

Abstract
Background and Purpose
Kinase inhibitors are a common treatment for cancer. Class I kinase inhibitors that target the ATP‐binding pocket are particularly prevalent. Many of these compounds are cardiotoxic and can cause arrhythmias. Spontaneous release of Ca2+ via cardiac ryanodine receptors (RyR2), through a process termed store overload‐induced Ca2+ release (SOICR), is a common mechanism underlying arrhythmia. We explored whether class I kinase inhibitors could modify the activity of RyR2 and trigger SOICR to determine if this contributes to the cardiotoxic nature of these compounds.
Experimental Approach
The impact of class I and II kinase inhibitors on SOICR was studied in HEK293 cells and ventricular myocytes using single‐cell Ca2+ imaging. A specific effect on RyR2 was confirmed using single channel recordings. Ventricular myocytes were also used to determine if drug‐induced changes in SOICR could be reversed using anti‐SOICR agents.
Key Results
Class I kinase inhibitors increased the propensity of SOICR. Single channel recording showed that this was due to a specific effect on RyR2. Class II kinase inhibitors decreased the activity of RyR2 at the single channel level but had little effect on SOICR. The promotion of SOICR mediated by class I kinase inhibitors could be reversed using the anti‐SOICR agent VK‐II‐86.
Conclusions and Implications
Part of the cardiotoxicity of class I kinase inhibitors can be assigned to their effect on RyR2 and increase in SOICR. Compounds with anti‐SOICR activity may represent an improved treatment option for patients.
Abbreviations
- AM
acetoxymethyl ester
- Cmax
peak plasma concentration
- FSOICR
release threshold for SOICR
- FTermi
termination threshold for SOICR
- KRH
Krebs–Ringer HEPES
- Po
channel open probability
- RyR2
cardiac ryanodine receptors type 2
- SOICR
store overload‐induced calcium release
- SR
sarcoplasmic reticulum
- Tc
channel closed time
- TES
tris[hydroxymethyl]methyl‐2‐aminoethanesulfonic acid
- To
channel open time
Introduction
Over the past decades, there has been a progressive development of new drugs to treat cancer. Leading the way are small compounds designed to inhibit protein kinases involved in metabolism, cell survival and cell cycle progression. There are around 500 protein kinases present in the human kinome, and around 150 of them have been linked to the progression of various diseases including inflammatory diseases, cardiovascular diseases, metabolic diseases and cancer (Cohen, 2001; Wilson et al., 2018). Several strategies are employed to inhibit kinases pharmacologically, the most common being those that target the active conformation of the ATP‐binding pocket; compounds termed class I kinase inhibitors. These compounds mimic the hydrogen bonds that normally form between ATP and the kinase. Given the conservation of this interaction of ATP and kinase amongst a range of kinases, these drugs are often relatively non‐selective for a specific kinase (Liu and Gray, 2006; Klaeger et al., 2017). A second class of inhibitors with potentially greater specificity are termed class II kinase inhibitors, which inhibit the kinase by interacting with the inactive form of the kinase. These compounds bind adjacently to the ATP‐binding pocket and modify it to prevent activation (Liu and Gray, 2006; Hu et al., 2015).
Although great advances have been made in treating cancer, an ongoing problem remains the off‐target toxicity of these compounds. An organ frequently adversely affected by these drugs is the heart, where several anti‐cancer compounds (both class I and class II) have been linked to electrical disturbances (Schmidinger et al., 2008; Shah and Morganroth, 2015). A common mechanism leading to electrical disturbances and arrhythmias is the disruption of intracellular Ca2+ release within individual cardiomyocytes (Bai et al., 2013). Under normal conditions, the controlled release of Ca2+ from the intracellular Ca2+ store [sarcoplasmic reticulum (SR)] through the cardiac ryanodine receptor (RyR2) provides the bulk of the Ca2+ required for contraction (Bers, 2002). However, the uncontrolled release of Ca2+ through RyR2 is pathogenic and linked to both arrhythmias and cardiomyopathies. A unifying mechanism underlying RyR2 dysfunction is store overload‐induced Ca2+ release (SOICR) (Liu et al., 2013), otherwise known as spontaneous Ca2+ release. We and others have shown that SOICR can be triggered due to an increase in SR Ca2+ content or due to an increase in the sensitivity of RyR2 to activation by SR Ca2+ (Kashimura et al., 2010; Zhang et al., 2014; Zhang et al., 2015). Pertinently, we have previously shown that the drug‐induced activation of RyR2 partially underlies the cardiotoxicity of anthracyclines, a different class of chemotherapeutic drugs (Hanna et al., 2014). Combined, this led us to hypothesize that part of the cardiotoxicity of class I and II kinase inhibitors is due to an increase in RyR2 activity and SOICR.
In this study, we examined the effect of two class I and two class II kinase inhibitors on the activity of RyR2 and SOICR. The class I inhibitors studied were CX‐4545 (silmitasertib) and sunitinib (sutent), inhibitors of protein kinase CK2 and cyclin‐dependent kinase 2 respectively (Mendel et al., 2003; Ferguson et al., 2011). The class II inhibitors examined were ponatinib (ICLUSIG) and nilotinib (Tasigna), both multi‐targeted kinase inhibitors with a primary target of BCR‐ABL TK (O'Hare et al., 2009). We found that both class I kinase inhibitors increased the incidence of SOICR by sensitizing RyR2 to activation by luminal Ca2+. However, neither class II kinase inhibitor enhanced RyR2 channel activity or the incidence of SOICR. These data suggest that part of the cardiotoxicity of class I kinase inhibitors can be attributed to a specific effect on RyR2, leading to an increase in SOICR, a precursor of arrhythmia and cardiomyopathies. Thus, this study identifies a novel mechanism that could underlie the cardiotoxicity of class I kinase inhibitors.
Methods
Single‐cell Ca2+ imaging (cytosolic Ca2+)
Measurements of cytosolic Ca2+ were conducted in stable, inducible HEK293 cells (RRID:CVCL_U427) expressing RyR2, generated as previously described (Waddell et al., 2016). RyR2 expression was induced ~16–18 h before the experiment via the addition of tetracycline (0.1 μg·mL−1). In order to visualize Ca2+ transients, cells were loaded with 2 μM of the acetoxymethyl ester (AM) form of the Ca2+ indicator, fluo‐4 (Life Technologies) in 0 Ca2+ Krebs–Ringer HEPES (KRH) buffer containing (mM): 125 NaCl, 5 KCl, 25 HEPES, 6 glucose, and 1.2 MgCl2 (pH 7.4) and BSA (1 mg·mL−1) for 10 min at room temperature. Cells were then incubated in the same solution plus each kinase inhibitor or DMSO as a vehicle control, for 10 min, in the absence of fluo‐4 to allow de‐esterification to take place. The cells were then mounted on an epi‐fluorescence microscope (Nikon Eclipse Ti, 20× plan‐fluor objective) and continuously superfused with KRH solution containing various concentrations of CaCl2 (0.1–1.0 mM) along with each kinase inhibitor or DMSO at room temperature. At the end of the experiments, 20 mM caffeine was applied to deplete the intracellular Ca2+ store. Fluo‐4 AM dye was excited at 470 nm (40 nm bandwidth) every 2 s with an exposure time of 100 ms using a CoolLED system (Coherent Scientific Pty. Ltd, Australia). Fluorescence of fluo‐4 was detected through a long‐pass dichroic mirror (495 nm) and a long‐pass emission filter (515 nm) by a Zyla 4.2 Plus sCMOS camera (Coherent Scientific Pty. Ltd). The cytosolic Ca2+ fluorescence is represented by F/F0, where F is the Ca2+ fluorescence intensity at any time and F0 is the average fluorescence intensity recorded in 0 Ca2+ KRH solution. Normalization to F0 was performed to control for variations in the amount of fluo‐4 loaded into each cell. The percentage of cells experiencing SOICR at each external Ca2+ concentration ([Ca2+]o) was determined using a custom image analysis script written in the Python programming language (code available on request). Measurements were carried out at 21 ± 2°C.
Single‐cell Ca2+ imaging (luminal Ca2+)
Stable, inducible HEK293 cells expressing RyR2 as described above were used with the additional transfection of 2 μg cDNA encoding for D1ER. D1ER transfection took place 24 h before RyR2 induction. The cells were incubated with each inhibitor (or DMSO) for 10 min before each experiment. The cells were then mounted on an epi‐fluorescence microscope (Nikon Eclipse Ti, 20× plan‐fluor objective, Coherent Scientific Pty. Ltd) and continuously superfused at room temperature with KRH containing various concentrations of Ca2+ (1 and 2 mM), tetracaine (2 mM to block RyR2) and caffeine (20 mM to deplete the SR store) along with either DMSO or kinase inhibitor. Fluorescent images of HEK293 cells were acquired every 2 s with an exposure time of 100 ms and excitation at 436 nm (20 nm bandwidth). The emissions of YFP and CFP were captured with the addition of a beamsplitter at 535 nm (40 nm bandwidth) and 480 nm (30 nm bandwidth) respectively. Two images were collected by a CoolSNAP HQ2 CCD camera (Photometrics, AZ, USA) using a dual‐channel imaging system (DV2). The amount of FRET was determined from the ratio of the emissions at 535 and 480 nm (YFP : CFP) as previously described (Waddell et al., 2016; Zhang et al., 2016). From each trace, several parameters were determined. The mean maximum ratio in the presence of tetracaine represents the maximum store level (Fmax), while the mean minimum ratio in the presence of caffeine represents a fully depleted store (Fmin). These values were calculated as the largest or smallest mean over a rolling 30 s period during the plateau phase of tetracaine and caffeine treatment. From this, the store size was calculated (Fmax − Fmin). The release threshold for SOICR (FSOICR) expressed as a percentage of store size was calculated from the mean of each of the maximum values before each of the final three SOICR events. Similarly, the termination of SOICR (FTermi), also expressed as percentage of store size, was calculated from the mean of each of the minimum points in the final three nadirs before application of tetracaine. Measurements were carried out at 21 ± 2°C.
Isolation and single‐cell imaging of rat ventricular myocytes
Individual myocytes were isolated by enzymatic digestion from 2‐ to 3‐month‐old specific pathogen‐free male Sprague–Dawley rats (RRID:MGI:5651135) as previously described (Xiao et al., 2007). The isolated cardiomyocytes were loaded with 2 μM fluo‐4 for 10 min in KRH containing 1 mM Ca2+. The cells were then mounted on an epi‐fluorescence microscope (Nikon Eclipse Ti, 20× plan‐fluor objective, Coherent Scientific Pty. Ltd) and continuously superfused at room temperature with KRH containing 1 mM Ca2+, 1 mM Ca2+ + DMSO or 10 μM of CX‐4945, ponatinib or nilotinib, or 1 μM sunitinib. Caffeine (20 mM) was applied at the end of each experiment to deplete the SR store and confirm cell viability. Cells were paced at 0.5 Hz apart from a 2 min window during which the number of spontaneous Ca2+ waves was counted. In those experiments examining the effect of VK‐II‐86, cells were pre‐incubated in 1 μM VK‐II‐86 for 30 min prior to the experiment. Fluo‐4 AM dye was excited at 470 nm (40 nm bandwidth) every 100 ms with an exposure time of 50 ms using a CoolLED system (Coherent Scientific Pty. Ltd). Fluorescence of fluo‐4 was detected through a long‐pass dichroic mirror (495 nm) and a long‐pass emission filter (515 nm) by an Andor Zyla 4.2 Plus sCMOS camera (Coherent Scientific Pty. Ltd). Measurements were carried out at 21 ± 2°C.
Single channel recordings
Sheep cardiac SR vesicles were incorporated into lipid bilayers in solutions containing (mM) cis: 230 CsCH3O3S, 20 CsCl, 1 CaCl2 and 10 N‐tris[hydroxymethyl]methyl‐2‐aminoethanesulfonic acid (TES; pH 7.4) and trans: 30 Cs CsCH3O3S, 20 CsCl, 1 CaCl2 and 10 TES (pH 7.4) as described previously (Beard et al., 2002; Richardson et al., 2017). Following SR vesicle incorporation, cis (cytoplasmic) Ca2+ was adjusted to 1 μM with BAPTA using a Ca2+ electrode. Trans [CsCH3O3S] was increased to 230 mM, while trans [Ca2+] was 1 mM throughout. Data were filtered at 1 kHz and sampled at 5 kHz. All electrical potentials are expressed as that of cytoplasmic solution relative to the luminal solution. Single channel recordings were obtained at +40 and −40 mV. These voltages were chosen to ensure a large current flow through the channel, which would maximize the signal‐to‐noise ratio, without easily breaking the bilayer. Single channel parameters from 30 s of recording were obtained using the Channel3 software (N.W. Laver, nic@niclaver.com; based on the original Channel2 software developed by P.W. Gage and M. Smith, John Curtin School of Medical Research, Canberra, Australia). The threshold levels for channel opening were set to exclude baseline noise at ∼20% of the maximum single channel conductance and open probability (P o), mean open time (T o) and mean closed time (T c) measured (Copello et al., 1997; Marengo et al., 1998; Smith et al., 2013). Currents at both positive and negative potentials were monitored to assess possible voltage‐dependent effects of the peptide. As no voltage‐dependent effects were observed, independent parameters measured at +40 and −40 mV were averaged for each channel. Control activity with 1 μM cytoplasmic Ca2+ was recorded for 3 to 5 min at the start of each experiment. Then, in the first series of experiments, 10 μM CX‐4945, ponatinib or nilotinib or 1 μM sunitinib were added to the cytoplasmic side of the channel (cis solution), and channel activity was recorded for 5 to 10 min. In a second series of experiments, 2 mM Na2ATP was added to the cis solution and channel activity recorded for 3 to 5 min before adding CX‐4945 or sunitinib. Measurements were carried out at 21 ± 2°C.
Experiments involving animals
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010). All experiments were approved (#DET29/16) and conducted within the guidelines of the Animal Ethics Committee of the University of Otago, New Zealand, and adhered to the New Zealand Animal Welfare Act 1999, which complies with the US National Institutes of Health Guide for the Care and Use of Laboratory Animals. This study used 2‐ to 3‐month‐old (~250 g) specific pathogen‐free male Sprague–Dawley rats, a strain and age commonly used for myocyte isolation, for example see study of Xiao et al. (2007). All animals were housed at least two animals per cage using standard conditions including filter topped cage with cage enrichment. All animals were killed using a lethal dose of pentobarbital (100 mg·kg−1).
Randomization, blinding and normalization
In all experiments, the order of drug treatments was randomized each day. All experiments were collected by one operator and analysed by a second using anonymized file names. Moreover, where possible, the analysis was automated preventing operator bias. Where possible, we have provided and analysed absolute values. Normalization was only applied to the myocyte and the supplementary Western blotting data. For myocytes, this was to control for unwanted differences in basal cell activity between animals. For Western blotting, the protein band density can vary dramatically between blots, so normalization was applied to remove unwanted sources of variation. For single channels, while statistical analysis was performed on absolute P o, the relative P o of some experiments is also reported in the text for easy comparison between treatments.
Unequal group sizes
The group size n was not equal in all experiments. For the HEK293 cell experiments, the data were analysed based on experimental number, but each experiment was an average of a large number of cells. Rather than setting an experimental n, we use the total number of cells to determine our group size. Cell density varies, so to provide the same number of cells requires a different number of experiments (leading to a variation in n). Additionally, for the HEK293 cell experiments, we performed at least two control experiments every day to ensure consistency of cells between days. For each drug examined, we also perform at least two experiments under that condition per day. It is not possible to test all drug conditions on each day; therefore, the n for controls is typically larger than any drug condition; this is particularly true where a large range of drug concentrations was studied. For single channel recordings, some channels did not provide stable recordings and were not included leading to a small variation in n. There is also a small difference in n for the myocyte‐based studies. This is due to the randomization of drug treatments across myocytes isolated from different animals and because some myocyte isolations produce fewer usable cells. This precluded the testing of all drugs in all animals.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018).
Results are presented as mean ± SEM. For statistical analysis of Figures 1 and 2, a two‐way ANOVA with a Dunnett's post hoc test was performed. For Figures 3, 4 and 7, a non‐parametric ANOVA (Kruskal–Wallis) with Dunn's post hoc test was performed. Absolute differences in channel properties (Figures 5 and 6 and Supporting Information Figure S4) were compared using a paired Student's t‐tests or paired ANOVA with a Tukey post hoc test. Normal distribution for parametric analysis was determined using a Shapiro–Wilk normality test. Post hoc tests were only performed if F was significant. All significance was determined as P < 0.05 (denoted by * or # in figures). All data analysis and data plotting were performed using GraphPad Prism 7 (RRID:SCR_002798).
Figure 1.
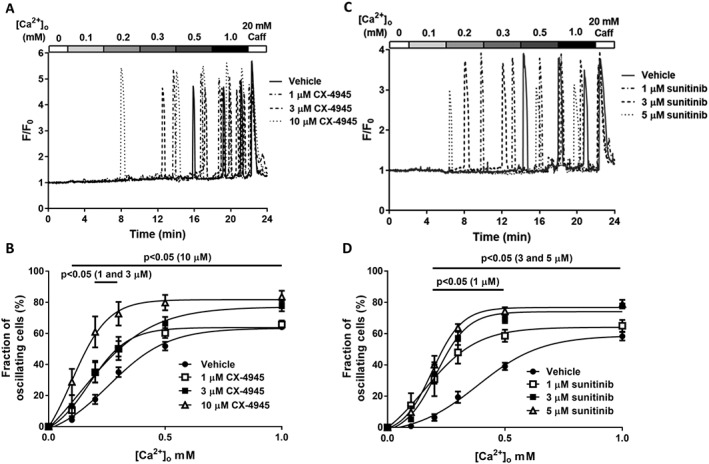
Class I kinase inhibitors increase the propensity for SOICR. HEK293 cells stably expressing RyR2 were loaded with fluo‐4 in KRH buffer. Cells were superfused with KRH containing 0, 0.1, 0.2, 0.3, 0.5 or 1 mM Ca2+. At the end of each experiment, cells were perfused with 20 mM caffeine. Representative fluo‐4 traces presented from cells treated with (A) 1, 3 or 10 μM CX‐4945 or (C) 1, 3 or 5 μM sunitinib. The fraction of cells experiencing SOICR at each [Ca2+]o when treated with each concentration of CX‐4945 or sunitinib (B and D respectively). Data shown are mean ± SEM. For CX‐4945, n = 25 (vehicle), 6 (1 μM), 5 (3 μM) or 8 (10 μM) independent experiments. For sunitinib, n = 6 (vehicle), 5 (1 μM), 5 (3 μM) or 6 (5 μM) independent experiments.
Figure 2.
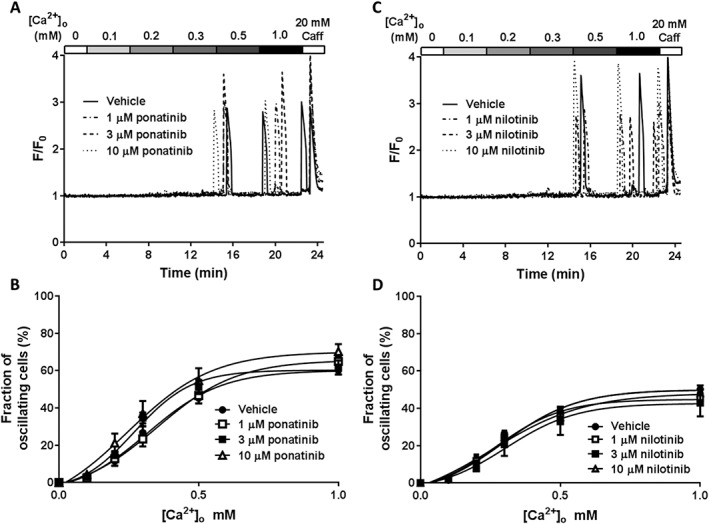
Class II kinase inhibitors do not affect the propensity for SOICR. HEK293 cells stably expressing RyR2 were loaded with fluo‐4 in KRH buffer. Cells were superfused with KRH containing 0, 0.1, 0.2, 0.3, 0.5 or 1 mM Ca2+. At the end of each experiment, cells were perfused with 20 mM caffeine. Representative fluo‐4 traces presented from cells treated with 1, 3 or 10 μM ponatinib (A) or nilotinib (C). (B) The fraction of cells experiencing SOICR at each [Ca2+]o when treated with vehicle, 1, 3 or 10 μM ponatinib. (D) The fraction of cells experiencing SOICR at each [Ca2+]o when treated with vehicle, 1, 3 or 10 μM nilotinib. Data shown are mean ± SEM. For ponatinib, n = 16 (vehicle), 8 (1 μM), 9 (3 μM) or 12 (10 μM) independent experiments. For nilotinib, n = 16 (vehicle), 12 (1 μM), 8 (3 μM) or 10 (10 μM) independent experiments.
Figure 3.
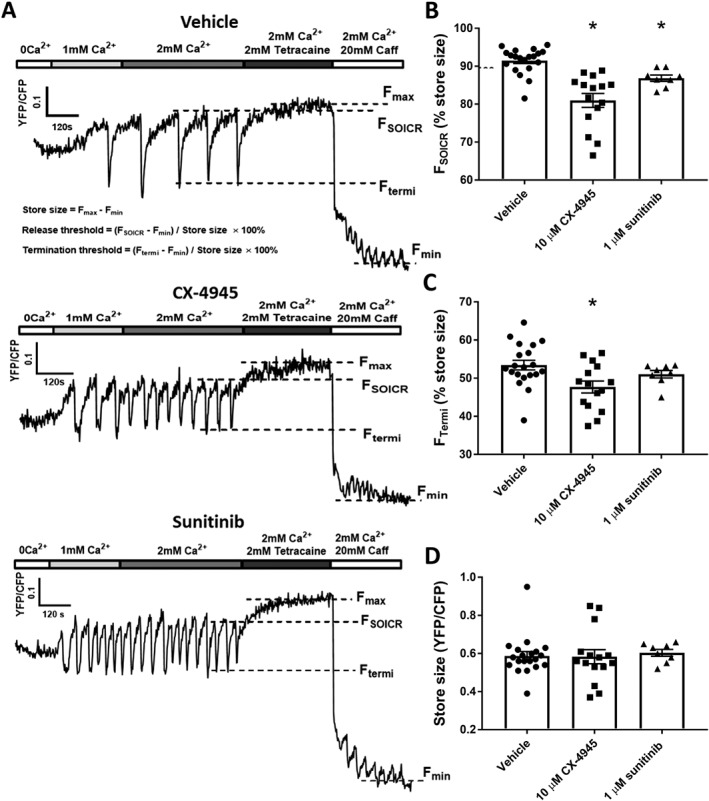
Class I kinase inhibitors reduce the release threshold for SOICR. HEK293 cells stably expressing RyR2 were transfected with the luminally expressed FRET‐based Ca2+ indicator, D1ER. The cells were superfused with KRH containing 1 or 2 mM Ca2+ with or without 10 μM CX‐4945 or 1 μM sunitinib. At the end of each experiment, cells were perfused with 2 mM tetracaine and 20 mM caffeine to determine the maximum and minimum store capacities respectively. (A) Representative traces from HEK293 cells treated with vehicle, 10 μM CX‐4945 or 1 μM sunitinib. Dashed lines represent the release threshold (FSOICR), termination threshold (FTermi) and maximum (Fmax) and minimum (Fmin) store capacities. Mean ± SEM values of FSOICR, FTermi and store size are shown in panels (B) to (D) and were calculated using the equations shown in the top trace of panel (A). n = 14, 15 and 9 independent experiments for vehicle, CX‐4945 and sunitinib respectively. *P < 0.05 versus vehicle
Figure 4.
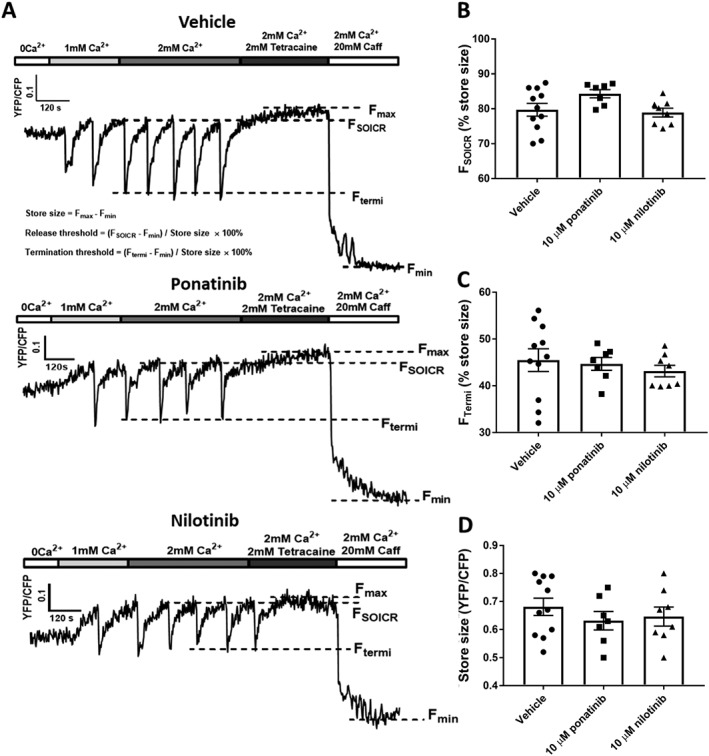
Effect of class II kinase inhibitors on SOICR dynamics. HEK293 cells stably expressing RyR2 were transfected with the luminally expressed FRET‐based Ca2+ indicator, D1ER. The cells were superfused with KRH containing 1 or 2 mM Ca2+ with or without 10 μM ponatinib or nilotinib. At the end of each experiment, cells were perfused with 2 mM tetracaine and 20 mM caffeine to determine the maximum and minimum store capacities respectively. (A) Representative traces from HEK293 cells treated with vehicle, 10 μM ponatinib or nilotinib. Dashed lines represent the release threshold (FSOICR), termination threshold (FTermi) and maximum (Fmax) and minimum (Fmin) store capacities. Mean ± SEM values of FSOICR, FTermi and store size are shown in panels (B) to (D) and were calculated using the equations shown in the top trace of panel A. n = 11, 8 and 7 independent experiments for vehicle, ponatinib and nilotinib respectively.
Figure 5.
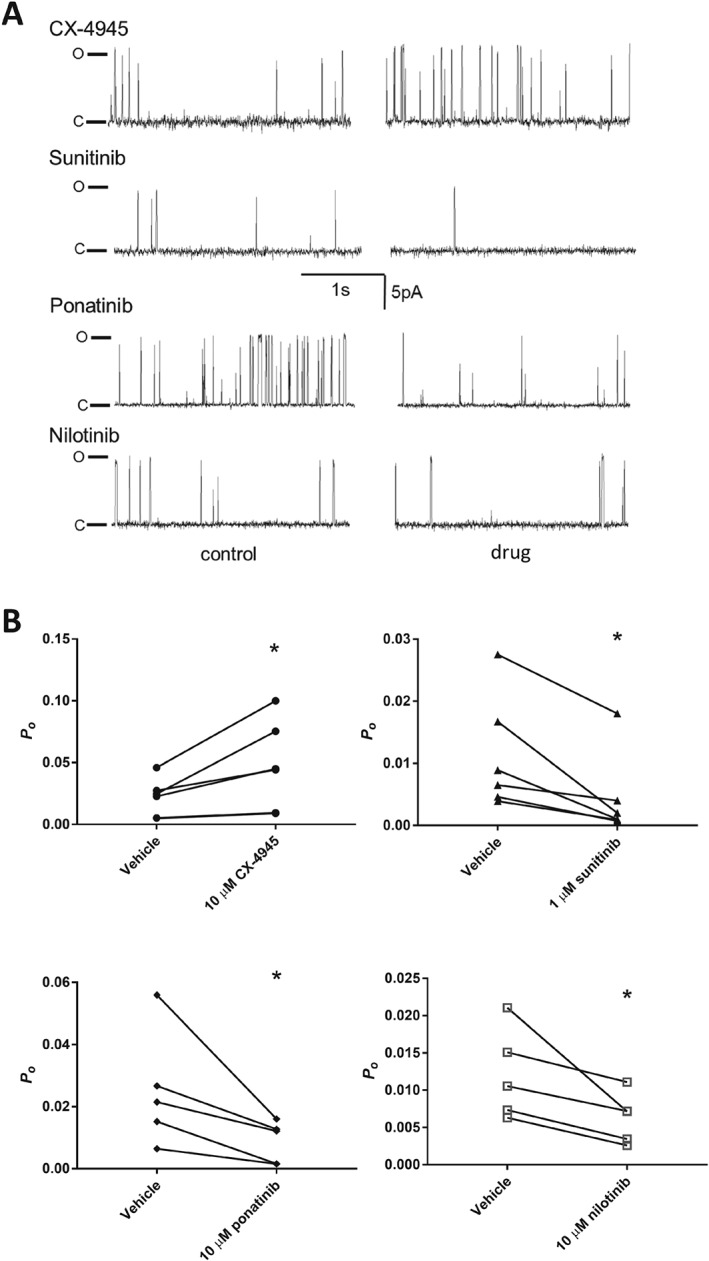
Effect of class I and II kinase on the activity of RyR2 single channels. (A) Representative 3 s continuous segments of channel currents recorded at +40 mV. The first column shows recordings before the application of each kinase inhibitor, and the second column shows the same channel following the application of each kinase inhibitor (10 μM CX‐4945, ponatinib, nilotinib or 1 μM sunitinib) to the cytosolic side of the channel. P o values shown were calculated over a full 30 s recording. (B) P o (mean ± SEM) for channels before and after the addition of each kinase inhibitor. n = 6, 6, 5 and 5 independent channels/experiments for CX‐4945, sunitinib, ponatinib and nilotinib respectively.
Figure 6.
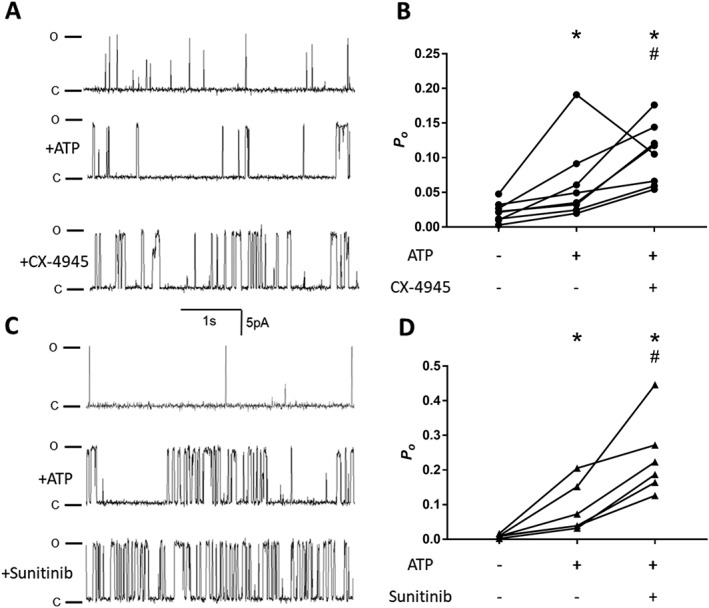
Synergistic effect of ATP and class I kinase inhibitors. (A and B) Representative 3 s continuous segments of channel currents recorded at +40 mV. The first row shows recordings before the application of ATP, the second row the same channel following the application of 2 mM ATP and the third row after the application of either 10 μM CX‐4945 (A) or 1 μM sunitinib (C). (B and D) P o (mean ± SEM) for channels before and after treatment with ATP and ATP plus each kinase inhibitor. n = 8 and 6 independent channels/experiments for CX‐4945 and sunitinib respectively. *P< 0.05 versus control, # P< 0.05 versus ATP.
Drugs, chemicals and reagents
NaCl, KCl, HEPES, glucose, MgCl2, CaCl2, caffeine, tetracaine, tetracycline, DMSO, CsCH3O3S, CsCl, TES, BAPTA, Na2ATP and BSA were obtained from Sigma‐Aldrich (Castle Hill, NSW, Australia). Fluo‐4 AM was obtained from ThermoFisher (Waltham, MA, USA). VK‐II‐86 was synthesized at the University of Otago according to the methods described by Zhou et al. (2011). CX‐4945 was obtained from Assay Matrix (Ivanhoe North, Australia), ponatinib and sunitinib from LC Laboratories (Woburn, MA, USA) and nilotinib from Santa Cruz Biotechnology (Dallas, TX, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Class I kinase inhibitors increase the propensity for SOICR
HEK293 cells expressing RyR2 have been well established to display SOICR with very similar characteristics to cardiac cells (Zhang et al., 2014; Waddell et al., 2016). In this study, HEK293 cells expressing RyR2 were used to investigate the impact of class I and II kinase inhibitors on the occurrence of SOICR. To assess the impact of the kinase inhibitors on SOICR, HEK293 cells were loaded with the Ca2+ indicator fluo‐4 AM and continuously superfused with KRH at room temperature. SOICR was induced by increasing the [Ca2+]o, and the occurrence of SOICR was monitored using single‐cell imaging. Figure 1A shows representative traces from cells treated with vehicle (DMSO), 1, 3 or 10 μM CX‐4945. Under all conditions, the cells experienced Ca2+ oscillations as the [Ca2+]o increased. As shown, cells treated with CX‐4945 typically experienced SOICR at lower [Ca2+]o than cells treated with vehicle. To quantify this, the cumulative fraction of caffeine‐responsive cells experiencing SOICR at each [Ca2+]o was determined at each CX‐4945 concentration. Figure 1B depicts the propensity of SOICR (mean ± SEM) in cells treated with vehicle, 1, 3 or 10 μM CX‐4945. Vehicle‐treated cells typically experienced no oscillations at lower [Ca2+]o. However, CX‐4945‐treated cells experienced SOICR at these lower [Ca2+]o. For example, at 0.2 mM [Ca2+]o, 61 ± 10% of cells treated with 10 μM CX‐4945 experienced SOICR compared to 17 ± 3% vehicle‐treated cells. Similarly, a significantly larger fraction of cells experienced SOICR at 0.3 mM [Ca2+]o, 1 μM (50 ± 5%), 3 μM (50 ± 7%) or 10 μM (73 ± 8%) CX‐4945 compared to vehicle (35 ± 3%).
Comparable to the effect of CX‐4945, sunitinib dramatically increased the fraction of cells experiencing SOICR at very low [Ca2+]o. Figure 1C shows representative traces of cells treated with vehicle, 1, 3 or 5 μM sunitinib. Data from HEK293 cells treated with 10 μM are not included as treatment with concentrations of sunitinib >5 μM led to a marked increase in basal Ca2+ and a loss of a caffeine response (loss of cell viability). Cells treated with sunitinib start to oscillate from 0.1 mM [Ca2+]o, whereas the majority of vehicle‐treated cells do not start to experience SOICR until 0.3 mM [Ca2+]o. Figure 1D depicts the propensity for SOICR under each condition. At 0.2 mM [Ca2+]o, 31 ± 11%, 34 ± 6% and 40 ± 6% of cells experienced SOICR when treated with 1, 3 or 5 μM sunitinib respectively. Only 6 ± 2% of vehicle‐treated cells experienced SOICR at the same [Ca2+]o. Likewise, at 0.3 mM [Ca2+]o, sunitinib markedly increased the % of cells experiencing SOICR [1 μM (48 ± 7%), 3 μM (57 ± 4%), 5 μM (64 ± 3%) vs. vehicle (19 ± 4%)]. Using these values, we calculated the EC50 for each drug. At physiological (1 mM) [Ca2+]o, the EC50 for CX‐4945 is 2.4 μM and is 0.8 μM for sunitinib (Supporting Information Figure S1).
Class II kinase inhibitors do not alter the propensity for SOICR
We next explored whether class II kinase inhibitors could also increase SOICR. Given class II inhibitors bind adjacent to the inactive ATP‐binding pocket of kinases, they likely have a different effect on RyR2. Figure 2A shows representative traces of HEK293 cells treated with vehicle, 1, 3 or 10 μM of the class II kinase inhibitor ponatinib. Figure 2A,B shows that, unlike class I kinase inhibitors, cells treated with ponatinib experience a similar propensity for SOICR as the vehicle control at all [Ca2+]o. To explore whether the lack of effect on SOICR was specific to ponatinib, we also tested a second class II kinase inhibitor, nilotinib. Consistent with ponatinib, 1, 3 nor 10 μM nilotinib impacted the propensity for SOICR at any [Ca2+]o (Figure 2C,D).
Class I but not class II kinase inhibitors decrease the release threshold for SOICR
Our previous work studying the effect of caffeine and oxidation has shown that a common mechanism by which the propensity of SOICR is increased is due to a reduction in the FSOICR (Kong et al., 2008; Waddell et al., 2016). Therefore, we next explored the effect of class I and class II kinase inhibitors on FSOICR (which is expressed as a percentage of the total store) using the luminally expressed Ca2+ sensor D1ER as previously described (Jones et al., 2008; Zhang et al., 2016). We also examined the termination threshold (the intracellular Ca2+ store level, expressed as a percentage of the total store), at which SOICR events terminate (FTermi), controlled by the inactivation of RyR2 due to partial depletion of SR Ca2+ (Zima et al., 2008; Tang et al., 2012) or luminal Ca2+‐dependent deactivation of RyR2 (Lukyanenko et al., 1998).
Figure 3A shows representative D1ER traces recorded from HEK293 cells expressing RyR2 treated with vehicle, 10 μM CX‐4945 or 1 μM sunitinib. FSOICR was determined by the mean FRET ratio immediately prior to each SOICR event (downward deflection), FTermi was determined by the mean FRET ratio of the nadir of SOICR and the maximum store was determined in the presence of 2 mM tetracaine (Fmax) and the minimum store in the presence of 20 mM caffeine (Fmin). Figure 3B–D shows the mean ± SEM for FSOICR, FTermi and store size in cells treated with vehicle, 10 μM CX‐4945 or 1 μM sunitinib. The presence of CX‐4945 significantly reduced both FSOICR (CX‐4945 81.0 ± 1.8, vehicle 91.5 ± 0.8) and FTermi (CX‐4945 47.7 ± 1.6, vehicle 53.4 ± 1.3) without altering the total Ca2+ store size. A similar effect on FSOICR was seen in cells treated with sunitinib (sunitinib 86.8 ± 2.881, vehicle 91.5 ± 0.8); however, sunitinib did not result in a significant reduction in FTermi (sunitinib 51.0 ± 1.0, vehicle 53.4 ± 1.3). In contrast to the class I kinase inhibitors, neither ponatinib nor nilotinib had any effect on FSOICR or FTermi (Figure 4A–C). Consistent with the class I kinase inhibitors, neither class II inhibitor had any effect on the total Ca2+ store size (Figure 4D).
Class I and II kinase inhibitors regulate RyR2 at the single channel level
We next explored whether the change in SOICR could be due to changes in the phosphorylation state of RyR2 as RyR2 phosphorylation of RyR2 at S2808 and S2814 has been previously shown to increase RyR2 activity (Xiao et al., 2007; Dobrev and Wehrens, 2014). Phosphorylation of RyR2 at S2808 and S2814 was determined in lysates prepared from HEK293 cells treated with each kinase inhibitor using specific phospho‐antibodies to each site. Supporting Information Figure S2 shows that the phosphorylation state of RyR2 remained unchanged when treated with any of the kinase inhibitors.
In the absence of change in the phosphorylation state of RyR2, we then determined if the change in FSOICR could be attributed to a specific effect of the drugs on single RyR2 channels. Single channel currents were recorded in RyR2 channels incorporated into artificial lipid bilayers from RyR2‐enriched cardiac SR vesicles. Figure 5A shows representative recordings of channels before and after the addition of 10 μM CX‐4945, ponatinib and nilotinib and 1 μM sunitinib to the cytoplasmic side of the channel. As shown in Figure 5B, the addition of CX‐4945 resulted in a significant increase in the open probability (P o) (0.02 ± 0.006 and 0.05 ± 0.01 for vehicle and CX‐4945‐treated channels respectively) and corresponding decrease in the open time (T o) (Supporting Information Figure S3). The increase in P o caused by CX‐4945 at this fixed luminal [Ca2+] likely underlies the decrease in FSOICR and corresponding increase in SOICR propensity observed in the previous experiments. Surprisingly, unlike CX‐4945, and in contrast to its effect on SOICR, sunitinib led to a decrease in P o (from vehicle: 0.01 ± 0.004 to sunitinib: 0.004 ± 0.003), which could be attributed to an increase in T c (Supporting Information Figure S3). The addition of both class II kinase inhibitors also led to a significant decrease in the P o (ponatinib: from 0.03 ± 0.008 to 0.009 ± 0.003; nilotinib: from 0.01 ± 0.003 to 0.006 ± 0.002).
Class I inhibitors increase RyR2 channel activity in the presence of ATP
In the previous set of experiments, 1 μM sunitinib unexpectedly inhibited RyR2 which is inconsistent with the increase in SOICR previously observed (Figures 1 and 3). It is well established that effective activation of RyR2 requires the presence of ATP which works synergistically with Ca2+ (Yang and Steele, 2000; Lindsay et al., 2018). Therefore, we next examined the effect of CX‐4945 and sunitinib in the presence of 2 mM ATP (Na2ATP), a concentration sufficient to elicit a maximal and physiological activation of RyR2 (Yang and Steele, 2000; Tencerová et al., 2012). As can be seen in Figure 6, the addition of ATP alone resulted in a modest increase in the P o of RyR2 (for T o and T c, see Supporting Information Figure S3). The addition of CX‐4945 further increased the P o of RyR2 (Figure 6B; pre‐ATP: 0.022 ± 0.005; +ATP: 0.063 ± 0.02; +CX‐4945: 0.105 ± 0.015). This suggests that CX‐4945 can increase RyR2 activity in the presence and absence of ATP but is more potent when combined with ATP (increase in relative P o in the absence of ATP was 2.16 ± 0.67 vs. 4.82 ± 0.7 when added in the presence of ATP). Interestingly, and in stark contrast to the effect observed in the absence of ATP, sunitinib dramatically increased the P o when added in the presence of ATP (Figure 6C; pre‐ATP: 0.01 ± 0.002; +ATP: 0.09 ± 0.03; +sunitinib: 0.24 ± 0.05). This is consistent with the increase in SOICR observed at the cell level suggesting that the effect of sunitinib is strongly ATP dependent. Although CX‐4945 and sunitinib behave differently in the absence of ATP, in the presence of ATP, both result in a comparable increase in relative P o (1.67 ± 0.69 and 2.63 ± 0.52 for CX‐4945 and sunitinib respectively). This is in keeping with the analogous increase in SOICR mediated by both drugs.
Class I kinase inhibitors but not class II kinase inhibitors increase the propensity for spontaneous Ca2+ waves in ventricular myocytes
Although HEK293 cells stably expressing RyR2 and single channel recordings provide excellent models to interrogate SOICR dynamics and specific effects on RyR2 function, they lack some of the regulatory proteins required for RyR2 function in situ. To determine the impact of class I and II kinase inhibitors on RyR2 in situ, the propensity for SOICR (Ca2+ waves) was measured in freshly isolated rat ventricular myocytes. Ventricular myocytes were loaded with the Ca2+ indicator fluo‐4 and paced at 0.5 Hz. Cells were superfused with KRH containing 1 mM [Ca2+]o and 10 μM CX‐4945, ponatinib and nilotinib and 1 μM sunitinib or vehicle control. After 10 min, pacing was stopped and the frequency of spontaneous Ca2+ waves determined over 2 min. Figure 7A shows the number of Ca2+ waves per minute following the cessation pacing normalized to vehicle control for cells isolated from the same animal. Normalization was performed to minimize variations in basal wave activity seen between cells isolated from different animals. CX‐4945 and sunitinib both increased the frequency of Ca2+ waves by 2 ± 0.3‐ and 2.2 ± 0.2‐fold respectively. Consistent with their lack of effect on SOICR occurrence in HEK293 cells, neither ponatinib nor nilotinib significantly altered the occurrence of Ca2+ waves (0.8 ± 0.3 and 0.9 ± 0.2 of vehicle respectively).
Figure 7.
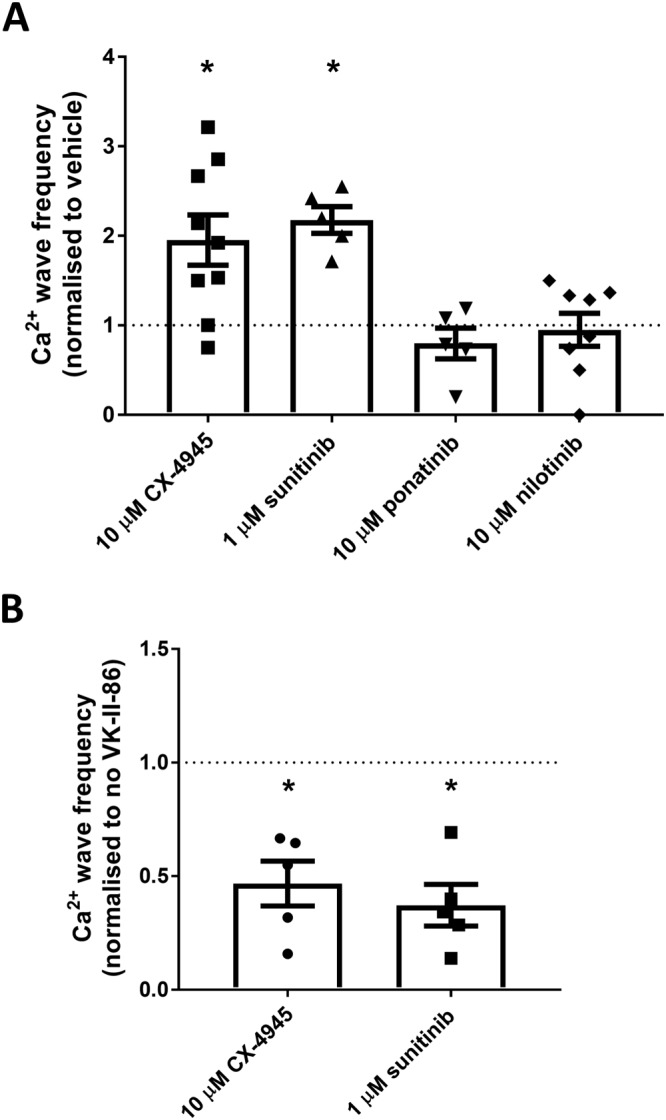
Class I kinase inhibitors increase Ca2+ wave generation in rat cardiomyocytes which can be prevented by VK‐II‐86. Freshly isolated rat ventricular myocytes were loaded with fluo‐4 in KRH buffer. (A) Cells were superfused with KRH containing 1 mM Ca2+ and paced at 0.5 Hz. Cells were treated with vehicle or 10 μM of CX‐4945, ponatinib or nilotinib or 1 μM sunitinib for 10 min before pacing was stopped for 2 min. The number of spontaneous Ca2+ waves occurring during the 2 min was recorded. At the end of each experiment, cells were perfused with 20 mM caffeine to confirm cell viability. Data shown are the Ca2+ wave frequency (mean ± SEM) normalized to vehicle‐treated cells from the same animal (10 μM CX‐4945 n = 7; 1 μM sunitinib n = 5; 10 μM ponatinib n = 7; 10 μM nilotinib n = 8, different animals); *P < 0.05 versus vehicle. (B) As with panel (A) except cells were pre‐incubated with 1 μM VK‐II‐86 or vehicle control (DMSO) for 30 min prior to imaging. Data shown are the Ca2+ wave frequency (mean ± SEM) normalized to non‐VK‐II‐86‐treated cells (vehicle control) from the same animal (n = 5 different animals); *P < 0.05 versus vehicle.
VK‐II‐86 can reverse the effect of class I kinase inhibitors by reducing Ca2+ wave generation in isolated rat cardiomyocytes
We have previously shown that a non‐β‐blocking carvedilol analogue, VK‐II‐86, is able to effectively suppress SOICR through a specific effect on RyR2 (Zhou et al., 2011). Therefore, we explored whether this compound could protect against the effect of class I kinase inhibitors on Ca2+ wave generation. Ventricular myocytes were pre‐inbubated with 1 μM VK‐II‐86 or vehicle control for 30 min before being exposed to the same protocol as described above (1 μM VK‐II‐86 was present throughout the assay). Figure 7B shows that VK‐II‐86 is able to markedly reduce the frequency of Ca2+ waves in myocytes treated with CX‐4945 (reduced to 0.5 ± 0.1 of cells treated with CX‐4945 alone) and sunitinib (reduced to 0.4 ± 0.1 of cells treated with sunitinib alone).
Discussion
This study examined the effect of class I and II kinase inhibitors on RyR2 function. It found that class I kinase inhibitors can increase the propensity for spontaneous Ca2+ release (SOICR) due to a drug‐induced increase in channel function (P o) in the presence of ATP. Although not observed at the cell level, class II kinase inhibitors tended to have an inhibitory effect on RyR2 through a decrease in P o. These data may explain the pro‐arrhythmogenic nature of several class I kinase inhibitors and perhaps the cardiotoxicity of class II compounds. We also show that the use of a known anti‐SOICR agent, designed to reverse the effect of RyR2 mutations on channel function, is able to protect against class I kinase inhibitor‐induced increases in SOICR.
Class I kinase inhibitors increase RyR2 single channel activity and propensity for SOICR
A key finding of this study is that class I kinase inhibitors can alter the function of RyR2. In particular, class I kinase inhibitors reduce the FSOICR resulting in a dramatic increase in the propensity for SOICR. It is well established that a reduction in the threshold for SOICR, combined with an increase in SR load, enhances the propensity for SOICR which is commonly linked to arrhythmias (Jones et al., 2008; Chen et al., 2014; Jones et al., 2017). We have found this to be a common mechanism contributing to how mutations within RyR2, oxidation, doxorubicin and methylxanthine derivatives such as aminophylline and theophylline all lead to arrhythmia (Jones et al., 2008; Kong et al., 2008; Hanna et al., 2014; Waddell et al., 2016). Although class I kinase inhibitors appear to have a strong agonist effect at the cell level, at the single channel, the effect is less clear cut. Of the two class I drugs examined, CX‐4945 increased the P o of RyR2 directly, whereas sunitinib required ATP to become a RyR2 agonist. Indeed, in the absence of physiological ATP, 1 μM sunitinib appears to inhibit RyR2 channels. How the presence of ATP converts sunitinib from an inhibitor to agonist is unclear. As our single channel data were recorded at a single [Ca2+], it could be argued that the effect of low concentrations of sunitinib in the absence of ATP is Ca2+ dependent. Indeed, it is likely that both CX‐4945 and sunitinib work synergistically with Ca2+, a mechanism shared by other regulators of RyR2. However, the finding that sunitinib increases SOICR across all of the [Ca2+]o examined in HEK293 cells suggests that the inhibitory effect observed in the absence of ATP is very unlikely to be due to the [Ca2+] used in the single channel recordings.
Although our data show a strong agonist effect of class I kinase inhibitors, the molecular mechanism by which either class I kinase inhibitor increases the activity of RyR2 in the presence of ATP remains unknown. The interaction of ATP and other regulators of RyR2 is complex with some, such as Ca2+, acting synergistically (Yang and Steele, 2000; Lindsay et al., 2018) and others in opposition (Dulhunty et al., 2005). Indeed, the precise molecular/structural mechanism by which ATP and other extensively studied regulators of RyR2 function, such as caffeine and catecholaminergic polymorphic tachycardia causative mutations, also remains poorly defined. We have previously speculated that they all act to induce changes in the local structure of RyR2 which are translated to the channel pore through long‐range allosteric interactions (Jones et al., 2017). This mechanism may also underlie the effects observed in this study. Future studies utilizing the increasingly detailed structures of RyR may finally allow these outstanding questions to be addressed.
Class II kinase inhibitors decrease RyR2 single channel activity
In stark contrast to the effect of class I kinase inhibitors, class II kinase inhibitors appear to reduce RyR2 function, at least at the single channel level. Channels treated with either ponatinib or nilotinib show a marked decrease in P o. Interestingly, the striking inhibitory effect at the single channel level does not appear to translate to a functional effect at the cell level as neither compound resulted in a significantly reduced propensity for SOICR in HEK293 cells or ventricular myocytes. Combined, these data suggest that class II kinase inhibitors may reduce single RyR2 channel function but not sufficiently to alter the activity of the channel in situ. Why this is the case remains to be determined. As we did not perform single channel recordings of class II kinase inhibitors in the presence of ATP, it is possible that this may explain the seemingly contradictory findings. However, as neither class II kinase inhibitor had an effect at the more physiologically relevant cell level, we did not explore this further.
Anti‐SOICR agents as a means of reversing the pro‐SOICR effect of class I kinase inhibitors
In this study, we found that both CX‐4945 and sunitinib are potent promotors of SOICR at or near the concentrations used in animal studies or clinically. In our HEK293 cell assays, we found that the EC50 to trigger SOICR for CX‐4945 and sunitinib were 2.3 and 0.8 μM respectively. CX‐4945 human trial data are not yet available, but in animal studies, the effective dose of CX‐4945 results in a peak plasma concentration (Cmax) of 15 μM (Siddiqui‐Jain et al., 2010). The Cmax for sunitinib in patients taking a typical oral dose of 50 mg·day−1 is 28 ng·mL−1 and increases threefold to fourfold over 10 days (equivalent of ~0.3 μM) (Kim et al., 2009). This indicates that SOICR is triggered by both drugs at clinically relevant concentrations. As the occurrence of SOICR is strongly linked to arrhythmias and cardiomyopathies, this suggests that some of the cardiotoxic effects of these drugs may be through this mechanism. The most common electrical disruption observed with both type I and II kinase inhibitors is a prolonged QT interval. Recently, mutations within RyR2 have also been linked to long QT syndrome (Valdivia et al., 2016; Fukuyama et al., 2017) suggesting that RyR2 dysfunction is, at least in part, associated with this disease. Characterization of one of the mutations found that it resulted in a gain‐of‐function (Valdivia et al., 2016) which is similar to the effect of class I kinase inhibitors reported here.
Given the clinical relevance of SOICR, there have been multiple studies offering mechanisms to prevent SOICR as a means of treating arrhythmia. We have previously shown that the β‐blocker carvedilol and its non‐β‐blocking derivative VK‐II‐86 are able to supress SOICR in HEK293 cells and myocytes and prevent arrhythmias in mice (Zhou et al., 2011). Here, we found that consistent with other models of SOICR, VK‐II‐86 is able to suppress class I kinase inhibitor‐mediated SOICR events. This suggests not only that carvedilol, VK‐II‐86 or derivatives may be future treatments for some of the cardiotoxicity of class I kinase inhibitors but also that these compounds are effective at preventing spontaneous Ca2+ release triggered by a multitude of mechanisms.
In summary, we demonstrate that class I kinase inhibitors increase the activity of RyR2 channels (P o) which causes a decrease in the FSOICR, ultimately increasing the propensity for SOICR. This may offer a novel insight into the cardiotoxic effects of these compounds. Excitingly, our data also suggest that the deleterious effects of these compounds may be reversed using agents known to display anti‐SOICR properties some of which, such as carvedilol, are already in clinical use.
Author contributions
A.D.C. and L.S. performed the majority of the HEK293 cell‐based experiments and the primary analysis. A.D.C., L.A.G. and M.L.M. collected and analysed the isolated myocyte data. C.T. and A.F.D. collected and analysed the single channel data. V.S. and A.B.G. synthesized VK‐II‐86 and advised on the interpretation of the data and pharmacology. A.D.C. performed and analysed the western blots. N.M. wrote the script to automatically detect and quantify the SOICR release data. P.P.J. designed the study, performed secondary analysis and wrote the paper with assistance from A.D.C., L.A.G. and A.F.D.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 EC50 for RyR2 activation by class I kinase inhibitors. HEK293 cells stably expressing RyR2 were loaded with fluo‐4 in KRH buffer. Cells were superfused with KRH containing 1 mM Ca2+ with various concentrations of either CX‐4945 or sunitinib. The percentage of cells experiencing SOICR at each drug concentration was normalised the maximum number of cells experiencing SOICR (at the highest drug concentration). The EC50 was calculated using the standard EC50 function in GraphPad Prism 7. For CX‐4945; N = 25 (vehicle), 6 (1 μM), 5 (3 μM), 5 (5 μM), 8 (10 μM), 15 (20 μM) and 18 (30 μM). For sunitinib; N = 17 (vehicle), 6 (0.3 μM), 9 (0.5 μM), 5 (1 μM), 5 (3 μM) and 6 (5 μM).
Figure S2 Class I or II kinase inhibitors do not alter the phosphorylation of RyR2 at S2808 or S2814. HEK293 cells expressing RyR2 were incubated with 10 μM CX‐4945, ponatinib, nilotinib or 1 μM sunitinib. A. Western blots were performed using antibodies to total RyR2, S2808 or S2814. B and C. The labelling of S2808 or S2814 was compared to total RyR2 in cells treated with each drug and normalised to the ratio in cells treated with vehicle control. n = 5 independent batches of HEK293 cells and significance was examined using a Kruskal‐Wallis (Dunn's post‐hoc) test.
Figure S3 Open and closed times for RyR2 channels treated with class I and II kinase inhibitors. The mean(±SEM) open (To) and closed (Tc) times (ms) of single RyR2 channels recorded over a 30 second period in the presence or absence of class I and II kinase inhibitors. Currents for each channel were recorded at ±40 mV and combined for each channel. For CX‐4945, n = 6; sunitinib, n = 6; ponatinib, n = 5; nilotinib, n = 5, individual channels for each drug. The effect of each drug on To and Tc was determined using a paired Student's t = test.
Figure S4 Effect of ATP and class I kinase inhibitors on To and Tc. The open (To) and closed (Tc) times (ms) of single RyR2 channels recorded over a 30 second period in the presence of absence of ATP and class I kinase inhibitors. For CX‐4945, n = 8; sunitinib, n = 6. * vs. control; # vs. ATP. The effect of each drug on To and Tc was determined using a paired ANOVA with Tukey post‐hoc test.
Acknowledgements
This work was supported in part by research grants from the Marsden Fund administered by the Royal Society of New Zealand (UOO1501), Health Research Council of New Zealand (18‐232) and the Heart Foundation of New Zealand to P.P.J. A.D.C. was a recipient of a PhD Scholarship from the University of Otago, and L.A.G. was the recipient of a Postdoctoral Research Fellowship from the Division of Health Sciences, University of Otago.
Chakraborty, A. D. , Gonano, L. A. , Munro, M. L. , Smith, L. J. , Thekkedam, C. , Staudacher, V. , Gamble, A. B. , Macquaide, N. , Dulhunty, A. F. , and Jones, P. P. (2019) Activation of RyR2 by class I kinase inhibitors. British Journal of Pharmacology, 176: 773–786. 10.1111/bph.14562.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Striessnig J, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b)). The Concise Guide to PHARMACOLOGY 2017/18: voltage‐gated ion channels. Br J Pharmacol 174: S160–S194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Y, Jones PP, Guo J, Zhong X, Clark RB, Zhou Q et al (2013). Phospholamban knockout breaks arrhythmogenic Ca2+ waves and suppresses catecholaminergic polymorphic ventricular tachycardia in mice. Circ Res 113: 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard NA, Sakowska MM, Dulhunty AF, Laver DR (2002). Calsequestrin is an inhibitor of skeletal muscle ryanodine receptor calcium release channels. Biophys J 82: 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation‐contraction coupling. Nature 415: 198–205. [DOI] [PubMed] [Google Scholar]
- Chen W, Wang R, Chen B, Zhong X, Kong H, Bai Y et al (2014). The ryanodine receptor store sensing gate controls Ca2+ waves and Ca2+ triggered arrhythmias. Nat Med 20: 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P (2001). The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur J Biochem 268: 5001–5010. [DOI] [PubMed] [Google Scholar]
- Copello JA, Barg S, Onoue H, Fleischer S (1997). Heterogeneity of Ca2+ gating of skeletal muscle and cardiac ryanodine receptors. Biophys J 73: 141–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D, Wehrens XHT (2014). Controversies in cardiovascular research: role of ryanodine receptor phosphorylation in heart failure and arrhythmias. Circ Res 114: 1311–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulhunty AF, Pouliquin P, Coggan M, Gage PW, Board PG (2005). A recently identified member of the glutathione transferase structural family modifies cardiac RyR2 substate activity, coupled gating and activation by Ca2+ and ATP. Biochem J 390: 333–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AD, Sheth PR, Basso AD, Paliwal S, Gray K, Fischmann TO et al (2011). Structural basis of CX‐4945 binding to human protein kinase CK2. FEBS Lett 585: 104–110. [DOI] [PubMed] [Google Scholar]
- Fukuyama M, Ohno S, Ichikawa M, Takayama K, Fukumoto D, Horie M (2017). Novel RYR2 mutations causative for long QT syndromes. Eur Heart J 38: P5860. [Google Scholar]
- Hanna AD, Lam A, Tham S, Dulhunty AF, Beard NA (2014). Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol Pharmacol 86: 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Furtmann N, Bajorath J (2015). Current compound coverage of the kinome. J Med Chem 58: 30–40. [DOI] [PubMed] [Google Scholar]
- Jones PP, Guo W, Chen SRW (2017). Control of cardiac ryanodine receptor by sarcoplasmic reticulum luminal Ca2+ . J Gen Physiol 149: 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PP, Jiang D, Bolstad J, Hunt DJ, Zhang L, Demaurex N et al (2008). Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store‐overload‐induced Ca2+ release. Biochem J 412: 171–178. [DOI] [PubMed] [Google Scholar]
- Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA et al (2010). In the RyR2(R4496C) mouse model of CPVT, β‐adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ Res 107: 1483–1489. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, Balis FM, Widemann BC (2009). Sorafenib and sunitinib. Oncologist 14: 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA et al (2017). The target landscape of clinical kinase drugs. Science 358. pii: eaan4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong H, Jones PP, Koop A, Zhang L, Duff HJ, Chen SR (2008). Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem J 414: 441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay C, Sitsapesan M, Chan WM, Venturi E, Welch W, Musgaard M et al (2018). Promiscuous attraction of ligands within the ATP binding site of RyR2 promotes diverse gating behaviour. Sci Rep 8: 15011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Gray NS (2006). Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2: 358–364. [DOI] [PubMed] [Google Scholar]
- Liu Y, Kimlicka L, Hiess F, Tian X, Wang R, Zhang L et al (2013). The CPVT‐associated RyR2 mutation G230C enhances store overload induced Ca2+ release and destabilizes the N‐terminal domains. Biochem J 454: 123–131. [DOI] [PubMed] [Google Scholar]
- Lukyanenko V, Wiesner TF, Gyorke S (1998). Termination of Ca2+ release during Ca2+ sparks in rat ventricular myocytes. J Physiol 507: 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marengo JJ, Hidalgo C, Bull R (1998). Sulfhydryl oxidation modifies the calcium dependence of ryanodine‐sensitive calcium channels of excitable cells. Biophys J 74: 1263–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G et al (2003). In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet‐derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9: 327–337. [PubMed] [Google Scholar]
- O'Hare T, Shakespeare WC, Zhu X, Eide CA, Rivera VM, Wang F et al (2009). AP24534, a pan‐BCR‐ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation‐based resistance. Cancer Cell 16: 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson SJ, Steele GA, Gallant EM, Lam A, Schwartz CE, Board PG et al (2017). Association of FK506 binding proteins with RyR channels – effect of CLIC2 binding on sub‐conductance opening and FKBP binding. J Cell Sci 130: 3588–3600. [DOI] [PubMed] [Google Scholar]
- Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C et al (2008). Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 26: 5204–5212. [DOI] [PubMed] [Google Scholar]
- Shah RR, Morganroth J (2015). Update on cardiovascular safety of tyrosine kinase inhibitors: with a special focus on QT interval, left ventricular dysfunction and overall risk/benefit. Drug Saf 38: 693–710. [DOI] [PubMed] [Google Scholar]
- Siddiqui‐Jain A, Drygin D, Streiner N, Chua P, Pierre F, O'brien SE et al (2010). CX‐4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res 70: 10288–10298. [DOI] [PubMed] [Google Scholar]
- Smith JJ, Vetter I, Lewis RJ, Peigneur S, Tytgat J, Lam A et al (2013). Multiple actions of phi‐LITX‐Lw1a on ryanodine receptors reveal a functional link between scorpion DDH and ICK toxins. Proc Natl Acad Sci U S A 110: 8906–8911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Tian X, Wang R, Fill M, Chen SR (2012). Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ Res 110: 968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tencerová B, Zahradníková A, Gaburjáková J, Gaburjáková M (2012). Luminal Ca2+ controls activation of the cardiac ryanodine receptor by ATP. J Gen Physiol 140: 93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia CR, Antunez‐Arguelles E, Hernandez J, Herron T, Villarreal T, Iturralde P et al (2016). Molecular and functional characterization of a novel RYR2 mutation linked to long QT syndrome. Circulation 134: A20155. [Google Scholar]
- Waddell HM, Zhang JZ, Hoeksema KJ, Mclachlan JJ, Mclay JC, Jones PP (2016). Oxidation of RyR2 has a biphasic effect on the threshold for store overload‐induced calcium release. Biophys J 110: 2386–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson LJ, Linley A, Hammond DE, Hood FE, Coulson JM, Macewan DJ et al (2018). New perspectives, opportunities, and challenges in exploring the human protein kinome. Cancer Res 78: 15–29. [DOI] [PubMed] [Google Scholar]
- Xiao B, Tian X, Xie W, Jones PP, Cai S, Wang X et al (2007). Functional consequence of protein kinase A‐dependent phosphorylation of the cardiac ryanodine receptor: sensitization of store overload‐induced Ca2+ release. J Biol Chem 282: 30256–30264. [DOI] [PubMed] [Google Scholar]
- Yang Y, Steele DS (2000). Effects of cytosolic ATP on spontaneous and triggered Ca2+‐induced Ca2+ release in permeabilised rat ventricular myocytes. J Physiol 523: 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JZ, Mclay JC, Jones PP (2014). The arrhythmogenic human HRC point mutation S96A leads to spontaneous Ca(2+) release due to an impaired ability to buffer store Ca(2+). J Mol Cell Cardiol 74: 22–31. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Waddell HM, Jones PP (2015). Regulation of RYR2 by sarcoplasmic reticulum Ca(2+). Clin Exp Pharmacol Physiol 42: 720–726. [DOI] [PubMed] [Google Scholar]
- Zhang JZ, Waddell HM, Wu E, Dholakia J, Okolo CA, Mclay JC et al (2016). FKBPs facilitate the termination of spontaneous Ca2+ release in wild‐type RyR2 but not CPVT mutant RyR2. Biochem J 473: 2049–2060. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A et al (2011). Carvedilol and its new analogs suppress arrhythmogenic store overload‐induced Ca(2+) release. Nat Med 17: 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zima AV, Picht E, Bers DM, Blatter LA (2008). Termination of cardiac Ca2+ sparks role of intra‐SR [Ca2+], release flux, and intra‐SR Ca2+ diffusion. Circ Res 103: e105–e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 EC50 for RyR2 activation by class I kinase inhibitors. HEK293 cells stably expressing RyR2 were loaded with fluo‐4 in KRH buffer. Cells were superfused with KRH containing 1 mM Ca2+ with various concentrations of either CX‐4945 or sunitinib. The percentage of cells experiencing SOICR at each drug concentration was normalised the maximum number of cells experiencing SOICR (at the highest drug concentration). The EC50 was calculated using the standard EC50 function in GraphPad Prism 7. For CX‐4945; N = 25 (vehicle), 6 (1 μM), 5 (3 μM), 5 (5 μM), 8 (10 μM), 15 (20 μM) and 18 (30 μM). For sunitinib; N = 17 (vehicle), 6 (0.3 μM), 9 (0.5 μM), 5 (1 μM), 5 (3 μM) and 6 (5 μM).
Figure S2 Class I or II kinase inhibitors do not alter the phosphorylation of RyR2 at S2808 or S2814. HEK293 cells expressing RyR2 were incubated with 10 μM CX‐4945, ponatinib, nilotinib or 1 μM sunitinib. A. Western blots were performed using antibodies to total RyR2, S2808 or S2814. B and C. The labelling of S2808 or S2814 was compared to total RyR2 in cells treated with each drug and normalised to the ratio in cells treated with vehicle control. n = 5 independent batches of HEK293 cells and significance was examined using a Kruskal‐Wallis (Dunn's post‐hoc) test.
Figure S3 Open and closed times for RyR2 channels treated with class I and II kinase inhibitors. The mean(±SEM) open (To) and closed (Tc) times (ms) of single RyR2 channels recorded over a 30 second period in the presence or absence of class I and II kinase inhibitors. Currents for each channel were recorded at ±40 mV and combined for each channel. For CX‐4945, n = 6; sunitinib, n = 6; ponatinib, n = 5; nilotinib, n = 5, individual channels for each drug. The effect of each drug on To and Tc was determined using a paired Student's t = test.
Figure S4 Effect of ATP and class I kinase inhibitors on To and Tc. The open (To) and closed (Tc) times (ms) of single RyR2 channels recorded over a 30 second period in the presence of absence of ATP and class I kinase inhibitors. For CX‐4945, n = 8; sunitinib, n = 6. * vs. control; # vs. ATP. The effect of each drug on To and Tc was determined using a paired ANOVA with Tukey post‐hoc test.