicanus genome (range: 86.93%-89.90%), with liquid nitrogen samples mapping on average 88.95% and RNAlater mapping 87.24%.
Filtering of the gene counts matrix to include only genes with ≥100 reads resulted in 15,515 genes being used for both clustering and differential expression analysis. Annotations were extracted from the Astyanax mexicanus annotation file (Astyanax_mexicanus.AstMex102.91.gtf). Distributions of raw and filtered gene expression counts are given in Figure S1.
The coefficients of variation between liquid nitrogen and RNAlater-preserved samples show a positive correlation (Figure S2, Kendall’s Tau, τ = 0.267, P < 2e-16), suggesting that the genes that are highly variable in the liquid nitrogen treatment are also highly variable in RNAlater storage. Thus, we do not expect that the storage methods significantly impact the ability to detect variation among individuals. However, there are slightly more genes with higher coefficients of variation in liquid nitrogen than in RNAlater (9,043 genes) than vice versa (6,472 genes), suggesting that RNAlater may reduce variation among individuals.
PCA and Differentially Expressed Genes
Principal components analysis showed that the major axis of differentiation among the samples was treatment (Figure 1). This corresponds to the first principal component, and explains 27.2% of the variation. Beyond the first principal component, the samples do not cluster into further discernable sub-groups, suggesting that the main axis of differentiation among these samples is their storage conditions (Figure 1).
A total of 2,708 (17.5%) genes were significantly differentially expressed between treatments at the 0.05 significance level (Figure 2). Of these, 1,635 exhibited significantly lower observed expression in RNAlater than liquid nitrogen, and 1,073 exhibited significantly higher observed expression in RNAlater than in liquid nitrogen.
Figure 2:
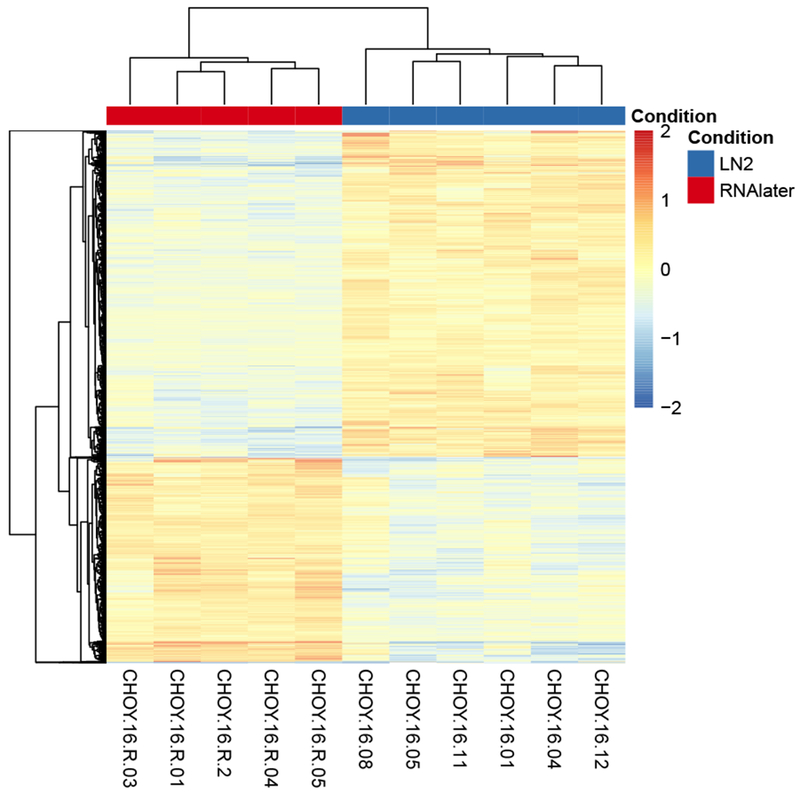
Clustering heatmap showing genes that are differentially expressed among RNAlater samples and liquid nitrogen samples. Gene expression values have been normalized by sample, then centred about 0 for each gene. This heatmap contains differentially expressed genes (after FDR correct with p < 0.05) including 1,073 genes that with higher expression values in the RNAlater treatment relative to the liquid nitrogen treatment, and 1,635 genes that exhibited lower expression values the RNAlater treatment.
Genomic Characters Contributing to Differential Expression
We identified four characteristics that contribute significantly to differential gene expression between treatments. Mean expression across samples, GC content, exon number, and interaction between GC content and SSR presence/absence were significant terms in the model (Table 2, Figure 3, Figure S3). GC content exhibited the largest coefficient. The coefficient for GC content is negative, suggesting that genes with higher GC content have a higher relative expression in liquid nitrogen than RNAlater (Figure S4). SSR presence also exhibited a non-significant association which resulted in higher relative expression in liquid nitrogen than RNAlater. Mean expression and exon number were significant, such that they exhibited a positive relationship with genes showing higher expression values in RNAlater (i.e., greater mean expression and more exons both related to higher expression in RNAlater). The small regression coefficients of these variable imply, however, that these factors have negligible impacts on differential gene expression observed between preservation methods. The interaction term between GC proportion and SSR presence/absence was also significant which we interpret to mean that SSR presence with high GC content is associated with higher expression in RNAlater. Despite the SSR term not being significant in the analysis of variance (Table 2), removing the term significantly impacted model fit.
Table 2:
Terms in the linear model that explain differences in expression between RNAlater storage and liquid nitrogen flash freezing and −80°C storage.
| Term | Sum Sq | Df | F-value | Estimate (SE) | P-value |
|---|---|---|---|---|---|
| Mean Expression | 1088.8 | 1 | 496.2719 | 0.155547 (0.007308) | <2e-16 |
| GC Proportion | 134.5 | 1 | 61.3069 | −5.277778 (1.358944) | 5.452e-15 |
| Exon Number | 584.9 | 1 | 266.6218 | 0.026825 (0.001670) | <2.2e-16 |
| SSR Presence | 0.2 | 1 | 0.0938 | −1.620474 (0.703584) | 0.75935 |
| GC Proportion : SSR Presence | 12.2 | 1 | 5.5619 | 3.269607 (1.386380) | 0.01838 |
Figure 3:
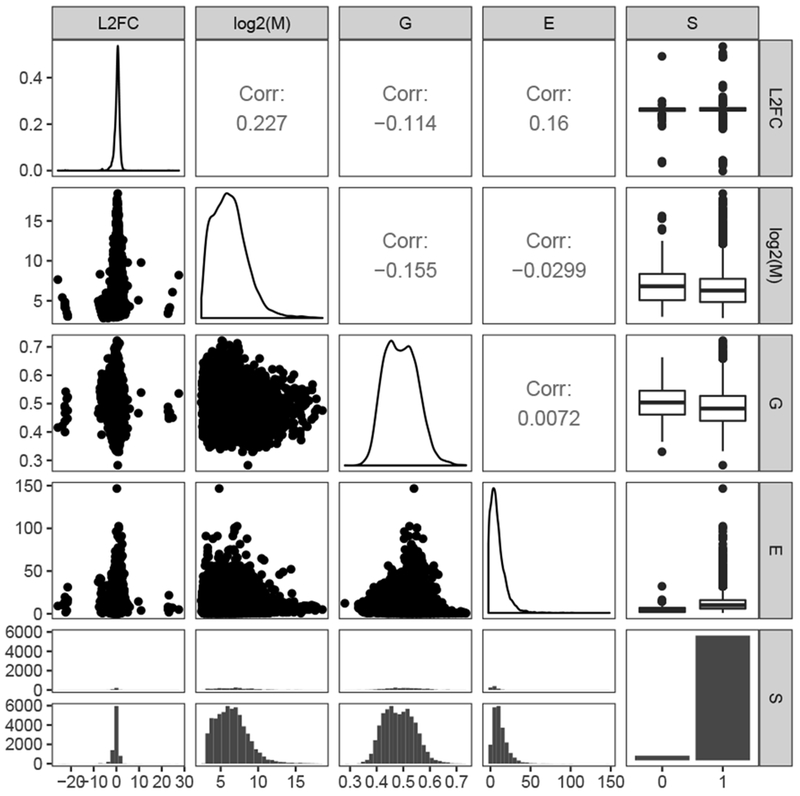
Relationships among the dependent variables retained in the best-fitting generalized linear model to explain the log2(RNAlater/liquid nitrogen) for each gene. L2FC: Log2(RNAlater/liquid nitrogen); log2(M): log2(mean expression across all samples); G: GC content; E: exon number; S: SSR presence (1) or absence (0). The panels along the diagonal show distributions of the individual explanatory variables with continuous variables displayed as density curves and categorical variables displayed as bar plots. Joint distributions or correlation coefficients are shown in the off-diagonal panels. Two continuous variables are shown as correlation coefficients and scatter plots. A continuous and categorical variable are shown as split boxplots and split histograms.
Annotation of differentially expressed genes
We expected little GO term enrichment as differences in gene expression would likely be due to differences in preservation techniques, not biological variation. The PANTHER suite annotation for genes that were significantly lower expressed in RNAlater compared to liquid nitrogen exhibited very few enriched functional categories (Supplemental Material). However, many categories were significantly enriched for genes that were more highly expressed in RNAlater than liquid nitrogen. The most enriched categories in Reactome pathways are involved in gene expression and processing of mRNA. Likewise, enriched PANTHER protein classes include RNA binding proteins, mRNA processing and splicing factors, and transcription factors. Enriched GO terms included RNA binding and RNA processing. Similar results were obtained with the GOrilla analyses (Figures S5 - S10). This consistent elevation of enrichment of functional categories for genes that are more abundant after an RNAlater treatment suggests that this treatment may be altering the physiology of the sample.
Discussion
Many sources contribute to variation in observed gene expression. Of these, most researchers are interested in assaying the variation that is due to a biological factor, such as genetic or physiological differences between samples. However, variation due to technical factors, such as noise in hybridization efficacy in microarray studies (Altman 2005) or noise in the number of reads that map to a particular gene in RNAseq studies are large sources of variability in observed gene expression, and can substantially influence results (Bryant et al. 2011; Marioni et al. 2008). For RNA-sequencing studies, the sources of technical variation are still being discovered, but can include many aspects of sample handling prior to actual measurement (McIntyre et al. 2011). Previous microarray studies have compared the sample handling procedures that were tested in our study, and have found no difference downstream, particularly in differential gene expression patterns (Dekairelle et al. 2007; Mutter et al. 2004). These studies, however, may not apply to the variance profile of RNA-sequencing studies (Romero et al. 2012).
Our results suggest that sample handling is an important factor in variation of observed gene expression. While the total percentages of reads mapped were generally similar between the two treatments, the treatments we tested had a significant impact on RNA quality. Our results suggest that preservation in RNAlater for extended periods of time, as opposed to flash freezing, non-randomly impacts gene expression values of over 20% of the transcriptome. Notably, other studies have found substantial RNA degradation for samples stored in RNAlater over extended periods, even when samples were stored at 4°C (Jones & Kennedy 2015) or −80°C (Riesgo et al. 2012b). In our study, samples that were stored in RNAlater exhibited lower average RIN scores than samples that were flash frozen in liquid nitrogen (Table S1), so our findings may be related to RNA degradation. Despite this, our RINs would be considered as acceptable for downstream applications, such as RNA-sequencing library preparation (Imbeaud et al. 2005).
Our results suggest genes with higher GC content, fewer exons, and lower expression are better preserved in liquid nitrogen. Conversely, our results suggest that genes with higher GC content, fewer exons, or lower mean expression may not be as well preserved with RNAlater (De Wit et al. 2012). The functional enrichment for genes exhibiting significantly higher observed expression in RNAlater than liquid nitrogen indicates that RNAlater may be substantially altering the physiology of the samples during fixation or that RNAlater preserves certain functional categories of genes better than liquid nitrogen. The latter seems unlikely as it is difficult to hypothesize a mechanism, and upregulation of genes associated with RNA metabolism and translation has been observed in other studies comparing RNAlater to liquid nitrogen preservation (Bray et al. 2010). Further, the converse does not appear to have extensive enrichment for certain functional categories (i.e., genes that experience presumably better preservation in liquid nitrogen than RNAlater often do not fall in particular functional categories).
Based on our results, we recommend that researchers use caution when comparing gene expression values derived from RNAseq datasets that may have variable storage conditions. This is especially important with the growth of genomics technologies and accessibility of public data in repositories such as the NCBI Sequence Read Archive. Many entries in these databases do not routinely report metadata such as storage conditions, posing a serious challenge for data utilization. Further, future work could expand on examination of storage in TRIzol (Fisher Scientific, Hampton, NH) as recent work indicates expression patterns might be substantially different from liquid nitrogen (Kono et al. 2016). Likewise, various taxonomic groups may be more susceptible to variation in storage conditions due to differences in tissue permeability or presence of secondary compounds (Riesgo et al. 2012a).
Several caveats are important in interpreting our study. While technical variation from storage condition is the dominant contributor to variation in our study, we acknowledge that biological variation also contributes to our observations. The samples in each storage condition are separate, whole individuals from the same clutch of fish. Fry at 30 dpf are too small to divide tissues equally into preservation treatments and obtain sufficient RNA quantity for RNAseq. Yet, even if a larger tissue sample was cut and divided, one might expect biological variation due to different cell populations. Additionally, juvenile fish tissue may interact with the RNAlater buffer in different ways from other organisms. However, other studies have demonstrated similar effects between RNAlater and flash freezing. For instance, between preservation methods over 5000 differentially regulated genes have been obtained from Arabidopsis thaliana tissue (c.f. (Kruse et al. 2017)). Though the Arabidopsis study did not assay systematic biases of particular gene attributes to preservation methods, many differentially regulated genes were related to osmotic stress, indicating a strong transcriptional response to RNAlater.
We also acknowledge that extraction batch was confounded with storage treatment. RNAlater samples were extracted in the same batch, while liquid nitrogen samples were extracted over several different batches (Table S1). The samples were part of a larger study, with 20 total RNA extraction batches of 169 liquid nitrogen samples and 1 extraction batch of the five RNAlater samples. Among the169 liquid nitrogen samples, lane of sequencing (which was randomized for RNAlater and liquid nitrogen samples in the this study, Table S1) and RNA extraction batch accounts for very little variation (Figure S11, Table S2). Though we cannot discount that the RNA extraction of the RNAlater-stored samples was different in some way and our results could potentially be due to RNA extraction batch, we view this as unlikely because the identical research, equipment, and reagents were used over a short window of time (e.g., 24 of the 169 liquid nitrogen samples were extracted on the same day as the RNAlater samples).
Finally, long-term storage temperature is confounded with liquid nitrogen and RNAlater treatments in our study and long-term storage temperature is known to drive RNA integrity (Gayral et al. 2013; Kono et al. 2016). Our goal was to replicate typical field experiments, where reliable refrigeration is not available for substantial amounts of time, and RNAlater is used as the predominant preservation method. Despite these caveats, our work demonstrates that differing preservation methods and storage conditions non-randomly impact gene expression, which may bias interpretation of results of RNA sequencing experiments. We look forward to future work that more thoroughly quantifies the impact on interpretation of biological signal derived solely from preservation methods (e.g. Bray et al. 2010).
Supplementary Material
Acknowledgements
We thank the University of Minnesota Genomics Center for their guidance and performing the cDNA library preparations and Illumina HiSeq 2500 sequencing. The authors acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing resources that contributed to the research results reported within this paper. URL: http://www.msi.umn.edu. Funding was supported by NIH (1R01GM127872-01 to SEM and ACK). CNP was supported by Grand Challenges in Biology Postdoctoral Program at University of Minnesota College of Biological Sciences. Institutional Animal Care and Use Committee at Florida Atlantic University (Protocol #A15-32).
Footnotes
Data accessibility
All reads are available in NCBI short read archive under accession numbers SRX3446133, SRX3446136, SRX3446135, SRX3446155, SRX3446156, SRS2736519, SRS2736520, SRS2736523, SRS2736524, SRS2736525, and SRS2736526. Scripts to perform all data handling and analysis tasks are available in a GitHub repository at https://github.com/TomJKono/CaveFish_RNAlater
References
- Altman N (2005) Replication, Variation and Normalisation in Microarray Experiments. Applied Bioinformatics 4, 33–44. [DOI] [PubMed] [Google Scholar]
- Alvarez M, Schrey AW, Richards CL (2015) Ten years of transcriptomics in wild populations: what have we learned about their ecology and evolution? Molecular ecology 24, 710–725. [DOI] [PubMed] [Google Scholar]
- Andrews S (2014) FastQC: a quality control tool for high throughput sequence data. Version 0.11. 2. Babraham Institute, Cambridge, UK: http://www.bioinformatics.babraham.ac.uk/projects/fastqc. [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray SE, Paulin FE, Fong SC, et al. (2010) Gene expression in colorectal neoplasia: modifications induced by tissue ischaemic time and tissue handling protocol. Histopathology 56, 240–250. [DOI] [PubMed] [Google Scholar]
- Bryant PA, Smyth GK, Robins-Browne R, Curtis N (2011) Technical variability is greater than biological variability in a microarray experiment but both are outweighed by changes induced by stimulation. PloS one 6, e19556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho-Sanchez M, Burraco P, Gomez-Mestre I, Leonard JA (2013) Preservation of RNA and DNA from mammal samples under field conditions. Molecular Ecology Resources 13, 663–673. [DOI] [PubMed] [Google Scholar]
- Cheviron ZA, Carling MD, Brumfield RT (2011) Effects of postmortem interval and preservation method on RNA isolated from field-preserved avian tissues. The Condor 113, 483–489. [Google Scholar]
- Choi S, Ray HE, Lai S-H, Alwood JS, Globus RK (2016) Preservation of multiple mammalian tissues to maximize science return from ground based and spaceflight experiments. PloS one 11, e0167391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdary D, Lathrop J, Skelton J, et al. (2006) Prognostic gene expression signatures can be measured in tissues collected in RNAlater preservative. The journal of molecular diagnostics 8, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet L, Hatjina F, Ioannidis P, et al. (2017) Stress indicator gene expression profiles, colony dynamics and tissue development of honey bees exposed to sub-lethal doses of imidacloprid in laboratory and field experiments. PloS one 12, e0171529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wit P, Pespeni MH, Ladner JT, et al. (2012) The simple fool’s guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis. Molecular Ecology Resources 12, 1058–1067. [DOI] [PubMed] [Google Scholar]
- Dekairelle A-F, Van der Vorst S, Tombal B, Gala J-L (2007) Preservation of RNA for functional analysis of separated alleles in yeast: comparison of snap-frozen and RNALater® solid tissue storage methods. Clinical Chemical Laboratory Medicine 45, 1283–1287. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, et al. (2005) BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature protocols 4, 1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florell SR, Coffin CM, Holden JA, et al. (2001) Preservation of RNA for functional genomic studies: a multidisciplinary tumor bank protocol. Modern pathology 14, 116. [DOI] [PubMed] [Google Scholar]
- Gayral P, Melo-Ferreira J, Glémin S, et al. (2013) Reference-free population genomics from next-generation transcriptome data and the vertebrate–invertebrate gap. PLoS genetics 9, e1003457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorokhova E (2005) Effects of preservation and storage of microcrustaceans in RNAlater on RNA and DNA degradation. Limnology and Oceanography: Methods 3, 143–148. [Google Scholar]
- Imbeaud S, Graudens E, Boulanger V, et al. (2005) Towards standardization of RNA quality assessment using user-independent classifiers of microcapillary electrophoresis traces. Nucleic Acids Research 33, e56–e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SP, Kennedy SW (2015) Feathers as a source of RNA for genomic studies in avian species. Ecotoxicology 24, 55–60. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nature methods 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono N, Nakamura H, Ito Y, Tomita M, Arakawa K (2016) Evaluation of the impact of RNA preservation methods of spiders for de novo transcriptome assembly. Molecular Ecology Resources 16, 662–672. [DOI] [PubMed] [Google Scholar]
- Kruse CP, Basu P, Luesse DR, Wyatt SE (2017) Transcriptome and proteome responses in RNAlater preserved tissue of Arabidopsis thaliana. PloS one 12, e0175943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Maury L, Marguerat S, Bähler J (2008) Tuning gene expression to changing environments: from rapid responses to evolutionary adaptation. Nature Reviews Genetics 9, 583. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaugh SE, Gross JB, Aken B, et al. (2014) The cavefish genome reveals candidate genes for eye loss. Nature communications 5, 5307–5307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre LM, Lopiano KK, Morse AM, et al. (2011) RNA-seq: technical variability and sampling. BMC genomics 12, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, et al. (2016) PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Research 45, D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutter GL, Zahrieh D, Liu C, et al. (2004) Comparison of frozen and RNALater solid tissue storage methods for use in RNA expression microarrays. BMC genomics 5, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL (2016) Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature protocols 11, 1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, et al. (2015) StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nature biotechnology 33, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesgo A, Perez-Porro AR, Carmona S, Leys SP, Giribet G (2012a) Optimization of preservation and storage time of sponge tissues to obtain quality mRNA for next-generation sequencing. Molecular Ecology Resources 12, 312–322. [DOI] [PubMed] [Google Scholar]
- Riesgo A, PÉREZ-PORRO AR, Carmona S, Leys SP, Giribet G (2012b) Optimization of preservation and storage time of sponge tissues to obtain quality mRNA for next-generation sequencing. Molecular Ecology Resources 12, 312–322. [DOI] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y (2012) Comparative studies of gene expression and the evolution of gene regulation. Nature Reviews Genetics 13, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC (2014) R: A language and environment for statistical computing. In: R Foundation for Statistical Computing. [Google Scholar]
- Todd EV, Black MA, Gemmell NJ (2016) The power and promise of RNA-seq in ecology and evolution. Molecular ecology 25, 1224–1241. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nature Reviews Genetics 10, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille M, Yin H, Lundkvist Å, et al. (2018) RNAlater® is a viable storage option for avian influenza sampling in logistically challenging conditions. Journal of virological methods 252, 32–36. [DOI] [PubMed] [Google Scholar]
- Wolf JB (2013) Principles of transcriptome analysis and gene expression quantification: an RNA-seq tutorial. Molecular Ecology Resources 13, 559–572. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.