

Abstract
Background.
Coagulation Factor XI (FXI) contributes to the pathobiology of sepsis-associated thrombosis and is a target for new therapeutics. Through cleavage of disulfide bonds, FXI becomes reduced (rFXI), accelerating intrinsic coagulation cascade activation. The role of rFXI in human sepsis has never been studied.
Objectives.
We determined levels of total FXI and rFXI in critically-ill septic patients with and without overt disseminated intravascular coagulation (DIC, a dysregulated pro-thrombotic condition).
Patients and Methods.
Total FXI and rFXI plasma levels were measured on ICU admission in prospectively enrolled septic patients (n=32) from three academic medical centers and matched, healthy controls (n=15). In septic patients, hematologic and physiologic parameters and pathological thrombosis (presence or absence of overt DIC) were determined.
Results.
rFXI was higher in septic patients than controls (p<0.05). In septic patients, rFXI was significantly associated with platelet count (r = 0.3511, p<0.05) and APACHE II score (r = −0.359, p<0.05), indices of illness severity. rFXI was lower in patients with overt DIC (p=0.088), suggesting a consumptive coagulopathy. In contrast, while total FXI levels were reduced in sepsis, they failed to correlate with illness severity, thrombosis, or hematologic parameters.
Conclusions.
We establish, for the first time, that rFXI is increased in patients with sepsis and correlates with illness severity (APACHE II score and platelet count) and pathological coagulopathy (overt DIC). Total FXI levels, in contrast, are decreased in sepsis but fail to associate with any indices. These findings suggest that rFXI has unique activity in human sepsis.
Keywords: Sepsis, Coagulation, Factor XI, Outcomes
Introduction
Sepsis is a systemic inflammatory response to infection that carries significant morbidity and mortality, including thrombosis [1, 2]. Inflammation and coagulation in sepsis are tightly linked. Inflammation promotes coagulation in a process known as immunothrombosis, which in turn further enhances inflammatory responses [3]. In sepsis, uncontrolled activation of immunothrombosis is common and contributes to disseminated intravascular coagulation (DIC), a dysregulated pro-thrombotic state characterized by thrombin generation and consumption of platelets and coagulation factors. Factor XI (FXI) is a key player in the intrinsic coagulation system. FXI can be activated by activated Factor XII (FXIIa) and thrombin, enabling FXI to cleave and activate its substrate, Factor IX. FXI function is regulated by the reduction of specific disulfide bonds in its catalytic domain by thiol isomerases such as protein disulfide isomerase (PDI) [4,5]. The reduced form of FXI (rFXI) is converted more rapidly to activated FXI (FXIa), thereby accelerating activation of the coagulation cascade and enhancing thrombus formation [5]. FXI is being investigated as a therapeutic target in sepsis [6–8]. FXI-deficient mice show improved survival from peritoneal polymicrobial sepsis induced by cecal ligation and puncture (CLP), as well as blunted contact activation and inflammation [9,10]. Similar findings were observed when FXI was pharmacologically inhibited [11]. However, in another study FXI deficiency increased bacterial growth and mortality in experimental sepsis due to pneumonia [12], highlighting that uncertainty remains in pre-clinical models of FXI and sepsis. Clinical studies investigating FXI levels in sepsis are very limited. The only study we are aware of reported that FXI levels were ~50% decreased in 13 children with meningococcal septic shock [13]. Investigations of FXI in adult sepsis, which may not always recapitulate findings in pediatric patients, are absent. Moreover, the role of rFXI in sepsis remains completely unknown. As rFXI has greater pro-thrombotic activity, an improved understanding of rFXI in human sepsis is needed. In this study, we investigated total and rFXI levels in adult patients admitted to the intensive care unit (ICU) with a primary diagnosis of sepsis.
Methods
Human Subjects Enrollment
This study was performed as an ancillary study to an NIH-funded study investigating acute lung injury and acute respiratory distress syndrome (ALI/ARDS) in critically-ill patients [2]. Sepsis is a common cause of ALI/ARDS [3]. Inclusion and exclusion criteria mirrored those of the parent study. Patients with sepsis were eligible for inclusion in the study. Sepsis was defined according to guidelines that were current during the time of this study [4]. For inclusion in the study, sepsis was defined as systemic infection and at least 2 of the following SIRS (systemic inflammatory response syndrome) criteria: a) temperature > 38°C or < 36°C; b) heart rate >90 beats/min; c) respiratory rate >20 breaths/min or PaCO2 < 32mm Hg; d) white blood cell (WBC) count >12,000/mm3, <4000/mm3, or >10% bands.
Exclusion criteria were the following: age younger than 18 years; neuromuscular disease that impairs ability to ventilate without assistance, such as C5 or higher spinal cord injury, amyotrophic lateral sclerosis, Guillain-Barre syndrome or myasthenia gravis; pregnancy (a negative pregnancy test was required for women of child-bearing potential); severe chronic respiratory disease defined as any of the following: a) chronic hypercapnia with PaCO2 greater than 45 mmHg b) chronic hypoxemia with PaO2 less than 55 mmHg on FiO2=0.21) c) hospitalization within the last 6 months for respiratory failure (PaCO2>50 mmHg and/or PaO2 less than 55 mmHg on 0.21 FiO2) d) secondary polycythemia e) severe pulmonary hypertension (mean PAP greater than 40 mmHg), or f) chronic ventilator dependency; burns greater than 40% total body surface area; malignancy or other irreversible disease or condition for which 6 month mortality is estimated to be greater than 50%; bone marrow transplant within the last 5 years; patient does not want resuscitative or full life support; diffuse alveolar hemorrhage from vasculitis; severe congestive heart failure (which confounds assessments of ALI/ARDS from sepsis); patient, surrogate, or physician not committed to full support (patients were not excluded if he/she would receive all supportive care except for attempts at resuscitation from cardiac arrest); no consent/inability to obtain consent; or a moribund patient not expected to survive 24 hours.
Healthy participants were recruited as part of a separate, active prospective study and were matched to septic patients a priori based on age, gender, and race. Studies were IRB approved and all subjects provided informed consent. For septic patients who were sedated or otherwise could not provide informed consent, a legally-authorized representative (LAR) provided informed consent, in accordance with ethical guidelines.
Measurement of Clinical Labs
Clinical labs in septic patients and healthy controls were measured on freshly harvested plasma using a national, CLIA-certified clinical laboratory (www.aruplabs.com). Overt disseminated intravascular coagulation (DIC) was defined using the recommended scoring system from the International Society of Thrombosis and Haemostasis [5,6]. The DIC scoring system incorporates the platelet count, INR, d-dimer, and fibrinogen on a point-based scale. Overt DIC was defined as a score of 5 or greater, non-overt DIC was defined as a score of 3-4, and no DIC was defined as a score of 2 or lower.
Measurement of total FXI and rFXI levels
Total FXI levels were measured by ELISA on harvested plasma as previously described [5]. Measurements of rFXI levels were based on an assay that we previously established 1. Briefly, plasma samples containing 50ng FXI were incubated with lμl 3- (N-Maleimidopropionyl)-biocytin (MPB) (Cayman Chemical, MI, USA) for 30 minutes at room temperature in the dark with agitation and then diluted with 1ml Hepes buffer. FXI was precipitated by the addition of 250 μl kaolin solution, incubation for 15 minutes at 37°C in the dark, followed by brief centrifugation and re-suspension of the pellet in loading buffer. The samples were then subjected to electrophoresis in non-reducing conditions (10% Bis-Tris gels), and transferred to a PVDF membrane (Invitrogen). Untreated FXI (75 ng) and FXI reduced by PDI (50 ng) were loaded as a reference for total FXI and rFXI, respectively. FXI was detected with a goat anti-XI polyclonal antibody (1:1000). Avidin HRP (1:40,000) was used to detect rFXI. Band densitometry was measured by EzQuant (Israel). For each sample, the band’s area and density was calculated and rFXI was expressed as a ratio of each sample’s value to the total rFXI value. rFXI levels in each plasma sample were calculated as an average of three technical replicates.
Statistical Analyses
All data was examined for normality using skewness and kurtosis tests. As data in septic patients generally followed a non-normal distribution, comparisons of continuous data were performed using the Mann-Whitney U test. Comparisons of categorical data were performed using the Fisher’s Exact Test. Associations between continuous variables were examined using linear regression and reported with the Spearman correlation coefficient (r). A two-tailed t-test of <0.05 was considered statistically significant for all analyses. All analyses were performed in GraphPad Prism (v7.0a).
Results and Discussion
Septic patients and healthy controls did not differ significantly with regards to age, gender, or race (Table 1). In septic patients, white blood cells, fibrinogen, prothrombin time and D-dimer values were significantly higher, and hemoglobin, hematocrit, and platelet counts were significantly lower than healthy controls. The most common pathogens identified in septic patients were gram positive (e.g. S. aureus, Streptococcus, Enterococcus) (Table 1). The average (range) APACHE II score was 17 (4-42) and all patients had severe sepsis or septic shock.
Table 1.
Characteristics of the Study Cohorts.
| Critically-ill Septic Patients (n=32) | Healthy Control Participants (n=15) | p-value | |
|---|---|---|---|
| Age, yrs. | 49 ± 19 | 41 ± 16 | 0.22 |
| Male Sex, n (%) | 11 (34%) | 6 (40%) | 0.75 |
| Race/Ethnicity | |||
| Caucasian, n (%) | 31 (99%) | 16 (100%) | 0.99 |
| Hispanic/Latino, n (%) | 1 (1%) | 0 (0%) | -- |
| Laboratory Values | |||
| Hemoglobin, mg/dL | 10.3 ± 1.7 | 15.3 ± 1.7 | <0.05 |
| Hematocrit, % | 31 ± 6 | 44 ± 4 | <0.05 |
| White Blood Count, k/μL | 12.8 ± 5.0 | 6.6 ± 1.8 | <0.05 |
| Platelets, k/μL | 221 ± 117 | 300 ± 81 | <0.05 |
| Fibrinogen, mg/dL | 651 ± 220 | 381 ± 78 | <0.05 |
| Prothrombin, sec−1 | 17 ± 3.1 | 13 ± 0.6 | <0.05 |
| aPTT, sec−1 | 40 ± 10 | 31 ± 3 | <0.05 |
| D-Dimer | 2403 ± 2368 | 90.5 ± 67.6 | <0.05 |
| Identified Pathogens | |||
| Gram Positive, n (%) | 8 (25%) | -- | -- |
| Gram Negative, n (%) | 3 (9.4%) | -- | -- |
| Medical History and Outcomes | |||
| Admission APACHE II Score | 17 ± 9 | -- | -- |
| Overt DIC, n (%) | 7 (21.8%) | -- | -- |
| History of CVD, n (%) | 2 (6%) | 0 (0%) | -- |
| History of CHF, n (%) | 1 (3%) | 0 (0%) | -- |
| 28-day all-cause mortality, n (%) | 3 (9%) | -- | -- |
Data are shown as the mean (±SD) unless otherwise indicated. Abbreviations: APACHE II: Acute Physiology and Chronic Health Evaluation II; CVD: Cardiovascular Disease, specifically a known prior history of myocardial infarction, stroke, and/or transient ischemic attack; CHF: Congestive Heart Failure; DIC: Disseminated Intravascular Coagulation; aPTT: activated partial thromboplastin time. Values are the mean ± SD, unless otherwise indicated.
Total FXI levels were significantly lower in septic patients than controls (Figure 1A, p<0.05), consistent with increased consumption of FXI in sepsis. In contrast, the proportion of rFXI was significantly higher in septic patients (Figure 1B, p<0.05), suggesting a possible role for rFXI in the pathobiology of sepsis. Activation of FXI during sepsis may occur via FXII activation on negatively-charged bacterium and increased TF-initiated coagulation [7,10] Moreover, PDI released from platelets and endothelial cells during sepsis may catalyze the reduction of FXI that augment its activation, thus contributing to injurious coagulation responses and thrombosis. In murine sepsis, FXI activation promotes DIC [9,10]. Whether total FXI or rFXI levels are associated with DIC in septic patients has never been examined. We observed that the proportion of rFXI was lower in septic patients with overt DIC compared to patients with non-overt DIC, although differences did not achieve statistical significance (p=0.0880, Figure 1C). In comparison, total FXI were similar in overt and non-overt DIC (p=0.578, Figure 1D). This was not explained by conversion of FXI to rFXI, as these two forms of FXI were not correlated (Figure 1E).
Figure 1. Total FXI and rFXI Levels in Sepsis and Disseminated Intravascular Coagulopathy.
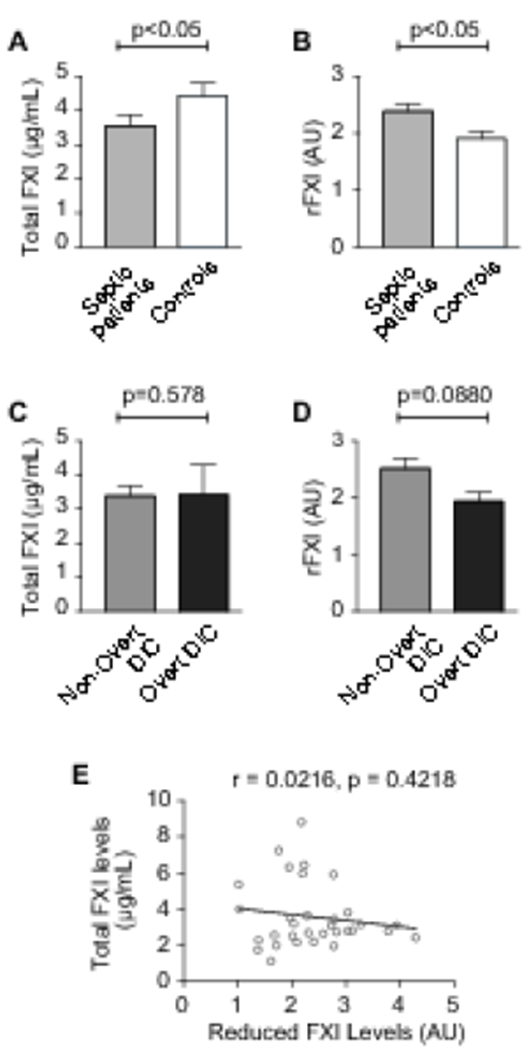
(A) Total FXI levels in septic patients (n=32) and matched healthy controls (n=15), as measured by ELISA. (B) The proportion of reduced FXI (rFXI) in septic patients (n=32) and healthy controls (n=15) was assessed by immunoblotting followed by densitometry (AU, arbitrary units). (C, D) Total FXI levels (C) and the proportion of rFXI levels (D) in septic patients with overt (n=7) vs. non-overt (n=22) disseminated intravascular coagulation (DIC). Note: in n=1 patient, there was no evidence of DIC and in n=2 patients, DIC could not be determined. (E) In septic patients, the proportion of rFXI did not correlate with total FXI levels.
We next asked whether rFXI in septic patients was associated with illness severity. Indeed, the proportion of rFXI and admission APACHE II scores (an accepted measurement of sepsis illness severity) were significantly and inversely associated (Figure 2A, p<0.05). FXI may regulate platelet trafficking and platelets are consumed in overt DIC [11,15]. Lower platelet counts in sepsis are also associated with adverse outcomes. Consistent with this, the proportion of rFXI and platelet counts were significantly and positively associated (Figure 2B, p<0.05). Total FXI levels did not correlate with either illness severity or platelet numbers (Figure 2C-D), indicating that rFXI, rather than total FXI levels, are associated with physiological and cellular responses during human sepsis. Reduced FXI levels did not correlate with age (r=0.101, p=0.58), fibrinogen (r=0.281, p=0.12), PT (r=−0.078, p=0.67), aPTT (r=0.051, p=0.78), D-dimer (r=−0.301, p=0.11), or hemoglobin (r=−0.125, p=0.50). Total FXI levels did not correlate with age (r=−0.110, p=0.55), fibrinogen (r=0.214, p=0.24), PT (r=0.003, p=0.99), aPTT (r=−0.321, p=0.07), or hemoglobin (r=−0.053, p=0.77), but did correlate negatively with D-dimer levels (r=−0.531, p=0.0032) perhaps reflecting increased consumption in DIC.
Figure 2. The Proportion of rFXI in Sepsis is Associated with Illness Severity and Platelet Count.
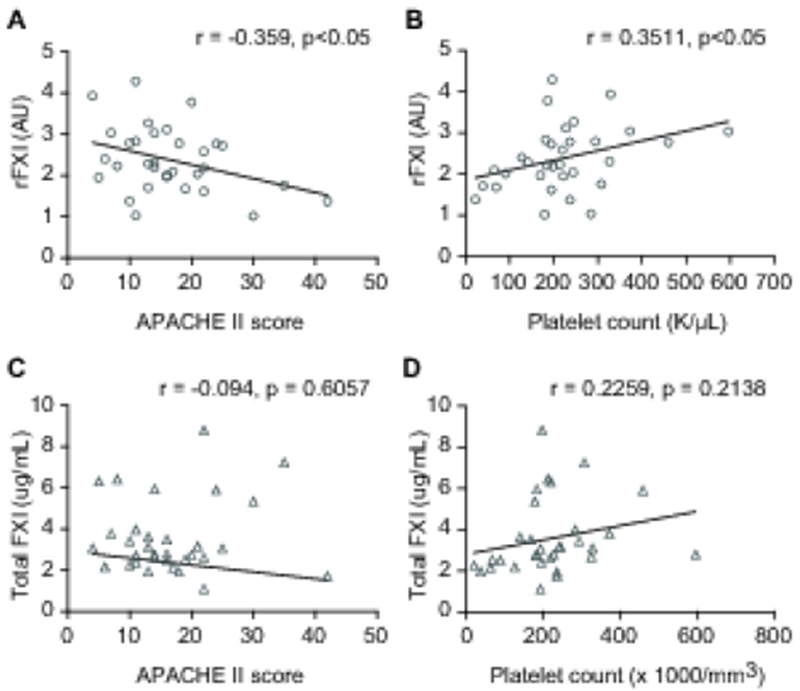
(A, B) Correlation plots between the proportion of rFXI and APACHE II score or platelet count in septic patients (n=32). (C, D) Total FXI levels did not correlate with either APACHE II score or platelet counts in sepsis (n=32).
Our finding that higher levels of rFXI in sepsis were associated with lower illness severity (Figure 1D) raises several possible explanations. It may be due to greater consumption of rFXI in more severely-ill septic patients, as rFXI is activated (and thus consumed) faster than non-reduced (e.g. total) FXI [5]. Alternatively, these data may suggest that rFXI has protective actions in sepsis. While this hypothesis must obviously be explored further, neutrophils from FXI deficient mice and human neutrophils in which FXI activation was inhibited displayed decreased phagocytosis of pneumonia bacterium [12]. Thus, it can be speculated that rFXI (which is rapidly activated), may augment host defenses to invading bacterial pathogens. We also found that while rFXI levels were higher in septic patients than controls, total FXI displayed the opposite pattern (e.g. lower in septic patients than controls). This may be due to the enhanced conversion of total FXI to rFXI by thiol isomerases such as PDI that are locally released from platelets and endothelial cells in sepsis [3].
In summary, we report on the first clinical study in sepsis to prospectively interrogate in parallel both total and reduced FXI. Our findings suggest that the proportion of rFXI may be a more informative marker than total FXI for identifying coordinate changes in FXI and pathophysiological host responses in sepsis. Moreover, as FXI is well-conserved between humans and mice, our findings may provide insights for murine models of polymicrobial sepsis, an important pre-clinical model for discoveries to improve the treatment of human sepsis syndromes. We also posit that rFXI may have previously-unrecognized roles in the pathobiology of sepsis syndromes. This hypothesis warrants further investigation, especially given current efforts studying the safety and efficacy of therapeutic targets against FXI in sepsis. While not the central focus of our study, we also identified that total FXI levels were significantly lower in septic patients as previously observed in pediatric patients with meningococcal septic shock [13]. Our data extend these clinical findings to adult patients with sepsis due to a heterogeneous mix of pathogens that reflects current clinical scenarios in many ICUs.
Key Points.
The reduced form of Factor XI catalyzes coagulation and thrombosis.
Total and reduced Factor XI levels were measured in septic patients and matched controls.
Reduced Factor XI was associated with platelet count, illness severity, and thrombosis.
These data indicate a unique role for reduced Factor XI in sepsis-associated thrombosis.
Acknowledgements
We appreciate the excellent assistance of Ms. Diana Lim with generation of figures and all subjects for participating in the study.
Disclosures of Potential Conflicts of Interest. The authors have no conflicts of interest to disclose. This work was supported by the National Heart, Lung, and Blood Institute (HL112311, HL130541, and HL126547 to MTR), the National Institute on Aging (AG048022 to MTR), the U.S. Department of Veterans Affairs (VA Merit Award I01CX001696 to MTR) and from a pilot award funded by the University of Utah (MTR). This material is the result of work supported with resources and the use of facilities at the George E. Wahlen VA Medical Center, Salt Lake City, Utah. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government. The research reported in this publication was supported (in part or in full) by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR001067. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Research involving Human Participants. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
References
- 1.Kaplan D, Casper TC, Elliott CG, Men S, Pendleton RC, Kraiss LW, Weyrich AS, Grissom CK, Zimmerman GA, Rondina MT. VTE Incidence and Risk Factors in Patients With Severe Sepsis and Septic Shock. Chest. 2015; 148: 1224–30. 10.1378/chest.15-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iba T, Levy JH. Inflammation and thrombosis: roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J Thromb Haemost. 2018; 16: 231–41. 10.1111/jth.13911. [DOI] [PubMed] [Google Scholar]
- 3.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. 2013; 13: 34–45. 10.1038/nri3345. [DOI] [PubMed] [Google Scholar]
- 4.Giannakopoulos B, Gao L, Qi M, Wong JW, Yu DM, Vlachoyiannopoulos PG, Moutsopoulos HM, Atsumi T, Koike T, Hogg P, Qi JC, Krilis SA. Factor XI is a substrate for oxidoreductases: enhanced activation of reduced FXI and its role in antiphospholipid syndrome thrombosis. J Autoimmun. 2012; 39: 121–9. 10.1016/j.jaut.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 5.Zucker M, Seligsohn U, Yeheskel A, Mor-Cohen R. An allosteric disulfide bond is involved in enhanced activation of factor XI by protein disulfide isomerase. J Thromb Haemost. 2016; 14: 2202–11. 10.1111/jth.13488. [DOI] [PubMed] [Google Scholar]
- 6.von dem Borne PA, Meijers JC, Bouma BN. Feedback activation of factor XI by thrombin in plasma results in additional formation of thrombin that protects fibrin clots from fibrinolysis. Blood. 1995; 86: 3035–42. [PubMed] [Google Scholar]
- 7.Vincent JL, Ramesh MK, Ernest D, LaRosa SP, Pachl J, Aikawa N, Hoste E, Levy H, Hirman J, Levi M, Daga M, Kutsogiannis DJ, Crowther M, Bernard GR, Devriendt J, Puigserver JV, Blanzaco DU, Esmon CT, Parrillo JE, Guzzi L, Henderson SJ, Pothirat C, Mehta P, Fareed J, Talwar D, Tsuruta K, Gorelick KJ, Osawa Y, Kaul I. A randomized, double-blind, placebo-controlled, Phase 2b study to evaluate the safety and efficacy of recombinant human soluble thrombomodulin, ART-123, in patients with sepsis and suspected disseminated intravascular coagulation. Crit Care Med. 2013; 41: 2069–79. 10.1097/CCM.0b013e31828e9b03. [DOI] [PubMed] [Google Scholar]
- 8.Delabranche X, Helms J, Meziani F. Immunohaemostasis: a new view on haemostasis during sepsis. Ann Intensive Care. 2017; 7: 117 10.1186/s13613-017-0339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamura M, Takeuchi T, Kawahara T, Hirose J, Nakayama K, Hosaka Y, Furusako S. Simultaneous targeting of CD14 and factor XIa by a fusion protein consisting of an anti-CD14 antibody and the modified second domain of bikunin improves survival in rabbit sepsis models. Eur J Pharmacol. 2017; 802: 60–8. 10.1016/j.ejphar.2017.02.045. [DOI] [PubMed] [Google Scholar]
- 10.Bane CE Jr., Ivanov I, Matafonov A, Boyd KL, Cheng Q, Sherwood ER, Tucker EI, Smiley ST, McCarty OJ, Gruber A, Gailani D. Factor XI Deficiency Alters the Cytokine Response and Activation of Contact Proteases during Polymicrobial Sepsis in Mice. PLoS One. 2016; 11: e0152968 10.1371/journal.pone.0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tucker EI, Gailani D, Hurst S, Cheng Q, Hanson SR, Gruber A. Survival advantage of coagulation factor XI-deficient mice during peritoneal sepsis. J Infect Dis. 2008; 198: 271–4. 10.1086/589514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJ, Gailani D, Gruber A. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012; 119: 4762–8. 10.1182/blood-2011-10-386185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wuillemin WA, Fijnvandraat K, Derkx BH, Peters M, Vreede W, ten Cate H, Hack CE. Activation of the intrinsic pathway of coagulation in children with meningococcal septic shock. Thromb Haemost. 1995; 74: 1436–41. [PubMed] [Google Scholar]
- 14.Levi M, Toh CH, Thachil J, Watson HG. Guidelines for the diagnosis and management of disseminated intravascular coagulation. British Committee for Standards in Haematology. Br J Haematol. 2009; 145: 24–33. 10.1111/j.1365-2141.2009.07600.x. [DOI] [PubMed] [Google Scholar]
- 15.Levi M, Scully M. How I treat disseminated intravascular coagulation. Blood. 2018; 131: 845–54. 10.1182/blood-2017-10-804096. [DOI] [PubMed] [Google Scholar]
- 16.Semeraro N, Ammollo CT, Semeraro F, Colucci M. Coagulopathy of Acute Sepsis. Semin Thromb Hemost. 2015; 41: 650–8. 10.1055/s-0035-1556730. [DOI] [PubMed] [Google Scholar]
- 17.Dellinger RP. Inflammation and coagulation: implications for the septic patient. Clin Infect Dis. 2003; 36: 1259–65. 10.1086/374835. [DOI] [PubMed] [Google Scholar]
- 18.DeLa Cadena RA, Suffredini AF, Page JD, Pixley RA, Kaufman N, Parrillo JE, Colman RW. Activation of the kallikrein-kinin system after endotoxin administration to normal human volunteers. Blood. 1993; 81: 3313–7. [PubMed] [Google Scholar]