


Abstract
Toxin–antitoxin (TA) systems are ubiquitous among bacteria, frequently expressed in multiple copies, and important for functions such as antibiotic resistance and persistence. Type I TA systems are composed of a stable toxic peptide whose expression is repressed by an unstable RNA antitoxin. Here, we investigated the functionalities, regulation, and possible cross-talk between three core genome copies of the pathogenicity island-encoded ‘sprG1/sprF1’ type I TA system in the human pathogen Staphylococcus aureus. Except for SprG4, all RNA from these pairs, sprG2/sprF2, sprG3/sprF3, sprG4/sprF4, are expressed in the HG003 strain. SprG2 and SprG3 RNAs encode toxic peptides whose overexpression triggers bacteriostasis, which is counteracted at the RNA level by the overexpression of SprF2 and SprF3 antitoxins. Complex formation between each toxin and its cognate antitoxin involves their overlapping 3′ ends, and each SprF antitoxin specifically neutralizes the toxicity of its cognate SprG toxin without cross-talk. However, overexpression studies suggest cross-regulations occur at the RNA level between the SprG/SprF TA systems during growth. When subjected to H2O2-induced oxidative stress, almost all antitoxin levels dropped, while only SprG1 and SprF1 were reduced during phagocytosis-induced oxidative stress. SprG1, SprF1, SprF2, SprG3 and SprF3 levels also decrease during hyperosmotic stress. This suggests that novel SprG/SprF TA systems are involved in S. aureus persistence.
INTRODUCTION
The Gram-positive Staphylococcus aureus bacteria is part of the human commensal flora, and can be found in the skin and nostrils of 20–30% of the population (1). But S. aureus is also a major pathogen that causes a wide spectrum of nosocomial and community-associated infections that can range from skin and soft tissue infections all the way to life-threatening diseases (2,3). In addition to expressing a large set of virulence factors, S. aureus antibiotic resistance is a major public health issue (4). The expression of S. aureus virulence factors is precisely tuned by regulatory proteins and small RNAs (sRNAs) (5,6). S. aureus expresses hundreds of these sRNAs, which have been compiled into the Staphylococcal regulatory RNA Database (SRD: http://srd.genouest.org/) (7–9). Initially detected by bioinformatic and transcriptome analyses (10), seven of these sRNAs are expressed from pathogenicity islands (PIs), so they were named SprA to SprG, for small pathogenicity island RNA. One of these, SprD, contributes to infection in a murine model of sepsis (11), while SprC attenuates S. aureus virulence (12). Several of the ‘Spr’ sRNAs have associated cis-antisense RNA (asRNAs). For instance, the SprA1/SprA1AS and SprG1/SprF1 RNA pairs have been identified as functional type I toxin–antitoxin (TA) systems (13,14).
TA systems are genetic modules that encode a stable toxic protein, whose overexpression leads to growth arrest or cell death, along with a labile antitoxin that neutralizes the toxin's activity (15). TA systems were initially discovered in the 1980s on plasmids, where they maintain daughter cells through post-segregational killing (16). They are currently classified into six types depending on the antitoxin's nature and mode of action (17). The antitoxins can be RNA (types I and III) or protein (types II, IV, V and IV). They act in different ways, by inhibiting toxin synthesis, sequestering toxins, promoting toxin degradation, or by counteracting toxic activity (17). Chromosomal TA systems possess various biological functions, including maintaining the stability of genetic elements, growth control in response to environmental stresses, and persister cell formation (18–21). Like other pathogens (22), S. aureus has genes that express types I, II, and III TA systems within its genome, and five have been characterized experimentally: sprA1/sprA1AS and sprG1/sprF1 (type I); and mazEF, yefM-yoeB and omega/epsilon/zeta (type II) (23,24).
Type I TA systems are frequently present in multiple copies in bacterial genomes (25). Three additional copies of the PI-encoded SprG1/SprF1 RNA pair were detected within the core genome of S. aureus strains (10,25). The sprG2/sprF2, sprG3/sprF3 and sprG4/sprF4 core genome copies were renamed srn_2230/srn_2240, srn_4100/srn_4090 and srn_2250/srn_2260 according to the SRD database classification (8). In this study, we investigated their functionalities, regulations, and cross-talk. Our experimental data confirmed the existence of these additional copies in the S. aureus HG003 strain, and demonstrated that they function as TA systems. SprG2 and SprG3 RNA encode toxic peptides whose overexpression trigger S. aureus stasis but not death, contrary to SprG1-encoded peptides (14). This toxicity is abolished by overexpressing SprF2 or SprF3 RNA antitoxins, which down-regulate, at the RNA level, SprG2 or SprG3 peptide translation. Each SprF antitoxin specifically neutralizes its cognate SprG toxin, without cross-talk. However, our results suggested cross-regulations between the SprG/SprF TA systems, with SprG/SprF pairs modulating the RNA levels of some other copies during S. aureus growth. All SprF antitoxin RNA levels drop during H2O2-induced oxidative stress, although only SprG1 and SprF1 levels are reduced during phagocytosis-induced oxidative stress. Furthermore, SprG1, SprF1, SprF2, SprG3 and SprF3 RNA levels also decrease during hyperosmotic stress. This suggests a biological function for SprG/SprF TA systems during stress adaptation.
MATERIALS AND METHODS
Strains, plasmids and genetic manipulations
Strains, plasmids, and primers are listed in Supplementary Tables S1–S3. Staphylococcus aureus strains were grown at 37°C, 160 rpm, in brain heart infusion broth (BHI, Oxoid). When necessary, chloramphenicol and erythromycin were added at 10 μg/ml, or at 5 μg/ml for strains harboring two plasmids. We used the high-copy-number plasmid pCN35 to overexpress SprF1–4, SprG2–3, SprG2-SprF2 and SprG3-SprF3, each under the control of its own promoter (26). For the flagged SprG2 construct, a sequence encoding for the 3xFLAG epitopes was added upstream from the termination codon, in-frame with the SprG2-encoded peptide (27). This fragment, with flanking BamHI/EcoRI sites, was then inserted into pCN35. The flagged SprG2 construct which also expresses SprF2 corresponds to a 706-bp fragment of 192 nucleotides (nts) upstream of SprG2 and 143 nts downstream from SprF2. To generate constructs that are inducible by anhydrotetracycline (aTc), we amplified S. aureus HG003 DNA fragments starting at the +1 nt of transcription and ending 38 (SprG2), 36 (SprG3) or 31 (SprG4) nts downstream from its 3′ end, then inserted these into pALC (28). To generate an inducible construct containing an ‘SprG2-STOP’ mutant, we created a premature TAA termination codon by replacing G88 by a thymine, and T89 and G90 by two adenines. For the ‘SprG3-STOP1–4’ mutants, the four initiation codons (TTG63, ATG87, ATG102 and ATG108) in-frame with the TAA termination codon were mutated one at a time to obtain termination codons, then inserted into pALC. All of the constructed plasmids were transformed into S. aureus RN4220 (29) and then into S. aureus HG003 (30). Overexpression strains were confirmed by northern blotting. To inactivate the sprF1/SprG1 gene pair, HG003 DNA fragments from 1007 bp upstream and 904 bp downstream of the sprF1/SprG1 gene were amplified by PCR then cloned onto the BamHI/PstI sites of the temperature-sensitive plasmid pBT2 (31). The resulting pBT2ΔsprF1/sprG1 plasmid was transformed into S. aureus strain RN4220, then into HG003. Mutants were enriched via growth at 42°C. Cells from stationary-phase cultures were plated onto trypticase soy agar (TSA) plates and incubated at 37°C. Colonies were imprinted on plates supplemented with 10 μg/mL chloramphenicol. Chloramphenicol-sensitive colonies were tested by PCR for the deletion of sprF1/sprG1, then confirmed by northern blots.
Race mapping
The primers used are presented in Supplementary Table S3. First, 5 μg total RNAs from S. aureus HG003 were treated with Terminator 5′-phosphate-dependent exonuclease (Epicentre) to degrade 5′-monophosphate RNA, then with polyphosphatase (Epicentre) to remove pyrophosphate from the terminal single-strand RNA (9). Race mapping (rapid amplification of cDNA ends) was performed for SprG2 and SprG3 RNAs as previously described (14).
RNA extraction, northern blots and RNA half-life determination
RNA extraction was performed as previously described (11). DNA probes for RNA detection are listed in Supplementary Table S3. We separated 10 μg each of total RNAs onto 8% polyacrylamide/8M urea gels and transferred them onto Hybond-N+ membranes (Amersham). Specific 32P-labeled probes were hybridized in ExpressHyb solution (Clontech), then washed, exposed, and scanned with a PhosphorImager (Molecular Dynamics). Quantifications were performed with ImageQuant and normalized to either transfer-messenger RNA (tmRNA) or 5S ribosomal RNA. For the RNA half-life measurements, S. aureus HG003 was cultured overnight, diluted to 1/100, grown for an additional 3 h at 37°C to the exponential growth phase, and finally incubated with 200 μg/ml rifampicin for 1–120 min. Samples of 2 ml each were taken before and then at 1, 3, 5, 7.5, 10, 15, 20, 30, 60 and 120 min after rifampicin addition. These samples were centrifuged, and pellets were frozen in liquid nitrogen then stored at −80°C. The RNA was then extracted and northern blot assays performed.
SprG induction, growth, and cell death experiments
For the kinetic assays, strains containing the relevant plasmids were grown overnight in BHI. The cultures were then diluted to 1/100 in BHI, then grown in 96-well microtiter plates for 135 min at 37°C until the exponential growth phase (14). To induce expression of SprG1, SprG2, SprG3 and SprG4, the cultures were then incubated with 1 or 2 μM of aTc. For the kinetics, the optical absorbance at 600 nm (OD600) was measured at 30-min intervals using a Synergy 2 multi-mode reader (Biotek). To determine the effects of SprG induction on bacterial growth, cultures were prepared by 2-fold serial dilutions of exponential phase cultures on BHI plates containing 1 or 2 μM aTc, then incubated for 24 h at 37°C. For the cell death experiments, cultures were incubated for 135 min, washed with 9‰ saline solution, then stained with a LIVE/DEAD bacterial viability kit (Invitrogen). Pictures of fluorescence-labeled cells were captured with a Leica DM RXA2 microscope and a CoolSNAP HQ charge-coupled device camera using Metavue software (Molecular Devices).
Protein extraction, cell fractionation and western blots
Protein extracts were recovered as previously described (13,32). Zinc staining was done on the samples using a Biorad zinc stain and destain kit to verify the protein amounts used for western blotting. Samples were separated onto 16% tricine SDS-PAGE gels and transferred onto Amersham Hybond-P PVDF membranes. After blocking, the membranes were incubated with mouse monoclonal anti-FLAG horseradish peroxidase (HRP) antibodies (Sigma). The membranes were revealed after incubation using an ECL Plus western blotting detection kit (Amersham) and scanned with a LAS4000 imager. In the case of western blots containing cell fractionation samples, the membranes were incubated with the following antibodies: anti-FLAG; rabbit polyclonal anti-Micrococcus luteus ATPase (33); rabbit anti-S. aureus SarA (produced in our laboratory); and mouse monoclonal anti-S. aureus Protein A (Sigma). After incubation with HRP-conjugated secondary antibodies, the membranes were revealed as detailed above.
In vitro transcription, RNA labeling and translation assays
All RNAs were transcribed from PCR-amplified templates using HG003 or N315 genomic DNA and forward primers containing a T7 promoter sequence (Supplementary Table S3). PCR-generated DNA was used as the template for transcription with a MEGAscript T7 kit (Ambion). RNAs were gel-purified, eluted, and ethanol precipitated. The 5′ ends of SprF1–4 purified in vitro were labeled as previously described (34). In vitro translation of [35S]-methionine was done (13) using an Escherichia coli S30 Extract System for Linear Templates (Promega).
Gel shift and toeprint assays
Gel shift assays were performed as previously described (34). For determination of the dissociation constant (Kd), 0.12 pmol labeled SprF RNA was incubated with varying concentrations (0.0025–0.2 or 0.025–2.0 pmol respectively) of unlabeled SprG2 or SprG3 RNA. Interaction analysis between the pairs SprG2/SprF2, SprG2/SprF4 and SprG3/SprF3 were performed with 0.5 pmol of complete or truncated SprG2 and SprG3 RNA incubated with 0.25 or 0.5 pmol of SprF2, SprF4, and SprF3 RNA. For the cross-regulation experiments, 0.01–0.1 pmol of labeled SprF RNA was incubated with 0.05–0.5 pmol of unlabeled SprG RNA. For gel shift assays, RNAs were incubated in binding buffer (80 mM K-HEPES pH 7.5, 4 mM MgCl2, 330 mM KCl) for 20 min at 30°C before native gel separation. For the toeprinting assays, annealing mixtures containing 4 pmol SprG3 RNA with 0.5 pmol ‘SprG3-AS-SRD’-labeled primer were incubated with a buffer (20 mM Tris–HCl, pH 7.5, 60 mM NH4Cl) for 2 min at 90°C. Renaturation was done in the presence of 10 mM MgCl2 for 20 min at 20°C. The ribosomes were reactivated for 15 min at 37°C and diluted in the reaction buffer. Various concentrations (0, 0.5, 1 or 2 pmol) of purified 70S ribosomes were added to each sample, and these were incubated for 5 min at 37°C, after which MgCl2 was adjusted to 10 mM. After 15 min at 37°C, 10 pmol of uncharged tRNAfMet was added and incubated for 5 min at 37°C. The cDNA were synthesized with 2 U of AMV RT (NEB) for 15 min. Reactions were stopped by the addition of 15 μl of loading buffer II (Ambion). The cDNAs were loaded and separated onto 8% polyacrylamide/8M urea gels. Sequencing ladders were generated with the primer labeled with ‘SprG3-AS-SRD’ (Supplementary Table S3).
Stress assays
Strains were grown overnight in tryptic soy broth (TSB) diluted to 1/10 with a TSBd buffer (2.5 g/l K2HPO4, 2.5 g/l glucose, 5 g/l NaCl). Cultures were diluted to an OD600 of 0.1 in TSBd and grown until the OD600 reached 0.5. To create hyperosmotic stress, sodium chloride was added to the culture to a final concentration of 2 M. For the oxidative stress, hydrogen peroxide was added to a final concentration of 10 mM. After the stress agents were added, the cultures were grown for 1 h at 37°C under agitation. Samples (40 ml) were collected by centrifugation, and the pellets stored at −80°C until RNA extraction was done.
Phagocytosis assays
107 THP-1 monocyte cells (ATCC) were plated into 175 cm2 flasks and cultured in RPMI-1640 medium (Invitrogen) containing 10% decomplemented fetal bovine serum (FBS, Biowest) at 37°C and 5% CO2. The monocytes were then differentiated into macrophages for 3 days using 120 ng/ml phorbol 12-myristate 13-acetate (PMA, Sigma). The macrophages were then cultured for another 3 days in complete RPMI medium without PMA. The phagocytes were infected at an MOI of 10:1 (bacteria:macrophages) with S. aureus HG003 in RPMI-1640 containing 5% human AB serum (hSAB, EFS Rennes) for 2 h at 37°C and 5% CO2. After three washes in phosphate-buffered solution (PBS) 1×, in order to eliminate extracellular bacteria the cells were treated with 10 μg/ml lysostaphin (Sigma) for 1 h at 37°C and 5% CO2. Cells were rewashed, lysed in PBS 1× containing 0.5% triton X-100 for 10 min at room temperature (RT), then centrifuged for 10 min at 4500 rpm, 4°C. The supernatant was eliminated and the cell lysate resuspended in RNA lysis buffer, after which the RNA extraction was performed as described (35). RNA preparations were treated twice in a row with amplification grade DNase I (ThermoFisher) for 20 min at RT, each time purified by phenol/chloroform extraction. Purified RNA were used to produce cDNA in a reverse transcription assay (High-Capacity cDNA Reverse Transcription Kit, Life Technologies) using either random or specific primers (Supplementary Table S3). The cDNA that corresponded to RNA expressed during phagocytosis were then amplified by qPCR using a RealMasterMIX SYBR Kit (Life Technologies). Their amounts were compared to those present in the inoculum before infection, using tmRNA as reference gene.
RESULTS
SprF and SprG end mapping, nucleotide overlaps and sequence alignments in the S. aureus HG003 strain
Up to three copies of the ‘sprG1/sprF1’ locus were detected in various S. aureus strains (10,14). They are all located in the core genome, with sprG2/sprF2 and sprG4/sprF4 next to each other. We worked with HG003, which has all of the gene pairs and is considered to be the reference strain for studying S. aureus regulation (30). We began with RACE mapping to compare the SprG2 and SprG3 5′ and 3′ ends in HG003 to those previously determined in the N315 strain (14). The positions of the 5′ and 3′ ends of the SprG and SprF RNAs in HG003, as well as their overlapping regions, are shown in Figure 1A. The sprF1 antitoxin gene shares 64%, 54% and 61% sequence identity with putative antitoxins sprF2, sprF3 and sprF4 respectively (Supplementary Table S4). The sprG1 toxin gene shares 56%, 46%, and 48% sequence identity with putative toxins sprG2, sprG3 and sprG4, respectively. The most similar copies are SprF2 and SprF4, which have 93% sequence identity (Supplementary Table S4). A region of 22 nucleotides is conserved between the four SprF copies (Figure 1B). This is located outside the complementary sequence with the SprG toxin mRNA, except in the case of SprF1. The sequence alignments of the sprG genes are more divergent, but each gene is able to encode for a peptide of 35, 26 or 19 amino acids for sprG2, sprG3 and sprG4 respectively (Figure 1C and D). These peptides possess C-terminal amino acid identity, especially between the SprG1- and SprG2-encoded peptides. In summary, the three core genome copies of the ‘sprG1/sprF1’ locus have overlapping sequences at their 3′ ends, and the SprF2 and SprF4 sequences are very similar.
Figure 1.

Genomic locations of the four copies of sprG and sprF, sequence alignments of the sprF and sprG gene copies and of the SprG-encoded peptides. (A) Genomic location, length determination by RACE, and nucleotide overlaps of SprG1–4 and SprF1–4 in Staphylococcus aureus HG003. (B and C) Sequence alignments of sprF and sprG including their promoters. TATA boxes are green; +1 of transcription are magenta boxes; asterisks indicate the nucleotides shared by the four sequences; and the mutation shared between a set of three common nucleotides is boxed in red. (C) The proposed Shine-Dalgarno sequences are boxed and underlined in dark gray; the first peptide initiation codon (M1 for SprG1) is boxed in red, and the second (M14 for SprG1) is boxed in blue. Sequences of SprG-encoded peptides are yellow until the stop codon, which is underlined and bold. (D) Sequence alignments of the SprG-encoded peptides. The peptide initiation codon is red, the amino acid residues common for two SprG-encoded peptides are blue, and those common to three SprG-encoded peptides are green.
SprG2/SprF2, SprG3/SprF3 and SprG4/SprF4 pairs are functional type I TA systems
Turning to the SprG1/SprF1 pair (14), we assayed the expression of SprF2, SprF3, and SprF4 in S. aureus to see how much they neutralized SprG2, SprG3, and SprG4 toxicity. Two sets of plasmids were used for this: pALC, which has promoter inducible by anhydrotetracycline (aTc) that can overexpress the putative three new SprG toxins; and multicopy pCN35, which has an endogenous promoter that might express the putative SprF cognate antitoxins. We used northern blots to validate the overexpression of SprF2, SprF3 and SprF4 RNA in HG003 containing pCN35ΩSprF2, pCN35ΩSprF3 or pCN35ΩSprF4 (Figure 2A). We did the same for SprG2, SprG3 and SprG4 expression after aTc induction in strains transformed with pALCΩSprG2, pALCΩSprG3 or pALCΩSprG4. These experiments also indicated that with aTc induction, SprG2 and SprG3 RNA levels decrease when SprF2 and SprF3 are overexpressed respectively (Figure 2A, last 2 lanes). Note that none of the overproducing strains designed have a growth phenotype without aTc induction (Supplementary Figure S1). In the presence of aTc, the strains containing pALC and pCN35ΩSprF2–4 (and therefore expressing SprF2–4) grew in the same way as those containing the two empty vectors (Figure 2B). The cells expressing SprG2–4 after aTc induction inhibited S. aureus growth, and this toxicity was fully rescued by overexpressing SprF2–4 (Figure 2B). When spotted on BHI plates in the presence of aTc (Figure 2C), serial dilutions of every S. aureus strain used in this experiment showed the same results as the growth kinetics in liquid cultures. Altogether, these experiments indicate that the SprG2/SprF2, SprG3/SprF3 and SprG4/SprF4 pairs are functional TA systems in S. aureus.
Figure 2.

SprG2, SprG3 and SprG4 toxin RNA overexpression triggers Staphylococcus aureus growth inhibition which is reversed by their respective antitoxins. (A) Northern blot analysis of SprG2, SprF2, SprG3, SprF3, SprG4 and SprF4 RNA expression levels in isogenic S. aureus HG003 strains containing the following plasmids: (left) pALC and pCN35; pALC and pCN35ΩSprF2; pALCΩSprG2 and pCN35; or pALCΩSprG2 and pCN35ΩSprF2; (middle) pALC and pCN35; pALC and pCN35ΩSprF3; pALCΩSprG3 and pCN35; or pALCΩSprG3 and pCN35ΩSprF3; or (right) pALC and pCN35; pALC and pCN35ΩSprF4; pALCΩSprG4 and pCN35; or pALCΩSprG4 and pCN35ΩSprF4. All samples after anhydrotetracycline (aTc) induction of the pALC vectors are blue, and tmRNA is the internal loading control. (B) Growth kinetics in liquid cultures and (C) serial dilutions of HG003 strains containing the following plasmids: (left) pALC and pCN35; pALC and pCN35ΩSprF2; pALCΩSprG2 and pCN35; or pALCΩSprG2 and pCN35ΩSprF2; (middle) pALC and pCN35ΩSprF3; pALCΩSprG3 and pCN35; or pALCΩSprG3 and pCN35ΩSprF3; or (right) pALC and pCN35ΩSprF4; pALCΩSprG4 and pCN35; or pALCΩSprG4 and pCN35ΩSprF4. The error bars indicate variations between three biological replicates.
SprF2-4 and SprG2-4 expression levels, relative amounts and stabilities in S. aureus HG003
Using probes specific to each SprG and SprF, we used northern blots to show that all SprF and SprG RNAs are expressed in rich medium except SprG4 (Figure 3A). We quantified the expression levels of these RNAs during growth as compared to tmRNA, used as a reference transcript (Figure 3B). We showed that SprG2, SprF2, SprF3 and SprF4 are mainly expressed during the exponential growth phase, while SprG3 was mainly expressed during the stationary phase (Figure 3A and B). We estimated the quantities of SprG2, SprF2 and SprF4, and of SprG3 and SprF3, by comparing their expressions to the concentrations of a range of purified synthetic RNA (Figure 3C and D). During the exponential and stationary growth phases (OD600 values of 1.6 and 9.3), there were respectively about 27 and 42 times more SprF2 than SprG2. We also had about 31 and 68 times more SprF2 than SprF4 during the exponential and stationary growth phases (Figure 3C). Meanwhile, SprG3 and SprF3 are present in equal amounts during the exponential growth phase (OD600 = 1.6), but during the stationary phase (OD600 = 9.3), SprG3 is about three times as abundant as SprF3 (Figure 3D). RNA half-life determinations (Figure 3E and F) showed that SprF2 (∼4 min) and SprF4 (∼6 min) have much shorter half-lives than SprG2 (>120 min), with SprF3 (∼7 min) also having a much shorter half-life than SprG3 (>120 min). So, we were able to demonstrate that the SprG2 and SprG3 toxins and SprF2, SprF3 and SprF4 antitoxins are expressed at various levels throughout S. aureus HG003 growth. Quantitative estimations and half-life determinations for each RNA indicated that the unstable SprF2 and SprF4 antitoxins are much more present in S. aureus than the stable SprG2 toxin, while at the end of growth, the stable SprG3 toxin is more present than its unstable SprF3 antitoxin.
Figure 3.

SprG2, SprG3, SprF2, SprF3 and SprF4 RNA expression survey, quantities, and stabilities in Staphylococcus aureus HG003. (A) Growth kinetics in liquid cultures of S. aureus HG003, and northern blots detecting SprG2, SprF2, SprG3, SprF3, SprG4 and SprF4 RNA at various times during growth, with tmRNA as the internal control. (B) SprF2, SprG2, SprF3, SprG3 and SprF4 RNA quantifications during S. aureus HG003 growth relative to tmRNA. (C) RNA quantifications of SprG2, SprF2, and SprF4 and (D) SprG3 and SprF3 relative to a range of increasing amounts of synthetic SprG2, SprF2, SprF4, SprG3 and SprF3 RNA. In vivo samples were taken during exponential (OD600 = 1.6) and stationary (OD600 = 9.3) growth phases. (E) SprG2, SprF2, SprG3, SprF3 and SprF4 expression levels during growth after rifampicin addition, relative to the 5S rRNA control. (F) Quantification of the RNA levels of SprG2 (black circles), SprF2 (white circles), SprG3 (white squares), SprF3 (black squares) and SprF4 (gray triangles) after rifampicin addition. AU, arbitrary units. The error bars in B and F indicate variations between three biological replicates.
SprG2 and SprG3 each express a peptide
We next decided to test whether SprG2 and SprG3 could encode and express peptides. In sprG2 and sprG3 sequences, we identified open reading frames (ORFs), with putative Shine-Dalgarno (SD) sequences ∼8 nts upstream from the potential initiation codon (Figure 1C, gray highlighting). SprG2 and SprG3 can therefore express peptides: 35 amino acids with a predicted molecular weight (MW) of ∼3.9 kDa for the SprG2 peptide, and 26 amino acids with a predicted MW of ∼2.9 kDa for the SprG3 peptide. To investigate whether SprG2 produces this peptide during growth, we generated a reporter construct by adding a sequence having a predicted MW of ∼2.75 kDa (3xFLAG) in-frame ahead of the predicted termination codon. In addition to endogenous SprG2 RNA, northern blots validated the expression of SprG2-FLAG during growth, an overexpression that caused a small difference in the amount of bacterial biomass formed as compared to the empty vector strain (Figure 4A). Using anti-FLAG antibodies, immunoblots revealed that a SprG2 fusion peptide was expressed during growth, and furthermore that this peptide was expressed at a higher level during the stationary growth phase (Figure 4B). We next studied the subcellular localization of the flagged SprG2-encoded peptide within S. aureus cells. HG003 cell wall, membrane, cytoplasm, and extracellular fractions were prepared and then analysed by western blotting. To control the purity of the cell fractionations, we used anti-ATPase, anti-SarA, and anti-protein A antibodies (Figure 4C). As expected, SarA was located in the cytoplasmic fraction, protein A was extracellular, and ATPase was mainly found at the membrane (Figure 4C). No peptide was detected for the S. aureus strain with an empty pCN35 tested as a negative control. Immunoblots with anti-FLAG indicated that the SprG2-encoded peptide was detected both in the membrane and extracellular fractions (Figure 4C), as with most type I toxins (36). Despite several attempts, we could not demonstrate SprG3-encoded peptide expression in vivo by immunoblotting with an anti-FLAG antibody. In fact, there are four predicted translation initiation codons within the SprG3 RNA sequence (Figure 4D, underlined). To evaluate the direct interaction between the ribosomes and the predicted translation initiation signals, we used toeprinting assays on the ternary initiation complexes of purified ribosomes, initiator tRNAfMet, and SprG3. In the SprG3 ORF, we detected a ribosome toeprint at position C103, 19 nts downstream from A+1 of the first predicted initiation codon (Figure 4D). We performed in vitro translation assays to demonstrate SprG3-encoded peptide production, with SprG1 and SprG2-encoded peptides as positive controls and SprB sRNA (10) as negative control (Figure 4E). This showed that synthetic SprG3 RNA produces a peptide with an apparent MW of 2.9 kDa. The results of all of these experiments show that SprG2 and SprG3 RNA do encode peptides, which could explain their toxicity.
Figure 4.

SprG2 and SprG3 both express a peptide. (A) Growth kinetics in liquid cultures of Staphylococcus aureus HG003 strains containing pCN35 (black) and pCN35ΩSprG2-FLAG (gray), plus northern blot analysis of SprG2 and SprG2-FLAG RNA during growth, with tmRNA as an internal loading control. (B) Western blot analysis of SprG2-encoded flagged peptide expression during HG003 growth. Zinc staining of the total proteins from each sample are the loading controls. (C) After cell fractionation, the expression of SprG2-encoded peptides was analysed by western blots in pCN35 and pCN35ΩSprG2-FLAG grown to the exponential phase. ATPase, SarA, and Protein A expression were monitored as membrane (M), cytosolic (C), extracellular (E) and wall (W) controls, respectively. (D) An experimentally-determined ribosomal toeprint on SprG3 (arrow) identifying the AUG internal initiation codon, recognized from the three AUG possible codons. U, C, G and A refer to the SprG3 sequencing ladder. The Shine-Dalgarno sequence is bold, and the termination codon is underlined. The predicted AUG codons 1, 2 and 3 are both bold and underlined. The AUG related to the toeprint and to nucleotide C103 are both highlighted in gray. (E) In vitro translation assays to monitor peptide expression by SprG1, SpG2 and SprG3 RNA, with SprB RNA used as a negative control.
The encoded peptides of both SprG2 and SprG3 inhibit S. aureus growth, triggering bacterial stasis
To determine whether SprG2 and SprG3 toxicity is linked to peptide production, we constructed ‘pALCΩSprG2-STOP’ and ‘pALCΩSprG3-STOP’ strains. These overexpress SprG2 or SprG3, and to abolish peptide production their initiation codon was replaced with a UAA termination codon (Figure 5A). SprG3’s RNA sequence contains several putative translation initiation codons, four of which were replaced by the STOP codons STOP1, -2, -3 and -4 (Figure 5A). We used northern blots to verify the production of each overexpressed RNA after aTc induction (Supplementary Figure S2). Without aTc, S. aureus cells containing each of the seven constructs grow similarly to the cells containing the empty vector (Supplementary Figure S2). In the presence of aTc, however, the induced transcription of SprG2, SprG3, or SprG3-STOP1 RNA inhibits S. aureus growth (Figure 5B). Growth inhibition is also observed after adding aTc to solid rich medium followed by serial dilutions (Figure 5C). S. aureus growth is restored almost to isogenic strain levels in both liquid and solid cultures after aTc induction of SprG2-STOP and SprG3-STOP2 (Figure 5, panels B and C). SprG2 and SprG3 also slightly impaired growth independently of the translation product (Figure 5B and C) implying toxicity of the SprG RNAs themselves. This provides experimental evidence that both SprG2 and SprG3 inhibit S. aureus growth since they each express a peptide, with translation initiation at G87UG89 and A85UG87, respectively. After aTc induction, strains overexpressing SprG3-STOP-3 or SprG3-STOP4 had similar growth rates as the isogenic strain having an empty vector. The translation initiation codons A100UG102 and A106UG108 in SprG3 are in-frame with the A85UG87 initiation codon, which is the translation initiation codon used to produce the peptide encoded by SprG3. This suggests that peptide toxicity is linked to the last 18 residues of the SprG3-encoded peptide. These residues are the most conserved between the sequences of the peptides encoded by SprG1–4 (Figure 1D). To determine whether the SprG2- and SprG3-encoded peptides induce S. aureus death, bacteria containing an empty pALC, pALCΩSprG2 and pALCΩSprG2-STOP mutant, or pALCΩSprG3 and pALCΩSprG3-STOP2 mutant were stained with fluorescent dyes to discriminate between living (green = intact membranes) or dead cells (red = disrupted membranes). These were then viewed by fluorescent microscopy. Cells transformed with an empty pALC vector are intact 1 h after aTc induction (Figure 5D). Cells transformed with pALCΩSprG2 and pALCΩSprG2-STOP or with pALCΩSprG3 and pALCΩSprG3-STOP2 were also mostly green, indicating the presence of living cells. This was in contrast to those treated with isopropanol or transformed with pALCΩSprG1, which were stained red (∼70% dead cells for isopropanol and ∼40% for SprG1) (Figure 5D). This suggests that contrary to the two SprG1-encoded peptides, SprG2- and SprG3-encoded peptides do not trigger S. aureus death but instead induce stasis (14).
Figure 5.

Determination of SprG2 and SprG3 internal coding sequences, with evidence that both trigger Staphylococcus aureus stasis but not death. (A) Schematic showing ‘pALCΩSprG2-STOP’ and ‘pALCΩSprG3-STOP’ strain constructs overexpressing either SprG2 or SprG3, with stop codons instead of the predicted initiation codons in order to abolish peptide production. (B) Growth kinetics in liquid cultures and (C) serial dilutions of exponentially grown HG003 bacteria containing pALC (yellow), pALCΩSprG2 or pALCΩSprG3 (blue), pALCΩSprG2-STOP (green), pALCΩSprG3-STOP1 (black), pALCΩSprG3-STOP2 (green), pALCΩSprG3-STOP3 (gray) or pALCΩSprG3-STOP4 (orange) RNA after induction with anhydrotetracycline (aTc). Error bars indicate the variations between three biological replicates. (D) Pictures of S. aureus cells labeled with a SYTO 9 fluorescent probe after LIVE/DEAD experiments demonstrate that overexpression of SprG2- and SprG3-encoded peptides induce stasis. HG003 bacteria containing pALCΩSprG1 are shown as a positive control for S. aureus death, as well as HG003 containing empty pALC treated with 70% isopropanol.
SprF2 and SprF3 reduce SprG2 and SprG3 RNA levels by a direct (cis)-interaction involving their sequence overlaps
We have previously (14) demonstrated that the SprF1 antitoxin interacts with SprG1 RNA to trigger its degradation and prevent translation. The SprG2/SprF2 and SprG3/SprF3 pairs have nucleotide complementarities at their 3′ ends, involving 57 nts for SprG2/SprF2 and 97 nts for SprG3/SprF3 (Figure 1A). Native gel shift assays performed between each pair member revealed an RNA complex (Figure 6A) with an apparent binding constant (Kd) of about 1.0 ± 0.4 nM between SprG2 and SprF2, and ∼1.9 ± 0.4 nM between SprG3 and SprF3. Binding is specific, because a 2,000-fold molar excess of unrelated RNA does not remove SprF2 or SprF3 from its preformed complex, but a 20-fold excess of SprF2 and a 10-fold excess of SprF3 will. To study which SprG2 and SprG3 domains interact with SprF2 and SprF3 RNA, we divided SprG2 and SprG3 into two. The SprG2- and SprG3–5′ ORF parts contain the 5′ domains with their ORFs, while the other sections, SprG2–3′ and SprG3–3′, include the 3′ domains that overlap with their respective antitoxins (Figure 6B). Gel shift assays indicated that SprF2 and SprF3 interact with the corresponding SprG2 or SprG3 via the toxin's 3′ end (Figure 6B). Therefore, SprG2/SprF2 and SprG3/SprF3 RNA interact in vitro with affinity and specificity. We next investigated whether these interactions affect RNA levels in vivo. Strains overexpressing SprF2, SprG2, SprG2/SprF2, SprF3, SprG3 and SprG3/SprF3 were constructed by cloning the genes with their endogenous promoters. Northern blots indicated that when either SprF2 or SprF3 is overexpressed, SprG2 and SprG3 are undetectable during growth (Figure 6C). Similarly, when SprG2 or SprG3 is overexpressed, there is a reduction in SprF2 and SprF3 RNA levels. On the other hand, when both SprF and SprG are overexpressed from the same plasmid, there is no imbalance in the expression of either RNA and no impact on SprF or SprG RNA levels. SprF2 and SprF3 overexpression therefore clearly reduces SprG2 and SprG3 RNA levels in vivo, and vice-versa. To determine whether SprF2 affects SprG2-encoded peptide expression, we designed a construct to produce the flagged peptide in the presence of an excess of SprF2. SprG2-FLAG and SprF2 RNA overexpression were verified by northern blots (Supplementary Figure S3). Immunoblot analysis with an anti-FLAG antibody showed that SprF2 expression does inhibit production of a SprG2-flagged peptide during growth (Figure 6D). However, in vitro translation experiments demonstrated that SprF2 does not directly inhibit the translation of the SprG2-encoded peptide (Figure 6E). Our results demonstrate that SprF2 and SprF3 antitoxins downregulate SprG2 and SprG3 RNA expression, respectively, at the RNA level, and vice-versa.
Figure 6.

SprG2/SprF2 and SprG3/SprF3 regulation at the RNA and peptide levels in Staphylococcus aureus HG003. (A) Native gel shift assays of purified labeled SprF2 or SprF3 RNA against unlabeled SprG2 or SprG3 RNA. The complexes were revealed using a PhosphorImager. Competition assays were done with a 2,000-fold molar excess of polyuridine (PolyU) RNA or with a 20- or 10-fold molar excess of unlabeled SprF2 and SprF3, respectively. The apparent binding constant (Kd) was inferred from the results of three independent experiments. (B) On the left side, native gel shift assays of SprF2 with SprG2, its SprG2 5′ sequence with open reading frame (SprG2–5′ORF), and its 3′ sequence (SprG2–3′); on the right side, the same assays respectively but using SprF3 with SprG3. The RNA complexes were revealed using a PhosphorImager, and labeled RNAs are indicated with asterisks. (C) Northern blot analysis of SprG2, SprF2, SprG3, and SprF3 RNA expression levels during growth in HG003 carrying an empty pCN35, and in isogenic strains containing pCN35ΩSprF2, pCN35ΩSprG2, pCN35ΩSprG2SprF2, pCN35ΩSprF3, pCN35ΩSprG3 or pCN35ΩSprG3SprF3. The internal loading control is tmRNA. (D) Western blot analysis of SprG2-encoded flagged peptide expression in HG003 carrying an empty pCN35, pCN35ΩSprG2-FLAG or pCN35ΩSprG2-FLAG-SprF2. Zinc stainings of the total proteins from each sample are the loading controls. (E) In vitro translation assays and quantification of detected peptides expressed by SprG2 RNA, with or without SprF2 RNA.
Cross-talk between the SprG/SprF pairs expressed by S. aureus HG003
We began by exploring whether an SprF antitoxin could influence the activity of a non-cognate SprG toxin. We used HG003 strains overexpressing each SprG toxin under the control of the aTc-inducible promoter, transformed with pCN35 overexpressing each SprF antitoxin. The strain overexpressing SprG1 was transformed with pCN35ΩSprF2, pCN35ΩSprF3 or pCN35ΩSprF4, and overexpression was monitored by northern blots (Supplementary Figure S4). In the same manner, we transformed the strain overexpressing SprG2 with pCN35ΩSprF1, pCN35ΩSprF3 or pCN35ΩSprF4, and the strain overexpressing SprG3 with pCN35ΩSprF1, pCN35ΩSprF2 or pCN35ΩSprF4, and checked them all with northern blots (Supplementary Figure S4). We then performed growth kinetics in the presence of aTc (Figure 7). Cells expressing SprG1–3 RNAs after aTc induction had inhibited S. aureus growth, and this was specifically rescued by overexpressing SprF1 (for SprG1/SprF1), SprF2 (for SprG2/SprF2), or SprF3 (for SprG3/SprF3) (Figure 7). SprG2 toxicity was not rescued by SprF4 overexpression, even under the control of the Pveg promoter (Figure 7B and D). Therefore, the cells did not grow when an SprG toxin was overexpressed in the presence of a non-cognate SprF antitoxin (Figure 7), and no cross-talk was detected in terms of SprG toxicity neutralization. To explore the specificity of these TA systems, we first investigated the potential regulatory interplay between all of the ‘sprG/sprF’ modules. We performed native gel shift assays between SprG1, SprG2, and SprG3 RNAs and the four radiolabeled SprF RNAs. No RNA complexes were detected between SprG1 and SprF2, SprF3 or SprF4, but we found them between SprG3 and SprF1, and between SprG2 and SprF4 (Figure 8A). Indeed, IntaRNA software (37) had predicted interactions between these same two pairs (Supplementary Figures S5B and S6B). However, the complex formation between SprG3 and SprF1 lacks specificity, because a 2,000-fold molar excess of polyuridine (PolyU) RNA displaces the preformed SprF1-SprG3 complex (Supplementary Figure S5A). We specifically analysed the duplex formation between SprF4 and SprG2 RNA because of the high sequence identity (93%) between SprF2 and SprF4. The binding turned out to be specific (Supplementary Figure S6A), and a 2000-fold molar excess of PolyU RNA did not displace either SprF4 or SprG2 RNA from preformed SprF4-SprG2 complexes, whereas a 10-fold excess of unlabeled SprF4 RNA does. SprF4 binds SprG2 with an apparent Kd of about 3.4 ± 1.6 nM (Supplementary Figure S6A), an affinity three times lower than for SprF2 and SprG2 RNA. To challenge the in vitro results, we used strains overexpressing SprF1, SprG1/SprF1, SprF2, SprG2, SprG2/SprF2, SprF3, SprG3, SprG3/SprF3 and SprF4 (all cloned in the pCN35 vector with their own promoters). To monitor the impact of putative interactions during S. aureus growth, these were compared to HG003 transformed with an empty pCN35 vector. Northern blot analysis showed that SprF1 overexpression decreases SprG2 RNA levels during all growth phases (to about 0.54, 0.68, and 0.55 times the RNA levels, respectively), and that it decreases (∼0.75) SprG3 during stationary growth (Figure 8B and Supplementary Figure S7A). At the beginning of growth, SprG2 overexpression decreases SprF1 and SprF3 RNA levels (by about 0.38 and 0.53, respectively), and SprG2/SprF2 overexpression increases SprG1 RNA levels by about 1.74 times at the exponential growth phase (Figure 8C and Supplementary Figure S7B). Overexpression of SprF3, SprG3, and SprG3/SprF3 do not affect SprG1, SprF1, SprG2, SprF2 or SprF4 RNA levels (Supplementary Figure S7C and D). Finally, SprF4 overexpression (Figure 8D and Supplementary Figure S7E) decreases SprG1 and SprG2 RNA levels during the stationary growth phase (about 0.64 and 0.36 times, respectively). To confirm these RNA level variations, we monitored the RNA levels in the HG003 strain deleted in either the sprG1/sprF1 or sprG2/sprF2 locus, yielding ΔsprG1/sprF1 and ΔsprG2/sprF2, respectively (38). Northern blot analysis indicated that the decreased SprG2 and SprG3 RNA levels observed during SprF1 overexpression is abolished in the ΔsprG1/sprF1 strain. Moreover, the modulation of SprF1, SprF3, and SprG1 RNA levels observed during SprG2 and SprG2/SprF2 overexpression is also reversed in the ΔsprG2/sprF2 strain (Figure 8E and Supplementary Figure S7F). Taken together, these results indicate that the control of SprG toxicity is specific to the cognate pair in each SprG/SprF TA system, but suggest that cross-regulations do indeed occur at the RNA level between the SprG/SprF TA systems during S. aureus growth.
Figure 7.

Specific neutralization of SprG-induced toxicity by SprF cognate antitoxin. (A–C) Growth kinetics in liquid culture of S. aureus HG003 after induction with anhydrotetracycline (aTc). (A) Strains shown: pALC and pCN35 (green); pALCΩSprG1 and pCN35 (blue); pALCΩSprG1 and pCN35ΩSprF1 (yellow); pALCΩSprG1 and pCN35ΩSprF2 (gray); pALCΩSprG1 and pCN35ΩSprF3 (orange); and pALCΩSprG1 and pCN35ΩSprF4 (black). (B) Strains shown: pALC and pCN35 (green); pALCΩSprG2 and pCN35 (blue); pALCΩSprG2 and pCN35ΩSprF1 (yellow); pALCΩSprG2 and pCN35ΩSprF2 (gray); pALCΩSprG2 and pCN35ΩSprF3 (orange); and pALCΩSprG2 and pCN35ΩSprF4 (black). (C) Strains shown: pALCΩSprG3 and pCN35 (blue); pALCΩSprG3 and pCN35ΩSprF1 (yellow); pALCΩSprG3 and pCN35ΩSprF2 (gray); pALCΩSprG3 and pCN35ΩSprF2 (orange); and pALCΩSprG3 and pCN35ΩSprF4 (black). (D) Growth kinetics in liquid cultures of HG003 deleted for SprG2 and SprF2, containing the following: pALC and pCN35 (green); pALCΩSprG2 and pCN35 (blue): pALC and pCN35ΩPvegSprF4, in which the Pveg promoter controls SprF4 expression (purple); and pALCΩSprG2 and pCN35ΩPvegSprF4 (red). The error bars indicate variations between three biological replicates.
Figure 8.

Crosstalk regulation between the four SprG/SprF toxin–antitoxin pairs. (A) Native gel shift assays of purified labeled SprF1, SprF2, SprF3 and SprF4 RNA with unlabeled SprG1, SprG2, or SprG3 RNAs. The complexes were revealed with a PhosphorImager, and labeled RNA are indicated with asterisks. (B, C and D) Charts showing SprG/SprF RNA quantifications during the pre-exponential (PE), exponential (E) and stationary (S) growth phases in Staphylococcus aureus. Quantifications are relative to tmRNA and then normalized to the pCN35 empty vector levels, considered as equal to 1. Welch's t-test results: *P < 0.05; **P < 0.01. (E) Chart of SprG/SprF RNA quantifications during the same growth phases in S. aureus HG003 ΔsprG1/sprF1 and ΔsprG2/sprF2 strains. Quantifications are relative to tmRNA, then normalized to levels of the wild-type HG003 strain, considered as equal to 1.
Modulation of SprG and SprF RNA levels upon oxidative and hyperosmotic stress
Several publications (17,22) have reported the involvement of TA systems in the adaptation of bacteria to their environment. To investigate the biological functions of the SprG/SprF TA systems, we began by trying to identify the conditions that might influence SprG and/or SprF RNA levels. HG003 was subjected to 1 h of either oxidative stress (by adding 10 mM hydrogen peroxide), or hyperosmotic stress (2M sodium chloride). Afterwards, northern blots revealed that the RNA levels of SprF antitoxins always dropped during oxidative stress (Figures 9A and Supplementary Figure S8). Our results also show that the RNA levels of SprG1, SprF1, SprF2, SprF3 and SprG3 decrease in the presence of hyperosmotic stress (Figures 9A and Supplementary Figure S8). On the other hand, the stress experiments did not highlight a particular condition for the detection of SprG4 expression (Supplementary Figure S8). Overall, our findings show that the SprG/SprF TA systems could be activated by stresses regularly encountered by bacteria during infection. We wondered whether SprG1/SprF1, SprG2/SprF2 and SprG3/SprF3 TA systems are activated in the context of S. aureus internalization by human phagocytes. Indeed, phagolysosomes are known to create an oxidative environment (39), and this could potentially reduce SprF antitoxin expression. We therefore performed an assay to monitor HG003 phagocytosis by the PMA-induced THP-1 macrophages, using RT-qPCR to quantify SprG and SprF RNA levels after 2 h of phagocytosis. During these experiments, SprF3 RNA levels were too low for quantitative PCR. Only SprF1 levels significantly decrease after S. aureus phagocytosis (Figure 9B), while all SprF antitoxins decrease after exposure to H2O2 (Figure 9A). Interestingly, a significant drop in SprG1 RNA levels is also observed after phagocytosis (Figure 9B), suggesting that the environmental conditions encountered in phagocytes, other than oxidative stress, contribute to reducing SprG1 RNA levels.
Figure 9.
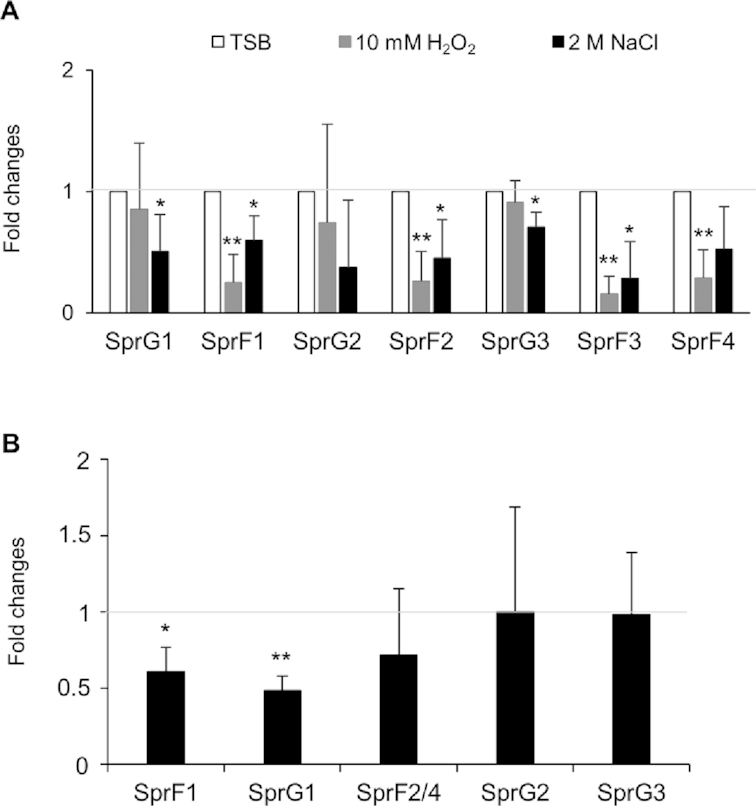
Modulation of SprG and SprF RNA levels upon oxidative and hyperosmotic stress and Staphylococcus aureus internalization in human macrophages. (A) Quantification of SprG and SprF RNA levels by northern blot during S. aureus growth under environmental stress. Each stress was performed for 1 h. Data are expressed as mean fold changes relative to the SprG and SprF RNA levels found without stress, measured as the growth in tryptic soy broth (TSB) arbitrarily set to a value of 1. (B) Quantification of SprG and SprF RNA levels by RT-qPCR during S. aureus internalization in PMA-induced THP-1 macrophages. Data are expressed as mean fold changes relative to the SprG and SprF RNA levels found in the inoculum before phagocytosis, arbitrarily set to a value of 1. Error bars correspond to variations between four independent experiments. Welch's t-test results: *P < 0.05; **P < 0.01.
DISCUSSION
Identifying a full set of expressed and functional TA systems in bacteria is a serious challenge, especially because there can be so many. Mycobacterium tuberculosis has up to 79 TA systems, with at least 50 in the VapBC family and at least 10 in the MazEF family (40). In S. aureus, only five functional TA systems have been reported (23). For the S. aureus type II TA systems, only one MazEF and two paralogues YefM/YoeB (‘yefM-yoeB-sa1’ and ‘yefM-yoeB-sa2,’ also referred to as ‘axe-txe-1’ and ‘axe-txe-2’), have been described (41). As for S. aureus type I TA systems, both SprA1/SprA1AS and SprG1/SprF1 have additional copies in the core genome (13,14). Here we report on the functionality, mechanisms, and cross-talk between four SprG/SprF type I TA pairs that are expressed in most S. aureus strains (10,14). One is expressed from a pathogenicity island (SprG1/SprF1), while the others are encoded in the core genome (SprG2–4/SprF2–4). Only SprF2 and SprF4 can be considered as bona fide gene copies, with 93% sequence identity (Supplementary Table S4). For SprG1, SprG2, SprG3 and SprG4, and as well as SprF1 and SprF3, their sequence identities are much weaker, which suggests that SprG1/SprF1, SprG2/SprF2, SprG3/SprF3 and SprG4/SprF4 are homologous TA systems (Supplementary Table S4). In Figure 3, half-life determinations indicate low half-lives and rapid turnovers for the four SprF antitoxins, whereas their cognate toxic mRNAs have much longer half-lives (up to 2 h). Although it is expressed in S. aureus N315, COL, and Newman strains, SprG4 expression is undetectable in various growth conditions (14).
The reason for the abundance of TA systems (either as multiple copies or as homologues in bacterial genomes) is unclear, but one possibility is that they could have acquired specific functions. Some bacterial TA systems can behave as non-functional pseudogenes due to sequence insertions, mutations or gene promoter defects. Several copies of the aapA/IsoA type I TA module are expressed in Helicobacter pylori (42). For that module, the IsoA2 RNA antitoxin is expressed, but the lack of a promoter upstream from the AapA2 mRNA prevents transcription, which is similar to what happens with SprG4/SprF4 in S. aureus HG003. The E. coli K1217 genome has five inactive homologues of the type I Hok toxin (43) which harbor point mutations, insertions, or large rearrangements (44). Multiple homologues are also observed in type II TA systems. For example, RelBE has seven homologues in Vibrio cholerae within the superintegron on chromosome II, with only six effective TA systems (45). Here, the sprG2/sprF2 and sprG3/sprF3 loci encode functional TA systems, with the exception of the sprG4/sprF4 locus, probably due to the lack of a functional promoter in the HG003 strain.
Mutations at the internal initiation codons show that toxic peptide translation controls the toxicity of both SprG2 and SprG3 RNA (Figure 5). Type I toxins are divided into two groups depending on whether they are located at the membrane or within the cytoplasm (36). SprG2 accumulates at the bacterial membrane and is also extracellular, like the SprG1-encoded peptides (14). SprG3-encoded peptides, however, were only detected in vitro, suggesting that SprG3 RNA folding must impede internal translation. Arnion et al. have demonstrated the importance of mRNA toxin maturation for translation, most notably because RNA structures block ribosomal access to the Shine-Dalgarno sequence. Toxin mRNA can be cleaved into shorter fragments with translatable conformations (42). A conformational change in SprG3 might be necessary before translation, as is the case with AapA1, TisB, and Hok, all of which require structural shifts for toxin translation (46,47). In S. aureus, ClpP protease is essential for the degradation of the MazE, Axe1 and Axe2 type II antitoxins (48). Although nothing has been described in the literature about the degradation of type I and type II toxins in S. aureus, it could be interesting to study the translation of the SprG3-encoded peptide in a context of clpP deletion. Toxic peptides encoded by SprG2 and SprG3 RNAs induce bacterial stasis stimulated by their expression. In S. aureus, overexpression of the sequence-specific MazF RNase, the type II mazEF locus toxin, also induces bacterial stasis but not cell death (49). This shows that at least three toxins (MazF, SprG2 and SprG3-encoded peptides) can induce stasis, as opposed to the SprA1- and SprG1-encoded peptides that trigger cell death (14,32). The mechanisms of action of SprG-encoded peptides are still unknown, but our results suggest that SprG2- and SprG3-encoded peptides should possess different biological functions than PepG131 and PepG144 cytolytic peptides. Peptide sequence alignments suggest that SprG2- and SprG3-encoded peptides are hydrophobic and contain cationic residues (Figure 1D), like the SprA1- and SprG1-encoded cytolytic peptides (13,14). The PepA1 helical toxin expressed from S. aureus SprA1 induces cell death by accumulating into and permeating bacterial and eukaryotic membranes (32). Further studies are needed to investigate the putative ability of SprG2- and SprG3-encoded peptides to lyse competitive bacteria and host erythrocytes, as SprG1-encoded peptides are known to do.
During S. aureus growth, SprG2 and SprG3 toxin expression is repressed at the RNA level by their cognate SprF2 and SprF3 antitoxins to prevent stasis. SprF2 and SprF3 both interact in cis with the overlapping 3′ ends of their respective toxins, as occurs for the SprF1/SprG1 pair (14). In type I TA systems, antitoxins can act by inhibiting toxic mRNA translation, by inducing mRNA degradation of the toxin by specific double-stranded RNases (50), or by doing both (14,51). The SprA1AS antitoxin forms a complex with SprA1 RNA, preventing toxin production in trans by occluding the translation initiation signals (13). The SprF1 antitoxin acts by a pairing mechanism with SprG1 toxin RNA to trigger its degradation and prevent its translation (14). In the case of SprG2/SprF2 and SprG3/SprF3, SprF2 and SprF3 antitoxins could trigger mRNA degradation and decay by recruiting RNases and/or by relaxing the toxic mRNA’s conformation, making it less stable. This was shown in Bacillus subtilis for both the txpA/RatA (52) and the bsrE/SR5 toxin–antitoxin systems (53).
Cross-regulation and cross-talk between TA homologues is mostly unknown, with very few examples for type II TA pairs. In 2016, Walling and Butler identified W48 and F52, two important residues in the Haemophilus influenzae type II VapB2 antitoxin which are required to neutralize VapC2 toxicity. If the VapB1 antitoxin is genetically modified to add just the tryptophan residue (T47W mutant) required for the VapB2-VapC2 interaction, both toxins VapC1 and VapC2 are neutralized (54). Cross-talk between TA systems was also studied within the V. cholerae N16961 superintegron, which contains 18 type II TA systems (55). Among these TA systems, no cross-talk was detected during toxin induction in the presence of a non-cognate antitoxin. This suggests that there is specificity between type II antitoxins and their cognate toxins. In addition, there was no cross-talk found between the S. aureus type II ‘yefM-yoeB-sa1’ and ‘yefM-yoeB-sa2’ systems, whether in terms of toxin activity neutralization, nor in transcriptional auto-regulation of the TA modules (56). Nothing is currently known about cross-talk in type I toxin–antitoxin systems. We therefore analysed all of the possible cross-interactions between the four antitoxins and their cognate toxins. For every pair, not a single instance of cross-talk prevents toxicity, even for those interacting in vitro (SprG1/SprF3 and SprG2/SprF4). This implies that each set of RNA pairs acts independently of the others when dealing with its own toxicity and regulations. However, heterologous cross-regulations between the RNAs from non-cognate pairs can occur via pairing interactions and/or can influence steady state expression levels. For instance, overexpression of the SprF1 antitoxin decreases the RNA levels not only of SprG1 (14) but also of SprG2 and SprG3 (Figure 8B). Moreover, overexpression of the SprG2 toxin decreases SprF antitoxin RNA levels and increases those of the SprG1 toxin (Figures 6C and 8C). In addition, when SprF4 is overexpressed, a drop in SprG1 and SprG2 RNA levels is observed (Figure 8D). This indicates both flexibility and interactions between the pairs, with probable additional effects on TA module functions unrelated to toxicity.
It is well established that TA systems are involved in the adaptation of bacteria to their environments (17,22). The bsrG/SR4 type I TA system, located on the SPβ B. subtilis prophage, is a stress-response system, because a temperature shock of at least 48°C increases RNA degradation of the BsrG toxin (57). Another stress-response TA system is hok/sok, which responds to high temperatures and to β-lactams by allowing bacteria to extend its lag phase for adaptation (58). The type I toxin, PepA1, also modulates its expression in response to decreased SprA1AS antitoxin RNA levels when undergoing oxidative and acidic stress (32). Activation of TA systems upon oxidative stress has also been demonstrated in Caulobacter crescentus for the type II systems ParDE2 and RelBE3 (59), and for the PasTI TA system of extraintestinal pathogenic E. coli (ExPEC) (60). Northern blot analysis provides evidence that SprG/SprF TA systems respond to stress, specifically hyperosmotic and oxidative stresses. In Figure 9A, we show decreased RNA levels of SprG1 and SprG3 toxins and SprF1, SprF2, and SprF3 antitoxins during hyperosmotic stress, and decreased levels of SprG1 toxin and all SprF RNA antitoxins during oxidative stress. However, when S. aureus was exposed to oxidative stress in the phagolysosome of THP-1 macrophages, only SprF1 and SprG1 were reduced. This last result suggests that during its internalization in human phagocytes, S. aureus favors inhibiting the expression of the SprG1/SprF1 TA system which induces its death, rather than the SprG2/SprF2 and SprG3/SprF3 ones that only induce stasis. This could be a way to promote its persistence in a stressful environment.
Activation of TA systems when bacteria encounter unfavorable growth conditions could explain entry into the persister state, promoting multi-drug tolerance and infection chronicity (21). Some bacteria enter metabolically inactive or dormant states to become persister cells, a slow-growing and drug-tolerant bacterial sub-population (61). In fact, the formation of persistent bacteria or stress adaptation are often mentioned as the biological roles of certain chromosomal TA systems. A period of stasis might allow bacteria to resume normal growth, and toxins from TA systems could be involved in this process (21,62). For type I TA systems, involvement in persister cell formation via Obg in response to nutrient starvation was demonstrated for the HokB toxin in E. coli (63). After Hok toxicity, depolarization of the membrane causes either stasis or bacterial death, depending on toxin concentrations, with persistence triggered by a physiological concentration of the Hok toxin (63). The TisB toxin of the tisB/IstR1 system in E. coli is induced during the SOS response, allowing bacterial persistence by decreasing the proton motive force to promote dormancy, thus indirectly promoting tolerance to antibiotics such as β-lactams and fluoroquinolones (20,64). Fourteen type II TA systems are important for Salmonella persistence because deletions of each of these systems show a reduction of persister formation after internalization in human macrophages (65–67). TA system redundancy on bacterial genomes implies that the deletion of one TA operon does not necessarily affect persistence or phenotype. In S. aureus, however, a knockout of all the type II TA systems has no persister phenotype (68). The physiological role of the MazEF type II TA system is not well understood in S. aureus, but recent studies reported that mazEF deletion increases sensitivity to β-lactam antibiotics (69) and leads to increased biofilm formation (70). The deletion of the SavRS TA system (probably the same as YefM-YoeB-sa2) (41) increases S. aureus hemolytic activity and virulence in a mouse subcutaneous abscess model (24). Our results suggest that SprG2/SprF2 and SprG3/SprF3 TA systems affect S. aureus persister cell formation during hyperosmotic or oxidative stress. This is in contrast to the SprG1/SprF1 (14), SprA1/SprA1AS (13), and SprA2/SprA2AS (71) TA systems, which trigger cell death and probably participate in S. aureus virulence through the liberation of hemolytic and/or antimicrobial peptides. Our findings and these studies suggest that although S. aureus type I TA systems have quite different toxicities, modes of action, regulation mechanisms, stress responses, and biological functions, they still all have something in common. In all case, the environmental stresses encountered by S. aureus during host infection affect how antitoxin RNA level decreases trigger the production of toxins.
In short, we identified two new functional type I TA systems in S. aureus: SprG2/SprF2 and SprG3/SprF3. These both induce bacterial stasis by expressing toxic peptides. Each set of SprG/SprF RNA pairs acts independently of each other while they deal with their own toxicity and regulations. However, heterologous cross-regulations between the RNAs from non-cognate pairs can occur via pairing interactions and/or by steady state expression level variations. The next challenge will be to unravel the biological functions of these new TA systems, particularly their involvement in S. aureus persistence.
Supplementary Material
ACKNOWLEDGEMENTS
We are thankful to Stéphane Dréano (IGDR, Rennes, France) for the DNA sequencing of all constructs in this study; to Wenfeng Liu and Philippe Bouloc (I2BC, Gif sur Yvette, France) for creating the HG003 strain deleted in the sprG2/sprF2 locus; and to Juliana Berland for help with editing. Thanks also to Noëlla Germain-Amiot, Yoann Augagneur, and Astrid Rouillon for sharing information about TA systems expressed by S. aureus, and for help with the manuscript.
Author contributions: C.R. and M.L.P.M. designed and performed the experiments, with phagocytosis experiments designed and performed by G.P. M.L.P.M. and B.F. supervised research. The data was analysed by C.R., M.L.P.M, G.P. and B.F. C.R. prepared the figures and wrote the manuscript, which was edited by M.L.P.M. and B.F.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministère Français de l’Enseignement Supérieur et de la Recherche [MENRT to C.R.]; School of Pharmacy and Medical Sciences of the University of Rennes 1; Agence Nationale pour la Recherche [ANR-15-CE12-0003-01 ‘sRNA-Fit’ to B.F.]; Fondation pour la Recherche Médicale [DBF20160635724 ‘Bactéries et champignons face aux antibiotiques et antifongiques’ to B.F.]; Institut National de la Santé et de la Recherche Médicale. Funding for open access charge: Inserm.
Conflict of interest statement. None declared.
REFERENCES
- 1. van Belkum A., Verkaik N.J., de Vogel C.P., Boelens H.A., Verveer J., Nouwen J.L., Verbrugh H.A., Wertheim H.F.. Reclassification of Staphylococcus aureus nasal carriage types. J. Infect. Dis. 2009; 199:1820–1826. [DOI] [PubMed] [Google Scholar]
- 2. Lowy F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998; 339:520–532. [DOI] [PubMed] [Google Scholar]
- 3. Tong S.Y., Davis J.S., Eichenberger E., Holland T.L., Fowler V.G. Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015; 28:603–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chambers H.F., Deleo F.R.. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009; 7:629–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caldelari I., Chao Y., Romby P., Vogel J.. RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb. Perspect. Med. 2013; 3:a010298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fechter P., Caldelari I., Lioliou E., Romby P.. Novel aspects of RNA regulation in Staphylococcus aureus. FEBS Lett. 2014; 588:2523–2529. [DOI] [PubMed] [Google Scholar]
- 7. Tomasini A., François P., Howden B.P., Fechter P., Romby P., Caldelari I.. The importance of regulatory RNAs in Staphylococcus aureus. Infect. Genet. Evol. 2014; 21C:616–626. [DOI] [PubMed] [Google Scholar]
- 8. Sassi M., Augagneur Y., Mauro T., Ivain L., Chabelskaya S., Hallier M., Sallou O., Felden B.. SRD: a Staphylococcus regulatory RNA database. RNA. 2015; 21:1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bronsard J., Pascreau G., Sassi M., Mauro T., Augagneur Y., Felden B.. sRNA and cis-antisense sRNA identification in Staphylococcus aureus highlights an unusual sRNA gene cluster with one encoding a secreted peptide. Sci. Rep. 2017; 7:4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pichon C., Felden B.. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:14249–14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chabelskaya S., Gaillot O., Felden B.. A Staphylococcus aureus small RNA is required for bacterial virulence and regulates the expression of an immune-evasion molecule. PLoS Pathog. 2010; 6:e1000927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Le Pabic H., Germain-Amiot N., Bordeau V., Felden B.. A bacterial regulatory RNA attenuates virulence, spread and human host cell phagocytosis. Nucleic Acids Res. 2015; 43:9232–9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sayed N., Jousselin A., Felden B.. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat. Struct. Mol. Biol. 2011; 19:105–112. [DOI] [PubMed] [Google Scholar]
- 14. Pinel-Marie M.L., Brielle R., Felden B.. Dual toxic-peptide-coding Staphylococcus aureus RNA under antisense regulation targets host cells and bacterial rivals unequally. Cell Rep. 2014; 7:424–435. [DOI] [PubMed] [Google Scholar]
- 15. Van Melderen L., Saavedra De Bast M.. Bacterial toxin–antitoxin systems: more than selfish entities. PLoS Genet. 2009; 5:e1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gerdes K., Bech F.W., Jørgensen S.T., Løbner-Olesen A., Rasmussen P.B., Atlung T., Boe L., Karlstrom O., Molin S., von Meyenburg K.. Mechanism of postsegregational killing by the hok gene product of the parB system of plasmid R1 and its homology with the relF gene product of the E. coli relB operon. EMBO J. 1986; 5:2023–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lobato-Márquez D., Díaz-Orejas R., García-del Portillo F.. Toxin-antitoxins and bacterial virulence. FEMS Microbiol. Rev. 2016; 40:592–609. [DOI] [PubMed] [Google Scholar]
- 18. Patel S. Drivers of bacterial genomes plasticity and roles they play in pathogen virulence, persistence and drug resistance. Infect. Genet. Evol. 2016; 45:151–164. [DOI] [PubMed] [Google Scholar]
- 19. Durand S., Jahn N., Condon C., Brantl S.. Type I toxin–antitoxin systems in Bacillus subtilis. RNA Biol. 2012; 9:1491–1497. [DOI] [PubMed] [Google Scholar]
- 20. Dörr T., Vulic M., Lewis K.. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010; 8:e1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Page R., Peti W.. Toxin-antitoxin systems in bacterial growth arrest and persistence. NCHEMBIO. 2016; 12:208–214. [DOI] [PubMed] [Google Scholar]
- 22. Fernández-García L., Blasco L., Lopez M., Bou G., García-Contreras R., Wood T., Tomas M.. Toxin-Antitoxin systems in clinical pathogens. Toxins. 2016; 8:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schuster C.F., Bertram R.. Toxin-Antitoxin systems of Staphylococcus aureus. Toxins. 2016; 8:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wen W., Liu B., Xue L., Zhu Z., Niu L., Sun B.. Autoregulation and virulence control by the Toxin-Antitoxin system SavRS in Staphylococcus aureus. Infect. Immun. 2018; 86:e00032–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fozo E.M., Makarova K.S., Shabalina S.A., Yutin N., Koonin E.V., Storz G.. Abundance of type I toxin–antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res. 2010; 38:3743–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Charpentier E., Anton A.I., Barry P., Alfonso B., Fang Y., Novick R.P.. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004; 70:6076–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zeghouf M., Li J., Butland G., Borkowska A., Canadien V., Richards D., Beattie B., Emili A., Greenblatt J.F.. Sequential Peptide Affinity (SPA) system for the identification of mammalian and bacterial protein complexes. J. Proteome Res. 2004; 3:463–468. [DOI] [PubMed] [Google Scholar]
- 28. Bateman B.T., Donegan N.P., Jarry T.M., Palma M., Cheung A.L.. Evaluation of a tetracycline-inducible promoter in Staphylococcus aureus in vitro and in vivo and its application in demonstrating the role of sigB in microcolony formation. Infect. Immun. 2001; 69:7851–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kreiswirth B.N., Lofdahl S., Betley M.J., O’Reilly M., Schlievert P.M., Bergdoll M.S., Novick R.P.. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983; 305:709–712. [DOI] [PubMed] [Google Scholar]
- 30. Herbert S., Ziebandt A.K., Ohlsen K., Schafer T., Hecker M., Albrecht D., Novick R., Gotz F.. Repair of global regulators in Staphylococcus aureus 8325 and comparative analysis with other clinical isolates. Infect. Immun. 2010; 78:2877–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brückner R. Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol. Lett. 1997; 151:1–8. [DOI] [PubMed] [Google Scholar]
- 32. Sayed N., Nonin-Lecomte S., Réty S., Felden B.. Functional and structural insights of a Staphylococcus aureus apoptotic-like membrane peptide from a toxin–antitoxin module. J. Biol. Chem. 2012; 287:43454–43463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Downer R., Park P.W., Mecham R.P., Foster T.J.. The elastin-binding protein of Staphylococcus aureus (EbpS) is expressed at the cell surface as an integral membrane protein and not as a cell wall-associated protein. J. Biol. Chem. 2002; 277:243–250. [DOI] [PubMed] [Google Scholar]
- 34. Antal M., Bordeau V., Douchin V., Felden B.. A small bacterial RNA regulates a putative ABC transporter. J. Biol. Chem. 2005; 280:7901–7908. [DOI] [PubMed] [Google Scholar]
- 35. Raynaud S., Le Pabic H., Felden B.. Selective recovery of RNAs from bacterial pathogens after their internalization by human host cells. Methods. 2018; 143:4–11. [DOI] [PubMed] [Google Scholar]
- 36. Brielle R., Pinel-Marie M.L., Felden B.. Linking bacterial type I toxins with their actions. Curr. Opin. Microbiol. 2016; 30:114–121. [DOI] [PubMed] [Google Scholar]
- 37. Busch A., Richter A.S., Backofen R.. IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics. 2008; 24:2849–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Le Lam T.N., Morvan C., Liu W., Jaszczyszyn Y., Bouloc P.. Finding sRNA-associated phenotypes by competition assays: an example with Staphylococcus aureus. Methods. 2017; 117:21–27. [DOI] [PubMed] [Google Scholar]
- 39. Horn J., Stelzner K., Rudel T., Fraunholz M.. Inside job: Staphylococcus aureus host-pathogen interactions. Int. J. Med. Microbiol. 2018; 308:607–624. [DOI] [PubMed] [Google Scholar]
- 40. Sala A., Bordes P., Genevaux P.. Multiple toxin–antitoxin systems in Mycobacterium tuberculosis. Toxins. 2014; 6:1002–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Donegan N.P., Cheung A.L.. Regulation of the mazEF toxin–antitoxin module in Staphylococcus aureus and its impact on sigB expression. J. Bacteriol. 2009; 191:2795–2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arnion H., Korkut D.N., Gelo S.M., Chabas S., Reignier J., Iost I., Darfeuille F.. Mechanistic insights into type I toxin antitoxin systems in Helicobacter pylori: the importance of mRNA folding in controllong toxin expression. Nucleic Acids Res. 2017; 45:4782–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Steif A., Meyer I.M.. The hok mRNA family. RNA Biol. 2012; 9:1399–1404. [DOI] [PubMed] [Google Scholar]
- 44. Pedersen K., Gerdes K.. Multiple hok genes on the chromosome of Escherichia coli. Mol. Microbiol. 1999; 32:1090–1102. [DOI] [PubMed] [Google Scholar]
- 45. Wang Y., Wang H., Hay A.J., Zhong Z., Zhu J., Kan B.. Functional RelBE-family toxin–antitoxin pairs affect biofilm maturation and intestine colonization in Vibrio cholerae. PLoS One. 2015; 10:e0135696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Darfeuille F., Unoson C., Vogel J., Wagner E.G.H.. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell. 2007; 26:381–392. [DOI] [PubMed] [Google Scholar]
- 47. Thisted T., Nielsen A.K., Gerdes K.. Mechanisms of post-segregational killing: translation of Hok, SrnB and Pnd mRNAs of plasmids R1, F and R483 is activated by 3’-end processing. EMBO J. 1994; 13:1950–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Donegan N.P., Thompson E.T., Fu Z., Cheung A.L.. Proteolytic regulation of toxin–antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 2010; 192:1416–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fu Z., Tamber S., Memmi G., Donegan N.P., Cheung A.L.. Overexpression of MazFsa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J.Bacteriol. 2009; 191:2051–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brantl S., Jahn N.. sRNAs in bacterial type I and type III toxin–antitoxin systems. FEMS Microbiobiol. Rev. 2015; 39:413–427. [DOI] [PubMed] [Google Scholar]
- 51. Jahn N., Brantl S.. One antitoxin-two functions: SR4 controls toxin mRNA decay and translation. Nucleic Acids Res. 2013; 41:9870–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Silvaggi J.M., Perkins J.B., Losick R.. Small untranslated RNA antitoxin in Bacillus subtilis. J. Bacteriol. 2005; 187:6641–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Meiβner C., Jahn N., Brantl S.. In vitro characterization of the type I toxin–antitoxin system bsrE/SR5 from Bacillus subtilis. J. Biol. Chem. 2016; 291:560–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Walling L.R., Butler J.S.. Structural determinants for antitoxin identity and insulation of cross talk between homologous toxin–antitoxin systems. J. Bacteriol. 2016; 198:3287–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Iqbal N., Guérout A.-M., Krin E., Le Roux F., Mazel D.. Comprehensive functional analysis of the 18 Vibrio cholerae N16961 toxin–antitoxin systems substantiates their role in stabilizing the superintegron. J. Bacteriol. 2015; 197:2150–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yoshizumi S., Zhang Y., Yamaguchi Y., Chen L., Kreiswirth B.N., Inouye M.. Staphylococcus aureus YoeB homologues inhibit translation initiation. J. Bacteriol. 2009; 191:5868–5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jahn N., Preis H., Wiedemann C., Brantl S.. BsrG/SR4 from Bacillus subtilis – the first temperature-dependent type I toxin–antitoxin system. Mol. Microbiol. 2012; 83:579–598. [DOI] [PubMed] [Google Scholar]
- 58. Chukwudi C.U., Good L.. The role of the hok/sok locus in bacterial response to stressful growth conditions. Microb. Pathogen. 2015; 79:70–79. [DOI] [PubMed] [Google Scholar]
- 59. Fiebig A., Castro Rojas C.M., Siegal-Gaskins D., Crosson S.. Interaction specificity, toxicity and regulation of a paralogous set of ParE/RelE-family toxin–antitoxin systems. Mol. Microbiol. 2010; 77:236–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Norton J.P., Mulvey M.A.. Toxin-antitoxin systems are important for niche-specific colonization and stress resistance of uropathogenic Escherichia coli. PLoS Pathog. 2012; 8:e1002954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Fisher R.A., Gollan B., Helaine S.. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017; 15:453–464. [DOI] [PubMed] [Google Scholar]
- 62. Hall A.M.J., Gollan B., Helaine S.. Toxin-antitoxin systems: reversible toxicity. Curr. Opin. Microbiol. 2017; 36:102–110. [DOI] [PubMed] [Google Scholar]
- 63. Verstraeten N., Knapen W.J., Kint C.I., Liebens V., Van den Bergh B., Dewachter L., Michiels J.E., Fu Q., David C.C., Fierro A.C. et al. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell. 2015; 59:9–21. [DOI] [PubMed] [Google Scholar]
- 64. Gurnev P.A., Ortenberg R., Dorr T., Lewis K., Bezrukov S.M.. Persister-promoting bacterial toxin TisB produces anion-selective pores in planar lipid bilayers. FEBS Lett. 2012; 586:2529–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Helaine S., Cheverton A.M., Watson K.G., Faure L.M., Matthews S.A., Holden D.W.. Internalization of Salmonella by macrophages induces formation of nonreplicating persisters. Science. 2014; 343:204–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cheverton A.M., Gollan B., Przydacz M., Wong C.T., Mylona A., Hare S.A., Helaine S.. A Salmonella toxin promotes persister formation through acetylation of tRNA. Mol. Cell. 2016; 63:86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rycroft J.A., Gollan B., Grabe G.J., Hall A., Cheverton A.M., Larrouy-Maumus G., Hare S.A., Helaine S.. Activity of acetyltransferase toxins involved in Salmonella persister formation during macrophage infection. Nat. Commun. 2018; 9:1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Conlon B.P., Rowe S.E., Gandt A.B., Nuxoll A.S., Donegan N.P., Zalis E.A., Clair G., Adkins J.N., Cheung A.L., Lewis K.. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016; 1:16051. [DOI] [PubMed] [Google Scholar]
- 69. Schuster C.F., Mechler L., Nolle N., Krismer B., Zelder M.E., Götz F., Bertram R.. The MazEF toxin–antitoxin system alters the β-lactam susceptibility of Staphylococcus aureus. PLoS One. 2015; 10:e0126118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kato F., Yabuno Y., Yamaguchi Y., Sugai M., Inouye M.. Deletion of mazF increases Staphylococcus aureus biofilm formation in an ica-dependent manner. Pathog. Dis. 2017; 75:doi:10.1093/femspd/ftx026. [DOI] [PubMed] [Google Scholar]
- 71. Germain-Amiot N., Augagneur Y., Camberlein E., Nicolas I., Lecureur V., Rouillon A., Felden B.. A novel Staphylococcus aureus cis-trans type I toxin–antitoxin module with dual effects on bacteria and host cells. Nucleic Acids Res. 2018; doi:10.1093/nar/gky1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.