

Abstract
Objective:
Recombinant inbred (RI) mouse strains generated from a LG/J and SM/J intercross offer a unique resource to study complex genetic traits such as osteoarthritis (OA). We determined the susceptibility of 14 strains to various phenotypes characteristic of post-traumatic OA (PTOA). We hypothesize that phenotypic variability is associated with genetic variability.
Methods:
10-week old male mice underwent destabilization of the medial meniscus (DMM) to induce PTOA. Mice were sacrificed at 8-week post-surgery and knee joints were processed for histology to score cartilage degeneration and synovitis. Micro-CT was used to analyze trabecular bone parameters including subchondral bone plate thickness and synovial ectopic calcification. Gene expression in the knees was assessed by QuantiGene Plex assay.
Results:
Broad-sense heritability ranged from 0.18–0.58 suggesting that the surgical responses are moderately heritable. LGXSM-33, LGXSM-5, LGXSM-46, and SM/J were highly susceptible to OA while LGXSM-131b, LGXSM-163, LGXSM-35, LGXSM-128a, and LG/J were relatively OA resistant. Our study allows, for the first time, the measurement of genetic correlations of phenotypes that are characteristic of PTOA: cartilage degeneration was significantly positively associated with synovitis (r = 0.83–0.91) and subchondral bone plate thickness was negatively correlated with ectopic calcification (r = −0.59). Moreover, we show that 40 of the 78 genes tested were significantly correlated with various (OA) phenotypes. Unlike the OA phenotypes, there was no evidence for genetic variation in differences in gene expression levels in DMM and SHAM knees.
Conclusion:
For these mouse strains, various characteristics of PTOA varied with genetic composition demonstrating a genetic basis for PTOA susceptibility. The heritability of PTOA was established. Phenotypes exhibited various degrees of correlations: (1) cartilage degeneration is positively correlated with synovitis, but (2) not with the formation of ectopic calcification. Further investigation of the regions of genome containing genes implicated in OA and expression data will be useful for studying mechanisms of OA and identifying therapeutic targets.
Keywords: Recombinant inbred mice, Genetics, Synovitis, Ectopic calcification, Gene expression
Osteoarthritis (OA) is a complex genetic disease with poorly defined etiopathogenesis. As a complex polygenic disease, OA has a considerable hereditary component, with a genetic contribution accounting for 39–78% of OA variation (1). Numerous other risk factors, such as aging, obesity, female sex and joint trauma influence OA development and progression (2). At least 12% of the overall prevalence of OA is attributable to post-traumatic OA (PTOA) (3) that stems from intra-articular injuries such as cartilage damage and ligament and meniscus tears over a time-course of 10–15 years (4). Clinical evidence suggests that 50% of individuals with ligament and/or meniscus tears are protected from developing PTOA while 50% are susceptible to it (5). This suggests that genetics is one of possible variations in susceptibility to PTOA as there could also be other presently unknown variables at the time of or subsequent to joint injury such as intra-articular bleeding and inflammation etc.
Major difficulties affect genetic studies of OA in human populations. First, OA is a complex disease affected by many genes of small effect so that large numbers of participants are required for genome-wide association studies (GWAS) with sufficient power to detect significant effects. Second, OA patients are not often compared to a similar number of control, non-OA patients, thus protective alleles may be missed. Third, the history of trauma is unreliable, therefore patients with prior trauma are included with patients without trauma. Recent progress has been made using large databases to identify genetic loci for high body mass index as potentially causal for OA (6). However, in spite of the progress and availability of genome screening tools, specific genetic polymorphisms causing and/or protecting individuals from OA remain elusive (1). Therefore, we began to elucidate alleles that are involved in PTOA susceptibility by turning to a population genetics approach in recombinant inbred (RI) mouse crosses.
Individual mice within RI strains are genetically identical but each strain is a different recombination of the parental genotypes, therefore any between-strain phenotypic differences would indicate potentially mappable genetic differences. The within-strain variation is “environmental”. However, it is random, unspecified environment that is responsible for that variation and it is not possible to state what environmental factors affecting phenotypic variations are involved. Thus, unraveling the genetic contribution to the susceptibility or protection from OA in mice may improve our understanding of OA pathogenesis and help identify patients who are at risk for developing OA as well as provide personalized treatment options. While mice are not the same as humans, they provide an excellent and much used experimental system, especially for obtaining data not available in humans. While specific murine and human populations are not likely to segregate at all the same loci, if a gene affects traumatic OA response in humans, it will also likely do so in mice. Murine studies can be used to identify genes participating in the human disease process, even if that gene is not a cause of disease in any specific human population.
Genetic variability in response to injury and PTOA has been described in a number of mouse strains. MRL/MpJ, a super-healer mouse, along with its close relative LG/J possesses remarkable ability to regenerate a wide range of body tissues (7–9). In contrast, C57BL/6J and SM/J mouse strains bear much less healing potential. An RI cross generated from LG/J (healer) and SM/J (non-healer) strains shows variation in a number of phenotypes (ear wound, cartilage healing, and OA) depending on the genetic contribution derived from the parental strains (10–13). Unlike unrelated mouse strains, RI strains can be utilized to measure and map the genetic variations that confer OA resistance such as in the MRL/MpJ and LG/J mouse strains. Based on early findings on non-related strains and two RI strains, we expanded our research using a population genetics approach to screen additional RI strains for their susceptibility to PTOA by investigating a number of phenotypes often associated with the development of OA, including cartilage degeneration, synovitis and bone changes.
MATERIALS AND METHODS
Mice
Washington University Animal Care and Use Committee approved this study. Parent (LG/J, SM/J) and their intercross mice were housed in individually ventilated cages under pathogen-free conditions with ad libitum access to standard mouse chow and water and a 12:12 hour light:dark cycle. All mice were housed in groups in cages (5 mice/cage). As some sexual dimorphism has been reported for these strains (10), only male mice were used. In total, we included 3–10 mice from each strain as shown in Supplemental Table 1.
DMM surgery
PTOA was induced in 10-wk old mice by destabilization of the medial meniscus (DMM surgery) as described previously (13, 14). Briefly, mice were anesthetized using an intra-peritoneal injection of ketamine (100 mg/kg), xylazine (20 mg/kg), and acepromazine (10 mg/kg). No other medication was given to the mice during DMM surgery. The joint capsule immediately medial to the patellar tendon was opened and the anterior attachment of medial meniscotibial ligament (MMTL) was transected with micro-surgical tools. Joints were closed with 6–0 absorbable polypropylene sutures and skin was closed by Vetclose skin-glue. The left knee−in which capsular incision was made but MMTL was left intact−served as a SHAM. Mice were sacrificed 8-wk after surgery by CO2 asphyxiation. Mice from each strain were evaluated at the same time over the course of the experiments and all surgeries were done by the same person. Although factors such as time or season may influence the potential variation on PTOA, it was difficult to completely match these conditions because our RI strains were obtained by repeated brother-sister mating. Therefore, all surgeries were performed in the well-controlled animal room to keep the same conditions during surgery and all mice were housed in an animal facility with controlled lighting hours and temperature before and after surgery, we think we could exclude such seasonal factors successfully.
Knee joints were decalcified using 12% formic acid and then embedded in paraffin. Eight coronal sections (5 µm thick) were taken from each joint at eight levels separated by 80 µm intervals. From each level we stained three sections with toluidine blue to perform scoring for cartilage degeneration and synovitis (15). In each mouse, the same number and depth of sections were evaluated. Briefly, slides were immersed for 5 min in 0.04% toluidine blue solution prepared in 0.1 M sodium acetate (pH 4.0). Following three-time washing with running tap water, twice with absolute ethanol, and twice with xylene, slides were cover-slipped. Cartilage damage was scored using the Osteoarthritis Research Society International (OARSI) scoring system (16, 17) to semi-quantitatively evaluate cartilage damage. Cartilage damage score was measured in all four quadrants of the joint (lateral and medial femoral condyles and lateral and medial tibial plateaus) at all sectioned levels (16). Summed OARSI scores representing whole joint changes and maximum OARSI scores representing the highest score within all sectioned levels of the knee (13, 15, 16) were recorded. Synovial pathology (synovitis) was measured from all four quadrants by examining the enlargement of the synovial lining cell layer and density of the cells using a previously established scoring system (15, 18). Two scorers blinded to sample identity scored all sections from all strains independently and sections from different RI strains were randomized to make sure differences in PTOA were not due systematic interobserver differences in scoring.
Micro-CT
Prior to decalcification, joints were scanned using a vivaCT 40 in vivo micro-CT scanner (SCANCO Medical) to analyze the 3-dimensional structure of bone using several parameters as reported previously (13, 15, 19, 20). The following ASBMR morphometric parameters of the tibial bone were assessed for trabecular compartments: bone volume fraction (BV/TV), trabecular number (Tb.N), thickness (Tb.Th), and separation (Tb.Sp), structure model index (SMI), and connectivity density index (CDI). Subchondral bone plate thickness was outlined separately from trabecular bone at the tibial side using contouring methods on the cortical regions as described elsewhere (15).
Ectopic calcification
We have recently shown that mice develop ectopic calcifications (i.e. calcified nodules in and around the synovium) after DMM (21) or mechanical loading (18, 22). Here, we quantified these nodules after joints were scanned by micro-CT scanner as described before (22). Briefly, after visualization by generating 3-dimensional images in OsiriX imaging software v.7 (OsiriX), these nodules were counted by including all mineralized areas in and around the joint space except for patella, anterior horns of the menisci and sesamoid bones.
Gene expression
We examined the expression pattern of 78 genes (Panel #21545; Supplemental Table 2) considered candidates because of previous QTL mapping of OA phenotypes in the LGXSM advanced intercross. Gene expression in the formalin-fixed paraffin-embedded sections was measured as described previously using an Affymetrix QuantiGene Plex assay (21, 23). Briefly, tissue homogenates were prepared by macro-dissecting the whole joint area (subchondral bone, cartilage, meniscus, growth plate, joint capsule, and synovium) using the Affymetrix QuantiGene Plex sample processing kit (Panomics Inc.). We used 25 sections per knee from 3–5 mice per strain. The mRNA expression level of each candidate gene in the sample of mouse strains was normalized by regressing log-transformed raw mRNA values against those of the Gapdh and Hprt1 housekeeping genes as independent variables to control for variation in tissue amounts. Residuals from the regression were averaged for each individual mouse’s pair of knees. For each mouse, the residual expression level of the SHAM knee was then subtracted from that of the DMM-operated knee to obtain a relative mRNA expression level reflecting response to medial meniscus destabilization. Details for generating gene expression data are in Rai et al.(21).
Statistical analyses.
Data are presented as mean ± 95% confidence interval unless indicated otherwise. With the design that we have used, we can detect effects of 0.865 within-group standard deviations at 80% power and most of our tests were significant, so this sample size proved sufficient.
Analysis of phenotypes:
Each individual was scored for a DMM-operated knee and a contralateral SHAM-operated knee. Hence, the basic unit of analysis is the individual, for each individual and trait we analyze (DMM minus SHAM). By pairing the SHAM and DMM knees from the same individuals we realize increased statistical power to detect the effects of DMM surgery and the effect of genotype (RI strain) on response to DMM surgery. There were 14 strains included in the analysis represented by approximately 4 individuals each. Univariate tests utilized the F-statistic while multivariate tests utilized the F-statistic associated with Wilk’s λ.
We first assessed the effects of DMM surgery by testing whether the (DMM-SHAM) difference is significantly greater or less than zero. The direction of the one-tailed test was based on expectations for the effects of DMM surgery. We expected positive values for maximum OARSI, summed OARSI, and synovitis scores, subchondral bone plate thickness, and number of ectopic calcifications. Trabecular architecture traits should show decreased bone in the DMM knees, with negative differences for BV/TV, CDI, Tb.N, and Tb.Th and positive differences for structural model index (SMI) and Tb.Sp. ANOVA and paired t-tests assume that the data is normally distributed. We tested for normality with the Lillefors test. All pairwise differences between SHAM and DMM knees fit a normal distribution except for summed OARSI and synovitis scores, which were slightly skewed left. For these two traits, we also tested for statistical significance with non-parametric rank order tests, Mann-Whitney U tests for differences in means and Kruskal-Wallis for ANOVA. In all instances, differences reported as significant using the parametric paired t-tests and ANOVA were confirmed at the 0.05 level with the appropriate non-parametric test.
The effects of genotype were tested with a series of MANOVAs or ANOVAs where appropriate with the difference between DMM and SHAM surgeries as the dependent variable and RI strain as the independent variable. RI strain is a random effect so we estimated the genetic variance [V(G); between strains], the environmental variance [V(E); residual variance within strains], and broad-sense Heritability (H2) [V(G)/(V(G) + V(E))]. The genetic variance is the magnitude of differences between mice due to differences in their genes. The environmental variance is the magnitude of differences between mice due to all other causes of differences between animals. Furthermore, within-strain variance is the residual variance in the analysis so it includes all kinds of factors, such as measurement error, variations in the surgical preps, etc. If we had a measured environment, it could be included. RI strains are especially good for measuring specific environmental effects as we get identical genotypes expressed under different conditions (25, 28). The broad-sense heritability is the proportion of all differences between animals attributed to differences in their genes. It is standardized, varying between 0.0 and 1.0 to compare between traits and populations with different variances. A higher heritability indicates that most of the differences between animals in the trait specified are due to genetic differences than for traits with lower heritability. These estimates are specific to the LGXSM cross population (cross different substrains of B6 from JAX (C57BL/6J) and NIH (C57BL/6N) and one would be likely to observe much lower heritabilities because the substrains have many, many fewer genetic polymorphisms but the same amount of “environmental” differences). They are also affected by differences in environments between labs. When data from multiple labs are combined, the greater environmental variation leads to lower heritability. For heritability, Type 1 error was controlled by using multivariate tests (MANOVA) of the null hypothesis. This test accounts for inter-trait correlations. There were three such tests so the Bonferroni correction places the 5% significance level at 0.016. The probabilities of all three tests are lower than this threshold.
Genetic correlations were calculated from the between-strain and within-strain variance/covariance matrices for trait sets analyzed as MANOVAs (maximum OARSI, summed OARSI, and synovitis scores), (BV/TV, CDI, SMI, Tb.N, Tb.Th, Tb.Sp), and (subchondral bone plate thickness, number of ectopic calcifications). For the 11 paired t-tests the Bonferroni 5% significance level is 0.0045. All significant differences reported here are lower than the threshold. Genetic correlations between these sets were calculated as correlations of strain means. The number of dependent variables included in the MANOVA is limited to five by the sample sizes. Correlations between strain means within these sets are very similar to those obtained through MANOVA with a slight negative bias for the correlations of strain means. Significance tests were congruent between the two methods.
Analysis of gene expression:
Gene expression level differences between DMM and SHAM knees (DMM-SHAM) were analyzed as for the phenotypes except that a 2-tailed probability was obtained as we did not have a priori expectations for the direction of differences between the DMM and SHAM knees. Heritabilities were also calculated as above but none of the differences in gene expression between paired DMM and SHAM knees were significantly heritable. Sample sizes for these tests were 45. It was not possible to obtain reliable estimates of genetic correlations between differences in gene expression levels and phenotypes from either MANOVA or by correlating strain means because of the lack of heritability for the differences in gene expression levels. Therefore, phenotypic correlations were analyzed. Because of missing data for some of the phenotypes for animals in which gene expression was analyzed, the sample size for these correlations was 39.
The 78 genes provided a −33 degree-of-freedom in a multivariate test. There were too many genes for our sample size but this is almost universal for gene expression studies. The fact is that heritability is low for gene expression traits, lower than for the skeletal phenotypes. We had sufficient power for the skeletal traits but not enough for low heritability traits (e.g. gene expression traits). Heritabilities had to be greater than 20% to be statistically significant given our design. These heritability estimates for gene expression levels are low, whether they are statistically significant or not. If the heritability is that low, genetic differences make only a minor contribution to differences between individuals.
RESULTS
Genetic variation in the response to surgery
DMM-operated knees had increased cartilage degeneration (as evidenced by loss of staining, cartilage fibrillation etc.) than SHAM-operated knees, with changes mainly confined to medial compartment (Fig. 1A-C). Synovitis was measured from all four quadrants of the knee joint (Fig. 1D). We observed increased synovitis in DMM-operated knees compared to SHAM-operated knees. Representative images of synovitis are shown in Fig. 1E. Synovial ectopic calcification was present in DMM-operated knees and less so in the SHAM-operated knees (Fig. 1F-G).
Figure 1:

Strain differences in cartilage degeneration, synovitis, and ectopic calcification. Representative images of the area we investigated show cartilage degeneration on the medial tibial plateau in both A) DMM-operated and B) SHAM-operated knees. Representative images (C) showing cartilage degeneration on the medial tibial plateau in various mouse strains (arrows show cartilage degeneration). (D) Synovitis was measured from all four quadrants in a blinded fashion by examining the enlargement of the synovial lining cell layer and density of the cells using previously established scoring system. (E) Representative images showing synovitis among RI strains (arrows show synovitis). Representative micro-CT images of both DMM-operated and SHAM-operated knee joint (F) without ectopic calcification and with (G) ectopic calcification from anterior and posterior view are shown. Scale bar = 100 µm; MFC = medial femoral condyle; LFC = lateral femoral condyle; MTP = medial tibial plateau; LTP = lateral tibial plateau; LM = lateral meniscus; MM = medial meniscus.
Nearly all traits tested were significantly different from zero in the expected direction and survived Bonferroni correction (threshold = 0.05/11 = 0.004). Cartilage damage, synovitis, and trabecular bone measures varied in the expected directions. Only trabecular thickness (Tb.Th) and subchondral bone plate thickness failed to show a significant difference between DMM and SHAM surgeries (Supplemental Table 3).
We observed that RI strains LGXSM-33, LGXSM-5, LGXSM-46, and SM/J were all highly susceptible to OA as a result of DMM surgery while LGXSM-131b, LGXSM-163, LGXSM-35, LGXSM-128a, and LG/J were relatively OA resistant (Fig. 2A-B). The two parent strains had opposite responses to DMM surgery; however, there was transgressive segregation in that the most extreme susceptible and resistant strains are more extreme than the parent strains. This suggests that each parent strain contains a mixture of susceptible and resistant alleles. The parent strains did not differ in synovitis scores in response to DMM surgery but LGXSM-46 and LGXSM-5 were relatively susceptible while LGXSM-131b and LGXSM-163 were relatively resistant to synovitis in response to DMM surgery (Fig. 2C). For subchondral bone plate thickness, LGXSM-33 and LGXSM-122b were relatively susceptible to DMM surgery while LGXSM-6 and LGXSM-19 were resistant to these effects (Fig. 2D). With regard to the amount of trabecular bone, RI strains LGXSM-5, LGXSM-6, LGXSM-19, and LGXSM-128a were relatively resistant to bone loss while LGXSM-135, LGXSM-131, LGXSM-46 and SM/J were relatively sensitive to bone loss due to the DMM surgery.
Figure 2:
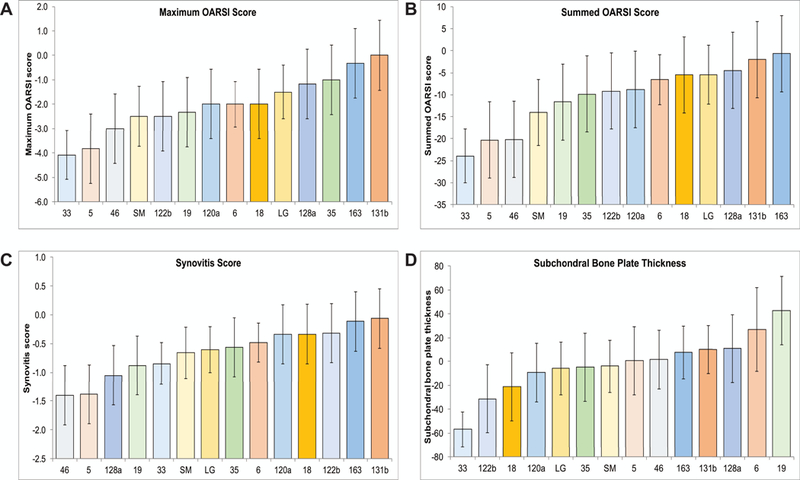
The strain mean values for maximum OARSI, summed OARSI, synovitis score, and subchondral bone plate thickness. RI strains LGXSM-33, LGXSM-5, LGXSM-46, and SM/J were all highly susceptible to OA as a result of DMM surgery while LGXSM-131b, LGXSM-163, LGXSM-35, LGXSM-128a, and LG/J are relatively OA resistant (A-B. LG/J and SM/J did not differ in synovitis score in response to DMM surgery but LGXSM-46 and LGXSM-5 were relatively susceptible while LGXSM-131b and LGXSM-163 were relatively resistant to synovitis in response to DMM surgery (C). For subchondral bone plate thickness, LGXSM-33 and LGXSM-122b were relatively susceptible to DMM surgery while LGXSM-6 and LGXSM-19 were resistant to these effects (D). Each bar represents a strain (x-axis) with mean and 95% confidence interval (y-axis).
Broad-sense heritabilities
Broad-sense H2 ranged from 0.18 to 0.58 suggesting that surgical responses are moderately heritable. Only CDI and the number of ectopic calcifications failed to be statistically significantly heritable at the 5% level, although both traits did show marginally significant results (P < 0.10). These results showed that there are genetic differences segregating in the LG/J by SM/J cross that affect response to PTOA (Table 1).
Table 1:
Genetic variances and broad-sense heritabilities
| Trait | V(G) | V(E) | H2 | P |
|---|---|---|---|---|
| Maximum OARSI score | 0.962 | 1.589 | 0.38 | 0.002 |
| Summed OARSI score | 38.917 | 58.765 | 0.40 | 0.001 |
| Synovitis score | 0.094 | 0.207 | 0.31 | 0.009 |
| Multivariate | 0.003 | |||
| BV/TV | 0.001 | 0.001 | 0.58 | 7.2 × 10−6 |
| CDI | 163.620 | 607.321 | 0.21 | 0.0518 |
| SMI | 0.174 | 0.165 | 0.51 | 7.6 × 10−5 |
| Tb.N | 0.076 | 0.105 | 0.42 | 0.001 |
| Tb.Th | 0.00001 | 0.00001 | 0.37 | 0.003 |
| Tb.Sp | 0.0002 | 0.0001 | 0.57 | 1.0 × 10−5 |
| Multivariate | 2.2 × 10−8 | |||
| Subchondral bone plate thickness | 296.363 | 636.668 | 0.32 | 0.008 |
| # of ectopic calcifications | 1.733 | 7.985 | 0.18 | 0.078 |
| Multivariate | 0.004 | |||
V(G) = genetic variance (between strains); V(E) = the environmental variance (within strains); H2 = broad-sense heritability.
Genetic and phenotypic correlation
Genetic correlations between phenotypes are strong and nearly universally significant within groups analyzed by MANOVA. The average R2 within the OARSI and synovitis group is 0.83, within the trabecular architecture group it is 0.69, and between subchondral bone plate thickness and the number of ectopic calcifications it is 0.35. Only one of these correlations, between Tb.Th and SMI, is not significant at the 5% level. In sharp contrast, the average R2 for all of the between group values is 0.10 with only the correlations between subchondral bone plate thickness and CDI and between number of ectopic calcifications and SMI are significantly different from zero at the 5% level. Raw correlation coefficients are shown in Table 2. In the description above, we used average R-squared values, rather than average raw correlation values, to prevent natural positive correlations, like BV/TV with CDI, and negative relationships, like BV/TV with SMI, from cancelling out when calculating a group mean.
Table 2:
Genetic correlations between (SHAM-DMM) values for various phenotypes
| Trait | MaxOARSI | SumOARSI | SynScore | BV/TV | CDI | SMI | Tb.N | Tb.Th | Tb.Sp | SCB | #Ectop |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MaxOARSI | 1.00 | ||||||||||
| SumOARSI | 0.98 | 1.00 | |||||||||
| SynScore | 0.83 | 0.92 | 1.00 | ||||||||
| BV/TV | −0.11 | 0.07 | −0.35 | 1.00 | |||||||
| CDI | −0.13 | −0.02 | −0.48 | 0.91 | 1.00 | ||||||
| SMI | 0.17 | −0.03 | 0.23 | −0.93 | −0.85 | 1.00 | |||||
| Tb.N | −0.19 | −0.03 | −0.30 | 0.99 | 0.72 | −0.99 | 1.00 | ||||
| Tb.Th | 0.04 | 0.16 | −0.12 | 0.73 | 0.76 | −0.39 | 0.59 | 1.00 | |||
| Tb.Sp | 0.24 | 0.13 | 0.39 | −0.97 | −0.83 | 0.99 | −0.99 | −0.59 | 1.00 | ||
| SCB | 0.41 | 0.38 | −0.08 | 0.47 | 0.53 | −0.42 | 0.44 | 0.26 | −0.42 | 1.00 | |
| #Ectop | 0.30 | 0.10 | 0.31 | −0.49 | −0.19 | 0.55 | −0.49 | −0.24 | 0.37 | −0.59 | 1.00 |
Correlations between sets of traits are between strain means. Bold-faced values are significantly different from 0.0 at a 5% level. MaxOARSI = maximum OARSI score; SumOARSI = summed OARSI score; SynScore = synovitis score; BV/TV = bone volumes total volume fraction; CDI = connectivity density index; SMI = structural model index; Tb.N = trabecular number; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation; SCB = subchondral bone plate thickness; #Ectop = # of ectopic calcifications.
Gene expression analysis
In order to examine strain-specific gene expression in response to DMM surgery, we used material from the histology slides to probe a panel of 78 candidate genes compiled previously for our work on cartilage repair (23) and ectopic calcification (21). The list of candidate genes is in Supplemental Table 2. Seventeen of the 78 gene expression traits were significantly different between paired DMM and SHAM knees (Table 3). Sixteen of these showed higher expression levels in the DMM surgically altered knees (Copz2, Col2a1, Col1a1, Timp3, Acan, Wnt16, Igf1, Tgfb2, Comp, Sgce, Bcl3, Ank, Bmp2, Postn, Myo1e, and Aff3) while one gene, Pcna, showed significantly lower expression levels in DMM knees. Only one gene, Comp, was significantly different between DMM and SHAM knees at the Bonferroni-corrected 5% level (P<0.000641) but the 17 genes significant at the pointwise 0.05 level were much higher than the number expected by chance (N=4).
Table 3:
Gene expression differences between DMM and paired SHAM knees
| Gene | Mean | 95% confidence interval |
2- tailed P value |
Gene | Mean | 95% confidence interval |
2-tailed P value |
||
|---|---|---|---|---|---|---|---|---|---|
|
Lower
limit |
Upper
limit |
Lower
limit |
Upper
limit |
||||||
| Copz2 | 17.43 | 4.66 | 30.19 | 0.0104 | Fam63b | −13.73 | −42.95 | 15.49 | 0.3622 |
| Csmd1 | 0.06 | −1.98 | 2.10 | 0.9562 | Bcl3 | 15.70 | 1.82 | 29.58 | 0.0318 |
| Axin2 | −1.73 | −20.20 | 16.74 | 0.8551 | Fam81a | 0.39 | −1.48 | 2.26 | 0.6834 |
| Kif26b | 0.95 | −1.04 | 2.95 | 0.3536 | Ihh | 0.16 | −0.73 | 1.05 | 0.7310 |
| Vegfa | 28.68 | 0.05 | 57.32 | 0.0560 | Htr1a | −0.52 | −2.16 | 1.12 | 0.5369 |
| Rasl11b | −0.85 | −5.58 | 3.87 | 0.7253 | Scfd2 | 1.28 | −3.26 | 5.82 | 0.5826 |
| Pnpo | −56.31 | −161.78 | 49.16 | 0.3011 | Gmds | 3.19 | −0.34 | 6.72 | 0.0836 |
| Snx11 | −0.68 | −4.97 | 3.62 | 0.7588 | Fam105b | 0.06 | −7.39 | 7.51 | 0.9869 |
| Col2a1 | 856.13 | 172.89 | 1539.38 | 0.0181 | Lnx1 | 4.72 | 0.08 | 9.35 | 0.0523 |
| Col1a1 | 2443.09 | 630.69 | 4255.49 | 0.0114 | Casd1 | 2.07 | −4.01 | 8.15 | 0.5078 |
| Gdf5 | 0.65 | −0.71 | 2.01 | 0.3554 | Pon1 | 0.30 | −0.85 | 1.45 | 0.6074 |
| Rrp15 | −0.86 | −3.52 | 1.80 | 0.5279 | Smyd3 | 0.18 | −0.91 | 1.27 | 0.7490 |
| Lrrc46 | −0.63 | −2.01 | 0.75 | 0.3734 | Fip1l1 | −1.03 | −6.16 | 4.10 | 0.6949 |
| Timp3 | 88.83 | 35.44 | 142.21 | 0.0021 | Oxgr1 | −7.30 | −20.25 | 5.64 | 0.2748 |
| Acan | 52.50 | 21.19 | 83.81 | 0.0020 | Scrn2 | −0.16 | −1.11 | 0.80 | 0.7473 |
| Wnt16 | 3.17 | 0.66 | 5.68 | 0.0172 | Il1r2 | 1.43 | −6.87 | 9.73 | 0.7367 |
| Spata17 | 0.30 | −1.15 | 1.75 | 0.6897 | Sp6 | −1.48 | −5.35 | 2.40 | 0.4584 |
| G0s2 | −11.25 | −31.16 | 8.66 | 0.2740 | Fam105a | 5.10 | −9.02 | 19.22 | 0.4831 |
| Skap1 | −0.53 | −1.87 | 0.82 | 0.4470 | Hs6st3 | −0.14 | −1.69 | 1.41 | 0.8611 |
| Igf1 | 13.18 | 4.89 | 21.48 | 0.0032 | Tle3 | −0.39 | −5.95 | 5.16 | 0.8901 |
| D1pas1 | −0.56 | −1.78 | 0.66 | 0.3708 | Ank | 33.47 | 7.46 | 59.49 | 0.0153 |
| Teddm1 | −0.36 | −6.54 | 5.83 | 0.9106 | Cdk5rap3 | 3.77 | −5.89 | 13.43 | 0.4486 |
| Cbx1 | −0.30 | −9.71 | 9.11 | 0.9497 | Zfp648 | −0.39 | −2.11 | 1.34 | 0.6617 |
| Mrpl10 | 1.30 | −3.64 | 6.24 | 0.6088 | Gpatch2 | 3.97 | −3.14 | 11.07 | 0.2799 |
| Casp3 | 0.60 | −14.62 | 15.83 | 0.9386 | Map4k4 | 33.98 | −22.75 | 90.72 | 0.2467 |
| Grid2 | 0.14 | −1.91 | 2.19 | 0.8945 | Bmp2 | 11.34 | 2.82 | 19.87 | 0.0124 |
| Trpm3 | 0.76 | −1.16 | 2.68 | 0.4402 | Tgfb1 | 0.54 | −20.94 | 22.02 | 0.9611 |
| Tbx21 | −0.88 | −1.89 | 0.14 | 0.0971 | Il1rl2 | 0.79 | −2.98 | 4.55 | 0.6844 |
| Mtor | 6.03 | −0.77 | 12.83 | 0.0892 | Sp2 | −4.68 | −13.39 | 4.02 | 0.2976 |
| Nfe2l1 | 17.66 | −4.78 | 40.10 | 0.1302 | Postn | 120.21 | 8.70 | 231.71 | 0.0403 |
| Stab2 | −10.92 | −23.92 | 2.07 | 0.1067 | Ppp1r9a | −0.57 | −2.15 | 1.01 | 0.4854 |
| Hoxb9 | 0.51 | −0.52 | 1.55 | 0.3380 | Ulk1 | −1.85 | −5.40 | 1.71 | 0.3141 |
| Pon2 | −1.09 | −6.76 | 4.58 | 0.7075 | Sorcs1 | −0.37 | −2.53 | 1.78 | 0.7364 |
| Tgfb2 | 12.04 | 4.29 | 19.80 | 0.0039 | Xrcc2 | −1.37 | −4.49 | 1.75 | 0.3938 |
| Syn3 | 0.00 | −1.08 | 1.07 | 0.9942 | Myo1e | 11.51 | 2.86 | 20.16 | 0.0124 |
| Comp | 758.90 | 382.73 | 1135.06 | 0.0003 | Aff3 | 5.76 | 1.94 | 9.58 | 0.0050 |
| Pcna | −91.91 | −168.16 | −15.66 | 0.0226 | Tmem2 | 9.72 | −0.69 | 20.12 | 0.0739 |
| Sgce | 4.60 | 1.79 | 7.41 | 0.0025 | Uaca | 23.48 | −3.95 | 50.92 | 0.1005 |
| Gda | −13.97 | −52.99 | 25.04 | 0.4864 | Adam10 | −31.61 | −71.11 | 7.88 | 0.1239 |
Statistically significant genes are in bold-face while borderline significant genes are italicized.
Heritabilities were calculated for all 78 gene expression difference traits. Heritability estimates varied from 0.00 to 0.24 and none were significantly different from zero. Unlike the OA phenotypes, there was no evidence for genetic variation in differences in gene expression levels in DMM and SHAM knees.
Correlation between gene expression and phenotypes
Since there was no heritability for gene expression differences between DMM and SHAM knees, it was not possible to estimate genetic correlations between PTOA phenotypes and gene expression level differences. However, we can present phenotypic correlations with gene expression. Table 4 lists gene expression level differences significantly correlated with differences in OA phenotypes. We would expect to see 4 statistically significant correlations between DMM-SHAM differences for each PTOA phenotype and the 78 gene expression level differences at the 5% level even when there is actually no such association. PTOA phenotype differences in Tb.N, Tb.Sp, and subchondral bone plate thickness showed four or fewer significant correlations with gene expression differences, as expected when there are truly no associations. Maximum OARSI (11), Summed OARSI (20), Synovitis score (27), BV/TV (20), CDI (9), Tb.Th (19), and Number of Ectopic Calcifications (8) all have at least double the number of significant associations expected under the null hypothesis of no associations. For most significant gene level differences, the DMM surgery increases gene expression levels relative to SHAM surgeries. Genes overexpressed in DMM-induced OA include Copz2, Rasl11b, Col1a1, Wnt16, Igf1, Tgfb2, Sgce, Cd5rap3, Gpatch2, Postn, Myo1e, Aff3, and Uaca. Genes where OA development leads to lower expression include Pcna, G0s2, and Xrcc2. The number of ectopic calcifications is positively associated with Col2a1, Col1a1, Timp3, Acan, Wnt16, Comp, and Ank and negatively associated with Stab2. Note that many of these same genes showed differences in expression between DMM and SHAM knees.
Table 4:
Significant associations between differences in gene expression levels and differences in phenotypes between DMM and SHAM knees.
| Trait | Positive association with disease | Negative association with disease |
|---|---|---|
| Maximum OARSI score | Copz2, Csmd1, Col1a1, Wnt16, Casp3, Ank, Postn, Uaca | Rrp15, G0s2, Skap1 |
| Summed OARSI score |
Copz2, Csmd1, Kif26b, Col1a1, Wnt16, Tgfb2, Sgce, Bcl3,
Scfd2, Lnx1, Ank, Cdk5rap3, Gpatch2, Postn, Myo1e, Uaca |
Rrp15, G0s2, Skap1, Stab2 |
| Synovitis score |
Copz2, Csmd1, Kif26b, Col1a1, Timp3, Wnt16, Igf1, Trpm3,
Nfe2l1, Tgfb2, Comp, Sgce, Bcl3, Scfd2, Gmds, Lnx1, Ank, Cdk5rap3, Gpatch2, Il1rl2, Postn, Myo1e, Aff3, Tmem2, Uaca |
G0s2, Pcna |
| BV/TV |
Copz2, Rasl11b, Col1a1, Timp3, Acan, Wnt16, Igf1, Mrpl10,
Nfe2l1, Tgfb2, Comp, Sgce, Cdk5rap3, Gpatch2, Postn, Myo1e, Aff3 |
Pcna, Ulk1, Xrcc2 |
| CDI | Copz2, Rasl11b, Col1a1, Tgfb2, Myo1e, Aff3 | Pon2, Pcna, Ulk1 |
| SMI |
Copz2, Rasl11b, Col2a1, Col1a1, Timp3, Acan, Igf1, Tgfb2,
Comp, Sgce, Cdk5rap3, Postn, Myo1e, Aff3 |
Tbx21, Pcna, Xrcc2 |
| Tb.N | Rasl11b, Mrpl10 | Xrcc2 |
| Tb.T |
Copz2, Rasl11b, Col1a1, Timp3, Wnt16, Igf1, Mrpl10, Nfe2l1,
Tgfb2, Sgce, Fip1l1, Cdk5rap3, Gpatch2, Postn, Myo1e, Uaca |
Tbx21, Ulk1, Xrcc2 |
| Tb.Sp | Rasl11b, Myo1e, Aff3 | Xrcc2 |
| Subchondral bone plate thickness | None | None |
| # of ectopic calcifications | Col2a1, Col1a1, Timp3, Acan, Wnt16, Comp, Ank | Stab2 |
For trabecular architecture traits BV/TV, CDI, Tb.N, and Tb.Th, positive associations with disease are numerically negative associations as DMM is expected to reduce bone in trabeculae under the DMM operated knee. OARSI = Osteoarthritis Research Society International; BV/TV = bone volume fraction; CDI = connectivity density index; SMI = structure model index; Tb.N = trabecular number; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation.
DISCUSSION
This is the first study that reports screening of a large number of genetically related mouse strains for their susceptibility to PTOA. Specifically, 12 RI strains generated from LGXSM intercross were used in a population genetics approach to demonstrate that the phenotypic differences among them depend on their genetic composition. LG/J and SM/J strains and RI strains generated from their intercross, have been widely used to investigate the role of genetics in the pathogenesis of a number of conditions such as fatty liver disease (24), diabetes and obesity (25–27), and osteoporosis (27). In a prior study, we found that LGXSM-6 exhibited greater protection from PTOA compared to LGXSM-33 (12, 13). The current study establishes heritability for PTOA phenotypes, that is, these phenotypic traits show significant genetic variation between strains in the population of RI strains: cartilage degeneration (maximum and summed OARSI scores), synovitis and various bone parameters. Importantly, our study allows, for the first time, the measurement of genetic correlations of phenotypes that are characteristic of PTOA. For instance, cartilage degeneration was significantly positively associated with synovitis (r=0.83 with summed OARSI; r=0.91 with maximum OARSI) and subchondral bone plate thickness was negatively correlated with ectopic calcification (r=−0.59). Moreover, we show that 40 of the 78 genes tested were significantly correlated with various (OA) phenotypes.
This study is a logical extension of our previous work, where we studied the susceptibility to cartilage degeneration using only two RI strains (13), which suggested genetic differences. Now using 14 strains and additional analyses such as synovitis, ectopic calcification and gene expression, this study does provide new information: that is, while two of our strains suggested differences, using 14 strains statistically defines the genetic differences, and the correlation with other phenotypes, viz. synovitis and ectopic bone formation. Thus, we established the contribution of genetics to the variation in response to injury.
We did not map these strains because mapping only 14 strains could result in “ghost QTLs” (28), parts of the genome remote from the locus of the actual genetic effect making interpretation difficult by placing a single effect in multiple genetic locations usually on different chromosomes. Future studies using even more RI strains or advanced intercross strains could further explore underlying genetics.
We imply that these mouse strains could be used to map the genetic differences, as it is their relatedness that allows mapping. There are 4 times more recombination events in a RI strain set compared to a standard F2 intercross mapping population. However, this is still a fairly large level of linkage disequilibrium (LD) compared to human historically-based LD. There are certainly long stretches of LD across chromosomes (but less than in a F2). It is LD amongst unlinked loci that cause a problem in mapping. This is due to too few strains; not lack of recombination and we have used related strains to map and narrow causal loci (28).
For the first time, we report the degree of synovitis and its heritability in RI strains following DMM-surgery. Cartilage degeneration and synovitis were strongly coupled (29–31) across strains, although the correlations were not absolute. Clinically, it is reported that synovitis is related to the severity of pain in OA (2, 32). However, the severity of pain does not have a strong correlation with structural changes in the joint (33). In addition, we performed SHAM-surgery in the contra-lateral knee in all mice to match the condition between RI strains. Our results did not show strong inflammatory findings in SHAM-operated knees when compared to DMM-operated knees as reported elsewhere (34). It has been reported that about 40% of patients with severe radiographic changes in knee joints are symptom-free (35), indicating the mismatch between the severity of synovitis and degree of radiographic findings. Here, we found significant correlations of synovitis with summed and maximum OARSI scores, suggesting that significant variance is shared between cartilage degeneration and synovitis. In contrast, some clinical data show a weaker correlation between these two phenotypes (35). This similarity between the mouse and human data indicate that the mouse DMM-induced PTOA can serve generally, as a model for OA in humans.
We found variability in the development of synovial ectopic calcification. Previously, using an advanced intercross of LG/J and SM/J, we established QTLs for ectopic calcification that strongly implicate variants of genes such as Aff3 and Ank. Here, we confirmed variability in synovial ectopic calcification across RI strains, which strengthens our previous results. In humans, two other genes have been associated with calcifications, osteoprotegerin causes OA with chondrocalcinosis in two families with familial generalized OA (36) and the calcification inhibitor matrix GLA protein is associated with hand OA (37). A weak negative correlation between OARSI scores and ectopic calcification and between synovitis and ectopic calcification suggests that ectopic calcification occurs independently from severity of cartilage damage. The relationship between synovitis and chondrocalcinosis in humans has recently been examined showing no differences in synovitis scores at baseline and at 4 years between the chondrocalcinosis and the control groups (38), suggesting their independence.
Our data revealed that 40 of the 78 genes tested were significantly correlated with various phenotypes. Postn was significantly correlated with OARSI score, synovitis and a couple of bone parameters (e.g. Tb.Sp). Postn influences matrix homeostasis, bone remodeling and OA development (39, 40). Wnt16 and Comp also showed similar correlations, which is in keeping with their known role in OA development (21, 23, 41–46). The Wnt-pathway is involved in bone and cartilage homeostasis and Wnt16 expression is high in OA cartilage and barely detectable in preserved cartilage (46). Comp is an essential component of the cartilage extracellular-matrix with roles in cellular proliferation and apoptosis in cartilaginous tissue (46, 47). TGFβs are well known to be associated with bone and cartilage homeostasis and are involved in OA development (48), and were also correlated here with OARSI score, synovitis, and several bone parameters. Previously, we reported that the expression of four genes that represent DNA repair (Xrcc2, Pcna) and Wnt signaling (Axin2, Wnt16) pathways were significantly correlated with both ear wound and cartilage healing, suggesting a common genetic basis for these two regenerative phenotypes. Continued genetic and molecular investigation with the wide variety of LGXSM mouse resources utilized here represent important tools for further revealing not only the mechanism of OA but also the mechanism of tissue regeneration in humans.
This study has some limitations. The mice used in this study were only 10 weeks of age at the time of DMM surgery, which is standard in the field. The differences among the strains in severity of cartilage damage (OARSI score) could have been due to differences in maturation. Although some authors consider age of skeletal maturity at 12 weeks or older (49), others have recommended that the minimum age for skeletal maturity in mice is 10 weeks for preclinical modes of OA to obtain the most meaningful insights into human OA (50). Prior work examining long bone growth and trabecular architecture in these and other strains has been performed at 10 weeks, when epiphyses have fused with skeletal maturation. Although growth in length continues indefinitely even into 5 or 6 months (51, 52), not all growth plates close completely. On the other hand, work by Zhang et al., (53) has demonstrated very early degenerative changes in the intervertebral disks of these mice. If we waited too long to operate on the animals they might also show lower variability in the presence of disease. Another limitation is the use of contralateral limb as SHAM-operated because this approach may question the interpretation of strain differences especially when RI strains have differences in pain-perception that might change load-bearing and pose issues such as gait modification and movement provoked pain. Yet another limitation was that only male mice were included in our study. Thus the similarity between the mouse and human data indicates that the mouse DMM-induced PTOA can serve generally, as a model for OA in at least 50% of humans. Lastly, we used only 3 mice for some strains, although this sample size appeared low, most of our tests were significant.
In conclusion, we identified a number of new mouse strains that are either protected or susceptible to OA and established the heritability of OA. Given the recombinant inbred nature of these strains, this variability in response to meniscus destabilization is caused by their genetic composition. Further investigation into the particular genomic regions that orchestrate these changes combined with molecular mechanisms underlying these changes will help predict the individuals who are at risk for developing OA.
Supplementary Material
Acknowledgement
The authors would like to thank Dr. Lei Cai, Dr. Xin Duan, Crystal Idleburg, Samantha Coleman and Daniel Leib for their technical assistance. We also thank Dr. Diane Bender (from the Bursky Center for Human Immunology and Immunotherapy Programs) for her technical assistance with QuantiGene Plex assay.
Funding information
This study was supported by R01 AR063757 (Sandell, Cheverud) and by P30 AR057235 (Musculoskeletal Research Center, P.I. Sandell.) from the National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH). Dr. Rai was supported through NIH Pathway to Independence Award (R00 AR064837) from NIAMS. The content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the NIAMS or the NIH.
Footnotes
Competing Interest
The authors have no competing interest to declare.
Financial conflict of interest
All other authors have nothing to disclose.
REFERENCES
- 1.Sandell LJ. Etiology of osteoarthritis: genetics and synovial joint development. Nat Rev Rheumatol 2012;8(2):77–89. [DOI] [PubMed] [Google Scholar]
- 2.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012;64(6):1697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown TD, Johnston RC, Saltzman CL, Marsh JL, Buckwalter JA. Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J Orthop Trauma 2006;20(10):739–44. [DOI] [PubMed] [Google Scholar]
- 4.Roos H, Adalberth T, Dahlberg L, Lohmander LS. Osteoarthritis of the knee after injury to the anterior cruciate ligament or meniscus: the influence of time and age. Osteoarthritis Cartilage 1995;3(4):261–7. [DOI] [PubMed] [Google Scholar]
- 5.Anderson DD, Chubinskaya S, Guilak F, Martin JA, Oegema TR, Olson SA, et al. Post-traumatic osteoarthritis: improved understanding and opportunities for early intervention. J Orthop Res 2011;29(6):802–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zengini E, Hatzikotoulas K, Tachmazidou I, Steinberg J, Hartwig FP, Southam L, et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat Genet 2018;50(4):549–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fitzgerald J, Rich C, Burkhardt D, Allen J, Herzka AS, Little CB. Evidence for articular cartilage regeneration in MRL/MpJ mice. Osteoarthritis Cartilage 2008;16(11):1319–26. [DOI] [PubMed] [Google Scholar]
- 8.Heydemann A The super super-healing MRL mouse strain. Front Biol (Beijing) 2012;7(6):522–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leferovich JM, Bedelbaeva K, Samulewicz S, Zhang XM, Zwas D, Lankford EB, et al. Heart regeneration in adult MRL mice. Proc Natl Acad Sci U S A 2001;98(17):9830–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blankenhorn EP, Bryan G, Kossenkov AV, Clark LD, Zhang XM, Chang C, et al. Genetic loci that regulate healing and regeneration in LG/J and SM/J mice. Mamm Genome 2009;20(11–12):720–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hrbek T, de Brito RA, Wang B, Pletscher LS, Cheverud JM. Genetic characterization of a new set of recombinant inbred lines (LGXSM) formed from the inter-cross of SM/J and LG/J inbred mouse strains. Mamm Genome 2006;17(5):417–29. [DOI] [PubMed] [Google Scholar]
- 12.Rai MF, Hashimoto S, Johnson EE, Janiszak KL, Fitzgerald J, Heber-Katz E, et al. Heritability of articular cartilage regeneration and its association with ear wound healing in mice. Arthritis Rheum 2012;64(7):2300–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto S, Rai MF, Janiszak KL, Cheverud JM, Sandell LJ. Cartilage and bone changes during development of post-traumatic osteoarthritis in selected LGXSM recombinant inbred mice. Osteoarthritis Cartilage 2012;20(6):562–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 2007;15(9):1061–9. [DOI] [PubMed] [Google Scholar]
- 15.Takebe K, Rai MF, Schmidt EJ, Sandell LJ. The chemokine receptor CCR5 plays a role in post-traumatic cartilage loss in mice, but does not affect synovium and bone. Osteoarthritis Cartilage 2015;23(3):454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glasson SS, Chambers MG, Van Den Berg WB, Little CB. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010;18 Suppl 3:S17–23. [DOI] [PubMed] [Google Scholar]
- 17.Lewis JS, Hembree WC, Furman BD, Tippets L, Cattel D, Huebner JL, et al. Acute joint pathology and synovial inflammation is associated with increased intra-articular fracture severity in the mouse knee. Osteoarthritis Cartilage 2011;19(7):864–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duan X, Rai MF, Holguin N, Silva MJ, Patra D, Liao W, et al. Early changes in the knee of healer and non-healer mice following non-invasive mechanical injury. J Orthop Res 2016. [DOI] [PMC free article] [PubMed]
- 19.Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 2010;25(7):1468–86. [DOI] [PubMed] [Google Scholar]
- 20.Odgaard A, Gundersen HJ. Quantification of connectivity in cancellous bone, with special emphasis on 3-D reconstructions. Bone 1993;14(2):173–82. [DOI] [PubMed] [Google Scholar]
- 21.Rai MF, Schmidt EJ, Hashimoto S, Cheverud JM, Sandell LJ. Genetic loci that regulate ectopic calcification in response to knee trauma in LG/J by SM/J advanced intercross mice. J Orthop Res 2015;33(10):1412–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rai MF, Duan X, Quirk JD, Holguin N, Schmidt EJ, Chinzei N, et al. Post-Traumatic Osteoarthritis in Mice Following Mechanical Injury to the Synovial Joint. Sci Rep 2017;7:45223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rai MF, Schmidt EJ, McAlinden A, Cheverud JM, Sandell LJ. Molecular insight into the association between cartilage regeneration and ear wound healing in genetic mouse models: targeting new genes in regeneration. G3 (Bethesda) 2013;3(11):1881–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Partridge CG, Fawcett GL, Wang B, Semenkovich CF, Cheverud JM. The effect of dietary fat intake on hepatic gene expression in LG/J AND SM/J mice. BMC Genomics 2014;15:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheverud JM, Ehrich TH, Kenney JP, Pletscher LS, Semenkovich CF. Genetic evidence for discordance between obesity- and diabetes-related traits in the LGXSM recombinant inbred mouse strains. Diabetes 2004;53(10):2700–8. [DOI] [PubMed] [Google Scholar]
- 26.Ehrich TH, Kenney JP, Vaughn TT, Pletscher LS, Cheverud JM. Diet, obesity, and hyperglycemia in LG/J and SM/J mice. Obes Res 2003;11(11):1400–10. [DOI] [PubMed] [Google Scholar]
- 27.Reich MS, Jarvis JP, Silva MJ, Cheverud JM. Genetic relationships between obesity and osteoporosis in LGXSM recombinant inbred mice. Genet Res (Camb) 2008;90(5):433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheverud JM, Ehrich TH, Hrbek T, Kenney JP, Pletscher LS, Semenkovich CF. Quantitative trait loci for obesity- and diabetes-related traits and their dietary responses to high-fat feeding in LGXSM recombinant inbred mouse strains. Diabetes 2004;53(12):3328–36. [DOI] [PubMed] [Google Scholar]
- 29.Zheng W, Zhang H, Jin Y, Wang Q, Chen L, Feng Z, et al. Butein inhibits IL-1beta-induced inflammatory response in human osteoarthritis chondrocytes and slows the progression of osteoarthritis in mice. Int Immunopharmacol 2017;42:1–10. [DOI] [PubMed] [Google Scholar]
- 30.Ribeiro M, Lopez de Figueroa P, Nogueira-Recalde U, Centeno A, Mendes AF, Blanco FJ, et al. Diabetes-accelerated experimental osteoarthritis is prevented by autophagy activation. Osteoarthritis Cartilage 2016;24(12):2116–25. [DOI] [PubMed] [Google Scholar]
- 31.Wu P, Holguin N, Silva MJ, Fu M, Liao W, Sandell LJ. Early response of mouse joint tissue to noninvasive knee injury suggests treatment targets. Arthritis Rheumatol 2014;66(5):1256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haywood L, McWilliams DF, Pearson CI, Gill SE, Ganesan A, Wilson D, et al. Inflammation and angiogenesis in osteoarthritis. Arthritis Rheum 2003;48(8):2173–7. [DOI] [PubMed] [Google Scholar]
- 33.Murphy NJ, Eyles JP, Hunter DJ. Hip Osteoarthritis: Etiopathogenesis and Implications for Management. Adv Ther 2016;33(11):1921–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fahlgren A, Messner K, Aspenberg P. Meniscectomy leads to an early increase in subchondral bone plate thickness in the rabbit knee. Acta Orthop Scand 2003;74(4):437–41. [DOI] [PubMed] [Google Scholar]
- 35.Davis MA, Ettinger WH, Neuhaus JM, Barclay JD, Segal MR. Correlates of knee pain among US adults with and without radiographic knee osteoarthritis. J Rheumatol 1992;19(12):1943–9. [PubMed] [Google Scholar]
- 36.Ramos YF, Bos SD, van der Breggen R, Kloppenburg M, Ye K, Lameijer EW, et al. A gain of function mutation in TNFRSF11B encoding osteoprotegerin causes osteoarthritis with chondrocalcinosis. Ann Rheum Dis 2015;74(9):1756–62. [DOI] [PubMed] [Google Scholar]
- 37.den Hollander W, Boer CG, Hart DJ, Yau MS, Ramos YFM, Metrustry S, et al. Genome-wide association and functional studies identify a role for matrix Gla protein in osteoarthritis of the hand. Ann Rheum Dis 2017;76(12):2046–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han BK, Kim W, Niu J, Basnyat S, Barshay V, Gaughan JP, et al. Association of Chondrocalcinosis in Knee Joints With Pain and Synovitis: Data From the Osteoarthritis Initiative. Arthritis Care Res (Hoboken) 2017;69(11):1651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Attur M, Yang Q, Shimada K, Tachida Y, Nagase H, Mignatti P, et al. Elevated expression of periostin in human osteoarthritic cartilage and its potential role in matrix degradation via matrix metalloproteinase-13. FASEB J 2015;29(10):4107–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang R, Fang H, Chen Y, Shen J, Lu H, Zeng C, et al. Gene expression analyses of subchondral bone in early experimental osteoarthritis by microarray. PLoS One 2012;7(2):e32356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dell’accio F, De Bari C, Eltawil NM, Vanhummelen P, Pitzalis C. Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt-16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum 2008;58(5):1410–21. [DOI] [PubMed] [Google Scholar]
- 42.Recklies AD, Baillargeon L, White C. Regulation of cartilage oligomeric matrix protein synthesis in human synovial cells and articular chondrocytes. Arthritis Rheum 1998;41(6):997–1006. [DOI] [PubMed] [Google Scholar]
- 43.Li G, Han N, Li Z, Lu Q. Identification of transcription regulatory relationships in rheumatoid arthritis and osteoarthritis. Clin Rheumatol 2013;32(5):609–15. [DOI] [PubMed] [Google Scholar]
- 44.Yuan Q, Sun L, Li JJ, An CH. Elevated VEGF levels contribute to the pathogenesis of osteoarthritis. BMC Musculoskelet Disord 2014;15:437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Tian Y. MiR-15a-5p regulates viability and matrix degradation of human osteoarthritis chondrocytes via targeting VEGFA. Biosci Trends 2017;10(6):482–8. [DOI] [PubMed] [Google Scholar]
- 46.Das BR, Roy A, Khan FR. Cartilage oligomeric matrix protein in monitoring and prognostication of osteoarthritis and its utility in drug development. Perspect Clin Res 2015;6(1):4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verma P, Dalal K. Serum cartilage oligomeric matrix protein (COMP) in knee osteoarthritis: a novel diagnostic and prognostic biomarker. J Orthop Res 2013;31(7):999–1006. [DOI] [PubMed] [Google Scholar]
- 48.Bauge C, Girard N, Lhuissier E, Bazille C, Boumediene K. Regulation and Role of TGFbeta Signaling Pathway in Aging and Osteoarthritis Joints. Aging Dis 2014;5(6):394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang H, Huang L, Welch I, Norley C, Holdsworth DW, Beier F, et al. Early Changes of Articular Cartilage and Subchondral Bone in The DMM Mouse Model of Osteoarthritis. Sci Rep 2018;8(1):2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Poole R, Blake S, Buschmann M, Goldring S, Laverty S, Lockwood S, et al. Recommendations for the use of preclinical models in the study and treatment of osteoarthritis. Osteoarthritis Cartilage 2010;18 Suppl 3:S10–6. [DOI] [PubMed] [Google Scholar]
- 51.Sanger TJ, Norgard EA, Pletscher LS, Bevilacqua M, Brooks VR, Sandell LJ, et al. Developmental and genetic origins of murine long bone length variation. J Exp Zool B Mol Dev Evol 2011;316B(2):146–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res 2007;22(8):1197–207. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Xiong C, Kudelko M, Li Y, Wang C, Wong YL, et al. Early onset of disc degeneration in SM/J mice is associated with changes in ion transport systems and fibrotic events. Matrix Biol 2018. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.