

Abstract
Ionotropic glutamate receptors (iGluRs) mediate excitatory neurotransmission within the mammalian central nervous system. iGluRs exist as three main groups: N-methyl-d-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) and kainate receptors. The past decades have witnessed a remarkable development of PET tracers targeting different iGluRs including NMDARs and AMPARs, and several of the tracers have advanced to clinical imaging studies. Here, we assess the recent development of iGluR PET probes, focusing on tracer design, brain kinetics, and performance in PET imaging studies. Furthermore, this Perspective will not only present challenges in the tracer development, but also provide novel approaches in conjunction with most recent drug discovery efforts on these iGluRs, including subtype-selective NMDAR and transmembrane AMPAR regulatory protein modulators, and positive allosteric modulators (PAMs) of AMPARs. These approaches, if successful as PET tracers, may provide fundamental knowledge to understand the roles of iGluR receptors under physiological and pathological conditions.
Graphical Abstract
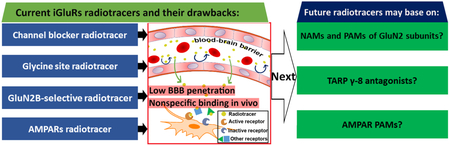
1. INTRODUCTION
L-Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system (CNS).1 Following its release from the presynaptic neuron, glutamate mediates two classes of receptor: ligand-gated ion channel ionotropic glutamate receptors (iGluRs) and G protein-coupled metabotropic glutamate receptors (mGluRs).2, 3 Based on their pharmacological properties, iGluRs can be further divided into three main distinct families: N-methyl-d-aspartate receptors (NMDARs), α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) and kainate receptors (KARs).4 These receptors are responsible for a majority of fast excitatory transmission in the brain and contribute to synaptic plasticity, which underpins learning and memory.1, 3 Furthermore, iGluRs play critical roles in both neural development and neurodegeneration, and their aberrant activity is implicated in a broad spectrum of brain dysfunctions including epilepsy, ischemic stroke and neurodegenerative diseases.3, 5 Therefore, iGluRs are targets of high therapeutic interest and there have been many ongoing drug development programs for these receptors.3, 6-9 Since positron emission tomography (PET) offers quantitative analysis by non-invasive imaging, which allows for tracking biological processes in vivo, a plethora of radiotracers have been developed and play indispensable roles in quantifying the in vivo concentration of iGluRs, as well as investigate the distribution and pharmacology of these receptors in physiological and pathological conditions.10-14 The PET tracers for NMDARs are the most studied, followed by the ones for AMPARs, and only one PET tracer has been reported to date for KARs. Complementary to several reviews on this topic,10-14 we assess the most recent development by providing not only an overview of iGluR PET probes, but also a brief introduction in recent pharmacological development, such as new subtype selective and/or allosteric iGluR modulators, which would offer a new direction for PET tracer development. In this review, we will first introduce ion channel and glycine binding site based NMDAR tracers, followed by GluN2B-subtype selective NMDAR tracers, as well as radioligands for AMPARs or KARs. Since there are no clinically approved PET tracers for iGluRs, we will also focus on new opportunities for the tracer development in alignment with recent drug discovery campaigns.
2. DEVELOPMENT OF RADIOTRACERS TARGETING NMDARs
Having crucial roles in CNS function, NMDARs are widely found in nerve cells of mammalian species,15 and are involved in a range of neurological and neurodegenerative disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and schizophrenia. These receptors are heteromeric complexes that contain three subunits: glycine-binding GluN1, glutamate-binding GluN2 (further divided into four subtypes GluN2A-GluN2D), and most rarely, glycine-binding GluN3 (further divided into two subtypes GluN3A and GluN3B) subunits.3, 6, 16-18 In most cases, NMDARs are considered as a dimer of dimers: two GluN1 and two GluN2 subunits. In GluN3-expressing cells, ternary GluN1/GluN2/GluN3 tetrameric complexes also exist.10, 15, 19 GluN1 subunits are widely distributed throughout the brain, while GluN2 subunits display different regional and developmental expression patterns, and most of them (except GluN2D subunit) are found in cerebral cortex, forebrain or cerebellum. In addition, GluN3A subunit is present in the cortex and brainstem, and GluN3B subunit is distributed in the cerebellum.6, 10, 19 NMDAR subunits all share a typical membrane topology containing three components: (1) two large extracellular domains, including amino-terminal domain (ATD), which are responsible for allosteric regulation, and ligand-binding domain (LBD) that binds glutamate (GluN2 subunits) and glycine (GluN1 and GluN3 subunits); (2) a transmembrane domain (TMD) comprising three transmembrane helices plus a re-entrant pore loop, which acts as the ion selectivity filter; and (3) a cytoplasmic C-terminus with the function of cellular trafficking and intracellular signaling.6, 15, 20 Current pharmacology discoveries have identified three classes of ligand binding sites: channel blockers,21, 22 competitive antagonists at the glutamate or glycine site,23 and most recently in drug development, the extracellular sites for non-competitive allosteric modulators.6, 10, 24
2.1. Channel blocker radiotracers
The flow of ions such as Na+, K+ and Ca2+ through the NMDAR ion channel can be inhibited by the binding of Mg2+ within the channel. Phencyclidine (PCP), thienylcyclohexyl piperidine (TCP), ketamine, memantine and MK-801 are channel blockers that were first used as therapeutic drugs, of which the working mechanism is thought to bind and block the ion channels in the open state. The radiolabeling of these compounds has led to the first generation of NMDAR PET tracers, and their preclinical evaluations have been reviewed by Waterhouse25 and Sobrio et al..10 So, in this section, we just give a brief review of several representative channel blocker radiotracers to clarify their development and drawbacks.
Two PCP derivatives [11C]1 (log P = 1.92) and [11C]2 (Figure 1, log P = 2.13) were labeled with carbon-11, and both tracers could readily enter the rat brain with the brain uptakes of 1.45% ([11C]1) and 1.38% ID/g ([11C]2) in cerebral cortex at 1 min post-injection (p.i.), but unfortunately, the distribution was uniform in different brain regions, indicating nonspecific binding in the brain.26 The radiolabeling of TCP derivatives generated seven 18F-labeled and one 11C-labeled PET tracers.10 Among them, [18F]3 had a Ki value of 38 nM to NMDARs (in a [3H]TCP assay), and the Bmax/Kd values (binding potential, a combined measure describing the ultimate binding force of a receptor to its ligand, and values greater than 5-10 are usually observed for successful radiotracers;10, 27-29 Bmax denotes the density of available binding sites in a sample of tissue, and Kd is for equilibrium dissociation constant of a radioligand) measured with [3H]3 in different rat brain regions were < 15.30, 31 In preliminary PET imaging in monkeys, N-methyl-d-aspartate (NMDA) could increase the uptake of [18F]3 in mesiotemporal regions, indicating that this tracer would be an index of the activation state of NMDARs, more than an index of their change in density.31 An analogue of compound 3 was radiolabeled with fluorine-18 to afford [18F]4 (Figure 1). The tracer displayed poor brain uptake in rat (0.10% ID/g at 30 min p.i.). PET imaging with [18F]4 in a rhesus monkey showed a rapid clearance and no different distribution between brain regions.32 MK-801 is an NMDAR blocker in PCP site with high affinity and high selectivity,33 and its derivatives have been labeled with carbon-11, fluorine-18 and iodine-123/125.10 Probe [11C]MKC ([11C]5, Figure 1, Kd = 8.2 nM in rat forebrain) was labeled in a high molar activity of 200-600 GBq/μmol, and showed a moderate Bmax/Kd ratio of 20 in rat forebrain membrane. PET imaging of [11C]5 in rhesus monkey exhibited rapid uptake in cortices, striatum and thalamic regions, but no substantial specific binding was detected upon the administration of MK-801 or ketamine.34 To develop single photon emission computed tomography (SPECT) probes, MK-801 was radiolabeled with iodine-123 and −125, and (±)-3-[123/125I]MK-801 ([123/125I]6, Figure 1) was obtained. Tracer [125I]6 demonstrated a desirable Bmax/Kd value of 28 in thin brain sections of rat. Furthermore, [123I]6 has entered the clinical study for SPECT imaging, but no significant specific uptake was observed in either cerebral ischemia patients or healthy age-matched controls.35, 36 These disappointing results of radiolabeled MK-801 analogs caused by high nonspecific binding may be attributed to their high lipophilicity.10 Ketamine is a non-competitive NMDAR blocker of PCP site with high specificity but poor affinity,37 and (S)-[11C]ketamine ([11C]7, Figure 1, Ki = 1.2 μM in pig forebrain38) has been used in clinic. In PET imaging studies of healthy volunteers, the heterogeneous uptake of [11C]7 in the brain was observed with 2.5-fold higher radioactivity in thalamus than in white matter, but rapidly declined into homogenous pattern at 20 min.39 Besides, a study in patients with medial temporal lobe epilepsy showed that no binding potential changes were observed after injection of [11C]7,40 which suggested the limited clinical usefulness of [11C]7 in assessing NMDARs. Memantine could readily penetrate the blood-brain barrier (BBB) and was used to treat the symptoms of moderate to severe AD. A 18F-labeled tracer [18F]fluoromemantine ([18F]8, Figure 1) has been used to detect NMDARs.41 The peak brain uptake of [18F]8 in mice was observed to be 3.7% ID/g occurred at 30 min p.i..42 However, [18F]8 failed to reflect the regional concentrations of NMDARs in the brains of neither rhesus monkey or human,41 which could be explained by its multi-target bindings to brain receptors such as dopamine D2 and sigma receptors.42, 43
Figure 1.

Chemical structures of selected channel blocker radiotracers.
Following the pharmacological studies of N-(2,5-disubstituted phenyl)-N´-(3-substituted phenyl)-N´-methylguanidines,44 several novel radiotracers binding the NMDAR ion channel (9-11, Figure 1) were developed.10 Among them, [11C]CNS 5161 ([11C]9, Figure 1, Ki = 1.87 nM in a [3H]MK-801 assay, log P = 2.68 45) has been translated into humans.45, 46 The results showed that [11C]9 had regionally heterogeneous uptakes in the brain with the lowest radioactivity in the cerebellum and the highest in the putamen and thalamus, but the specific binding was not demonstrated, and rapid plasma metabolism was observed.47, 48 In 2010, by introducing a S-fluoroalkyl substituent on the N-aryl group of 9, a 18F-labeled PET probe [18F]GE-179 ([18F]10, Figure 1) was reported.49 Probe 10 had high affinity to NMDAR at PCP site (Ki = 2.4 nM), and showed high selectivity toward 60 other neuroreceptors, channels, and transporters. PET imaging with nine healthy participants suggested that [18F]10 had high brain uptake and rapid brain extraction, with a relatively homogeneous distribution in gray matter and low between-subject variability (26.1% - 29.2%).50 A recent report from Sander and Hooker et al. revealed that, in PET imaging of both rats and nonhuman primates, [18F]10 failed to show any significant difference in brain signals during NMDA-specific modulation with drug challenges from GE-179, MK801, PCP, ketamine and ifenprodil.51 In addition, [123I]CNS 1261 ([123I]11, Figure 1, Ki = 4.2 nM in a [3H]MK-801 with rat brain synaptic membranes, log P = 2.19) was developed for SPECT,52 and turned out to be the most advanced ion channel radiotracer for imaging NMDARs to date.53 In healthy volunteers, [123I]11 presented different regional distribution in the brain with total distribution volume (VT, defined as radiotracer concentration in tissue of interest relative to that of unchanged tracer in plasma, and could be considered as proportional to binding potential and independent of blood flow10, 54, 55) values following: thalamus > striatum > cortical regions > white matter.54, 56 The selectivity of [123I]11 toward PCP site of NMDARs was confirmed by ketamine-displacement studies in human.57 In human studies, [123I]11 could monitor the schizophrenia and psychoactive effects caused by (S)-ketamine.58, 59 However, a recent study pointed out that [123I]11 had limited capability in detecting small changes of NMDARs.53
In summary, since the first radiolabeling of MK-801 in 1988, dozens of tracers have been developed for in vivo imaging of the NMDAR system. Channel blockers account for a substantial proportion, and can be termed as the first-generation radiotracers. Six radiolabeled probes including [123I]6, [11C]7, [18F]8, [11C]9, [18F]10 and [123I]11 have been tested in clinical studies. However, only the SPECT probe [123I]11 showed limited success in the clinical study of patients suffering from schizophrenia.60 Most of the channel blocker radioligands displayed excellent in vitro properties, such as high affinity (Ki < 10 nM), proper log P value (2 - 3.5) for BBB penetration,61, 62 and good selectivity in in vitro autoradiography. However, these tracers encountered impediments in in vivo PET imaging by presenting uptake patterns inconsistent with NMDAR distribution. These disappointing results may be attributed to 1) poor brain penetration, and 2) most importantly, high nonspecific bindings in vivo. In addition, since the channel blocker derived tracers bind to the NMDARs in an open state, the estimation of the total available sites remains challenging and unreliable, which makes it difficult to establish an appropriate kinetic model for PET quantification. Furthermore, since the PCP sites at NMDARs are at the intracellular side of the ion channels, cell internalization may slow receptor binding, and externalization from cells may slow receptor dissociation kinetics, which possibly complicate data analysis.25
2.2. Glycine site radiotracers
In the extracellular domain of GluN1 and GluN3 subunits, glycine or D-serine binds to LBD and serve as co-agonists of glutamate to activate the ion channel of NMDARs. Based on L-703717 (12, Figure 2, IC50 = 4.5 nM in rat membrane with [3H]L-689560 assay), an antagonist of NMDAR glycine site, a series of 11C-labeled quinolone derivatives were developed.10 [11C]L-703717 ([11C]12, Figure 2) displayed high binding ability to plasma albumin (8.37% ID/g in blood of ddY mice at 1 min p.i.), which led to its low BBB penetration in ddY mice with the uptake of 0.32% ID/g in cerebrum and 0.36% ID/g in cerebellum at 1 min p.i..63, 64 Radiolabeling of pro-drug acetyl L-703717 generated [11C]AcL703 ([11C]13, Figure 2) with two-fold increase in BBB penetration when compared with that of [11C]12.65 PET imaging in healthy human showed that [11C]13 had insufficient brain uptake (1.30% ID at 1.5 min p.i.), and the distribution pattern in brain was inconsistent with the expression of NMDARs.66 Another radiolabeled quinolone derivative [11C]4HQ ([11C]14, Figure 2, log P = 1.23) displayed moderate affinity (Ki = 170 nM) in a [3H]MDL-105519 binding assay,67 and was studied in the PET imaging of nonhuman primates, but failed because of low BBB permeability (0.53% ID/g in rat cerebrum 1 min p.i.) and high nonspecific binding.68 Continued research has resulted in several new radiotracers with high affinity to glycine site, such as [11C]GV150526X ([11C]15, Figure 2, Ki = 1 nM in [11C]glycine binding assay, no biological data reported)69 and [11C]3MPICA ([11C]16, Figure 2, Ki = 4.8 nM in [3H]MDL-105519 binding assay with rat cortical membranes)70. The uptake of [11C]16 in rat at 2 min p.i. was high in blood (6.1% ID/g), much lower in frontal cortex (0.12% ID/g), hippocampus (0.13% ID/g), thalamus (0.18% ID/g) and cerebellum (0.19% ID/g).70 According to the binding models revealed by Tikhonova and Leeson et al.,71, 72 a compound binding to glycine site usually have a negatively charged residue and an aromatic ring. The charged nature of these molecules may make them unlikely to cross the BBB. Therefore, low BBB penetration and high nonspecific binding impeded further PET imaging studies of these radiotracers, which represents a major obstacle for the development of glycine site radiotracers. Furthermore, since glycine is one of the major endogenous agonists of NMDARs, its presence in large quantity and regional variation in vivo makes it difficult to image the glycine binding site with PET.73
Figure 2.

Chemical structures of selected glycine site radiotracers.
The glutamate site is located at the extracellular domain of GluN2 subunits, which acts as orthostatic binding site for both agonists and antagonists. According to the structure-activity relationship studies, the agonists at glutamate site are featured by the presence of at least three charged groups, which poses major obstacles for them to penetrate BBB.74, 75 The antagonists are developed by making minor modifications to the structures of agonists, so it is not surprising that they also have low BBB permeability.75 Thus, there are no successful radioligands were reported to interact with glutamate site.13
2.3. GluN2B subtype-selective radiotracers
GluN2B subunit displays developmental expression patterns in the brain, which is maintained at high levels around birth, and then becomes progressively restricted to the forebrain, but barely detectable in the cerebellum.15 The GluN2B-ATD is the major site for subunit-selective allosteric binding, which harbors negative allosteric modulators (NAMs, e.g. ifenprodil), and might also show overlap with positive allosteric modulators (PAMs, e.g., Mg2+ and polyamines).6, 15 GluN2B subunit has been found to play essential roles in neurological and psychiatric disorders such as pain, AD, PD and schizophrenia, and gained increased attention as a novel therapeutic approach.15 Broad-spectrum NMDAR antagonists such as memantine, ketamine or amantadine are mainly used in clinic, but these drugs often induce a range of cognitive and motor side effects, which are probably attributed to a very narrow therapeutic window.3, 6 In comparison, GluN2B-selective antagonists, like ifenprodil (17, Figure 3) and its derivatives, Ro 256981, RGH-896 and MK-0657 (26, Fig 3), are well tolerated with few undesired effects, paving a promising way in developing subtype selective NMDAR drugs.6, 76, 77 PET imaging with GluN2B-selective radiotracers can be a useful tool for studying diagnosis and treatment intervention of related neurological diseases, and further supporting the drug development.
Figure 3.

Chemical structures of ifenprodil (17) and GluN2B-selective PET tracers (18-32).
Ifenprodil is the first member of ‘prodil’ drugs that are used as GluN2B-selective antagonists, and the radiolabeling of these drugs produced several PET probes. In 2002, Haradahira et al. reported the first ‘prodil’ PET tracer [11C](±)-methoxy-CP-101606 ([11C]18, Figure 3) with moderate affinity (IC50 = 14 nM) to GluN2B subunit.78 In vitro autoradiography revealed highly specific binding of [11C]18 in the forebrain of rat brain slices with nonspecific binding < 5%. However, in vivo evaluations, including biodistribution in mice and PET imaging in nonhuman primates, showed that no specific localization of the radioactivity was observed in any of the brain regions. In addition, [11C]18 showed good biostability in mice brain with 70% remaining unchanged at 90 min p.i.. However, it was postulated that polyamines and cations behaved as endogenous inhibitors for the probe binding of [11C]18, which caused the loss of the specific binding in vivo. In 2003, Roger et al. reported compound 19 with both high affinity (IC50 = 5.3 nM) and selectivity to GluN2B subunit (IC50: GluN2A = 35 μM, GluN2C > 1 mM). However, the radio-labelled counterpart [11C]19 displayed low uptake (0.07% ID/mL at 30 min) and uniform distribution in rat brain with the highest uptake in the cerebellum (0.1% ID/mL) and the lowest in the striatum (0.04% ID/mL), which is inconsistent with the GluN2B distribution pattern. In competition studies with ifenprodil, an increase in brain uptake of [11C]19 was observed, which failed to indicate its specific binding in brain.79 In 2004, by modifying the structure of 19, the same research group reported [11C]EMD-95885 ([11C]20, Figure 3, clog P = 4.0) with better affinity to GluN2B subunit (IC50 = 3.9 nM).80 Similar to [11C]19, [11C]20 displayed low and uniform uptake in different regions of rat brain (0.4%-0.6% ID/mL at 5 min p.i.), in which the highest and lowest uptakes were found in the cerebellum and striatum, respectively. In pretreatment studies, both compounds 17 and 20 significantly reduced the brain uptake of [11C]20 by a degree of 40-60%. Pretreatment of rats with DTG (a sigma receptor agonist), MDL105519 (a glycine site antagonist) and MK801 had no inhibitory effect on the uptake of [11C]20. Use of haloperidol (an antagonist for D2, D3, and D4 receptors and an inverse agonist of sigma-1 receptor) as a blocking drug led to homogeneous inhibition of [11C]20 uptake by 66-60%, which indicated significant off-target binding. In 2009, [11C]21 with high affinity to GluN2B subunit (IC50 = 5 nM) was reported by Labas et al. as an analog of 20.81 However, no sufficient BBB penetration of [11C]21 was observed in the PET imaging on rats.
In 2003, Merck Research Laboratories reported a novel series of benzamidines as GluN2B-subtype selective NMDA antagonists,82 and four PET tracers were reported thereafter. A 18F-labeled compound [18F]22 (Figure 3, log P = 0.9) showed high affinity to GluN2B subunit with the Ki value of 1.5 nM, but no further biological evaluation was reported.83 In 2006, three benzamidine analogs were radiolabeled with carbon-11 ([11C]23-25, Figure 3, log P = 1.02, 2.05 and 1.47, respectively), and their biological properties were reported.84, 85 These probes displayed high affinity to GluN2B subunit with Ki values < 6 nM. In vitro autoradiography in rat brain sections suggested that [11C]25 had comparable binding patterns with [3H]ifenprodil by exhibiting high radioactivity in caudate putamen and thalamus while much lower in the cerebellum. The specific signals can be blocked by both ifenprodil and 25 itself. Furthermore, the initial brain uptake of [11C]23-25 was measured in mice, and the results showed that they had limited brain uptake (< 2% ID/g at 5 min p.i.) with slow brain washout (brain5min/brain40min < 3). Besides, [11C]23-25 metabolized rapidly in mice brain, leading to 8%, 14% and 49% intact compounds were remained at 40 min p.i., respectively. In conclusion, though high affinity to GluN2B subunit, the use of these benzamidine PET tracers have been impeded by low brain uptake and rapid metabolism in the brain.
Different from aforementioned ‘prodil’ molecules, MK-0657 (26, Figure 3) is a non-phenol containing GluN2B antagonist (IC50 = 3.6 nM), and has been tested in clinical trials for the treatment of patients with PD and major depression.6, 86, 87 The related PET tracers, [18F]trans-26 and [18F]cis-26 (Figure 3, log P = 2.66 and 2.80, respectively), were developed by Koudih et al. in 2012.88 As demonstrated by autoradiography, both radioligands showed extremely high specific binding in GluN2B subtype rich regions of rat brain sections with the highest density detected in the hippocampus and cortex, followed by striatum and thalamus, and the lowest in the olfactory tubercle and cerebellum. Furthermore, both tracers showed moderate to high Bmax/Kd ratios (>10) in different brain regions, and [18F]trans-26 (Bmax/Kd = 17-37) showed higher values than that of [18F]cis-26 (Bmax/Kd = 8-19). In the subsequent PET studies, although [18F]trans-26 and [18F]cis-26 exhibited reasonable brain-to-blood ratios of 1.97 and 1.88 at 90 min p.i., respectively, no significant difference of radioactivity uptake was observed within different brain regions. Also, co-injection of non-radioactive 26 did not significantly affect the regional cerebral uptakes, which demonstrated possible high nonspecific binding in vivo. Metabolite analysis showed that both tracers displayed good biostability in rat brain with 79% ([18F]trans-26) and 71% ([18F]cis-26) of parent tracers at 90 min p.i..89 Despite promising in vitro properties (high affinity, good selectivity to GluN2B subunit in brain sections, and proper log P values), the limited brain uptake and homogeneous distribution have impeded [18F]trans-26 and [18F]cis-26 as proper PET tracers for the imaging of GluN2B subunit.
To pursue optimal PET tracers for imaging GluN2B subtype, several new chemical scaffolds were proposed. In 2004, Dolle et al. developed a pyridine tracer [11C]Ro-647312 ([11C]27, Figure 3) with high affinity of 8 nM (Ki in [3H]Ro-256981 assay).90 However, biodistribution results in rat indicated that [11C]27 distributed homogenously in all brain regions, which was not consistent with the known expression of GluN2B subunit. In addition, throughout the PET scan (0-60 min), plasma radioactivity was observed higher than brain in any period. In 2014, Christiaans et al. reported [11C]28 (Figure 3) as a PET probe for GluN2B subunit. Probe [11C]28 had moderate affinity to GluN2B subunit (Ki = 12.4 nM in [3H]ifenprodil assay).91 In vitro autoradiography suggested that [11C]28 had homogenous distribution in rat brain sections with low specific binding. Biodistribution results in mice showed that [11C]28 could readily penetrate BBB, and washed out rapidly without significant regional differences at 30 min after intraperitoneal injection. In ex vivo autoradiography, brain uptake of [11C]28 in mice was reduced by pretreatment of Ro-256981, but the reduction was inconsistent with the expression patterns of GluN2B subunit, which may be caused by the high level of sigma-1 receptor binding. In 2017, [11C]Me-NB1 ([11C]29, Figure 3) was generated by 11C-labeling of a GluN2B antagonist, WMS-1405,92 at the methoxy group.93 This probe displayed high affinity to the GluN2B-ATD binding site with the Ki value of 5.3 nM, and moderate affinity to the sigma-1 receptor (Ki = 182 nM).92 In vitro autoradiography revealed that [11C]29 could bind to the whole brain with nearly homogenous distribution, and the binding could be blocked by non-radioactive 29 (100 μM), eliprodil (10 μM, an NMDAR antagonist) and Ro-256981 (100 μM, a GluN2B NAM), but not by glutamate (1 mM) and haloperidol (10 μM). In addition, ex vivo biodistribution results revealed that [11C]29 distributed in different brain regions with standardized uptake values (SUVs) of 3.8 for midbrain, 3.7 for cortex, 2.8 for cerebellum and 2.7 for olfactory bulb. During eliprodil blocking studies, these values were reduced by > 40% for midbrain, brain stem, cortex and striatum, and to a less extent, for hippocampus (31.9%) and cerebellum (34.5%). PET imaging results suggested that the binding of [11C]29 in rat brain was blocked by cold compound of 29 (1 mg/kg), eliprodil (2 mg/kg) and Ro-256981 (7.5 mg/kg). However, the specific binding in PET imaging was abolished by (+)-pentazocine (2.5 mg/kg, sigma-1 receptor agonist) and haloperidol (0.13 mg/kg), which may be caused by in vivo indirect effect of sigma-1 receptor on [11C]29 binding. Furthermore, [11C]29 was successfully used in the determination of receptor occupancy by eliprodil. The results revealed that the half-maximal receptor occupancy blocked by eliprodil was observed at 1.5 μg/kg, and at a dose with reported neuroprotective effects (1 mg/kg), more than 99.5% of binding sites were occupied. Thus, [11C]29 has great potential in in vivo imaging of GluN1/GluN2B receptors and the determination of receptor occupancy by GluN2B-ATD modulators. More recently, [18F]30, a 18F-labeled derivative of 29, was reported by Szermerski et al. as a GluN2B PET imaging probe.94 Compound 30 displayed 30-fold lower affinity (Ki = 162 nM) to GluN2B subunit compared with 29. In vitro autoradiography results in rat brain sections suggested that [18F]30 accumulated in brain regions of cortex, hippocampus, striatum, and hypothalamus, which are known to have high expression of GluN2B subunit. This binding was blocked by non-radioactive of 30 (100 μM), eliprodil (100 μM) and Ro-256981 (100 μM), which further confirmed the specific binding between [18F]30 and GluN2B subunit. However, no further in vivo results of [18F]30 were reported.
The aforementioned GluN2B-selective radiotracers are NAMs and possibly bind to GluN2B subunit as non-competitive antagonists. In addition, two alkylamines ([11C]31 and [11C]32, Figure 3) were radiolabeled as tracers that bind with GluN2B subunit as PAMs.95 In in vitro autoradiography, both probes exhibited similar distributions in rat brain slices with homogenous radioactivity in different brain regions. Since [11C]31 showed off-target binding to serotonin receptors, [11C]32 was selected for further evaluations. Biodistribution revealed that [11C]32 had poor uptake in different brain regions (0.74-0.89% ID/g at 10 min p.i.) in rodents, and further PET scans in rhesus monkey showed similar results with homogeneous regional distribution, which was likely affected by endogenous PAMs of GluN2B, such as polyamines and neurosteroids.
In summary, many GluN2B-selective radiotracers reported to date showed high affinity and high selectivity to GluN2B subunit, and most of them displayed in vitro specific binding on rat brain slices by autoradiography. However, undesirable in vivo properties, including low BBB penetration, uniform distribution in whole brain, rapid brain metabolism, and/or off-target binding to other brain receptors have led to the limited success of GluN2B PET tracer development.
3. DEVELOPMENT OF PET TRACERS FOR AMPARs
As another type of glutamate-gated ion channels, the AMPARs are primarily expressed on postsynaptic membranes of excitatory synapses, and mediate the majority of fast synaptic transmission in the CNS by the activation of glutamate.96, 97 AMPARs assemble as functional homotetramers of four subunits GluA1-GluA4,98 and directly or indirectly associate with numerous scaffolding proteins,99 including transmembrane AMPAR regulatory protein (TARP), cornichon (CNIH), and CKAMP44, to modify their trafficking, localization, gating as well as pharmacology.100-105 It has been revealed that AMPAR subunits have a widespread and varied distribution in rat brain: GluA1-GluA3 subunits have high expression in hippocampus, outer layers of the cortex, olfactory regions, lateral septum, basal ganglia and amygdala; and GluA4 subunit is found enriched in reticular thalamic nuclei and cerebellum.4, 106 Like all iGluR subunits, AMPAR subunits share a modular composition consisting of 1) a ATD that for the assembly and stability of tetrameric receptors98 (this is different from NMDARs, see Chapter 2.3), and may also harbor allosteric binding sites (some functions are still unknown);106 2) a LBD possessing binding sites for modulators (such as agonist, antagonist and PAM) of receptor desensitization and deactivation;98, 107, 108 3) a TMD that forms the ion channel;109 and 4) a cytoplasmic carboxy-terminal trafficking and anchoring domain. Studies have indicated that AMPARs not only play an important part in learning and memory,3, 110 but also are involved in the pathology of several CNS disorders including epilepsy, schizophrenia, multiple sclerosis as well as PD.111-113 Thus, modulation of AMPARs are attractive therapeutic approaches, and much attention has been focused on AMPAR antagonists for neuroprotection,114 and AMPAR potentiators for cognitive enhancement.115
3.1. Antagonist-type AMPAR tracers
Non-invasive in vivo PET imaging studies offers an ideal opportunity for quantification of AMPARs in the living brain under normal and disease conditions, which would facilitate and advance drug development, as well as enable pharmacokinetic profiling of candidate drugs. While considerable efforts have been devoted to imaging NMDARs, the development of AMPAR PET tracers is still in its infancy. To date, the focus has been dedicated to AMPAR antagonist-type PET ligands. In 2006, based on N-acetyl-1-aryl-6,7-dimethoxyl-1,2,3,4-tetrahydroisoquinoline scaffold, Gao et al. disclosed the synthesis of several 11C- and 18F-labeled AMPAR antagonists ([11C]33-38 and [18F]39, Figure 4).116 However, except that [11C]37 was further described by the Årstad et al.,117 no other biological data were reported. Tracer [11C]37 had a moderate Kd value of 14.8 nM in rat cortex membranes, and the Bmax was 148 fmol/mg protein. In addition, [11C]37 showed good initial brain uptake at 1 min p.i. in rats with the highest in inferior colliculi (3.12% ID/g), following by superior colliculi, cingulate cortex, olfactory tubercles, thalamus and prefrontal cortex around 2.4% ID/g, and the lowest in hippocampus (1.84% ID/g). Furthermore, [11C]37 displayed good biostability in rat brain with 86% of the tracer was intact at 30 min p.i.. However, the authors claimed that the binding patterns of [11C]37 in the brain is likely the combination of regional blood flow and unspecific binding. Another class of AMPAR antagonist-type PET ligands was based on perampanel (Fycompa™) scaffold, which is the only AMPAR drug that has been approved by the U.S. Food and Drug Administration (FDA) for the treatment of epilepsy.7-9 In 2015, Lee et al. developed a novel radiosynthetic protocol for the synthesis of [11C]aryl nitriles, and translated it into the preparation of 11C-labeled perampanel [11C]40, but no in vitro or in vivo evaluations were reported to date.118 In the same year, Oi et al. developed a series of perampanel derivatives as AMPAR antagonists, and twelve 11C-labeled and one 18F-labeled radioprobes were successfully prepared.119 Among them, five radiotracers ([11C]41-44 and [18F]45, Figure 4) were highlighted because of their higher affinity (Ki = 6, 10, 5. 20 and 22 nM, respectively) and desirable lipophilicity (log D = 1.67, 2.57, 1.95. 1.70 and 2.54 nM, respectively). In in vitro autoradiography with rat brain slices, [11C]41-44 displayed excellent specific binding with the radioactivity signals concentrated at brain regions of hippocampus and cortex, versus brain stem, which is consistent with the expression of AMPARs. These specific signals were well blocked by the corresponding unlabeled compounds (10 μM). On the contrary, no specific binding was observed from the autoradiographic results of [18F]45, which was further confirmed by the PET imaging in rats. The in vivo performance of [11C]41-44 were assessed with PET imaging in both rats and monkeys. In PET imaging of both species, [11C]42 displayed moderate brain uptake, and showed notable radioactivity retention in AMPARs-rich brain regions like hippocampus and cortex, which contrasted with the relatively rapid clearance from brain stem. Furthermore, the uptake was blocked by pretreatment with unlabeled 42 in a dose-dependent manner. Besides, [11C]41 and [11C]43 showed low brain uptake in both rat and monkey. Although [11C]44 showed higher brain uptake than [11C]42 in rat, no specific binding was observed with PET imaging in both species, which may caused by the insufficient affinity. Further human PET studies using [11C]42 demonstrated that this tracer had low binding potential less than 0.15 (here, a value > 0.5 is desirable for reliable quantification120), and only some specific binding in the cerebral cortex was observed, so this tracer may not be optimal for AMPARs PET imaging.121 In addition, in 2016, Liang and his coworkers demonstrated the feasibility to label compound 44 with fluorine-18. The radiosynthesis of [18F]44 was achieved via a one-pot two-step approach that utilized a spirocyclic iodonium ylide (SCIDY) mediated radio fluorination with K18F, followed by cross-coupling with an amide, in a decay-corrected radiochemical yield of 15%.122 PET imaging results in mice demonstrated that [18F]44 could penetrate BBB and reach a maximum whole brain activity of 2.3 SUV at ~70 s p.i.. However, no information regarding specific binding of [18F]44 was reported.
Figure 4.

Chemical structures of reported PET tracers for AMPARs (32-48 and 50) and KARs (51), and an AMPARs potentiator (49).
3.2. Potentiator-type AMPAR tracers
Compared with the existence of antagonist-type PET ligands, fewer tracers of potentiator-type were reported. AMPAR-enhancing Ampakines™ drugs were developed as PAMs, and three compounds CX-465, CX-546 and CX-516 (46-48, respectively, Figure 4) were labeled with carbon-11 in the carbonyl position.123 The biodistribution of [11C]46-48 in rats was determined by rapid PET screening. The PET imaging results in rats showed that [11C]46 could be extracted into the brain rapidly with the uptake of ~15% ID/mL tissue at 1 min, but rapidly decreased to the level of extracerebral tissue at 10 min. When co-injected with CX-465 (self-blocking, intraperitoneal injection), the brain uptake of [11C]46 was decreased and the washout rate was slowed down. Tracer [11C]47 displayed improved cerebral retention (half-time of 20 min) than [11C]46, and the retention time also became longer in self-blocking experiments. The brain uptake of [11C]48 reached a peak at 2-3 min p.i., and followed by a decrease then a gradual increased uptake at later time point, which may be caused by polar brain penetrant radiometabolites. Another potentiator-type tracer was developed by 18F-labeling of a LY395153 (49, Figure 4)124 derivative ([18F]50).125 However, unlike LY395153, cold compound 50 failed to potentiate the [3H]AMPA binding on frozen brain slices. Besides, in in vitro assay with rat brain slices, [18F]50 showed a uniform distribution and high non-specific binding, which hence hampered subsequent in vivo imaging evaluation.
Despite these advances, all reported AMPAR PET tracers suffered from at least one of the following disadvantages, including low BBB penetration, high non-specific binding or low binding potential, which in turn rendered them unsuitable for preclinical and clinical imaging studies. Furthermore, it has been found that AMPARs have a low density in the brain, which suggests that a ligand with very high affinity, even in a sub-nanomolar range, might be required to precisely image the AMPARs in vivo.
4. PET TRACER DEVELOPMENT TOWARDS IMAGING KARs
Among the three main types of iGluRs, KARs possess the lowest density in the CNS, and the study of their biological and physiological function is still in early stage. KARs are similar to NMDARs and AMPARs in homologs, and can be further divided into five subtypes named GluK1-GluK5, which co-assemble as a tetramer in various combinations to exert either pre- or post-synaptic functions.126 Unlike NMDARs and AMPARs, KARs do not act predominantly as modulators of excitatory postsynaptic signaling. KARs principally associate to metabotropic signaling pathways to regulate synaptic transmission and neuronal excitability. These modulatory functions have positioned KARs as potential therapeutic targets, and thus KAR antagonists have emerged for the amelioration of several neurological disorders such as epilepsy, migraine and chronic pain.127-129 Compared with the development of other iGluR PET tracers, the progress of imaging KARs has lagged behind, possibly due to unavailability of suitable high-affinity and high-selectivity ligands. The only candidate KAR PET tracer [11C]51 (Figure 4) to date was reported by Suzuki in 2011, which undesirably showed higher affinity to AMPARs rather than to KARs. Thus, no utility of [11C]51 as a PET probe has been established.130
5. PERSPECTIVES FOR iGluRs PET TRACER DEVELOPMENT
5.1. Opportunities in developing PET tracers for GluN2A, GluN2C and GluN2D subunits
While traditional non-subtype selective NMDAR drugs elicited a range of side effects, including hallucination, catatonia and anesthesia,19, 75 recent efforts have been shifted to subtype-selective NMDAR modulators via allosteric binding sites targeting GluN2B or other NMDAR subunits.24, 72, 75 These allosteric agents often could minimize off-target activity and detrimental side-effects. Among them, NAMs could avoid excessive blockade by inhibiting less than 100% of the agonist response at high concentrations, and this makes them superior to other pan NMDAR drugs including channel blockers and competitive antagonists, which at high concentrations can block all pharmacological effects. Furthermore, potentiating NMDARs could be beneficial for the treatment of cognitive disorders and schizophrenia that are caused by the receptor hypofunction. However, direct activation of NMDARs by glutamate agonists may potentially cause excitotoxicity. PAMs may offer distinct advantages in the treatment of such NMDAR hypofunction-related diseases because they could selectively increase the activity of weakly-activated NMDAR signals.72, 131
Several GluN2B-selective agents, including RGH-896 and CP-101606, have been developed and entered clinical trials. In synergy with drug development, major efforts in exploring NMDAR subunit-selective PET probes have been focused on GluN2B subunit. In addition, the recent development of other subtype-selective agents, and NMDAR NAMs and PAMs offer unique opportunities for a new series of subtype-selective NMDAR PET tracers that are not limited to GluN2B subunit.72, 108, 131 In 2015, Sephton et al. reported a 18F-labeled quinoxaline derivative, [18F]FP-PEAQX ([18F]52, Figure 5), as a potential PET imaging probe for GluN2A subunit.132 Compound 52 displayed high binding affinity (2 nM) to GluN2A subunit. In in vitro autoradiography studies, the distribution of [18F]52 in rat brain showed high brain uptake in regions such as cortex and hippocampus, which was consistent with the known expression of GluN2A.19 Furthermore, by blocking experiment, these radioactivity signals were completely blocked by NVP-AAM077 (a GluN2A antagonist), while no significant change was observed when eliprodil was used. Although high specificity to GluN2A subunit in vitro, the log D7.4 value of [18F]52 was −1.11, which indicated that the probe may have poor BBB penetration.61 So, further tuning of lipophilicity based on [18F]52 may lead to optimal brain penetrant probes for GluN2A subunit. In 2016, Tamborini et al. reported several radiolabeled amino acids as GluN1/2A tracers with poor affinity and low selectivity toward GluN1/2B.133 Ex vivo autoradiography suggested that [125I]53 (Figure 5, estimated Ki: 250 and 270 nM to GluN1/2A and GluN1/2B, respectively; clog P = −3.0) failed to penetrate BBB, while the injection of its prodrug [125I]54 (Figure 5, clog P = 1.1) generated brain region-specific uptake that was consistent with mRNA expression of GluN1/2A. Unfortunately, biodistribution and PET imaging of [125I]54 and [11C]55 (Figure 5, clog P = 0.3) revealed that none of them are brain permeable (< 1% ID/g across all the brain regions). More recently, Lind et al. reported a fluorinated analog ST3 (56, Figure 5), which displayed an improved binding affinity and selectivity with Ki values of 52 nM and 782 nM for GluN1/2A and GluN1/2B receptors, respectively.134 This work could serve as an excellent starting point for a new series of PET tracers that can be radiolabeled by fluorine-18.
Figure 5.

Chemical structures of reported radiotracers (52-55) and an antagonist (56) for GluN1/2A subunit, and NAMs (57-59 and 62) and PAMs (60, 61, 63 and 64) for GluN2A-2D subunits.
Ifenprodil is a GluN2B-selective NAM, and its pharmacology has been well described.6 Recently, NMDAR allosteric agents have received increasing attention in drug discovery, and several subtype-selective NAMs and PAMs have been reported.135 TCN-201 (57, Figure 5, clog P = 3.39) was the first identified subtype-selective NAM that had low affinity to GluN2A (IC50, 340 nM), and > 300-fold selectivity over other GluN2 subunits.136 MPX-004 and MPX-007 (58 and 59, Figure 5, clog P = 1.02 and 0.69, respectively) are sulfonamide compounds that act as GluN2A-selective NAMs. They showed improved affinity to GluN2A subunit with the IC50 values of 79 and 27 nM, respectively, and displayed potent selectivity.137 In addition, GluN2A-selective PAMs were also reported, and GNE-0723 (60, Figure 5) is the most successful compound to date. GNE-0723 displayed a EC50 value of 21 nM to GluN2A, and ~300-fold selectivity over both GluN2C and GluN2D subunits, > 200-fold selectivity over AMPARs, and yet acted more selectively toward GluN2B.138 Another generation of compound GNE-5729 (61, Figure 5) showed improved pharmacokinetic profile, such as oral bioavailability and in vivo mouse clearance.139 However, the affinity to GluN2A subunit (EC50 = 37 nM) and the selectivity to GluN2C and GluN2D subunits (127- and 256-fold, respectively) were both decreased. Furthermore, NAMs and PAMs with subtype-selectivity for other subunits including GluN2C/2D and GluN2C have been reported. NAB-14 (62, Figure 5) is a GluN2C/2D-selective NAM with the potency (IC50) of 3.7 and 2.2 μM to GluN2C and GluN2D, respectively, and showed high selectivity over GluN2A and GluN2B (> 800-fold).140 In addition, a series of tetrahydroisoquinoline derivatives were reported as PAMs of GluN2C and GluN2D subunits. Compound 63 is the most potent one with the EC50 value of 0.3 μM for both GluN2C and GluN2D subunits.141 PYD-106 (64, Figure 5) is a notable PAM because it can distinguish GluN2C from GluN2D.142 Compound 64 had no effect on GluN2A, GluN2B and GluN2D, as well as AMPARs and KARs, and acted on GluN2C subunit to stabilize an open channel state of the receptor complex. The new class of subtype-selective compounds that are featured by these NAMs and PAMs still belong to the starting stage, and only have moderate to poor affinity to respective subunits. But with future efforts, more subtype-selective compounds with improved solubility, brain penetration, and pharmacokinetic/toxicity properties would be developed,135 which should offer important opportunities to drug discovery. Thus, related PET tracers could help to explore their pharmacological properties, and further understand the structure-function characteristics in their allosteric sites.
5.2. Opportunities for the development of subtype-selective AMPAR PET tracers
Considering the widespread distribution of AMPARs throughout the CNS, general antagonism or potentiation may exert beneficial pharmacological effects yet with severe adverse effects, including ataxia, sedation, and dizziness.8 Development of subtype-selective agonists or antagonists for AMPARs seems to be a very difficult task because the orthosteric binding sites have high degree of sequence identity among the subunits.143 Gratifyingly, one of the recent drug discovery efforts towards this target entails subtype-selective TARP modulation. TARPs have been found to dramatically enhance AMPAR trafficking and gating in a brain region-specific manner. TARPs belong to claudin protein family and include several subtypes, including type I TARPs (γ2, γ3, γ4 and γ8), type II TARPs (γ5 and γ7), and other candidate AMPAR auxiliary proteins.144 Among them, TARP γ-8 is highly expressed in the hippocampus but very low in the cerebellum, and acts as an exciting target in the treatment of pathologic disorders featured by hyperactivity within forebrain.145 In 2016, several TARP γ-8 dependent antagonists were developed, and among them, JNJ-55511118 (Ki = 26 nM in [3H]JNJ-56022486 assay with rat hippocampus membranes)146 and LY3130481 147 (65 and 66, respectively, Figure 6) displayed promising pharmacokinetic properties. Compound 65 showed high binding affinity to human GluA1i/γ-8 with the pIC50 value of 7.91, and act as a reversible TARP γ-8 antagonist with potential therapeutic utility as an anticonvulsant or neuroprotectant. In rat hippocampus membranes, 65 showed binding affinity (Kd) of 27 nM, and the Bmax is 3.8 pmol/mg protein. In vivo clearance and volume of distribution (VD, also known as apparent volume of distribution, is a theoretical volume that calculated as a ratio of the dose present in the body and its plasma concentration, when the distribution of the drug between the tissues and the plasma is at equilibrium) in rats were 4.8 mL/min/kg and 1.8 L/kg, respectively.146 Compound 66 also had high potency to GluA1/γ-8 with the IC50 value of 65.3 nM, and the VD and total clearance in rat are 1280 mL/kg and 11.4 mL/min/kg. It has been demonstrated that 66 displayed an effective neuroprotective effect toward pentylenetetrazole-induced convulsions in rats without motor impairment.147 These compounds could provide informative molecular scaffolds in the exploration of subtype-selective PET tracers for AMPARs. Moreover, TARP γ-2, the first discovered TARP member, is highly expressed in the cerebellum, where it plays a crucial role in the biological function of AMPARs in cerebellar granule neurons.144 However, no γ-2-selective antagonist was reported to date. Furthermore, AMPAR PAMs show selectivity for AMPAR subpopulations of different subunit compositions. These PAMs could potentiate AMPARs, and showed cognition- and memory-enhancing, and antidepressant-like effects in the preclinical models, demonstrating therapeutic effects on cognitive impairment, schizophrenia, depression and PD.113 A variety of PAMs have been developed, and several of which have developed into clinical trials, such as CX-516 (48, Figure 4) and LY404187 (67, Figure 6).113, 148 Furthermore, recent drug discovery efforts led to a few new AMPAR PAMs with excellent safety and tolerability in humans, such as LY451395,149, 150 PF-04958242151 and UoS12258152, 153 (68-70, respectively, Figure 6). Thus, AMPAR-TARP subtype-selective antagonists and AMPAR PAMs may hold great potential for new CNS drug discovery, and in parallel, the future direction towards PET imaging ligand development of AMPAR may be shifted to seek subtype-selective radiotracers, thereby facilitating target engagement studies and monitoring treatment efficacy of AMPAR related pharmacotherapy.154, 155
Figure 6.

Chemical structures of TARP γ-8-dependent antagonists (65 and 66) and AMPAR PAMs (67-70).
Currently, the development of iGluR PET agents usually proceeds by synthesizing compounds based on the scaffolds of drugs with therapeutic activity. However, the pharmacokinetic requirements of a PET agent, like of high initial brain uptake, rapid brain extrusion and minimum brain nonspecific binding, are not requirements of neuroactive therapeutic drugs. In addition, the doses of therapeutic agents are far higher than imaging agents, allowing their interaction with receptors in lower affinity. High-level of specific binding is very important for PET imaging agents and the corresponding quantification process. The nonspecific binding of a radioligand may be caused by high lipophilicity and/or insufficient binding affinity to the target. Therefore, while a therapeutic lead may provide a good entry point for PET tracer development, the proposed imaging scaffold must focus on the improvement/fine tuning of binding affinity (in subnanomolar level) to achieve high specific binding. Furthermore, their molecular properties such as topological polar surface area (tPSA) and log D, should be scrutinized in silico against a training set of successful neuroimaging agents. In addition, BBB permeability is increasingly amenable to investigation as a screen for compounds to be tested in vivo. Thus, these methods would help to improve success rate in developing useful imaging agents.
6. CONCLUSION
Given the critical role of these iGluRs in various neurological and neurodegenerative disorders, imaging these receptors by PET or SPECT would provide invaluable information on disease diagnosis and therapeutic intervention. During the past decades, significant advances have been made in the field of developing radioligands for iGluRs. Several tracers have been transitioned into clinical research studies, among which [123I]CNS 1261 ([123I]11) is the only one that has gained promising yet limited success in studying NMDAR-related disorders, such as ketamine-induced blockade, psychoactive effects of (S)-ketamine and schizophrenia. Most of the PET tracers displayed inferior in vivo results, including low brain uptake, rapid metabolism, and non-specific binding. In addition, limited progress has been achieved in the development of radiotracers for AMPARs and KARs, and no subtype-selective tracer has been reported to date. Furthermore, recent advances in the protein structures of AMPAR and KAR subtypes in the closed, active and desensitized states have promoted molecular level understanding of receptor-drug interactions, which may present new opportunities for PET tracer development.156, 157
ACKNOWLEDGMENTS
We thank the Division of Nuclear Medicine and Molecular Imaging, Department of Radiology, MGH and HMS for general support and Prof. Thomas J. Brady for helpful discussion. This study was financially supported by the National Natural Science Foundation of China (81501534), Fourth Round Fujian Health Education Joint Research Projects (WKJ2016-2-08), Fundamental Research Funds for the Central Universities (20720180050), Fujian Province Young Teacher Research Program (JA15010) and Scientific Research Foundation of State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics (2016ZY002). S.H.L is a recipient of NIH career development award (DA038000) and Early Career Award in Chemistry of Drug Abuse and Addiction (ECHEM, DA043507) from the National Institute on Drug Abuse.
ABBREVIATIONS USED
- AD
Alzheimer’s disease
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
AMPA receptor
- ATD
amino-terminal domain
- CNIH
cornichon
- CNS
central nervous system
- FDA
Food and Drug Administration
- iGluRs
ionotropic glutamate receptors
- KARs
kainate receptors
- LBD
ligand-binding domain
- mGluRs
metabotropic glutamate receptors
- NAM
negative allosteric modulators
- NMDA
N-methyl-d-aspartate
- NMDAR
NMDA receptor
- PAM
positive allosteric modulators
- PCP
phencyclidine
- PD
Parkinson’s disease
- PET
positron emission tomography
- p.i.
post-injection
- SCIDY
spirocyclic iodonium ylide
- SPECT
single photon emission computed tomography
- SUV
standardized uptake value
- TARP
transmembrane AMPAR regulatory protein
- TCP
thienylcyclohexyl piperidine
- TMD
transmembrane domain
- tPSA
topological polar surface area
- VD
volume of distribution
- VT
total distribution volume
Biography
Hualong Fu obtained his B.S. in Applied Chemistry from Lanzhou University in 2012. He continued his scientific education as a Ph.D. candidate in Beijing Normal University and received his Ph.D. degree in Inorganic Chemistry in 2017. He is now a research fellow in the Division of Nuclear Medicine and Molecular Imaging, Department of Radiology, Massachusetts General Hospital (MGH) and Harvard Medical School (HMS). His research interests include development of small molecules as fluorescent probe/radiotracers for biological targets in the central nervous system, including β-amyloid plaques of Alzheimer’s disease, and N-methyl-d-aspartate (NMDA) receptors in the ionotropic glutamate receptor system.
Zhen Chen received his B.S. in Applied Chemistry from Tianjin University in 2013. He then joined the research group of Professor Jun-An Ma at Tianjin University as a Ph.D. candidate of Organic Chemistry. In 2017, He moved to Division of Nuclear Medicine and Molecular Imaging, Department of Radiology, HMS/MGH as an exchange Ph.D. student. His research interests include design and synthesis of new fluorine-containing molecules, novel transformation of diazo compounds, C-H functionalization and asymmetric synthesis, as well as application of these reactions in the synthesis of PET tracers for the central and peripheral nervous system.
Lee Josephson obtained his B.S. in Chemistry from the University of Wisconsin, and his Ph.D. in Chemical Biology at Stony Brook University (1975). He did a postdoctoral fellowship in the Department of Molecular and Cellular Biology at Harvard University. He was a cofounder of AMAG Pharmaceuticals (AMAG, AMEX Exchange) where he served as Chief Scientific Officer. He then joined the Department of Radiology at MGH, where he pioneered the use of magnetic nanoparticles, and was a cofounder of T2 Biosystems (TTOO, NASDQ). His research interests include magnetic nanoparticles, imaging biomarkers for cell death and cell health, and peptide PEGylation. He is currently Associate Professor at HMS, and a partner in MedChem Imaging LLC which supplies radiotracers to the pharmaceutical industry.
Zijing Li is an Assistant Professor of radiopharmaceutical sciences at Xiamen University, China. She received a BS degree in Chemistry (2009) and a PhD degree (2014) in Inorganic Chemistry from Beijing Normal University, China. Dr. Li did part of her doctoral research at the New York University School of Medicine. Dr. Li’s research at Xiamen University is primarily focused on radiopharmaceuticals for PET/SPECT and molecular imaging.
Steven H. Liang obtained his B.S. at Tianjin University in 2003, followed by his Ph.D. in Chemistry with Professor Marco Ciufolini in the University of British Columbia in 2010. Then he started as a NSERC fellow with Professor EJ Corey at Harvard University. In 2012, Dr. Liang accepted a junior faculty position at HMS and MGH. Dr. Liang is currently the Director of Radiochemistry and Biomarker Development, Nuclear Medicine and Molecular Imaging at MGH and Assistant Professor of Radiology at HMS. Dr. Liang’s research interests include radiochemistry, imaging biomarker and radiotherapy development, as well as clinical translation.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Madden DR The structure and function of glutamate receptor ion channels. Nat. Rev. Neurosci 2002, 3, 91–101. [DOI] [PubMed] [Google Scholar]
- 2.Nicoletti F; Bockaert J; Collingridge GL; Conn PJ; Ferraguti F; Schoepp DD; Wroblewski JT; Pin JP Metabotropic glutamate receptors: from the workbench to the bedside. Neuropharmacology 2011, 60, 1017–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Traynelis SF; Wollmuth LP; McBain CJ; Menniti FS; Vance KM; Ogden KK; Hansen KB; Yuan H; Myers SJ; Dingledine R Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev 2010, 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollmann M; Heinemann S Cloned glutamate receptors. Annu. Rev. Neurosci 1994, 17, 31–108. [DOI] [PubMed] [Google Scholar]
- 5.Lau A; Tymianski M Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010, 460, 525–542. [DOI] [PubMed] [Google Scholar]
- 6.Mony L; Kew JN; Gunthorpe MJ; Paoletti P Allosteric modulators of NR2B-containing NMDA receptors: molecular mechanisms and therapeutic potential. Br. J. Pharmacol 2009, 157, 1301–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.French JA; Krauss GL; Steinhoff BJ; Squillacote D; Yang H; Kumar D; Laurenza A Evaluation of adjunctive perampanel in patients with refractory partial-onset seizures: results of randomized global phase III study 305. Epilepsia 2013, 54, 117–125. [DOI] [PubMed] [Google Scholar]
- 8.Zwart R; Sher E; Ping X; Jin X; Sims JR; Chappell AS; Gleason SD; Hahn PJ; Gardinier K; Gernert DL; Hobbs J; Smith JL; Valli SN; Witkin JM Perampanel, an antagonist of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors, for the treatment of epilepsy: studies in human epileptic brain and nonepileptic brain and in rodent models. J. Pharmacol. Exp. Ther 2014, 351, 124–133. [DOI] [PubMed] [Google Scholar]
- 9.Ko D; Yang H; Williams B; Xing D; Laurenza A Perampanel in the treatment of partial seizures: time to onset and duration of most common adverse events from pooled Phase III and extension studies. Epilepsy Behav. 2015, 48, 45–52. [DOI] [PubMed] [Google Scholar]
- 10.Sobrio F; Gilbert G; Perrio C; Barre L; Debruyne D PET and SPECT imaging of the NMDA receptor system: an overview of radiotracer development. Mini. Rev. Med. Chem 2010, 10, 870–886. [DOI] [PubMed] [Google Scholar]
- 11.Sobrio F Radiosynthesis of carbon-11 and fluorine-18 labelled radiotracers to image the ionotropic and metabotropic glutamate receptors. J. Labelled Compd. Radiopharm 2013, 56, 180–186. [DOI] [PubMed] [Google Scholar]
- 12.Majo VJ; Prabhakaran J; Mann JJ; Kumar JS PET and SPECT tracers for glutamate receptors. Drug Discov. Today 2013, 18, 173–184. [DOI] [PubMed] [Google Scholar]
- 13.Fuchigami T; Nakayama M; Yoshida S Development of PET and SPECT probes for glutamate receptors. Sci. World J 2015, 2015, 716514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kassenbrock A; Vasdev N; Liang SH Selected PET radioligands for ion channel linked neuroreceptor imaging: focus on GABA, NMDA and nACh receptors. Curr. Top. Med. Chem 2016, 16, 1830–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paoletti P; Bellone C; Zhou Q NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci 2013, 14, 383–400. [DOI] [PubMed] [Google Scholar]
- 16.Johnson JW; Ascher P Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–531. [DOI] [PubMed] [Google Scholar]
- 17.Alexander SP; Mathie A; Peters JA Guide to receptors and channels (GRAC), 5th edition. Br. J. Pharmacol. 2011, 164 Suppl 1, S1–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furukawa H; Singh SK; Mancusso R; Gouaux E Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185–192. [DOI] [PubMed] [Google Scholar]
- 19.Paoletti P; Neyton J NMDA receptor subunits: function and pharmacology. Curr. Opin. Pharmacol 2007, 7, 39–47. [DOI] [PubMed] [Google Scholar]
- 20.Salussolia CL; Prodromou ML; Borker P; Wollmuth LP Arrangement of subunits in functional NMDA receptors. J. Neurosci 2011, 31, 11295–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kew JN; Kemp JA Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl) 2005, 179, 4–29. [DOI] [PubMed] [Google Scholar]
- 22.Kashiwagi K; Masuko T; Nguyen CD; Kuno T; Tanaka I; Igarashi K; Williams K Channel blockers acting at N-methyl-D-aspartate receptors: differential effects of mutations in the vestibule and ion channel pore. Mol. Pharmacol 2002, 61, 533–545. [DOI] [PubMed] [Google Scholar]
- 23.Childers WE Jr.; Baudy RB N-methyl-D-aspartate antagonists and neuropathic pain: the search for relief. J. Med. Chem 2007, 50, 2557–2562. [DOI] [PubMed] [Google Scholar]
- 24.Nikam SS; Meltzer LT NR2B selective NMDA receptor antagonists. Curr. Pharm. Des 2002, 8, 845–855. [DOI] [PubMed] [Google Scholar]
- 25.Waterhouse RN Imaging the PCP site of the NMDA ion channel. Nucl. Med. Biol 2003, 30, 869–878. [DOI] [PubMed] [Google Scholar]
- 26.Haradahira T; Sasaki S; Maeda M; Kobayashi K; Inoue O; Tomita U; Nishikawa T; Suzuki K Synthesis and brain distribution of carbon-11 labeled analogs of antagonists for the NMDA receptor coupled PCP-binding site. J. Labelled Compd. Radiopharm 1998, 41, 843–858. [Google Scholar]
- 27.Patel S; Gibson R In vivo site-directed radiotracers: a mini-review. Nucl. Med. Biol 2008, 35, 805–815. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y; Fox GB PET imaging for receptor occupancy: meditations on calculation and simplification. J. Biomed. Res 2012, 26, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eckelman WC; Kilbourn MR; Mathis CA Discussion of targeting proteins in vivo: in vitro guidelines. Nucl. Med. Biol 2006, 33, 449–451. [DOI] [PubMed] [Google Scholar]
- 30.Kiesewetter DO; Rice KC; Matson MV; Finn RD Radiochemical synthesis of [18F]‐fluorothienylcyclohexylpiperidine ([18F]FTCP). J. Labelled Compd. Radiopharm 1989, 27, 277–286. [Google Scholar]
- 31.Ferrarese C; Guidotti A; Costa E; Miletich RS; Rice KC; de Costa BR; Fulham MJ; Di Chiro G In vivo study of NMDA-sensitive glutamate receptor by fluorothienylcyclohexylpiperidine [correction of fluorothienylcycloexylpiperidine], a possible ligand for positron emission tomography. Neuropharmacology 1991, 30, 899–905. [DOI] [PubMed] [Google Scholar]
- 32.Ouyang X; Mukherjee J; Yang Z-Y Synthesis, radiosynthesis, and biological evaluation of fluorinated thienylcyclohexyl piperidine derivatives as potential radiotracers for the NMDA receptor-linked calcium ionophore. Nucl. Med. Biol 1996, 23, 315–324. [DOI] [PubMed] [Google Scholar]
- 33.Keana JF; Scherz MW; Quarum M; Sonders MS; Weber E Synthesis and characterization of a radiolabelled derivative of the phencyclidine/N-methyl-D-aspartate receptor ligand (+) MK-801 with high specific radioactivity. Life Sci. 1988, 43, 965–973. [DOI] [PubMed] [Google Scholar]
- 34.Sihver S; Sihver W; Andersson Y; Murata T; Bergstrom M; Onoe H; Matsumura K; Tsukada H; Oreland L; Langstrom B; Watanabe Y In vitro and in vivo characterization of (+)-3-[11C]cyano-dizocilpine. J. Neural. Transm. (Vienna) 1998, 105, 117–131. [DOI] [PubMed] [Google Scholar]
- 35.Owens J; Wyper DJ; Patterson J; Brown DR; Elliott AT; Teasdale GM; McCulloch J First SPET images of glutamate (NMDA) receptor activation in vivo in cerebral ischaemia. Nucl. Med. Commun 1997, 18, 149–158. [DOI] [PubMed] [Google Scholar]
- 36.Brown DR; Wyper DJ; Owens J; Patterson J; Kelly RC; Hunter R; McCulloch J 123Iodo-MK-801: a spect agent for imaging the pattern and extent of glutamate (NMDA) receptor activation in Alzheimer's disease. J. Psychiatr. Res 1997, 31, 605–619. [DOI] [PubMed] [Google Scholar]
- 37.Lodge D; Johnson KM Noncompetitive excitatory amino acid receptor antagonists. Trends Pharmacol. Sci 1990, 11, 81–86. [DOI] [PubMed] [Google Scholar]
- 38.Smith DJ; Bouchal RL; DeSanctis CA; Monroe PJ; Amedro JB; Perrotti JM; Crisp T Properties of the interaction between ketamine and opiate binding sites in vivo and in vitro. Neuropharmacology 1987, 26, 1253–1260. [DOI] [PubMed] [Google Scholar]
- 39.Hartvig P; Valtysson J; Lindner KJ; Kristensen J; Karlsten R; Gustafsson LL; Persson J; Svensson JO; Oye I; Antoni G; et al. Central nervous system effects of subdissociative doses of (S)-ketamine are related to plasma and brain concentrations measured with positron emission tomography in healthy volunteers. Clin. Pharmacol. Ther 1995, 58, 165–173. [DOI] [PubMed] [Google Scholar]
- 40.Kumlien E; Hartvig P; Valind S; Oye I; Tedroff J; Langstrom B NMDA-receptor activity visualized with (S)-[N-methyl-11C]ketamine and positron emission tomography in patients with medial temporal lobe epilepsy. Epilepsia 1999, 40, 30–37. [DOI] [PubMed] [Google Scholar]
- 41.Compans B; Choquet D; Hosy E Review on the role of AMPA receptor nano-organization and dynamic in the properties of synaptic transmission. Neurophotonics 2016, 3, 041811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ametamey SM; Samnick S; Leenders KL; Vontobel P; Quack G; Parsons CG; Schubiger PA Fluorine-18 radiolabelling, biodistribution studies and preliminary PET evaluation of a new memantine derivative for imaging the NMDA receptor. J. Recept. Signal Transduct Res 1999, 19, 129–141. [DOI] [PubMed] [Google Scholar]
- 43.Samnick S; Ametamey S; Leenders KL; Vontobel P; Quack G; Parsons CG; Neu H; Schubiger PA Electrophysiological study, biodistribution in mice, and preliminary PET evaluation in a rhesus monkey of 1-amino-3-[18F]fluoromethyl-5-methyl-adamantane (18F-MEM): a potential radioligand for mapping the NMDA-receptor complex. Nucl. Med. Biol 1998, 25, 323–330. [DOI] [PubMed] [Google Scholar]
- 44.Hu LY; Guo J; Magar SS; Fischer JB; Burke-Howie KJ; Durant GJ Synthesis and pharmacological evaluation of N-(2,5-disubstituted phenyl)-N'-(3-substituted phenyl)-N'-methylguanidines as N-methyl-D-aspartate receptor ion-channel blockers. J. Med. Chem 1997, 40, 4281–4289. [DOI] [PubMed] [Google Scholar]
- 45.Dumont F; Sultana A; Waterhouse RN Synthesis and in vitro evaluation of N,N'-diphenyl and N-naphthyl-N'-phenylguanidines as N-methyl-D-aspartate receptor ion-channel ligands. Bioorg. Med. Chem. Lett 2002, 12, 1583–1586. [DOI] [PubMed] [Google Scholar]
- 46.Walters MR; Bradford AP; Fischer J; Lees KR Early clinical experience with the novel NMDA receptor antagonist CNS 5161. Br. J. Clin. Pharmacol 2002, 53, 305–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asselin MC; Hammer A; Turton D; Osman S; Koepp M; Brooks D Initial kinetic analysis of the in vivo binding of the putative NMDA receptor ligand [11C]CNS 5161 in humans. NeuroImage, 2004, 22, T137–138. [Google Scholar]
- 48.Hammers A; Asselin MC; Brooks DJ; Luthra K; Hume SP; Thompson PJ; Turton DR; Duncan JS; Koepp MJ Correlation of memory function with binding of [11C]CNS 5161, a novel putative NMDA ion channel PET ligand. NeuroImage, 2004, 22, T54–55. [Google Scholar]
- 49.Robins EG; Zhao Y; Khan I; Wilson A; Luthra SK; Rstad E Synthesis and in vitro evaluation of 18F-labelled S-fluoroalkyl diarylguanidines: novel high-affinity NMDA receptor antagonists for imaging with PET. Bioorg. Med. Chem. Lett 2010, 20, 1749–1751. [DOI] [PubMed] [Google Scholar]
- 50.McGinnity CJ; Hammers A; Riano Barros DA; Luthra SK; Jones PA; Trigg W; Micallef C; Symms MR; Brooks DJ; Koepp MJ; Duncan JS Initial evaluation of 18F-GE-179, a putative PET Tracer for activated N-methyl D-aspartate receptors. J. Nucl. Med 2014, 55, 423–430. [DOI] [PubMed] [Google Scholar]
- 51.Shi Y; Suh YH; Milstein AD; Isozaki K; Schmid SM; Roche KW; Nicoll RA Functional comparison of the effects of TARPs and cornichons on AMPA receptor trafficking and gating. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 16315–16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Owens J; Tebbutt AA; McGregor AL; Kodama K; Magar SS; Perlman ME; Robins DJ; Durant GJ; McCulloch J Synthesis and binding characteristics of N-(1-naphthyl)-N'-(3-[125I]-iodophenyl)-N'-methylguanidine ([125I]-CNS 1261): a potential SPECT agent for imaging NMDA receptor activation. Nucl. Med. Biol 2000, 27, 557–564. [DOI] [PubMed] [Google Scholar]
- 53.Knol RJ; de Bruin K; van Eck-Smit BL; Pimlott S; Wyper DJ; Booij J In vivo [123I]CNS-1261 binding to D-serine-activated and MK801-blocked NMDA receptors: a storage phosphor imaging study in rats. Synapse 2009, 63, 557–564. [DOI] [PubMed] [Google Scholar]
- 54.Bressan RA; Erlandsson K; Mulligan RS; Gunn RN; Cunningham VJ; Owens J; Cullum ID; Ell PJ; Pilowsky LS A bolus/infusion paradigm for the novel NMDA receptor SPET tracer [123I]CNS 1261. Nucl. Med. Biol 2004, 31, 155–164. [DOI] [PubMed] [Google Scholar]
- 55.Laruelle M Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J. Cereb. Blood. Flow. Metab 2000, 20, 423–451. [DOI] [PubMed] [Google Scholar]
- 56.Erlandsson K; Bressan RA; Mulligan RS; Gunn RN; Cunningham VJ; Owens J; Wyper D; Ell PJ; Pilowsky LS Kinetic modelling of [123I]CNS 1261-a potential SPET tracer for the NMDA receptor. Nucl. Med. Biol 2003, 30, 441–454. [DOI] [PubMed] [Google Scholar]
- 57.Stone JM; Erlandsson K; Arstad E; Bressan RA; Squassante L; Teneggi V; Ell PJ; Pilowsky LS Ketamine displaces the novel NMDA receptor SPET probe [123I]CNS-1261 in humans in vivo. Nucl. Med. Biol 2006, 33, 239–243. [DOI] [PubMed] [Google Scholar]
- 58.Bressan RA; Erlandsson K; Stone JM; Mulligan RS; Krystal JH; Ell PJ; Pilowsky LS Impact of schizophrenia and chronic antipsychotic treatment on [123I]CNS-1261 binding to N-methyl-D-aspartate receptors in vivo. Biol. Psychiatry 2005, 58, 41–46. [DOI] [PubMed] [Google Scholar]
- 59.Stone JM; Erlandsson K; Arstad E; Squassante L; Teneggi V; Bressan RA; Krystal JH; Ell PJ; Pilowsky LS Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy: a [123I]CNS-1261 SPET study. Psychopharmacology (Berl) 2008, 197, 401–408. [DOI] [PubMed] [Google Scholar]
- 60.Stone JM Imaging the glutamate system in humans: relevance to drug discovery for schizophrenia. Curr. Pharm. Des 2009, 15, 2594–2602. [DOI] [PubMed] [Google Scholar]
- 61.Ametamey SM; Honer M; Schubiger PA Molecular imaging with PET. Chem. Rev 2008, 108, 1501–1516. [DOI] [PubMed] [Google Scholar]
- 62.Clark DE Computational prediction of blood-brain barrier permeation In Annual Reports in Medicinal Chemistry; Doherty AM, Bock MG, Desai MC, Overington J, Plattner JJ, Stamford A, Wustrow D, Young H, Eds.; Academic Press: Essex, 2005; pp 403–415.. [Google Scholar]
- 63.Opacka-Juffry J; Morris H; Ashworth S; Osman S; Hirani E; Macleod AM; Luthra SK; Hume SP CHAPTER 45 - Preliminary evaluation of the glycine site antagonists [11C]L 703,717 and [3H]MDL 105,519 as putative PET ligands for central NMDA receptors: in vivo studies in rats In Quantitative Functional Brain Imaging with Positron Emission Tomography; Carson RE, Daube-Witherspoon ME, Herscovitch P, Eds.; Academic Press: San Diego, 1998; pp 299–303. [Google Scholar]
- 64.Haradahira T; Zhang M; Maeda J; Okauchi T; Kawabe K; Kida T; Suzuki K; Suhara T A strategy for increasing the brain uptake of a radioligand in animals: use of a drug that inhibits plasma protein binding. Nucl. Med. Biol 2000, 27, 357–360. [DOI] [PubMed] [Google Scholar]
- 65.Haradahira T; Zhang MR; Maeda J; Okauchi T; Kida T; Kawabe K; Sasaki S; Suhara T; Suzuki K A prodrug of NMDA/glycine site antagonist, L-703,717, with improved BBB permeability: 4-acetoxy derivative and its positron-emitter labeled analog. Chem. Pharm. Bull. (Tokyo) 2001, 49, 147–150. [DOI] [PubMed] [Google Scholar]
- 66.Matsumoto R; Haradahira T; Ito H; Fujimura Y; Seki C; Ikoma Y; Maeda J; Arakawa R; Takano A; Takahashi H; Higuchi M; Suzuki K; Fukui K; Suhara T Measurement of glycine binding site of N-methyl-D-asparate receptors in living human brain using 4-acetoxy derivative of L-703,717, 4-acetoxy-7-chloro-3-[3-(4-[11C] methoxybenzyl) phenyl]-2(1H)-quinolone (AcL703) with positron emission tomography. Synapse 2007, 61, 795–800. [DOI] [PubMed] [Google Scholar]
- 67.Fuchigami T; Haradahira T; Fujimoto N; Okauchi T; Maeda J; Suzuki K; Suhara T; Yamamoto F; Sasaki S; Mukai T; Yamaguchi H; Ogawa M; Magata Y; Maeda M Difference in brain distributions of carbon 11-labeled 4-hydroxy-2(1H)-quinolones as PET radioligands for the glycine-binding site of the NMDA ion channel. Nucl. Med. Biol 2008, 35, 203–212. [DOI] [PubMed] [Google Scholar]
- 68.Haradahira T; Suhara S; Okauchi T; Maeda J; Arai T; Sasaki S Developments of PET radioligands for NMDA receptors. World J. Nucl. Med 2002, 1, S183–184. [Google Scholar]
- 69.Westerberg G; Carr RM; Langstrom B; Sutherland D Synthesis of (E)-3-[2-([1-11C]phenylcarbamoyl)ethenyl]-4-6-dichloroindole-2-carboxylic acid ([11C]GV150526X)- a glycine antagonist for the NMDA-receptor complex. J. Labelled Compd. Radiopharm 1997, 40, 656–657. [Google Scholar]
- 70.Waterhouse RN; Sultana A; Laruelle M In vivo evaluation of [11C]-3-[2-[(3-methoxyphenylamino)carbonyl]ethenyl]-4,6-dichloroindole-2-carboxylic acid ([11C]3MPICA) as a PET radiotracer for the glycine site of the NMDA ion channel. Nucl. Med. Biol 2002, 29, 791–794. [DOI] [PubMed] [Google Scholar]
- 71.Tikhonova IG; Baskin II; Palyulin VA; Zefirov NS CoMFA and homology-based models of the glycine binding site of N-methyl-D-aspartate receptor. J. Med. Chem 2003, 46, 1609–1616. [DOI] [PubMed] [Google Scholar]
- 72.Leeson PD; Carling RW; Moore KW; Moseley AM; Smith JD; Stevenson G; Chan T; Baker R; Foster AC; Grimwood S; Kemp JA; Marshall GR; Hoogsteen K 4-Amido-2-carboxytetrahydroquinolines. Structure-activity relationships for antagonism at the glycine site of the NMDA receptor. J. Med. Chem 1992, 35, 1954–1968. [DOI] [PubMed] [Google Scholar]
- 73.Haradahira T; Okauchi T; Maeda J; Zhang MR; Nishikawa T; Konno R; Suzuki K; Suhara T Effects of endogenous agonists, glycine and D-serine, on in vivo specific binding of [11C]L-703,717, a PET radioligand for the glycine-binding site of NMDA receptors. Synapse 2003, 50, 130–136. [DOI] [PubMed] [Google Scholar]
- 74.Curtis DR; Watkins JC The excitation and depression of spinal neurones by structurally related amino acids. J. Neurochem 1960, 6, 117–141. [DOI] [PubMed] [Google Scholar]
- 75.Bonaccorso C; Micale N; Ettari R; Grasso S; Zappala M Glutamate binding-site ligands of NMDA receptors. Curr. Med. Chem 2011, 18, 5483–5506. [DOI] [PubMed] [Google Scholar]
- 76.Layton ME; Kelly MJ 3rd; Rodzinak KJ Recent advances in the development of NR2B subtype-selective NMDA receptor antagonists. Curr. Top. Med. Chem 2006, 6, 697–709. [DOI] [PubMed] [Google Scholar]
- 77.Chazot PL The NMDA receptor NR2B subunit: a valid therapeutic target for multiple CNS pathologies. Curr. Med. Chem 2004, 11, 389–396. [DOI] [PubMed] [Google Scholar]
- 78.Haradahira T; Maeda J; Okauchi T; Zhang MR; Hojo J; Kida T; Arai T; Yamamoto F; Sasaki S; Maeda M; Suzuki K; Suhara T Synthesis, in vitro and in vivo pharmacology of a C-11 labeled analog of CP-101,606, (+/−)threo-1-(4-hydroxyphenyl)-2-[4-hydroxy-4-(p-[11C]methoxyphenyl)piperidino]-1-propanol, as a PET tracer for NR2B subunit-containing NMDA receptors. Nucl. Med. Biol 2002, 29, 517–525. [DOI] [PubMed] [Google Scholar]
- 79.Roger G; Lagnel B; Besret L; Bramoulle Y; Coulon C; Ottaviani M; Kassiou M; Bottlaender M; Valette H; Dolle F Synthesis, radiosynthesis and in vivo evaluation of 5-[3-(4-benzylpiperidin-1-yl)prop-1-ynyl]-1,3-dihydrobenzoimidazol-2-[11C]one, as a potent NR1A/2B subtype selective NMDA PET radiotracer. Bioorg. Med. Chem 2003, 11, 5401–5408. [DOI] [PubMed] [Google Scholar]
- 80.Roger G; Dolle F; De Bruin B; Liu X; Besret L; Bramoulle Y; Coulon C; Ottaviani M; Bottlaender M; Valette H; Kassiou M Radiosynthesis and pharmacological evaluation of [11C]EMD-95885: a high affinity ligand for NR2B-containing NMDA receptors. Bioorg. Med. Chem. Lett 2004, 12, 3229–3237. [DOI] [PubMed] [Google Scholar]
- 81.Labas R; Sobrio F; Bramoullé Y; Hérard A-S; Guillermier M; Hantraye P; Dollé F; Barré L Radiosynthesis of N-[4-(4-fluorobenzyl)piperidin-1-yl]-N'-(2-[11C]oxo-1,3-dihydrobenzimidazol-5-yl)oxamide, a NR2B-selective NMDA receptor antagonist. J. Labelled Compd. Radiopharm 2010, 53, 63–67. [Google Scholar]
- 82.Claiborne CF; McCauley JA; Libby BE; Curtis NR; Diggle HJ; Kulagowski JJ; Michelson SR; Anderson KD; Claremon DA; Freidinger RM; Bednar RA; Mosser SD; Gaul SL; Connolly TM; Condra CL; Bednar B; Stump GL; Lynch JJ; Macaulay A; Wafford KA; Koblan KS; Liverton NJ Orally efficacious NR2B-selective NMDA receptor antagonists. Bioorg. Med. Chem. Lett 2003, 13, 697–700. [DOI] [PubMed] [Google Scholar]
- 83.Hamill GH; McCauley JA; Burns HD The synthesis of a benzamidine‐containing NR2B‐selective NMDA receptor ligand labelled with tritium or fluorine‐18. J. Labelled Compd. Radiopharm 2005, 48, 1–10. [Google Scholar]
- 84.Thominiaux C; de Bruin B; Bramoulle Y; Hinnen F; Demphel S; Valette H; Bottlaender M; Besret L; Kassiou M; Dolle F Radiosynthesis of (E)-N-(2-[11C]methoxybenzyl)-3-phenyl-acrylamidine, a novel subnanomolar NR2B subtype-selective NMDA receptor antagonist. Appl. Radiat. Isot 2006, 64, 348–354. [DOI] [PubMed] [Google Scholar]
- 85.Arstad E; Platzer S; Berthele A; Pilowsky LS; Luthra SK; Wester HJ; Henriksen G Towards NR2B receptor selective imaging agents for PET-synthesis and evaluation of N-[11C]-(2-methoxy)benzyl (E)-styrene-, 2-naphthyl- and 4-trifluoromethoxyphenylamidine. Bioorg. Med. Chem. Lett 2006, 14, 6307–6313. [DOI] [PubMed] [Google Scholar]
- 86.Addy C; Assaid C; Hreniuk D; Stroh M; Xu Y; Herring WJ; Ellenbogen A; Jinnah HA; Kirby L; Leibowitz MT; Stewart RM; Tarsy D; Tetrud J; Stoch SA; Gottesdiener K; Wagner J Single-dose administration of MK-0657, an NR2B-selective NMDA antagonist, does not result in clinically meaningful improvement in motor function in patients with moderate Parkinson's disease. J. Clin. Pharmacol 2009, 49, 856–864. [DOI] [PubMed] [Google Scholar]
- 87.Ibrahim L; Diaz Granados N; Jolkovsky L; Brutsche N; Luckenbaugh DA; Herring WJ; Potter WZ; Zarate CA Jr. A Randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. J. Clin. Psychopharmacol 2012, 32, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Koudih R; Gilbert G; Dhilly M; Abbas A; Barre L; Debruyne D; Sobrio F Synthesis and in vitro characterization of trans- and cis-[18F]-4-methylbenzyl 4-[(pyrimidin-2-ylamino)methyl]-3-fluoropiperidine-1-carboxylates as new potential PET radiotracer candidates for the NR2B subtype N-methyl-D-aspartate receptor. Eur. J. Med. Chem 2012, 53, 408–415. [DOI] [PubMed] [Google Scholar]
- 89.Koudih R; Gilbert G; Dhilly M; Abbas A; Barre L; Debruyne D; Sobrio F Radiolabelling of 1,4-disubstituted 3-[18F]fluoropiperidines and its application to new radiotracers for NR2B NMDA receptor visualization. Org. Biomol. Chem 2012, 10, 8493–8500. [DOI] [PubMed] [Google Scholar]
- 90.Dollé F; Valette H; Demphel S; Coulon C; Ottaviani M; Bottlaender M; Kassiou M Radiosynthesis and in vivo evaluation of [11C]Ro-647312: a novel NR1/2B subtype selective NMDA receptor radioligand. J. Labelled Compd. Radiopharm 2004, 47, 911–920. [Google Scholar]
- 91.Christiaans JA; Klein PJ; Metaxas A; Kooijman EJ; Schuit RC; Leysen JE; Lammertsma AA; van Berckel BN; Windhorst AD Synthesis and preclinical evaluation of carbon-11 labelled N-((5-(4-fluoro-2-[11C]methoxyphenyl)pyridin-3-yl)methyl)cyclopentanamine as a PET tracer for NR2B subunit-containing NMDA receptors. Nucl. Med. Biol 2014, 41, 670–680. [DOI] [PubMed] [Google Scholar]
- 92.Tewes B; Frehland B; Schepmann D; Schmidtke KU; Winckler T; Wunsch B Design, synthesis, and biological evaluation of 3-benzazepin-1-ols as NR2B-selective NMDA receptor antagonists. ChemMedChem 2010, 5, 687–695. [DOI] [PubMed] [Google Scholar]
- 93.Krämer SD; Betzel T; Mu L; Haider A; Herde AM; Boninsegni AK; Keller C; Szermerski M; Schibli R; Wünsch B; Ametamey SM Evaluation of 11C-Me-NB1 as a potential PET radioligand for measuring GluN2B-containing NMDA receptors, drug occupancy, and receptor cross talk. J. Nucl. Med 2018, 59, 698–703. [DOI] [PubMed] [Google Scholar]
- 94.Szermerski M; Borgel F; Schepmann D; Haider A; Betzel T; Ametamey SM; Wunsch B Fluorinated GluN2B receptor antagonists with a 3-benzazepine scaffold designed for PET studies. ChemMedChem 2018, 13, 1058–1068. [DOI] [PubMed] [Google Scholar]
- 95.Sasaki S; Kurosaki F; Haradahira T; Yamamoto F; Maeda J; Okauchi T; Suzuki K; Suhara T; Maeda M Synthesis of 11C-labelled bis(phenylalkyl)amines and their in vitro and in vivo binding properties in rodent and monkey brains. Biol. Pharm. Bull 2004, 27, 531–537. [DOI] [PubMed] [Google Scholar]
- 96.Bowie D Redefining the classification of AMPA-selective ionotropic glutamate receptors. J. Physiol 2012, 590, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Compans B; Choquet D; Hosy E Review on the role of AMPA receptor nanoorganization and dynamic in the properties of synaptic transmission. Neurophotonics 2016, 3, 041811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sobolevsky AI; Rosconi MP; Gouaux E X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 2009, 462, 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shaw AS; Filbert EL Scaffold proteins and immune-cell signalling. Nat. Rev. Immunol 2009, 9, 47–56. [DOI] [PubMed] [Google Scholar]
- 100.Hashimoto K; Fukaya M; Qiao X; Sakimura K; Watanabe M; Kano M Impairment of AMPA receptor function in cerebellar granule cells of ataxic mutant mouse stargazer. J. Neurosci 1999, 19, 6027–6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicoll RA; Tomita S; Bredt DS Auxiliary subunits assist AMPA-type glutamate receptors. Science 2006, 311, 1253–1256. [DOI] [PubMed] [Google Scholar]
- 102.Schwenk J; Harmel N; Zolles G; Bildl W; Kulik A; Heimrich B; Chisaka O; Jonas P; Schulte U; Fakler B; Klöcker N Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science 2009, 323, 1313–1319. [DOI] [PubMed] [Google Scholar]
- 103.Shi Y; Suh YH; Milstein AD; Isozaki K; Schmid SM; Roche KW; Nicoll RA Functional comparison of the effects of TARPs and cornichons on AMPA receptor trafficking and gating. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 16315–16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kato AS; Gill MB; Ho MT; Yu H; Tu Y; Siuda ER; Wang H; Qian Y-W; Nisenbaum ES; Tomita S; Bredt DS Hippocampal AMPA receptor gating controlled by both TARP and cornichon proteins. Neuron 2010, 68, 1082–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Engelhardt J. v.; Mack V; Sprengel R; Kavenstock N; Li KW; Stern-Bach Y; Smit AB; Seeburg PH; Monyer H CKAMP44: a brain-specific protein attenuating short-term synaptic plasticity in the dentate gyrus. Science 2010, 327, 1518–1522. [DOI] [PubMed] [Google Scholar]
- 106.Palmer CL; Cotton L; Henley JM The molecular pharmacology and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol. Rev 2005, 57, 253–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun Y; Olson R; Horning M; Armstrong N; Mayer M; Gouaux E Mechanism of glutamate receptor desensitization. Nature 2002, 417, 245–253. [DOI] [PubMed] [Google Scholar]
- 108.Jin R; Clark S; Weeks AM; Dudman JT; Gouaux E; Partin KM Mechanism of positive allosteric modulators acting on AMPA receptors. J. Neurosci 2005, 25, 9027–9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wollmuth LP; Sobolevsky AI Structure and gating of the glutamate receptor ion channel. Trends Neurosci. 2004, 27, 321–328. [DOI] [PubMed] [Google Scholar]
- 110.Kessels HW; Malinow R Synaptic AMPA receptor plasticity and behavior. Neuron 2009, 61, 340–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bleakman D; Lodge D Neuropharmacology of AMPA and kainate receptors. Neuropharmacology 1998, 37, 1187–1204. [DOI] [PubMed] [Google Scholar]
- 112.Konradi C; Heckers S Molecular aspects of glutamate dysregulation: implications for schizophrenia and its treatment. Pharmacol. Ther 2003, 97, 153–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Black MD Therapeutic potential of positive AMPA modulators and their relationship to AMPA receptor subunits. A review of preclinical data. Psychopharmacology (Berl) 2005, 179, 154–163. [DOI] [PubMed] [Google Scholar]
- 114.Mellor IR The AMPA receptor as a therapeutic target: current perspectives and emerging possibilities. Future Med. Chem 2010, 2, 877–891. [DOI] [PubMed] [Google Scholar]
- 115.O'Neill MJ; Bleakman D; Zimmerman DM; Nisenbaum ES AMPA receptor potentiators for the treatment of CNS disorders. Curr. Drug Targets CNS Neurol. Disord 2004, 3, 181–194. [DOI] [PubMed] [Google Scholar]
- 116.Gao M; Kong D; Clearfield A; Zheng Q-H Synthesis of carbon-11 and fluorine-18 labeled N-acetyl-1-aryl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivatives as new potential PET AMPA receptor ligands. Bioorg. Med. Chem. Lett 2006, 16, 2229–2233. [DOI] [PubMed] [Google Scholar]
- 117.Arstad E; Gitto R; Chimirri A; Caruso R; Constanti A; Turton D; Hume SP; Ahmad R; Pilowsky LS; Luthra SK Closing in on the AMPA receptor: synthesis and evaluation of 2-acetyl-1-(4'-chlorophenyl)-6-methoxy-7-[11C]methoxy-1,2,3,4-tetrahydroisoquinol ine as a potential PET tracer. Bioorg. Med. Chem 2006, 14, 4712–4717. [DOI] [PubMed] [Google Scholar]
- 118.Lee HG; Milner PJ; Placzek MS; Buchwald SL; Hooker JM Virtually instantaneous, room-temperature [11C]-cyanation using biaryl phosphine Pd(0) complexes. J. Am. Chem. Soc 2015, 137, 648–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Oi N; Tokunaga M; Suzuki M; Nagai Y; Nakatani Y; Yamamoto N; Maeda J; Minamimoto T; Zhang M-R; Suhara T; Higuchi M Development of novel PET probes for central 2-amino-3-(3-hydroxy-5-methyl-4-isoxazolyl)propionic acid receptors. J. Med. Chem 2015, 58, 8444–8462. [DOI] [PubMed] [Google Scholar]
- 120.Laruelle M; Slifstein M; Huang Y Relationships between radiotracer properties and image quality in molecular imaging of the brain with positron emission tomography. Mol. Imaging Biol 2003, 5, 363–375. [DOI] [PubMed] [Google Scholar]
- 121.Takahata K; Kimura Y; Seki C; Tokunaga M; Ichise M; Kawamura K; Ono M; Kitamura S; Kubota M; Moriguchi S; Ishii T; Takado Y; Niwa F; Endo H; Nagashima T; Ikoma Y; Zhang M-R; Suhara T; Higuchi M A human PET study of [11C]HMS011, a potential radioligand for AMPA receptors. EJNMMI Res. 2017, 7, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yuan G; Jones GB; Vasdev N; Liang SH Radiosynthesis and preliminary PET evaluation of 18F-labeled 2-(1-(3-fluorophenyl)-2-oxo-5-(pyrimidin-2-yl)-1,2-dihydropyridin-3-yl) benzonitrile for imaging AMPA receptors. Bioorg. Med. Chem. Lett 2016, 26, 4857–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rogers G, Thorell JO, Johnstrom P, Eriksson L, Ingvar M, Stone-Elander Ampakines S, labelling with 11C for PET distribution studies. J. Labelled Compd. Radiopharm 1997, 40, 645–647. [Google Scholar]
- 124.Nicoll RA; Tomita S; Bredt DS Auxiliary subunits assist AMPA-type glutamate receptors. Science 2006, 311, 1253–1256. [DOI] [PubMed] [Google Scholar]
- 125.Kronenberg UB; Drewes B; Sihver W; Coenen HH N-2-(4-N-(4-[18F]Fluorobenzamido)phenyl)-propyl-2-propanesulphonamide: synthesis and radiofluorination of a putative AMPA receptor ligand. J. Labelled Compd. Radiopharm 2007, 50, 1169–1175. [Google Scholar]
- 126.Contractor A; Mulle C; Swanson GT Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci. 2011, 34, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lerma J; Paternain AV; Rodriguez-Moreno A; Lopez-Garcia JC Molecular physiology of kainate receptors. Physiol. Rev 2001, 81, 971–998. [DOI] [PubMed] [Google Scholar]
- 128.Jane DE; Lodge D; Collingridge GL Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology 2009, 56, 90–113. [DOI] [PubMed] [Google Scholar]
- 129.Matute C Therapeutic potential of kainate receptors. CNS Neurosci. Ther 2011, 17, 661–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kanazawa M; Furuta K; Doi H; Mori T; Minami T; Ito S; Suzuki M Synthesis of an acromelic acid a analog-based 11C-labeled PET tracer for exploration of the site of action of acromelic acid a in allodynia induction. Bioorg. Med. Chem. Lett 2011, 21, 2017–2020. [DOI] [PubMed] [Google Scholar]
- 131.O'Hara PJ; Sheppard PO; Thogersen H; Venezia D; Haldeman BA; McGrane V; Houamed KM; Thomsen C; Gilbert TL; Mulvihill ER The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron 1993, 11, 41–52. [DOI] [PubMed] [Google Scholar]
- 132.Sephton SM.; Auberson Y; Cermak S; Mu L; Herde AM; Schibli R; Krämer SD; Ametamey SM Radiosynthesis and in vitro evaluation of [18F]FP-PEAQX as a potential PET radioligand for imaging the GluN2A subunit of the NMDA receptor. J. Labelled Compd. Radiopharm 2015, 58, S410. [Google Scholar]
- 133.Tamborini L; Chen Y; Foss CA; Pinto A; Horti AG; Traynelis SF; De Micheli C; Mease RC; Hansen KB; Conti P; Pomper MG Development of radiolabeled ligands targeting the glutamate binding site of the N-Methyl-D-aspartate receptor as potential imaging agents for brain. J. Med. Chem 2016, 59, 11110–11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lind GE; Mou TC; Tamborini L; Pomper MG; De Micheli C; Conti P; Pinto A; Hansen KB Structural basis of subunit selectivity for competitive NMDA receptor antagonists with preference for GluN2A over GluN2B subunits. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E6942–E6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Burnell ES; Irvine M; Fang G; Sapkota K; Jane DE; Monaghan DT Positive and negative allosteric modulators of N-Methyl-D-aspartate (NMDA) receptors: structure-activity relationships and mechanisms of action. J. Med. Chem 2018, 10.1021/acs.jmedchem.7b01640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bettini E; Sava A; Griffante C; Carignani C; Buson A; Capelli AM; Negri M; Andreetta F; Senar-Sancho SA; Guiral L; Cardullo F Identification and characterization of novel NMDA receptor antagonists selective for NR2A- over NR2B-containing receptors. J. Pharmacol. Exp. Ther 2010, 335, 636–644. [DOI] [PubMed] [Google Scholar]
- 137.Volkmann RA; Fanger CM; Anderson DR; Sirivolu VR; Paschetto K; Gordon E; Virginio C; Gleyzes M; Buisson B; Steidl E; Mierau SB; Fagiolini M; Menniti FS MPX-004 and MPX-007: new pharmacological tools to study the physiology of NMDA receptors containing the GluN2A subunit. PLoS One 2016, 11, e0148129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hackos DH; Lupardus PJ; Grand T; Chen Y; Wang TM; Reynen P; Gustafson A; Wallweber HJ; Volgraf M; Sellers BD; Schwarz JB; Paoletti P; Sheng M; Zhou Q; Hanson JE Positive allosteric modulators of GluN2A-containing NMDARs with distinct modes of action and impacts on circuit function. Neuron 2016, 89, 983–999. [DOI] [PubMed] [Google Scholar]
- 139.Villemure E; Volgraf M; Jiang Y; Wu G; Ly CQ; Yuen PW; Lu A; Luo X; Liu M; Zhang S; Lupardus PJ; Wallweber HJ; Liederer BM; Deshmukh G; Plise E; Tay S; Wang TM; Hanson JE; Hackos DH; Scearce-Levie K; Schwarz JB; Sellers BD GluN2A-selective pyridopyrimidinone series of NMDAR positive allosteric modulators with an improved in vivo profile. ACS Med. Chem. Lett 2017, 8, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Swanger SA; Vance KM; Acker TM; Zimmerman SS; DiRaddo JO; Myers SJ; Bundgaard C; Mosley CA; Summer SL; Menaldino DS; Jensen HS; Liotta DC; Traynelis SF A novel negative allosteric modulator selective for GluN2C/2D-containing NMDA receptors inhibits synaptic transmission in hippocampal interneurons. ACS Chem. Neurosci 2018, 9, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Santangelo Freel RM; Ogden KK; Strong KL; Khatri A; Chepiga KM; Jensen HS; Traynelis SF; Liotta DC Synthesis and structure activity relationship of tetrahydroisoquinoline-based potentiators of GluN2C and GluN2D containing N-methyl-D-aspartate receptors. J. Med. Chem 2013, 56, 5351–5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Khatri A; Burger PB; Swanger SA; Hansen KB; Zimmerman S; Karakas E; Liotta DC; Furukawa H; Snyder JP; Traynelis SF Structural determinants and mechanism of action of a GluN2C-selective NMDA receptor positive allosteric modulator. Mol. Pharmacol 2014, 86, 548–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Risgaard R; Hansen KB; Clausen RP Partial agonists and subunit selectivity at NMDA receptors. Chem. - Eur. J 2010, 16, 13910–13918. [DOI] [PubMed] [Google Scholar]
- 144.Jackson AC; Nicoll RA The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 2011, 70, 178–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tomita S; Chen L; Kawasaki Y; Petralia RS; Wenthold RJ; Nicoll RA; Bredt DS Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J. Cell Biol 2003, 161, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Maher MP; Wu N; Ravula S; Ameriks MK; Savall BM; Liu C; Lord B; Wyatt RM; Matta JA; Dugovic C; Yun S; Ver Donck L; Steckler T; Wickenden AD; Carruthers NI; Lovenberg TW Discovery and characterization of AMPA receptor modulators selective for TARP-gamma 8. J. Pharmacol. Exp. Ther 2016, 357, 394–414. [DOI] [PubMed] [Google Scholar]
- 147.Gardinier KM; Gernert DL; Porter WJ; Reel JK; Ornstein PL; Spinazze P; Stevens FC; Hahn P; Hollinshead SP; Mayhugh D; Schkeryantz J; Khilevich A; De Frutos O; Gleason SD; Kato AS; Luffer-Atlas D; Desai PV; Swanson S; Burris KD; Ding C; Heinz BA; Need AB; Barth VN; Stephenson GA; Diseroad BA; Woods TA; Yu H; Bredt D; Witkin JM Discovery of the first alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist dependent upon transmembrane AMPA receptor regulatory protein (TARP) gamma-8. J. Med. Chem 2016, 59, 4753–4768. [DOI] [PubMed] [Google Scholar]
- 148.Quirk JC; Nisenbaum ES LY404187: a novel positive allosteric modulator of AMPA receptors. CNS Drug Rev. 2002, 8, 255–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jhee SS; Chappell AS; Zarotsky V; Moran SV; Rosenthal M; Kim E; Chalon S; Toublanc N; Brandt J; Coutant DE; Ereshefsky L Multiple-dose plasma pharmacokinetic and safety study of LY450108 and LY451395 (AMPA receptor potentiators) and their concentration in cerebrospinal fluid in healthy human subjects. J. Clin. Pharmacol 2006, 46, 424–432. [DOI] [PubMed] [Google Scholar]
- 150.Trzepacz PT; Cummings J; Konechnik T; Forrester TD; Chang C; Dennehy EB; Willis BA; Shuler C; Tabas LB; Lyketsos C Mibampator (LY451395) randomized clinical trial for agitation/aggression in Alzheimer's disease. Int. Psychogeriatr 2013, 25, 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shaffer CL; Patel NC; Schwarz J; Scialis RJ; Wei Y; Hou XJ; Xie L; Karki K; Bryce DK; Osgood SM; Hoffmann WE; Lazzaro JT; Chang C; McGinnis DF; Lotarski SM; Liu J; Obach RS; Weber ML; Chen L; Zasadny KR; Seymour PA; Schmidt CJ; Hajos M; Hurst RS; Pandit J; O'Donnell CJ The discovery and characterization of the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor potentiator N-{(3S,4S)-4-[4-(5-cyano-2-thienyl)phenoxy]tetrahydrofuran-3-yl}propane-2-sulfona mide (PF-04958242). J. Med. Chem 2015, 58, 4291–4308. [DOI] [PubMed] [Google Scholar]
- 152.Ward SE; Harries M; Aldegheri L; Andreotti D; Ballantine S; Bax BD; Harris AJ; Harker AJ; Lund J; Melarange R; Mingardi A; Mookherjee C; Mosley J; Neve M; Oliosi B; Profeta R; Smith KJ; Smith PW; Spada S; Thewlis KM; Yusaf SP Discovery of N-[(2S)-5-(6-fluoro-3-pyridinyl)-2,3-dihydro-1H-inden-2-yl]-2-propanesulfonamide, a novel clinical AMPA receptor positive modulator. J. Med. Chem 2010, 53, 5801–5812. [DOI] [PubMed] [Google Scholar]
- 153.Ward SE; Beswick P; Calcinaghi N; Dawson LA; Gartlon J; Graziani F; Jones DN; Lacroix L; Selina Mok MH; Oliosi B; Pardoe J; Starr K; Woolley ML; Harries MH Pharmacological characterization of N-[(2S)-5-(6-fluoro-3-pyridinyl)-2, 3-dihydro-1H-inden-2-yl]-2-propanesulfonamide: a novel, clinical AMPA receptor positive allosteric modulator. Br. J. Pharmacol 2017, 174, 370–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schober DA; Gill MB; Yu H; Gernert DL; Jeffries MW; Ornstein PL; Kato AS; Felder CC; Bredt DS Transmembrane AMPA receptor regulatory proteins and cornichon-2 allosterically regulate AMPA receptor antagonists and potentiators. J. Biol. Chem 2011, 286, 13134–13142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gill MB; Bredt DS An emerging role for TARPs in neuropsychiatric disorders. Neuropsychopharmacology 2011, 36, 362–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hansen KB; Traynelis SF Glutamate receptors: mechanistic twists and turns. Nat. Chem. Biol 2014, 10, 698–699. [DOI] [PubMed] [Google Scholar]
- 157.Meyerson JR; Kumar J; Chittori S; Rao P; Pierson J; Bartesaghi A; Mayer ML; Subramaniam S Structural mechanism of glutamate receptor activation and desensitization. Nature 2014, 514, 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]