


Abstract
Bacteriophage λ encodes a DNA recombination system that includes a 5′-3′ exonuclease (λ Exo) and a single strand annealing protein (Redβ). The two proteins form a complex that is thought to mediate loading of Redβ directly onto the single-stranded 3′-overhang generated by λ Exo. Here, we present a 2.3 Å crystal structure of the λ Exo trimer bound to three copies of the Redβ C-terminal domain (CTD). Mutation of residues at the hydrophobic core of the interface disrupts complex formation in vitro and impairs recombination in vivo. The Redβ CTD forms a three-helix bundle with unexpected structural homology to phage λ Orf, a protein that binds to E. coli single-stranded DNA binding protein (SSB) to function as a recombination mediator. Based on this relationship, we found that Redβ binds to full-length SSB, and to a peptide corresponding to its nine C-terminal residues, in an interaction that requires the CTD. These results suggest a dual role of the CTD, first in binding to λ Exo to facilitate loading of Redβ directly onto the initial single-stranded DNA (ssDNA) at a 3′-overhang, and second in binding to SSB to facilitate annealing of the overhang to SSB-coated ssDNA at the replication fork.
INTRODUCTION
Bacteriophage λ encodes a simple ‘SynExo’ (synaptase-exonuclease) recombination system for repair of double stranded DNA (dsDNA) breaks by a single strand annealing (SSA) mechanism (Figure 1; 1). This system, called Red for recombination defective, consists of two proteins: a 5′-3′exonuclease, λ Exo, that binds to DNA ends and resects the 5′-strand (2,3), and a synaptase, Redβ, that binds the resulting 3′-overhang to anneal it to a complementary strand from another copy of the chromosome (Figure 1A; 4–6). In most cases, annealing is thought to occur at a target site that is exposed as ssDNA on the lagging strand of a replication fork (7). A third protein, Gam, binds and inactivates the host RecBCD enzyme, to prevent it from degrading the phage genome (8). Interest in λ Exo and Redβ stems in part from their distant homologies with the mammalian recombination proteins Dna2 (9) and Rad52 (10–14), respectively. The proteins have also been exploited in powerful methods for bacterial genome engineering, most notably recombineering (15,16) and Multiplex Automated Genome Engineering (17). Interestingly, λ Exo and Redβ bind to one another to form a complex that is required for recombination in vivo (18,19). While its exact role is unknown, the complex may serve to integrate the two steps of the SSA reaction, end-resection and annealing, by physically loading Redβ onto the 3′-overhang as it is generated by λ Exo. Such a function would be closely analogous to the loading of RecA onto 3′-overhangs generated by RecBCD (20,21).
Figure 1.
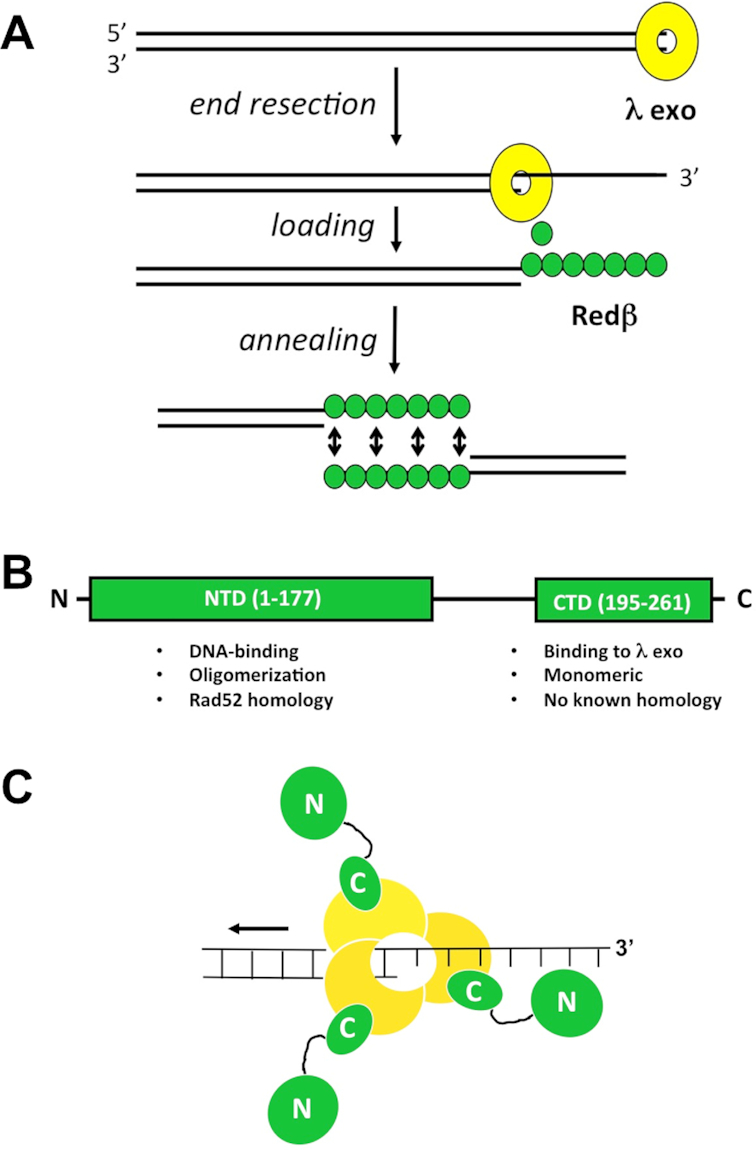
Model for phage λ Red recombination. (A) Overview of the activities of λ Exo and Redβ in the single-strand annealing pathway. (B) Domain structure of Redβ. The functions of each domain are listed. Residues 178–194 of Redβ likely form a flexible linker that connects the two domains. (C) Model of the complex, based on the α3β3 hexamer observed in the crystal, which did not include the Redβ N-terminal domains. The C-terminal domains of each Redβ monomer each bind to the λ Exo trimer, leaving the N-terminal domains free to bind the nascent 3′-overhang. According to the model, as the λ Exo trimer moves along the DNA from right to left, it loads Redβ monomers onto the nascent 3′-overhang, one after another.
Little is known about the structure of the λ Exo–Redβ complex. It has an apparent 1:1 stoichiometry, as initially observed by co-purification of the proteins from cell lysates (18). The dissociation constant of 7.9 μM measured by isothermal titration calorimetry is consistent with a transient interaction, as implied by the loading mechanism described above (22). Gel filtration chromatography and dynamic light scattering suggested a molecular weight ranging from 100 to 800 kDa for the complex (23,24). These differences may arise in part from the dynamic oligomerization behavior of Redβ. While λ Exo forms a stable trimer under all conditions tested, Redβ exists in a variety of different oligomeric states, ranging from monomers (25,26), to closed rings (27), split lock washers (12), and helical filaments (12,27).
Redβ consists of two domains connected by a flexible linker (Figure 1B, 25,28–29). The N-terminal domain (NTD), residues 1–177, shares distant homology with Rad52 (12–14) and is responsible for oligomerization and DNA binding (22,28). The C-terminal domain (CTD), residues 183–261, shares no significant homology with other proteins, exists as a monomer that does not bind to DNA, and forms the complex with λ Exo (29). Based on this apparent separation of function, we reasoned that a minimal complex formed by λ Exo and just the CTD of Redβ might be more amenable to crystallization, but still recapitulate the essential features of the intact complex. To this end, we have determined the crystal structure of λ Exo in complex with the Redβ CTD. The structure shows the residues of each protein at the interface, and thus provides a framework for mutational analysis, to test the role of the complex in promoting recombination in vivo. The structure also reveals an unexpected relationship between the CTD of Redβ and phage λ Orf (30), a protein that binds to SSB to function as a recombination mediator. Based on this relationship, we found that Redβ binds to full-length SSB and to a peptide corresponding to the SSB C-terminus. Through mutational analysis, we established that this interaction is required for recombination in vivo.
MATERIALS AND METHODS
Protein expression and purification
The gene fragment encoding Redβ CTD residues 183–261 was cloned between the NdeI and BamHI sites of pET14b to express an N-terminally 6His-tagged protein with an intervening site for thrombin cleavage (29). The gene for full-length λ Exo was cloned into a pST39 vector to express it as an untagged protein (31). The two plasmids were transformed into separate batches of BL21(AI) cells (Life Technologies), and grown in 3 × 1 L cultures at 37°C. When the optical density (O.D.) at 600 nm reached 0.5, expression was induced by addition of 0.2% arabinose (pET14b) or 0.2% arabinose and 0.2 mM IPTG (pET28b). After shaking at 37°C for 4 h post-induction, cells were collected by centrifugation at 10 000 × g, re-suspended in 20 ml of sonication buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0), and frozen at −80°C. After thawing, the cells expressing each protein were combined (∼50 ml total), lysozyme was added to 1 mg/ml, leupeptin and pepstatin were added to 1 μg/ml, and PMSF was added to 1 mM. After incubating at 4°C for 1 h, cells were lysed by sonication on ice for 3 × 3 min using a Branson sonifier at full power, 30% duty. The resulting mixed cell lysate was centrifuged at 48 000 × g three times for 30 min, and the final clarified supernatant was loaded at 0.5 ml/min onto a 2 × 5 ml HisTrap chelating FF column charged with nickel (GE Healthcare) at 4°C. After extensive washing with sonication buffer, the complex was eluted with a linear gradient from 10 to 500 mM imidazole in sonication buffer. Fractions containing purified complex were mixed with 100 units of thrombin (GE Healthcare) and dialyzed overnight at 22°C into 4 L of PBS buffer (20 mM NaH2PO4, 200 mM NaCl, pH 7.4). The resulting protein mixture was clarified by centrifugation at 10 000 × g and loaded back onto the HisTrap FF column. The untagged complex bound loosely to the column and was eluted with low (30 mM) imidazole. Pooled fractions containing purified complex were dialyzed at 4°C into 20 mM Tris, 1 mM DTT, pH 8.0, concentrated to 25 mg/ml, and frozen at −80°C in 100 μl aliquots.
Crystallization and x-ray structure determination
The complex was crystallized by hanging drop vapor diffusion using a reservoir solution consisting of 25% PEG 3350, 0.2M Li2SO4 and 0.1M Bis–Tris pH 6.0, and a hanging drop consisting of 1.5 μl of reservoir mixed with 1.5 μl of 10 mg/ml complex (diluted in 20 mM Tris, 1 mM DTT, pH 8.0). Prior to data collection, crystals were exchanged into a cryoprotectant solution consisting of reservoir solution with PEG 3350 increased to 27% and glycerol added to 10%. Crystals were mounted in nylon loops and flash frozen in a nitrogen cryostream at 100 K. X-ray diffraction data were collected to 2.30 Å resolution using a Rigaku Compact home source consisting of a MicroMax 003 X-ray generator and a PILATUS3 R 200K detector. Diffraction data were integrated and scaled with HKL3000 (32). Crystals belong to space group P3121 with one complete α3β3 hexamer complex per asymmetric unit. The structure was solved by molecular replacement using the MOLREP function (33) in CCP4 (34) and the λ Exo trimer (PDB code 1AVQ; 35) as a search model. After an initial round of restrained refinement with REFMAC5 version 5.8.0131 (36) at 2.3 Å resolution, the R- and free R-factors were reduced to 26.0 and 30.0%, respectively, and the resulting electron density maps showed clearly traceable density for three Redβ CTDs, one bound to each subunit of the λ Exo trimer. An atomic model for one of the Redβ CTDs was built using COOT (37) and transformed to the other two non-crystallographic symmetry-related positions. Multiple iterative rounds of refinement and model building reduced the Rwork and Rfree to 19.5 and 25.3%, respectively. Non-crystallographic symmetry restraints were used in early rounds of refinement, but completely released in later rounds. The weighting term in REFMAC5 was adjusted to achieve the lowest free R-factor, and eventually set to 0.05. Side chains for which electron density was weak or absent (at a 0.8σ contour level) were truncated to the Cβ atom. The final model consists of 6910 protein atoms, 604 water molecules (one for every 1.4 protein residues), and 16 sulfate anions. Structural figures were prepared with PyMOL (38). Coordinates and structure factors have been deposited in the Protein Data Bank under accesssion ID 6M9K.
Ni-spin pull-down assay to test for in vitro protein-protein interaction
Mutations for the 15 protein variants (5 in λ Exo, 10 in Redβ) were introduced into expression vectors for N-terminally 6His tagged λ Exo in pET-14b (39) and full-length untagged Redβ in pET-9a (29), using the QuikChange method (Agilent Technologies). To detect if the mutations affected complex formation, Ni-spin pull-down assays were performed on proteins from mixed cell lysates, as described previously (29). Briefly, proteins were overexpressed separately in BL21-AI cells in 50 ml cultures grown in LB at 37°C for 4 h post-induction with 0.2% arabinose. Cells from each culture were harvested by centrifugation, re-suspended in 2 ml of sonication buffer, and frozen at −80°C. Cell suspensions for each protein variant were thawed and mixed with cells expressing the WT version of the partner protein, treated with 1mg/ml of lysozyme and protease inhibitors as described above, and lysed by sonication using a microtip at 30% duty, 70% power, on ice. After centrifugation at 48 000 × g, 2 ml of each combined cell lysate was loaded onto a Ni-NTA spin column (Qiagen) pre-equilibrated in sonication buffer. Columns were then washed four times with 600 μl of sonication buffer containing 30 mM imidazole, and then eluted twice with 200 μl of sonication buffer containing 500 mM imidazole. Fractions for whole cell lysate, whole cell lysate after centrifugation (soluble fraction), flow through, wash, and elution were analyzed by 12.5% SDS-PAGE with Coomassie blue staining.
This Ni-spin pull-down assay was also used to assess the interaction between full-length Redβ and SSB. Proteins were overexpressed separately in BL21-AI cells using pET28b (or pET28a) to express N-terminally 6His-tagged Redβ (or SSB), and pET9a to express the untagged proteins. Site directed mutations were introduced into pET9a-Redβ and pET28a-SSB, using the QuikChange method. As a positive control, 6His-tagged Escherichia coli exonuclease I (ExoI), expressed from pET28b in BL21-AI cells (40), was used to pull down untagged SSB expressed from pET9a.
In vivo recombination assays
The activities of variants of Redβ and λ Exo were compared using dsDNA and ssDNA recombination assays, as described previously (29,41–43). Briefly, all recombination experiments were performed in GB2005 E. coli cells, a DH10B derivative that is recA− (41). Lambda red functions (λ Exo, Redβ, and Gam) were expressed from a temperature-sensitive pSC101-BAD-γβα-tet vector, which is arabinose inducible and carries resistance to tetracycline (41). Mutations in the genes for Redβ and λ Exo were introduced by the QuikChange method and confirmed by DNA sequencing. Deletions of λ Exo and Redβ were generated previously by inserting stop codons in place of the codons for Trp-24 and Glu-14, respectively (29,42). The resulting pSC101 plasmid for each mutant was co-transformed into GB2005 cells, together with the corresponding target DNA, either a p15A-CmR plasmid for dsDNA recombination, or MII-BAC-CmR-neo* for ssDNA recombination and selected on LB-agar plates containing 10 μg/ml chloramphenicol and 4 μg/ml tetracycline (Cm10Tet4). To initiate recombination experiments, single colonies from the selection plates were grown in 1.2 ml of LB containing Cm10Tet4 for 18 h at 30°C. All cultures were grown in 1.5 ml microcentrifuge tubes (with pierced caps) in an Eppendorf thermomixer set at 1000 rpm. The next morning, 30 μl of each overnight culture was added to 1.2 ml of fresh LB containing Cm10Tet4 to give a starting OD at 600 nm of 0.09, and grown at 30°C for 2 h. Red functions were then induced with 0.2% l-arabinose and grown at 37°C for an additional 40 min. Cultures were then immediately chilled on wet ice, centrifuged at 9000 rpm at 4°C in a refrigerated Eppendorf microcentrifuge, washed with 1 ml of ice-cold ddH2O and centrifuged at 10 000 rpm, and finally washed again with 1 ml cold ddH2O and centrifuged at 11 000 rpm. Cell pellets were then re-suspended in 50 μl of ice-cold ddH2O, and 100 ng of the appropriate DNA was added, which was either a dsDNA EM7- PCR product (amplified from a linear p15A plasmid), or a single-stranded 100-mer oligonucleotide, both of which were prepared as described (29). After adding the DNA, the 50 μl of cells was transferred to a pre-chilled 0.1 cm electroporation cuvette (Research Products International) and electroporated at 1.45 kV using a BioRad GenePulser. 1 ml of LB was added and the cells were transferred to a microcentrifuge tube and allowed to recover for 70 min with shaking in the thermomixer at 1000 rpm at 37°C, after which an appropriate amount of each culture was plated on LB agar with 10 μg/ml chloramphenicol and 15 μg/ml kanamycin (Cm10Kan15). After incubation of each plate at 37°C for ∼15 h, colonies were counted and reported as the number of colonies per 108 viable cells, as determined by plating on LB-agar with no antibiotics. Experiments for each mutant were performed in triplicate, as three separate cultures started from three different colonies, and on at least two separate days, such that reported values are based on at least six individual experiments.
Western blots
The expression of λ Exo and Redβ proteins was evaluated by Western blot for each of the 15 pSC101 plasmids containing different λ Exo and Redβ mutations. Cell lysates were prepared from cells grown and induced with arabinose as described above for the ssDNA recombineering experiments, except that the culture volume was 50 ml instead of 1.2 ml. After induction for 40 min at 37°C, the cells were collected by centrifugation, re-suspended in 2.5 ml of buffer containing 50 mM NaH2PO4, 500 mM NaCl, 10 mM imidazole, 10% glycerol, pH 8.0, and frozen at −80°C. Frozen cell suspensions were thawed, lysed by sonication and then centrifuged at 48 000 × g to isolate 2.5 ml of each soluble lysate, which was stored at −80°C. Gel samples were prepared by mixing 80 μl of each thawed lysate with 20 μl of 2.5× SDS-PAGE loading buffer, 17 μl of which was run on 13.5% SDS-PAGE gels at 22°C, and transferred to nitrocellulose membranes (Bio-Rad) in transfer buffer (25 mM Tris pH 8.3, 192 mM glycine, 0.05% SDS, 20% methanol) for 70 min at 90 V (constant voltage) at 4°C. Membranes were blocked with Fluorescent Blocker (MilliporeSigma) for 1.5 h at 22–25°C, incubated with anti-Redβ-anti-λ Exo primary antibody (generously provided by Dr Kenan Murphy) in Fluorescent Blocker overnight at 4°C, washed three times for 15 min in 1× PBS, 0.1% Tween® 20, incubated with IRDye 680RD Donkey anti-Rabbit secondary antibody (Li-COR) in Fluorescent Blocker for 45 min at 22–25°C, and washed again for 3 × 15 min in 1× PBS, 0.1% Tween® 20. Fluorescence was detected and visualized using an Odyssey Blot Imager (LI-COR).
Exonuclease assays
Briefly, the L91A and F94A variants of λ Exo were purified as N-terminally 6His-tagged proteins as described (39), dialyzed into 20 mM Tris pH 8.0, 1 mM DTT, concentrated to > 20 mg/ml, and stored at −80°C in 50 μl aliquots. Exonuclease assays were performed as described (39). Briefly, BamHI-linearized pUC19 plasmid (1.2 nM DNA ends) was pre-equilibrated at 37°C in reaction buffer (67 mM glycine–KOH, pH 9.4, 2.5 mM MgCl2, 50 μg/ml BSA), and the reactions (100 μl total) were initiated by adding a limiting amount (0.05 nM) of λ Exo trimer. Aliquots (10 μl) were removed at the indicated time points, loaded onto a 0.8% agarose gel, and electrophoresed at 200 V in TAE buffer to separate the dsDNA substrate from the ssDNA product. Gels were visualized by staining with SYBR Gold.
Fluorescence anisotropy assay for binding of Redβ to the SSB-Ct peptide
For measuring the interaction of purified Redβ with the SSB C-terminal peptide (SSB-Ct) by fluorescence anisotropy, full-length Redβ protein was purified as an untagged protein as described previously (29). The following control proteins were purified as described: ExoI (40), Redβ CTD (29), Redβ NTD (28), λ Exo (39) and Redβ 1–250 (29). The K253A and F249D variants of Redβ were purified as un-tagged proteins as described for untagged Redβ and Redβ 1–250 (29). The complex consisting of λ Exo and full-length untagged Redβ was purified as described above for the λ Exo – Redβ CTD complex, except that the N-terminal 6His tag was on λ Exo instead of Redβ. The complex was further purified by gel filtration chromatography on Sephacryl S-300 to remove excess λ Exo. Protein concentrations were determined by O.D. at 280 nm, using extinction coefficients calculated from the amino acid sequences. A stoichiometry of 1:1 was assumed for the purified λ Exo–Redβ complex for this calculation.
Fluorescently labeled peptides corresponding to the SSB-Ct (FAM-WMDFDDDIPF) and its F177A variant (FAM-WMDFDDDIPA) were synthesized using standard Fmoc chemistry, purified by reversed phase HPLC to > 95% homogeneity, authenticated by high-resolution mass spectrometry, and quantified by O.D. at 490 nM (ϵ = 70,000 cm−1 M−1). This peptide corresponds to the last nine residues of E. coli SSB, with additional N-terminal fluorescein (FAM) and tryptophan residue. Binding reactions (in 20 mM Tris pH 7.5, 150 mM NaCl, 0.1 mg/ml bovine serum albumin, 1 mM DTT) contained 20 nM fluorescein-labeled peptide and varying amounts of each binding protein. Fluorescence anisotropy measurements were performed in triplicate, in 20 μl volumes in Corning 384-well round-bottom black polystyrene plates using a Spectra Max M5 microplate reader at 25°C (Molecular Devices). Excitation and emission wavelengths were 490 and 515 nm, respectively. For measuring apparent Kd values quantitatively for ExoI and Redβ, three separate experiments for each protein were performed, and data for each experiment were fit to a 1:1 binding model (44) using KaleidaGraph software. The equation for this binding model is:
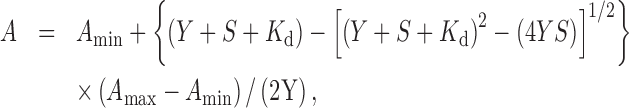 |
where A is the measured anisotropy, Amin is the minimum anisotropy (the anisotropy of the labeled peptide in the absence of protein), Amax is the maximum anisotropy (the anisotropy of the fully bound peptide), S is the concentration of unlabeled protein at each titration point, and Y is the concentration of the labeled peptide, which was 20 nM in all experiments. During the fitting procedure, S and Y are fixed at their input value, A is fixed at its measured value, and Amin, Amax and Kd are allowed to float (an initial approximate value of Kd is required). The mean and standard deviation reported for each Kd measurement were based on three independent titrations. Within each titration, the anisotropy value at each protein concentration was the mean of three replicates.
RESULTS
Crystal structure of a minimal λ Exo–Redβ complex
We have determined the crystal structure of a complex formed by the λ Exo trimer bound to three molecules of the Redβ CTD (Figure 2A). The complex was purified by Ni-affinity chromatography, using N-terminally 6His-tagged Redβ CTD (residues 183–261) to pull-down full-length, untagged λ Exo (residues 1–226). The structure, which contains an α3β3 hexamer in each asymmetric, was refined at 2.30 Å resolution to R- and free R-factors of 19.5 and 25.3%, respectively (Supplementary Figure S1, Supplementary Table S1). The λ Exo trimer itself does not undergo any significant conformational changes in the complex relative to the structures of free λ Exo (35) or its complex with DNA substrate (39). The Redβ CTD folds into a 3-helix bundle, as expected from secondary structure prediction and circular dichroism, which suggested a predominance of α-helix (29). Residues 183–194 of the CTD construct in the crystal are disordered in all three copies, in agreement with their being flagged as a low complexity region by sequence analysis (29). In full-length Redβ, these residues are presumably part of a flexible linker connecting the N- and C-terminal domains.
Figure 2.

Structure of the complex. (A) View from the back of the λ Exo trimer, looking into the channel from which the nascent 3′-overhang would emerge. The dsDNA substrate would enter the channel on the opposite side. The three subunits of λ Exo are colored yellow, cyan, and wheat. The three Redβ CTDs are shown in green, each bound to a single λ Exo subunit at the periphery of the complex. (B) Side view of the complex, from the left of panel A. The paths of the dsDNA substrate and ssDNA product are indicated. Notice that the C-terminal domains are bound on the side of the λ Exo trimer from which the 3′-overhang would emerge, and that the N-terminal domains, which would connect to Thr-195 (labeled for the CTD in front), would be well positioned to bind to the emerging ssDNA. (C) Close-up view of the interactions at the interface, zooming in on the boxed region of panel B from the lower right, as indicated by the arrow. Hydrogen bonds (within 3.5 Å) are shown as dashed lines. (D) View from the left of panel C, to show the interactions behind helix E, although Thr-95 of helix E of λ Exo is still occluded in this view.
Each Redβ CTD is bound to a single subunit of the λ Exo trimer, at the periphery of the complex, but biased towards the side of the trimer from which the 3′ overhang of a DNA substrate would be extruded (Figure 2B). The first ordered residue of each CTD (Thr-195), to which the N- terminal DNA binding domain would connect, also points to this side of the trimer. By contrast, the last residue of each CTD points to the opposite side of the trimer, the side at which the dsDNA substrate would enter. Thus, as the N-terminal DNA-binding domains of each Redβ monomer would be ideally positioned to bind to the nascent 3′-overhang as it emerges from the λ Exo trimer, the overall geometry of the complex is consistent with the loading mechanism proposed above (Figure 1C). Although no DNA is present in the crystal, modeling of a DNA duplex into the central channel of the λ Exo trimer shows that it does not overlap with any of the three copies of the CTD (Supplementary Figure S2), as expected from the proposed function.
A total solvent accessible surface area of 1,400 Å2 is buried at the interface between each Redβ CTD and its respective subunit of the λ Exo trimer. The interaction mainly involves helix E of λ Exo, which wedges into a groove between helices 2 and 3 of the CTD. Specifically, a hydrophobic ridge on the surface of helix E, formed by Thr-90, Leu-91, Phe-94 and Thr-95, inserts into a pocket formed by Leu-223, Ile-227, Phe-249 and Leu-250 of the CTD (Figure 2C). This central core of hydrophobic interactions is shielded on one side by a network of electrostatic interactions between Gln-226 and Arg-229 of the CTD, and Gln-83, Asp-87 and Thr-90 of λ Exo. On the other side, additional hydrogen bonds are formed, between Gln-252, Lys-253, and Glu-256 of the CTD and Leu-184, Phe-94 and Tyr-186 of λ Exo, respectively (Figure 2C, D). Lys-253 caps the C-terminal end of helix E to form a charge-dipole interaction that could be particularly stabilizing. A complete listing of all interacting residues is provided in Supplementary Table S2. Examination of this table shows that the details of the interactions are remarkably consistent across the three independent copies of the CTD in the crystal.
Interactions at the hydrophobic core of the interface are required for complex formation
To determine the importance of individual residues forming contacts at the interface, a Ni-spin pull-down assay was performed (29), in which 6His-tagged λ Exo was used to pull down full-length, un-tagged Redβ. Although this assay is qualitative, it allows us to test a large number of mutants without having to purify the proteins, and also tests the interaction in the presence of other cellular components. The two proteins were individually expressed in separate cell cultures, and supernatants from mixed cell lysates were loaded onto each Ni-spin column. A total of 5 λ Exo variants at 3 residues and 10 Redβ variants at 8 residues were chosen for analysis, based on their location at the interface. A non-interfacial residue, Lys-132 of the NTD of Redβ, was included as a control. Most of the mutations result in truncation of a side chain to alanine. For residues forming the central core of hydrophobic interactions, Leu-91 and Phe-94 of λ Exo and Leu-223 and Phe-249 of Redβ, mutations to both alanine and an acidic side chain (aspartate or glutamate) were introduced. This was intended to test for simple removal of each nonpolar side chain, as well as a more drastic alteration to a charged residue.
The results of the Ni-spin pull down for each variant are shown in Figure 3. All 15 mutants were expressed in soluble form at similar levels as the WT proteins (Figure 3, lanes labeled ‘S’). Although soluble expression does not confirm proper folding in the same way that circular dichroism would, it does provide a qualitative assessment of folding, as mis-folded proteins would likely be insoluble. Note that WT 6-His λ Exo protein and most of its variants were only marginally soluble. A few of the variants, in particular D87A, L91D and F94A, may have increased solubility, which for L91D and F94A could arise from truncation of exposed apolar side chains. However, the amount of soluble WT λ exo seen in the ‘S’ lane also varied in the experiments with different Redβ variants, indicating that it may just be sensitive to exact experimental conditions. This variation does not detract from the pull-down experiment however, as 6His- λ Exo protein is always enriched in the elution fractions.
Figure 3.

Effects of mutations on complex formation in vitro. The SDS-PAGE gels (stained with Coomassie) show the results of a Ni-spin assay in which 6His λ Exo was used to pull down un-tagged, full-length Redβ from mixed cell lysates. An experiment for the two WT proteins is shown at the top left, while results using variants of Redβ or λ Exo with the respective WT version of the partner protein are shown at the lower left and right, respectively. The individual lanes are: WC (whole cell lysate), S (soluble part of lysate after centrifugation), FT (flow through after spin-loading of S), W1, W4 (30 mM imidazole washes 1 and 4, 2–3 not shown), E1, E2 (500 mM imidazole elutions). The presence of a band for un-tagged Redβ in the elutions indicates an intact complex. Variants with little to no such band are defective for complex formation and indicated by an asterisk.
Strikingly, mutation of residues at the central hydrophobic core of the interface, including Leu-91 and Phe-94 of λ Exo, and Leu-223 and Phe-249 of Redβ, completely disrupts complex formation—there is no (or very little) band for untagged Redβ in the elution for these variants (Figure 3, lanes labeled ‘E1’ and ‘E2’). This was true for both the alanine and the acidic variants at each of these positions, indicating that simple removal of any one of these four nonpolar side chains is enough to disrupt the complex completely, at least as seen by this assay. By contrast, mutation of residues forming the more peripheral electrostatic interactions, including Asp-87 of λ Exo, and Gln-226, Arg-229, Gln-252, Lys-253 and Glu-256 of Redβ, did not have any observable effect on complex formation. In summary, these results show that while the central core of hydrophobic interactions is essential for complex formation, the surrounding network of electrostatic interactions is not.
Variants of λ Exo and Redβ that disrupt complex formation in vitro are defective for recombination in vivo
To determine if the complex is required for recombination in vivo, the 15 variants of λ Exo and Redβ described above were tested in dsDNA and ssDNA recombination assays developed previously (29,41–43). Due to a high degree of variance, even for WT, these assays are not good at detecting small differences in activity among protein variants, but they are good for identifying mutants with large effects. Briefly, DNA encoding kanamycin resistance was electroporated into E. coli cells expressing λ Exo and Redβ under the control of arabinose, and the number of colonies was counted to score for recombination efficiency. Western blot analysis confirmed that all 15 variants were expressed at similar levels as the WT proteins (Supplementary Figure S3). For dsDNA recombination, the electroporated DNA was a linear PCR-generated ∼2 kb neomycin cassette with 50 bp terminal homologies to a target site on a plasmid. For ssDNA recombination, a 100-mer oligonucleotide targeting the lagging strand was used to correct a 4 nt insertion within a neomycin resistance gene on a bacterial artificial chromosome. Whereas dsDNA recombination requires both λ Exo and Redβ for full activity (19,45), ssDNA recombination only requires Redβ (7,46). Therefore, we expected that mutations disrupting the complex would potentially disrupt dsDNA recombination, but not ssDNA recombination.
For dsDNA recombination, the WT proteins resulted in 8.4 × 103 recombinants per 108 viable cells (Table 1). Controls with no arabinose, no DNA, or a Redβ deletion gave zero colonies. Deletion of λ Exo however did not completely abolish recombination: the number of colonies was 6% of WT. This result is qualitatively consistent with previous data (42,47–48), and is presumably due to productive resection of the linear input dsDNA by one or more endogenous E. coli nucleases (47). Interestingly, variants of λ Exo that were defective for complex formation in vitro (L91A, L91D, F94A and F94E) resulted in only 0.6–3% of the activity of WT, an even greater reduction than the λ Exo deletion. Apparently, resection of the input dsDNA by λ Exo without coordinated loading of Redβ results in either degradation or folding of the nascent 3′-overhang, so as to block access of Redβ. However, it is conceivable that these mutations could also be affecting exonuclease activity, which could in and of itself reduce recombination, particularly if they were to bind and block the dsDNA ends. To rule out this possibility, the purified F94A and L91A variants of λ Exo were tested for their exonuclease activities, and found to be fully active (Supplementary Figure S4). Variants of Redβ that were defective in complex formation (L223A, L223D, F249A and F249D) resulted in similarly low levels of recombination, 0–0.7% of WT. Collectively, these results show that the protein-protein interaction is indeed required for dsDNA recombination, and further suggest that when λ Exo resects the input dsDNA without coordinated loading of Redβ, the nascent 3′-overhang is either unprotected and degraded, or folds up on itself to become inaccessible to annealing.
Table 1.
In vivo recombination of Redβ and λ Exo variants*
| DS | DS (%) | SS | SS (%) | Complex | |
|---|---|---|---|---|---|
| No DNA | 0 | 0 | 0 | 0 | |
| No arabinose | 0 | 0 | 0 | 0 | |
| Redβ - | 0 | 0 | 0 | 0 | |
| λexo - | 470 ± 250 | 5.6 | 10400 ± 4000 | 104 | |
| WT | 8400 ± 5000 | 100 | 10000 ± 4000 | 100 | Yes |
| K132A Redβ | 1600 ± 530 | 19 | 2000 ± 600 | 20 | Yes |
| L223A Redβ | 61 ± 81 | 0.72 | 350 ± 360 | 3.5 | No |
| L223D Redβ | 9 ± 15 | 0.11 | 68 ± 130 | 0.68 | No |
| Q226A Redβ | 8900 ± 9000 | 106 | 1300 ± 1100 | 13 | Yes |
| R229A Redβ | 6000 ± 4200 | 71 | 5800 ± 3200 | 58 | Yes |
| F249A Redβ | 18 ± 20 | 0.21 | 73 ± 69 | 0.73 | No |
| F249D Redβ | 0 | 0 | 41 ± 48 | 0.41 | No |
| Q252A Redβ | 7200 ± 5400 | 85 | 10100 ± 8500 | 101 | Yes |
| K253A Redβ | 17 ± 19 | 0.20 | 250 ± 260 | 2.5 | Yes |
| E256A Redβ | 5800 ± 2400 | 69 | 5500 ± 2300 | 55 | Yes |
| D87A λexo | 4600 ± 3000 | 55 | 17 900 ± 3300 | 179 | Yes |
| L91A λexo | 53 ± 81 | 0.63 | 10 300 ± 4200 | 103 | No |
| L91D λexo | 260 ± 220 | 3.1 | 11 000 ± 5800 | 110 | No |
| F94A λexo | 140 ± 90 | 1.6 | 18 300 ± 4400 | 183 | No |
| F94E λexo | 57 ± 62 | 0.68 | 17 700 ± 6200 | 177 | No |
*The values in the DS and SS columns gives the number of colonies observed, per 108 viable cells for the dsDNA and ssDNA recombination experiments. The values to the right of these columns give the % compared to WT. Experiments for WT and all variants were performed in triplicate, on at least two separate days, such that each value is the mean of at least six separate experiments. WT was performed on 9 separate days (27 individual measurements) for DS and six separate days (18 individual measurements) for SS. All data for each variant were included in the reported numbers, with the exception of seven deviant values that were discarded based on analysis of interquartile range. A value of zero indicates that no colonies were observed in any of the 6 experiments (the maximum amount plated was 1% of the final 1 ml culture after recovery). The far-right column indicates whether or not the variant forms the λ Exo–Redβ complex in vitro, as seen in Figure 3.
As for the variants of λ Exo and Redβ that retained the ability to form the complex, we expected that they would be fully active for dsDNA recombination. This was indeed the case for five of the variants, D87A in λ Exo and Q226A, R229A, Q252A and E256A in Redβ, which gave 55–106% of the activity as WT, within the limits of detection of the assay. However, one of the variants of Redβ that could still form the complex, K253A, gave surprisingly low activity, only 0.2% of WT. As will be discussed below, it is likely that this mutation disrupts an additional function of the CTD, one that is required for recombination, but distinct from its role in binding to λ Exo.
For ssDNA recombination, the WT proteins resulted in 1.0 × 104 recombinants per 108 viable cells, slightly higher than for dsDNA. Controls with no arabinose, no DNA, or a Redβ deletion again resulted in no recombinants. Deletion of λ Exo resulted in no significant reduction in recombination compared to WT, consistent with previous data showing that λ Exo is dispensable for ssDNA recombination (7,46). Accordingly, all five of the λ Exo variants tested were at least as active as WT (some even more so). Surprisingly however, over half of the Redβ variants tested, including all that were defective for complex formation, as well as K253A, resulted in dramatically lower levels of ssDNA recombination, only 0.4–3.5% of WT. This was unexpected, because the mere presence of λ Exo, let alone its interaction with Redβ, is not required for ssDNA recombination (the λ Exo deletion was fully active). Four of the Redβ variants tested, Q226A, R229A, Q252A and E256A, gave comparable levels of ssDNA recombination as WT, ranging from 13 to 101%. Notably, these four variants also retained high activity for dsDNA recombination.
Collectively, these results suggest that the complex is indeed required for dsDNA recombination, but not for ssDNA recombination. However, they also suggest the possibility that the site on the Redβ CTD used for binding to λ Exo overlaps with another function of the CTD, one that is necessary for both dsDNA and ssDNA recombination. In the following section, we present data suggesting that this other function may be binding to SSB.
Unexpected homology between the Redβ CTD and phage λ Orf
As described above, the Redβ CTD consists of ∼67 residues that form a 3-helix bundle. Sequence comparisons of the CTD do not reveal significant homology with other known proteins. However, a search for structural homologs using the Dali server (49) revealed an unexpected similarity with a core region of phage λ Orf (30). The two proteins align with one another to an rmsd of 2.4 Å over 64 residues sharing 9% sequence identity (Figure 4). Orf was the top hit from the Dali search, and the only hit for which a functional parallel was apparent. This relationship would have been difficult to detect by sequence analysis alone, due to two large insertions in Orf relative to Redβ.
Figure 4.

Structural similarity with λ Orf uncovers an interaction with SSB. (A) Structural superposition of the CTD with phage λ Orf (PDB code 1PC6, 30). The two subunits of Orf are colored light blue and magenta. The CTD (green) is shown superimposed on only one of the Orf subunits, but in the Dali search it superimposed equally well on the other. The three helices of the CTD, and the N- and C-termini of Orf, are indicated. The arrow shows the site on the CTD that is used to bind λ Exo. (B) Structure-based sequence alignment from the Dali server (49). Secondary structure elements above and below are for the CTD and Orf, respectively. The sequence of YbcN is also included in the alignment, based on a previous multiple sequence alignment of Orf homologs (51). Residues of Redβ that contact λ Exo are shown in bold red. Four of these residues in Orf, and three in YbcN, are conserved as apolar residues (also in bold red). These residues map out a hydrophobic surface that could putatively bind to the SSB C-tail.
Orf forms an asymmetric dimer that functions as a recombination mediator (30). Specifically, Orf binds to SSB and facilitates loading of RecA onto SSB-coated ssDNA in the absence of RecFOR (30,50–53). Although it is not known if the site for binding SSB on Orf is within the three-helix core that shares homology with Redβ, the structural similarity suggests the possibility that the Redβ CTD may have a similar function as Orf in binding to SSB, perhaps to facilitate access of the NTD to SSB-coated ssDNA. To test this idea, in vitro pull-down assays were performed using 6His-Redβ to pull down untagged SSB and vice versa (Figure 5). These experiments confirmed a significant interaction between the two proteins (Figure 5A, top two panels), comparable to the interactions of Redβ with λ Exo (Figure 3), and ExoI with SSB (Figure 5B), with the latter used as a positive control (54).
Figure 5.

Ni-spin pull down reveals an interaction between Redβ and SSB. (A) The top two gels, prepared and labeled as described in Figure 3, show pull downs of untagged Redβ by 6His-SSB, and vice versa. The next three gels show attempted pull-downs using variants of Redβ and SSB, as described in the text. The bottom gel shows a negative control in which 6His-SSB fails to pull down untagged λ Exo. (B) Positive control showing a pull down of untagged SSB with 6His-ExoI. A lysate overexpressing untagged SSB was included in the sample loaded for the upper gel, but not for the sample in the lower gel (the interaction with endogenous SSB is not detected by this assay, presumably due to its lower level of expression).
Based on our unexpected finding above that some of the mutations within the Redβ CTD significantly disrupted ssDNA recombination, we hypothesized that the site on the CTD used for binding to λ Exo may overlap with another essential function of the protein. To test if this other function is binding to SSB, Ni-spin pull downs were performed using 6His-SSB to pull down untagged Redβ variants with two of these mutations, K253A and F249D. As shown in Figure 5A, both variants completely failed to interact with SSB. This result suggests that the site on the CTD for binding to λ Exo overlaps with the site for binding to SSB. Many proteins that interact with SSB bind to its C-terminal tail (C-tail), particularly the last residue of the protein, Phe-177 (54–58). To test if this residue is also used for the interaction with Redβ, Ni-spin pull downs were performed using the F177A variant of 6His-SSB to pull down untagged Redβ. As shown in Figure 5A, the F177A mutation completely disrupts the interaction. All together, these results establish an interaction between Redβ and SSB that requires the Redβ CTD and the SSB C-tail.
To quantify the binding of Redβ to the SSB C-tail, fluorescence anisotropy titrations were performed with purified Redβ and a fluorescein-labeled peptide corresponding to the last nine residues of SSB (SSB-Ct). As a positive control, ExoI bound tightly to SSB-Ct with an apparent dissociation constant (Kd) of 98 ± 5 nM (Supplementary Figure S5A). Redβ also bound to SSB-Ct, but less tightly, with an apparent Kd of 10.6 ± 2.3 μM (Figure 6A and Supplementary Figure S5B). By contrast, λ Exo, used as a negative control, showed no interaction with SSB-Ct. Interestingly, neither the individual N- nor the C- terminal domains of Redβ showed the interaction with SSB-Ct, indicating that binding requires both domains (Figure 6A). The K253A and F249D variants of Redβ also failed to interact with the SSB-Ct peptide (Figure 6B), in agreement with the results from the Ni-spin pull downs. Moreover, a previously purified variant of Redβ lacking the C-terminal 11 residues, Redβ1–250 (29), also lost the ability to bind to SSB-Ct (Figure 6B). Based on the structure of the CTD this deletion would be expected to disrupt its folding, as the deleted residues form part of helix 3 (Figure 2). Finally, a variant of the SSB-Ct peptide with the F177A mutation was tested for binding to Redβ, and it completely failed to interact (Figure 6C). These results, together with the Ni-spin pull downs, suggest that the SSB C-tail binds to the Redβ CTD at a site that overlaps with λ Exo-binding site.
Figure 6.

Fluorescence anisotropy titrations of Redβ with SSB-Ct peptide. (A) Full-length Redβ, its N- and C- terminal domains, or λ Exo were titrated into 20 nM fluorescein-labeled SSB-Ct peptide (FAM-WMDFDDDIPF). While Redβ binds to the peptide with an apparent Kd of 11 μM, the other proteins show no significant binding. (B) Three variants of Redβ that are defective for ssDNA recombination (K253A, F249D and Redβ250) show no interaction with the SSB-Ct peptide. The Redβ-λ Exo complex binds with a similar apparent Kd (12 μM) as Redβ alone, but with lower anisotropy. (C) Redβ fails to bind to an SSB-Ct peptide with the F177A mutation.
If the binding sites for λ Exo and SSB on Redβ do in fact overlap, then one would expect the two proteins to compete for binding to Redβ. To test if this is the case, the complex of λ Exo with full-length Redβ was purified, and titrated into the SSB-Ct peptide to test for binding. Since λ Exo is present at equivalent levels as Redβ in this experiment (it is a 1:1 complex), and is thus in large excess over the 20 nM peptide, we expected it to block binding of the SSB-Ct peptide. However, an increase in anisotropy was still observed, although only to about half the level observed for Redβ alone. Thus, although λ Exo did not completely block binding of Redβ to the SSB-Ct peptide, it did have a significant effect on the interaction (Figure 6B). Fitting of the titration data for the complex resulted in a similar affinity as Redβ alone (Kd 12.8 ± 3.5 μM), even though the anisotropy level was reduced (Figure 6B and Supplementary Figure S6). The lower anisotropy suggests that the size of the Redβ-λ Exo complex is smaller than that of the oligomer formed by Redβ alone, or that the peptide is bound differently, with the fluorophore more exposed. We do not at present understand the significance of this result, but it is conceivable that the sites on the CTD for binding λ Exo and SSB are not completely overlapping, such that the SSB-Ct can still bind in the presence of λ Exo. Alternatively, given the high density of negative charge on the peptide, it is also possible that it binds to a new site that is formed in the complex, through non-specific electrostatic interactions.
In summary, these results reveal an interaction between Redβ and SSB that occurs through the Redβ CTD and the SSB C-tail. They further suggest that the site on Redβ for binding to the SSB C-tail involves both the N- and C-terminal domains of Redβ, and that the site on the CTD overlaps with the site for binding to λ Exo. As will be discussed below, this interaction suggests a secondary role of the CTD in helping the NTD to access SSB-coated ssDNA at the replication fork, presumably by binding and displacing SSB, as seen for several other E. coli proteins (54–58).
DISCUSSION
Homologs of λ Exo and Redβ are often encoded as pairs in the genomes of dsDNA bacteriophage, implying that their functions are coordinated. It is well established that both proteins are required for dsDNA recombination (19), which is the likely function for which the recombination system has evolved, presumably to generate concatemers of the genome for packaging (6). That the two proteins form a complex was first observed for phage λ (18) and subsequently for RecET of the E. coli rac prophage (19). The functional importance of the complex has largely been inferred from experiments showing that combining SynExo proteins from different sources as heterologous pairs, such as λ Exo and RecT or RecE and Redβ, does not result in successful recombination (19,59). However, due in part to a lack of structural information, mutations in λ Exo and Redβ that specifically disrupt the complex, without affecting other activities of the proteins, have not yet been identified, and thus the functional consequences of disrupting the complex have not been fully assessed.
In this work, we present the first structural data on the λ Exo-Redβ interaction. The structure clearly defines the residues of each protein at the interface and provides a framework for designing mutations that are specifically defective in complex formation. Based on this analysis, we have identified variants at two residues of λ Exo, Leu-91 and Phe-94, which are completely defective for complex formation, but fully active as exonuclease enzymes. The fact that these variants are strongly and specifically defective for dsDNA recombination supports the conclusion that the complex is essential for function. Mutations in Redβ designed by the same rationale were also defective for dsDNA recombination, but non-specifically: they also disrupted ssDNA recombination, presumably by affecting other activities of the protein, such as binding to SSB (see below).
The structure of the complex also permits mapping and interpretation of prior mutational data. Our group showed that deletion of the C-terminal 11 residues of Redβ (to make Redβ1–250) completely disrupts the interaction with λ Exo, as well as both dsDNA and ssDNA recombination (29). The structure shows that this truncation would remove about 1/3 of helix 3 of the CTD, which would very likely disrupt folding. Stewart and colleagues identified four individual mutations in the CTD (E191A, Q252A, E256A, K258A) that decreased dsDNA more than ssDNA recombination, implicating the corresponding residues as being at the interface with λ Exo (22). While two of these residues, Gln-252 and Glu-256, are indeed at the interface, the other two, Glu-191 and Lys-258, are not (Supplementary Figure S7A). Another mutation from this study, Q240A, disrupted dsDNA and ssDNA equally, and weakened (but did not abolish) the interaction with λ Exo. The structure shows that Gln-240 is not at the interface, but could be important for folding of the CTD, as it forms hydrogen bonds that bridge helices 1 and 3 (Supplementary Figure S7B).
Interestingly, the involvement of helix E of λ Exo in forming the complex with Redβ had been predicted, based on its exposed location and its homology with a helix in type II restriction endonucleases that is used for dimerization (60). That this is the key region of λ Exo that is used for the interaction with Redβ is quite remarkable and speaks to the power of using distant evolutionary relationships to predict protein function. Helix E was also identified as being involved in the interaction by a separate study that used pepSPOT™ analysis (24). However, several alanine mutations on this helix (L91A, F92A, E93A, F94A and T95A) had little if any effect on the complex in that study, as seen by co-immunoprecipitation, and only mild effects (at most a 2-fold reduction) on dsDNA recombination (24). By contrast, our data show that the L91A and F94A variants are completely defective in complex formation, and severely reduced in dsDNA recombination. We do not at present understand why the results from the two studies are different.
One limitation of our study is that the crystal structure used just the CTD of Redβ, as opposed to the full-length protein. In our structure, the minimal complex exists as an α3β3 hexamer, which would be 165 kDa for the full-length proteins. However, biophysical measurements of the size of the complex indicate that it may be considerably larger, perhaps as large as 800 kDa (23). The larger complexes could arise from interactions between the N-terminal domains of Redβ, which are known to oligomerize (22,28–29). The extent to which these larger complexes would exist under cellular conditions however, is unclear (26).
Also unclear is whether or not the N-terminal domain of Redβ participates at all in the interaction with λ Exo. Previous data from our lab by Ni-spin pull down showed a clear interaction between λ Exo and the Redβ CTD, but no interaction with the NTD (29). However, Stewart and colleagues identified a region within residues 20–38 of the NTD that affected complex formation (22). It is conceivable however that this region is involved in interactions between the N-terminal domains themselves, which could stabilize the complex indirectly, through cooperativity. The structure tends to support the conclusion that the NTD does not form direct interactions with λ Exo, because the first residue of each CTD (Thr-195), to which each NTD would connect, points away from the interface with λ Exo (Figure 2B). However, as the linker connecting the two domains (residues 178–194) is likely to be flexible, direct contacts between the N-terminal domains and λ Exo cannot be ruled out.
An unexpected result from our analysis was the number of mutations in the Redβ CTD that severely reduced ssDNA recombination. Since ssDNA recombination does not even require the presence of λ Exo (7,46), let alone its interaction with Redβ, and the mutations were designed to specifically disrupt the complex, the effect on ssDNA recombination was not expected. However, such mutations in the CTD are not unprecedented. We previously demonstrated that deletion of the last 11 residues of the CTD completely disrupts ssDNA recombination (29), and Stewart and colleagues showed that similar C-terminal truncations, as well as mutations at several positions within the CTD, affected ssDNA recombination (22). An interesting observation from our analysis is that the three residues of Redβ at which mutations significantly reduced ssDNA recombination (Leu-223, Phe-249 and Lys-253) cluster together on the surface of the CTD, whereas the four residues where there was little effect, Gln-226, Arg-229, Gln-252 and Glu-256, are more peripheral (Figure 7). The mutational data thus appear to identify a surface of the CTD, overlapping with the surface used for binding to λ Exo, that is used for another necessary function of the protein.
Figure 7.

Surface view of the Redβ CTD showing the binding site for λ Exo, which may overlap with a binding site for SSB-Ct. (A) Mapping of mutational data. The labeled residues constitute the surface of the CTD that contacts λ Exo in the complex. Residues mutated in this study are shown in cyan if the mutation disrupted ssDNA recombination, or magenta if not. The residues in cyan thus map out a putative site for binding SSB-Ct. Notice the deep pocket, to which Phe-94 of λ Exo binds, and to which Phe-177 of the SSB C-tail could putatively bind. (B) Surface electrostatics. The surface is colored red for negative and blue for positive (±75 kBT). The positive charge around Lys-253 binds to the C-terminal end of helix E of λ Exo, and putatively to the C-terminus of SSB.
What could this function be? Prior data from our lab demonstrated that the CTD has no interaction with ssDNA or dsDNA, no interaction with itself (it is a monomer), and no interaction with the NTD (29). The structure of the CTD reveals an unexpected homology with λ Orf, a protein that binds to SSB and facilitates binding of RecA to SSB-coated ssDNA (30). Based on this relationship, we hypothesized that Redβ may also bind to SSB, and found that it binds to both full-length SSB, by Ni-spin pull down, and to a peptide corresponding to the SSB C-tail, by fluorescence anisotropy. The interaction with the C-tail requires both domains of Redβ, implying that there is indeed a binding site for the SSB C-tail on the surface of the CTD. Interestingly, the surface on the CTD that is used for binding to λ Exo includes a hydrophobic pocket with a neighboring positively charged residue (Lys-253; Figure 7), which is similar to the surfaces used by several other proteins for binding the SSB C-tail (54–58). A key residue of the SSB C-tail in these structures is the terminal phenylalanine (Phe-177), which binds through hydrophobic interactions with its phenyl ring, and electrostatic interactions with its negatively charged carboxylate. One could envision this residue of SSB binding to the CTD through very similar interactions, by taking the place of Phe-94 of λ Exo to bind to the hydrophobic pocket and Lys-253 (Figure 7). The observation that the interaction between Redβ and SSB is completely disrupted by the K253A and F249D mutations in Redβ, and by the F177A mutation in SSB, is consistent with this hypothesis.
While Redβ displays a moderate affinity for the SSB-Ct peptide (Kd = 10.6 μM), especially as compared to ExoI (Kd = 98 nM), its affinity is comparable to that of other proteins that interact with SSB, such as RecQ and DNA Pol III, which bind to similar peptides with Kd values of 6 and 2.7 μM, respectively (61,62). The different affinities of these proteins for SSB could arise from their different biological functions, which may require a tighter or weaker interaction with SSB, depending on the context. It is also possible that some of these proteins interact with other parts of SSB, in addition to the C-tail, to enhance their affinities.
If this surface on the CTD of Redβ is indeed a site for binding the SSB C-tail, then one might expect the equivalent surface on λ Orf to also bind the SSB C-tail. However, mutational analyses have not been able to define the SSB-binding site on Orf, and one study concluded that the SSB C-tail is not required for the interaction (51,52). On the other hand, the Orf homolog YbcN from E. Coli cryptic prophage DLP12, does require the C-tail for its interaction with SSB (51), suggesting that this surface on these proteins may be functionally conserved. Along these lines, residues of the CTD that form the hydrophobic pocket at the center of this surface (Leu-223, Ile-227, Phe-249 and Leu-250), are predominantly conserved as apolar residues in λ Orf and YbcN (Figure 4B). However, this pocket on Orf is partially plugged by a β-hairpin from the other subunit of the dimer (Figure 4A), such that binding of the SSB C-tail to the equivalent pocket on Orf would likely require conformational changes.
If Redβ can bind and displace SSB on its own, as our data indicate, then what is the role of Orf in phage λ recombination? De Paepe et al. demonstrated that an Orf deletion causes a 10-fold decrease in Redβ dependent recombination with resident prophage during a phage λ infection. The authors suggested that in the absence of Orf, Redβ may be unable to access DNA coated by SSB (63). Orf is not present in the recombination assay that we used, and our data clearly show that a productive interaction between Redβ and SSB can occur in the absence of Orf. However, the potential effect of Orf on this recombination system has not yet been tested, and it is conceivable that Orf could bind to SSB and somehow modify it to allow for easier displacement by Redβ.
In summary, our results suggest a dual role for the CTD, first in binding to λ Exo to facilitate loading of Redβ directly onto the 3′-overhang of the first DNA molecule (Figure 1A), and second in binding to SSB to gain access to the ssDNA exposed on the second DNA molecule, likely at the lagging strand of a replication fork (Figure 8). While direct demonstration of these processes will require further biochemical studies, our mutational data suggest that both of these interactions are required for dsDNA recombination, while only the latter activity, binding to SSB, is required for ssDNA recombination. The crystal structure of the Redβ CTD and its complex with λ Exo now provide a framework for probing these activities in more detail.
Figure 8.

A revised model for phage λ Red recombination that includes an interaction with SSB. (1) Redβ (green) is loaded onto the 3′ overhang generated by λ Exo (dsDNA recombination), or binds directly to an electroporated oligonucleotide (ssDNA recombination). (2) The Redβ–ssDNA complex is targeted to the lagging strand of the replication fork through its interaction with SSB (blue). (3) The ssDNA is annealed to the target site as SSB is displaced, until the reaction is complete (4).
DATA AVAILABILITY
Coordinates and Structure Factors have been deposited in the Protein Data Bank under accession number 6M9K.
Supplementary Material
ACKNOWLEDGEMENTS
Fluorescence anisotropy experiments were performed at the Biophysical Interaction and Characterization Facility at the Ohio State University. X-ray diffraction data were collected at the Ohio State Campus Chemical Instrumentation Center. The authors thank Dr Lynn Thomason and Dr Donald Court for generously providing the anti-Redαβ antibody used in the Western blots, which was originally provided by Dr Kenan Murphy. The authors thank Dr Kotaro Nakanishi and Dr Ekram W. Abd El-Wahab for generously providing the plasmid for overexpression of N-terminally 6His-tagged SSB.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (NSF) [MCB-1616105 to C.E.B., DGE1745303 to G.T.F.]; National Institutes of Health [T32GM118291 to B.J.C, GM122459 to D.P.]; United States Department of Energy [DE-FG02-02ER63445 to G.T.F]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation, the National Institutes of Health, or the Department of Energy. Funding for open access charge: NSF [MCB-1616105].
Conflict of interest statement. None declared.
REFERENCES
- 1. Stahl M.M., Thomason L., Poteete A.R., Tarkowski T., Kuzminov A., Stahl F.W.. Annealing vs. invasion in phage λ recombination. Genetics. 1997; 147:961–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Little J.W. An exonuclease induced by bacteriophage λ II. Nature of the enzymatic reaction. J. Biol. Chem. 1967; 242:679–686. [PubMed] [Google Scholar]
- 3. Carter D.M., Radding C.M.. The role of exonuclease and β protein of phage λ in genetic recombination. II. Substrate specificity and the mode of action of λ exonuclease. J. Biol. Chem. 1971; 246:2502–2512. [PubMed] [Google Scholar]
- 4. Kmiec E., Holloman W.K.. β protein of bacteriophage λ promotes renaturation of DNA. J. Biol. Chem. 1981; 256:12636–12639. [PubMed] [Google Scholar]
- 5. Muniyappa K., Radding C.M.. The homologous recombination system of phage λ. Pairing activities of β protein. J. Biol. Chem. 1986; 261:7472–7478. [PubMed] [Google Scholar]
- 6. Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage λ. Microbiol. Mol. Biol. Rev. 1999; 63:751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ellis H.M., Yu D., DiTizio T., Court D.L.. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:6742–6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wilkinson M., Troman L., Wan Nur Ismah W.A., Chaban Y., Avison M.B., Dillingham M.S., Wigley D.B.. Structural basis for the inhibition of RecBCD by Gam and its synergistic antibacterial effect with quinolones. Elife. 2016; 5:e22963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou C., Pourmal S., Pavletich N.P.. Dna2 nuclease-helicase structure, mechanism and regulation by Rpa. Elife. 2015; 4:e09832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kagawa W., Kurumizaka H., Ishitani R., Fukai S., Nureki O., Shibata T., Yokoyama S.. Crystal structure of the homologous-pairing domain from the human Rad52 recombinase in the undecameric form. Mol. Cell. 2002; 10:359–371. [DOI] [PubMed] [Google Scholar]
- 11. Singleton M.R., Wentzell L.M., Liu Y., West S.C., Wigley D.B.. Structure of the single-strand annealing domain of human RAD52 protein. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:13492–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Erler A., Wegmann S., Ellie-Caille C., Bradshaw C.R., Maresca M., Seidel R., Habermann B., Muller D.J., Stewart A.F.. Conformational adaptability of Redβ during DNA annealing and implications for its structural relationship with Rad52. J. Mol. Biol. 2009; 391:586–598. [DOI] [PubMed] [Google Scholar]
- 13. Lopes A., Amarir-Bouhram J., Faure G., Petit M-A, Guerois R.. Detection of novel recombinases in bacteriophage genomes unveils Rad52, Rad51, and Gp2.5 remote homologs. Nucleic Acids Res. 2010; 38:3952–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsubara K., Malay A.D., Curtis F.A., Sharples G.J., Heddle J.G.. Structural and functional characterization of the Redβ recombinase from bacteriophage λ. PLoS One. 2013; 8:e78869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Copeland N.G., Jenkins N.A., Court D.L.. Recombineering: A powerful new tool for mouse functional genomics. Nat. Rev. Genet. 2001; 2:769–779. [DOI] [PubMed] [Google Scholar]
- 16. Muyrers J.P., Zhang Y., Stewart A.F.. Techniques: Recombinogenic engineering- new options for cloning and manipulating DNA. Trends Biochem. Sci. 2001; 26:325–331. [DOI] [PubMed] [Google Scholar]
- 17. Wang H.H., Isaacs F.J., Carr P.A., Sun Z.Z., Xu G., Forest C.R., Church G.M.. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009; 460:894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Radding C.M., Rosenweig J., Richards F., Cassuto E.. Separation and characterization of exonuclease, β protein, and a complex of both. J. Biol. Chem. 1971; 246:2510–2512. [Google Scholar]
- 19. Muyrers J.P.P., Zhang Y., Buchholz F., Stewart A.F.. RecE/RecT and Redα/Redβ initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev. 2000; 14:1971–1982. [PMC free article] [PubMed] [Google Scholar]
- 20. Anderson D.G., Kowalczykowski S.C.. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a χ-regulated manner. Cell. 1997; 90:77–86. [DOI] [PubMed] [Google Scholar]
- 21. Churchill J.J., Anderson D.G., Kowalczykowski S.C.. The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of χ, resulting in constitutive recombination activation. Genes Dev. 1999; 13:901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subramaniam S., Erler A., Fu J., Kranz A., Tang J., Gopalswamy M., Ramakrishnan S., Keller A., Grundmeier G., Müller D. et al. . DNA annealing by Redβ is insufficient for homologous recombination and the additional requirements involve intra- and inter-molecular interactions. Sci. Rep. 2016; 6:34525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tolun G. More Than the Sum of Its Parts: Physical and Mechanistic Coupling in the Phage λ Red Recombinase. 2007; Ph.D. thesisFL: University of Miami. [Google Scholar]
- 24. Bohorquez L.C. Defining Protein-Protein Interactions between Redα and Redβ from λ Bacteriophage. 2013; MSc ThesisGermany: Technische Universität Dresden. [Google Scholar]
- 25. Mythili E., Kumar K.A., Muniyappa K.. Characterization of the DNA-binding domain of β protein, a component of phage λ Red-pathway, by UV catalyzed cross-linking. Gene. 1996; 182:81–87. [DOI] [PubMed] [Google Scholar]
- 26. Ander M., Subramaniam S., Fahmy K., Stewart A.F., Schäffer E.. A single-strand annealing protein clamps DNA to detect and secure homology. PLoS Biol. 2015; 13:e1002213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Passy S.I., Yu X., Li Z., Radding C.M., Egelman E.H.. Rings and filaments of β protein from bacteriophage λ suggest a superfamily of recombination proteins. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:4279–4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu Z., Xing X., Wisler J.W., Dalton J.T., Bell C.E.. Domain structure and DNA binding regions of β protein from bacteriophage λ. J. Biol. Chem. 2006; 281:25205–25214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith C.E., Bell C.E.. Domain structure of the Redβ single-strand annealing protein: the C-terminal domain is required for fine-tuning DNA-binding properties, interaction with the exonuclease partner, and recombination in vivo. J. Mol. Biol. 2016; 428:561–578. [DOI] [PubMed] [Google Scholar]
- 30. Maxwell K.L., Reed P., Zhang R.G., Beasley S., Walmsley A.R., Curtis F.A., Joachimiak A., Edwards A.M., Sharples G.J.. Functional similarities between phage λ Orf and Escherichia coli RecFOR in initiation of genetic exchange. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:11260–11265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selleck W., Tan S.. Recombinant protein complex expression in E. coli. Curr. Protoc. Protein Sci. 2008; 52:5.21.1–5.21.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M.. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 2006; 62:859–866. [DOI] [PubMed] [Google Scholar]
- 33. Vagin A., Teplyakov A.. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 2001; 30:1022–1025. [Google Scholar]
- 34. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al. . Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kovall R., Matthews B.W.. Toroidal structure of λ-exonuclease. Science. 1997; 277:1824–1827. [DOI] [PubMed] [Google Scholar]
- 36. Murshudov G.N., Vagin A.A., Dodson E.J.. Refinement of macromolecular structures by the maximum-likelihood method. Acta Cryst. D Biol. Crystallogr. 1997; D53:240–255. [DOI] [PubMed] [Google Scholar]
- 37. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; D66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. The PyMOL Molecular Graphics System, Version 2.0. Schrödinger, LLC. [Google Scholar]
- 39. Zhang J., McCabe K.A., Bell C.E.. Crystal structures of λ exonuclease in complex with DNA suggest an electrostatic ratchet mechanism for processivity. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Korada S.K., Johns T.D., Smith C.E., Jones N.D., McCabe K.A., Bell C.E.. Crystal structures of Escherichia coli exonuclease I in complex with single-stranded DNA provide insights into the mechanism of processive digestion. Nucleic Acids Res. 2013; 41:5887–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fu J., Bian X., Hu S., Wang H., Huang F., Seibert P.M., Plaza A., Xia. L., Müller R., Stewart A.F. et al. . Full-length RecE enhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 2012; 30:440–446. [DOI] [PubMed] [Google Scholar]
- 42. Pan X., Smith C.E., Zhang J., McCabe K.A., Fu J., Bell C.E.. A structure-activity analysis for probing the mechanism of processive double-stranded DNA digestion by λ exonuclease trimers. Biochemistry. 2015; 54:6139–6148. [DOI] [PubMed] [Google Scholar]
- 43. Wang J., Sarov M., Rientjes J., Fu J., Hollak H., Kranz H., Xie W., Stewart A.F., Zhang Y.. An improved recombineering approach by adding RecA to λ Red recombination. Mol. Biotechnol. 2006; 32:43–53. [DOI] [PubMed] [Google Scholar]
- 44. Reid S.L., Parry D., Liu H.-H., Connolly B.A.. Binding and recognition of GATATC target sequences by the EcoRV restriction endonuclease: a study using fluorescent oligonucleotides and fluorescence polarization. Biochemistry. 2001; 40:2484–2494. [DOI] [PubMed] [Google Scholar]
- 45. Yu D., Ellis H.M., Lee E.C., Jenkins N.A., Copeland N.G., Court D.L.. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang Y., Muyrers J.P., Rientjes J., Stewart A.F.. Phage annealing proteins promote oligonucleotide-directed mutagenesis in Escherichia coli and mouse ES cells. BMC Mol. Biol. 2003; 4:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mosberg J.A., Gregg C.J., Lajoie M.J., Wang H.H., Church G.M.. Improving lambda red genome engineering in Escherichia coli via rational removal of endogenous nucleases. PLoS One. 2012; 7:344638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yu D., Sawitzke J.A., Ellis H., Court D.L.. Recombineering with overlapping single-stranded DNA oligonucleotides: testing a recombination intermediate. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:7207–7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holm L., Laakso L.M.. Dali server update. Nucleic Acids Res. 2016; 44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sawitzke J.A., Stahl F.W.. The phage λ orf gene encodes a trans-activating factor that suppresses Escherichia coli recO, recR, and recF mutations for recombination of λ but not of E. coli. J. Bacteriol. 1994; 176:6730–6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Curtis F.A., Malay A.D., Trotter A.J., Wilson L.A., Barradell-Black M.M., Bowers L.Y., Reed P., Hillyar C.R., Yeo R.P., Sanderson J.M. et al. . Phage Orf family recombinases: conservation of activities and involvement of the central channel in DNA binding. PLoS One. 2014; 9:e102454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Curtis F.A., Reed P., Wilson L.A., Bowers L.Y., Yeo R.P., Sanderson J.M., Walmsley A.R., Sharples G.J.. The C-terminus of phage λ Orf recombinase is involved in DNA binding. J. Mol. Recognit. 2011; 24:333–340. [DOI] [PubMed] [Google Scholar]
- 53. Sawitzke J.A., Stahl F.W.. Roles for λ Orf and Escherichia coli RecO, RecR and RecF in λ recombination. Genetics. 1997; 147:357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lu D., Keck J.L.. Structural basis of Escherichia coli single-stranded DNA-binding protein stimulation of exonuclease I. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:9169–9174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Petzold C., Marceau A.H., Miller K.H., Marqusee S., Keck J.L.. Interaction with single-stranded DNA-binding protein stimulates Escherichia coli ribonuclease HI enzymatic activity. J. Biol. Chem. 2015; 290:14626–14636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Marceau A.H., Bahng S., Massoni S.C., George N.P., Sandler S.J., Marians K.J., Keck J.L.. Structure of the SSB-DNA polymerase III interface and its role in DNA replication. EMBO J. 2011; 30:4236–4247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Shereda R.D., Reiter N.J., Butcher S.E., Keck J.L.. Identification of the SSB binding site on E. coli RecQ reveals a conserved surface for binding SSB’s C terminus. J. Mol. Biol. 2009; 386:612–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cadman C.J., McGlynn P.. PriA helicase and SSB interact physically and functionally. Nucleic. Acids. Res. 2004; 32:6378–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Datta S., Costantino N., Zhou X., Court D.L.. Identification and analysis of recombineering functions from gram-negative and gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:1626–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kovall R.A., Matthews B.W.. Structural, functional, and evolutionary relationships between λ-exonuclease and the type II restriction endonucleases. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:7893–7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shereda R.D., Bernstein D.A., Keck J.L.. A central role for SSB in Escherichia coli RecQ DNA helicase function. J. Biol. Chem. 2007; 26:19247–19258. [DOI] [PubMed] [Google Scholar]
- 62. Glover B.P., McHenry C.S.. The χψ subunits of DNA polymerase III holoenzyme bind to single-stranded DNA-binding protein (SSB) and facilitate replication of an SSB-coated template. J. Biol. Chem. 1998; 36:23476–23484. [DOI] [PubMed] [Google Scholar]
- 63. De Paepe M., Hutinet G., Son O., Amarir-Bouhram J., Schbath S., Petit M.A.. Temperate phages acquire DNA from defective prophages by relaxed homologous recombination: the role of Rad52-like recombinases. PLos Genet. 2014; 10:e1004181. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and Structure Factors have been deposited in the Protein Data Bank under accession number 6M9K.