


Abstract
Antimalarial resistance is a major obstacle in the eradication of the human malaria parasite, Plasmodium falciparum. Genome amplifications, a type of DNA copy number variation (CNV), facilitate overexpression of drug targets and contribute to parasite survival. Long monomeric A/T tracks are found at the breakpoints of many Plasmodium resistance-conferring CNVs. We hypothesize that other proximal sequence features, such as DNA hairpins, act with A/T tracks to trigger CNV formation. By adapting a sequence analysis pipeline to investigate previously reported CNVs, we identified breakpoints in 35 parasite clones with near single base-pair resolution. Using parental genome sequence, we predicted the formation of stable hairpins within close proximity to all future breakpoint locations. Especially stable hairpins were predicted to form near five shared breakpoints, establishing that the initiating event could have occurred at these sites. Further in-depth analyses defined characteristics of these ‘trigger sites’ across the genome and detected signatures of error-prone repair pathways at the breakpoints. We propose that these two genomic signals form the initial lesion (hairpins) and facilitate microhomology-mediated repair (A/T tracks) that lead to CNV formation across this highly repetitive genome. Targeting these repair pathways in P. falciparum may be used to block adaptation to antimalarial drugs.
INTRODUCTION
Major efforts have succeeded in eradicating malaria in North America and Europe, but have largely failed in Southeast Asia and Africa (1). Some of the remaining challenges include a lack of accessible treatments and the widespread development of drug resistance. Plasmodium falciparum, the protozoan parasite that causes the most severe form of malaria and the majority of malaria deaths, has developed resistance to all drug interventions thus far (2). Single nucleotide polymorphisms (SNPs) are the most commonly studied genetic contribution to antimalarial drug resistance. However, chromosomal size polymorphisms, including copy number variations (CNVs) that encompass the genes of antimalarial targets or drug transporters, also play a key role in parasite survival (3).
CNVs often carry strong fitness costs due to increased cellular burden for DNA replication and alterations of metabolic flux due to differing levels of enzyme expression (4). However, it has been proposed that in many organisms, including P. falciparum, the creation of redundant gene copies facilitates the accumulation of SNPs (5–8). Studies observing both types of mutations in Plasmodium provide evidence that CNVs appear to eventually be lost in favor of SNPs (9–11).
Two CNVs associated with clinical antimalarial resistance encompass the genes encoding the multiple drug resistance protein 1 (pfmdr1) and GTP-cyclohydrolase 1 (gch1) (12–17). Additionally, a number of resistance-associated CNVs across many chromosomes were detected in the P. falciparum genome following laboratory selections with novel antimalarials (8,9,13,16,18–25). CNVs have also been detected in clinical Plasmodium vivax isolates (18,26–29), providing evidence that this form of adaptation is not confined to P. falciparum.
Mechanisms leading to CNVs in Plasmodium are currently unknown. Due to a lack of significant sequence homology surrounding the CNV breakpoints, homologous recombination is not likely to be involved in the process. The most compelling evidence of a shared mechanism is the presence of long monomeric A/T tracks at CNV boundaries (8,17,27,30,31). In other organisms, there is precedence for polymerase pausing and DNA double-stranded breaks (DSBs) at long mononucleotide repeats or AT/TA dinucleotide repeats (32–35). However, in-depth characterization of multiple independently generated CNVs on chromosome 6 indicates an additional signal present that triggers amplification (8). Specifically, two distinct CNVs were found to share a common boundary on one end (C and F clones, Figure 1A), an event that is highly unlikely to occur by chance. The A/T track at this shared breakpoint is not significantly longer (37 bp long compared to a mean of 33 bp for all CNVs included in our analysis) and thus other factors must be driving this repeat occurrence. Abnormal DNA structures, including hairpins and stem-loops, have also been implicated in replication fork stalling and DSBs in yeast and humans (36–41). Therefore, we investigated whether sequences proximal to CNV breakpoints across the highly A/T-rich P. falciparum genome are enriched in these DNA structures.
Figure 1.

Highly stable DNA hairpins are found near pre-CNV boundaries. Resistant clones from various selections exhibit a range of CNV sizes but all have long A/T track breakpoints on their upstream and downstream ends (see Supplementary Table S3). Shared breakpoints are indicated with arrows and depicted in red (boxes and plots); unique breakpoints are shown for comparison and depicted in gray (boxes and plots). Although 2kb was analyzed, for simplicity, the insets show the ΔG of folding for each 50 bp window across 1 kb of sequence surrounding the A/T track breakpoint (vertical red/gray bar at 500 bp). The dotted line demarks the threshold for stable hairpin formation (ΔG of −5.8 kCal/mol, see ‘Materials and Methods’ section for how this was defined). (A) Each CNV from DSM1 resistant parasites (C, D, E and F) encompasses the gene for the target dihydroorotate dehydrogenase (DHODH, gray bar with star). The shared 3′ breakpoint from clones C and F is indicated (arrows). (B) Each CNV from Cladosporin resistant parasites (A, B and C) encompasses the gene for the target lysyl-tRNA synthetase (KRS1, gray bar with star). The shared 3′ breakpoint from CladoA and CladoC is indicated (arrows). (C) Each CNV from the MMV019662 and MMV028038 resistant parasites (1F4, 2G6, 3B6 and 2B6) encompasses the gene target Pf3D7_0107500, a member of the resistance-nodulation-division transporter family (gray bar with star). The shared breakpoints are indicated (arrows).
Here, we present evidence that DNA hairpin formation is likely an initiating event in the generation of CNVs in P. falciparum. First, we adapted a CNV-calling pipeline to achieve near single base pair resolution to study laboratory acquired CNVs in 35 total resistant parasite clones selected with eight different antimalarials (19 parasite clones with distinct CNVs). Sequence analysis of sensitive parent genomes (before CNV generation, termed pre-CNV) confirmed that long A/T tracks are found at nearly all breakpoint locations and identified four additional shared breakpoints (five in total, Fig. 1B and C). Computational predictions revealed stable hairpin structures in close proximity to all pre-CNV breakpoint locations. Especially stable hairpins sat close to the shared breakpoints, providing further support for a role of hairpin structures in alterations of copy number. We defined the relationship between these genomic features on a genome-wide scale and this association provided a map of CNV-capable sites available to the parasite during adaptation to countless antimalarials. These ‘trigger sites’ are found broadly throughout the parasite genome and would facilitate adaptation to most selective forces. In-depth analysis of breakpoints in resistant clones (termed post-CNV) suggests the action of two repair pathways that utilize the A/T tracks as short stretches of homology. These findings contribute to a growing model of the mechanisms that lead to enhanced generation of CNVs across highly repetitive genomes.
MATERIALS AND METHODS
Collecting genome and breakpoint sequences
We analyzed whole genome sequencing data to identify CNVs from in vitro haploid erythrocytic P. falciparum parasites that were selected with a number of different antimalarials (Table 1, see details on parent and resistant clones, antimalarial target, chromosome, CNV sizes and accession numbers in Supplementary Table S1 (8,16,42,43)). For clarity of procedures, we present a flow chart of our overall analysis methods (Supplementary Figure S1). Briefly, low quality bases and adapter sequences from Illumina-based whole genome sequencing of both the parent and resistant clones (Supplementary Table S1) were removed using BBTools (version 35.82, https://sourceforge.net/projects/bbmap/). Uncorrectable errors were assigned low quality scores and cleaned reads were evaluated using FastQC to check per base read qualities, sequence duplication levels, over-represented sequences, and read length distributions (Version 0.11.7, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (Supplementary Figure S1A).
Table 1.
Characteristics of Plasmodium falciparum CNVs used in this study
| Clone | Data source | CNV Chr. | CNV start (bp w/ 95% confidence interval) | CNV end (bp w/ 95% confidence interval) | # of supporting genomes |
|---|---|---|---|---|---|
| DSM1C | Guler et al., (8) | 6 | 79 067 ± 0 | 152 482 ± 0 | 1 |
| DSM1D | 6 | 64 578 ± 0 | 158 152 ± 0 | 1 | |
| DSM1E | 6 | 118 425a | 153 231a | 1 | |
| DSM1F | 6 | 113 523a | 152 482a | 1 | |
| HFGRII | Herman et al., (42) | 12 | 587 623 ± 61 | 612 922 ± 3 | 1 |
| HFGRIII | 12 | 589 189 ± 5 | 621 909 ± 1 | 1 | |
| CladoA | Manary et al., (60) | 13 | 2 000 221 ± 11 | 2 058 842 ± 1 | 1 |
| CladoB | 13 | 2 004 915 ± 2 | 2 055 159 ± 1 | 1 | |
| CladoC | 13 | 2 000 213 ± 4 | 2 022 803 ± 0 | 1 | |
| PQA11 | Cowell et al., (43) | 10 | 290 655 ± 0 | 308 771 ± 2 | 1 |
| F7 | 1 | 264 317 ± 0 | 359 349 ± 0 | 6 | |
| 3B6 | 1 | 264 317 ± 1 | 362 912 ± 0 | 1 | |
| 1F4 | 1 | 321 511 ± 5 | 373 058 ± 9 | 2 | |
| 2G9 | 1 | 321 511 ± 2 | 362 912 ± 10 | 2 | |
| 1E3 | 1 | 321 511 ± 2 | 362 913 ± 9 | 2 | |
| 33XC3 | 12 | 1 733 591 ± 3 | 1 768 713 ± 3 | 1 | |
| 3C3 | 12 | 1 718 154 ± 3 | 1 769 038 ± 3 | 6 | |
| R2B2 | 3 | 782 909 ± 45 | 845 526 ± 0 | 4 | |
| 1B2 ch10 | 10 | 285 731 ± 27 | 315 681 ± 3 | 1 | |
| 1B2 ch12 | 12 | 1 549 855 ± 5 | 1 567 426 ± 1 | 1 |
aWhole genome sequencing is not available for these two clones. Analysis was performed using locations identified by PCR sequencing.
For whole genome sequencing alignments, BWA-MEM was utilized to align reads with default settings to the 3d7 reference genome (PlasmoDB release 32, Supplementary Figure S1B) (44). Alignment quality of the resulting bam files were evaluated for mean read depth, mean mapping quality, and quartiles of paired read insert-size using Qualimap 2 (Supplementary Table S2) (45). Breakpoints of the CNVs, or locations where DNA recombination occurred to generate genome amplifications, were identified by adapting the Speedseq pipeline (46). We used the CNVnator algorithm for automated read-depth analysis and copy number estimation, the LUMPY algorithm for split-read and discordant read pair analysis, and a Bayesian analytical method to genotype structural variants and call precise breakpoints (47,48) (see more details below). CNVnator utilizes a read-depth mean-shift approach to CNV detection and applies additional corrections including those for GC-content bias of Illumina sequencing; for this analysis, we used default settings to calculate read-depth in 100 bp bins. This was recommended in the CNVnator manuscript for 30× and 100× coverage, which is the range observed in our analysis. The Speedseq pipeline extracts discordant read-pairs and split-reads that can be visualized to determine CNV orientation and type (i.e. inversion or translocation). LUMPY takes the discordant read-pairs and split-reads and calculates probability distributions of breakpoints spanning a putative DNA structural variant. As discordant read-pair and split-read analysis give greater breakpoint resolution than read depth, the resulting LUMPY breakpoint locations were evaluated for sample quality scores (>100), quantity of supporting reads (>3) and significant overlap with amplification boundaries from CNVnator and the published data (Supplementary Table S3). CNV calls were then manually verified and visualized using IGV 2.4.10 to determine CNV type and observe mutational signatures near CNV breakpoints (49). These breakpoint locations were used to obtain consensus sequence (2 kb in total, 1 kb upstream (5′end) and downstream (3′end)) from the parent line for secondary structure predictions (pre-CNV, see below). For clones in which whole genome sequencing was not available (DSM1E and F), previously published sequence from PCR amplification across the breakpoint was used to pinpoint precise breakpoint locations (8).
Calculating the likelihood of DNA hairpin formation
The probability of hairpin structure formation across the desired regions was predicted essentially as previously described (50,51). In brief, 50 bp windows were selected by shifting by 1 bp across a 2 kb stretch of sequence surrounding the pre-CNV breakpoint position in the parent genome. The 50 bp windows were chosen to ensure hairpin formation was possible within the Okazaki initiation zone during replication. The size of the Okazaki initiation zone is not known in Plasmodium but it is expected to be in the same range as other eukaryotes (300–1000 bp (52)). Next, the Gibbs free energy (ΔG), which predicts the stability of the sequence folding on itself, was determined for each window using Vienna 2.1.9 folding prediction software with Mathews 2004 DNA folding parameters and G-quadruplexes, GU pairing, and lonely base pairs were disallowed (53). Lonely base pairs are helices in a hairpin or stem-loop that are composed of only 1 bp and do not stack on other base pairs. These structures are not energetically favorable and cannot form and are therefore excluded from analyses. During this analysis, each 50 bp window was counted as a separate possible hairpin. Initially this analysis was confined to sequences from the parent genome prior to CNV generation (the pre-CNV breakpoint position). Predictions were subsequently performed on sequences from post-CNV breakpoint locations from resistant clones.
Defining stable hairpins
Due to a non-normal distribution of predicted hairpin ΔG-values, the ΔG cut-off of stable hairpins was determined using a randomization method: sequence from each chromosome was randomly shuffled using the EMBOSS shuffleseq function to maintain overall A/T composition and hairpins were again predicted (54). In this analysis, 50 kb of sequence on either chromosome end was trimmed to avoid highly repetitive telomeric sequences. The value of the resulting top 3% of shuffled hairpins was used as the stability cut-off for all analyses (−5.8 kCal/mol); sequences with values below this cut-off indicated a high probability of a ‘stable’ structure forming. This value is consistent with that utilized in previous P. falciparum investigations (51). Furthermore, this value is similar to the top 5% of non-shuffled hairpins (ΔG of −5.5 kcal/mol in our analysis), a threshold utilized in secondary structure studies of other organisms (55).
Determining the mean ΔG profile
The mean ΔG of folding in close proximity to CNV breakpoints (shared or all) was determined by setting the end of the A/T track breakpoint to distance zero and calculating the mean ΔG for each 50 bp window as the sequence is shifted by 1 bp. The 95% confidence interval of each position was calculated and then plotted using Graphpad PRISM 7 (www.graphpad.com). For comparison with sequences not associated with CNVs, this process was repeated with 36 randomly chosen A/T tracks between 20 and 40 bp in length from intergenic regions across the genome. This length was chosen for random analysis because these A/T tracks are similar to those associated with CNV breakpoints (mean of 33 bp, Supplementary Table S4 and see ‘Evaluating A/T track lengths across the genome’ section). Each random A/T track position was chosen using a random number generator to pick a line number from the bed file of all A/T tracks of this size across the genome (excluding telomeres). Due to unequal sample sizes and a non-normal distribution, the level of significance in differences was calculated using the Wilcoxon–Mann–Whitney test.
Evaluating A/T track lengths across the genome
A/T tracks were identified with the Phobos Repeat Finder (Version 3.3.11, http://www.rub.de/ecoevo/cm/cm_phobos.htm), which mapped the locations and lengths of long monomeric A/T tracks >9 bp across the 3d7 genome (Supplementary Figure S1C). The level of significance in differences between the two datasets was again calculated using Wilcoxon–Mann–Whitney test. This length of track was chosen based on a previous study that showed that those above 9 bp were over-represented on P. falciparum chromosome 2 (56). To determine if A/T tracks were observed solely due to the high A/T content of P. falciparum (80.6%), we calculated the probability of observing different A/T tracks lengths based purely on nucleotide composition. Frequencies of monomeric A/T tracks of length N were calculated as follows:
The observed frequency of A and T tracks of length N were obtained using the following equation:
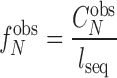 |
Where 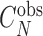 is the observed number of monomeric tracks of length N and
is the observed number of monomeric tracks of length N and 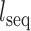 is the length of the chromosome sequence.
is the length of the chromosome sequence.
For each A or T track observed with length N, the corresponding expected frequency of mononucleotide A and T tracks was obtained from the following equation:
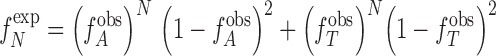 |
where 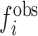 is the observed frequency of any base pair which corresponds to the overall percent base composition.
is the observed frequency of any base pair which corresponds to the overall percent base composition.
Maximum expected length for each chromosome was found using the following formula:
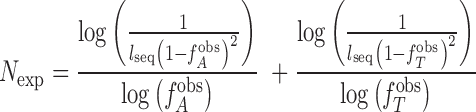 |
Investigating genome scale A/T track-hairpin relationships
In order to assess the hairpin and A/T track relationship on a larger scale, hairpins across the entire genome were predicted as described above. Where indicated, analyses were confined to tracks >20 bp as this reflects the lengths of A/T tracks found at observed CNV breakpoints (Supplementary Table S4). The relationship between hairpins and long A/T tracks was then determined in genic and intergenic regions separately. This was accomplished by taking gene annotations from the 3d7 reference genome and extracting A/T tracks from regions within or outside of gene annotations utilizing the ‘intersect’ and ‘subtract’ bedtools functions, respectively (Supplementary Figure S1C). Distance between genic or intergenic A/T tracks to the nearest stable hairpin (either upstream or downstream) was then calculated using the ‘closest’ function in bedtools (57). For this analysis, the positions of the local minima of hairpins had to be identified. First, we extracted all hairpins below our significance threshold (−5.8 kCal/mol, see ‘Defining stable hairpins’ section). Then, for each set of windows with contiguous positions below this threshold, we identified the window with the most negative value and created a data subset with these minima. If there were multiple contiguous windows with the same value, all matching windows were extracted and used for analysis. The level of significance in differences was calculated using the Wilcoxon–Mann–Whitney test. Visualization of the frequency of lengths of the A/T tracks compared to the distance to stable hairpins was performed using ggplot2 in R version 3.2.4 (58,59). The Kolmogorov–Smirnov non-parametric test was used to compare the equality of intergenic and genic distributions to determine significant differences.
RESULTS
CNV breakpoint features are conserved in Plasmodium falciparum
We obtained sequence from P. falciparum clones that had been selected for resistance to novel antimalarials in vitro (8,16,43,60) (Supplementary Table S1). After read alignment and CNV calling using an adapted Speedseq pipeline with stringent quality controls (see ‘Materials and Methods’ section), we selected sequence from 35 parasite clones that displayed high confidence CNV breakpoints for further analysis (Table 1 and Supplementary Table S2). Due to improved resolution, breakpoint locations were identified primarily through discordant- and split-read analysis extracted by LUMPY. This analysis identified 19 distinct CNVs for a total of 33 CNV breakpoints: 5 were conserved between different CNVs in multiple parasite clones (termed ‘shared’ breakpoints) and 28 were unique to their respective CNV (Table 1). In total, these breakpoints had a median of 27 supporting split and discordant reads (range of 3–1025 reads, Supplementary Table S3). Read depth changes detected by CNVnator further confirmed these general breakpoint locations and the orientation of reads confirmed the tandem duplications at these sites (Supplementary Figure S2).
Confidence in this analysis was bolstered by overall read depth and quality scores determined for each sequenced genome. Read depth across each chromosome, excluding telomeric regions, was >40-fold (median of 87-fold); coverage across CNV breakpoints was similar with a median of 107-fold for 2 kb surrounding breakpoints and a median of 57-fold for 100 bp surrounding breakpoints (Supplementary Table S2). The mean mapping quality scores across the genome was 57 out of a maximum score of 60 (44).
To determine whether DNA hairpins were associated with CNV breakpoints in P. falciparum, we went to the locations of the shared breakpoints in the pre-CNV parent genome. Two kilobases of proximal sequence were used to predict the probability of secondary structure formation nearby; a ΔG of <−5.8 kCal/mol indicated a high probability of a ‘stable’ structure forming from this sequence window (see ‘Materials and Methods’ section). From this focused analysis, we invariably detected extremely stable hairpins (the top 0.2% most stable structures across the entire genome, mean ΔG of <−9.7 kCal/mol) within a few hundred base pairs of the shared breakpoint A/T tracks (Figure 1A–C, mean distance of 165 ± 58 bp, Table 2). Stable hairpin structures were predicted to form by inverted repeats and AT dinucleotides present in the analyzed sequence (Table 2).
Table 2.
Hairpin stability and distance relationships at CNV breakpoints
| Breakpoint | ΔG of closest hairpin | Track-hairpin distancea | Hairpin forming sequence |
|---|---|---|---|
| DSM1F/C_3 | −10.9 | 88 | Inverted repeat |
| CladoA/C_5 | −9.1 | 222 | Inverted repeat |
| F7/3B6_5 | −8.1 | 218 | AT dinucleotide |
| 1F4/1E3_5 | −10.2 | 104 | AT dinucleotide |
| 3B6/1E3_3 | −10.2 | 194 | AT dinucleotide |
| Mean of shared | −9.7 ± 1.0 | 165 ± 58 | NA |
| DSM1C_5 | −6.7 | 216 | Inverted repeat |
| DSM1D_5 | −9.7 | 49 | Inverted repeat |
| DSM1D_3 | −7.1 | 2 | Inverted repeat |
| DSM1E_5b | −6.3 | 234 | AT dinucleotide |
| DSM1E_5b | −5.8 | 424 | Inverted repeat |
| DSM1F_5b | −8.4 | 172 | AT dinucleotide |
| HFGRII_5 | −6.1 | 0 | Inverted repeat |
| HFGRII_3 | −7.3 | 2 | Inverted repeat |
| HFGRIII_5 | −7.1 | 21 | Inverted repeat |
| HFGRIII_3 | −6.7 | 137 | AT dinucleotide |
| CladoA_3 | −7.9 | 59 | AT dinucleotide |
| CladoB5 | −6.6 | 105 | Inverted repeat |
| CladoB3 | −6.1 | 14 | AT dinucleotide |
| CladoC3 | −8.3 | 92 | AT dinucleotide |
| PQA11_5 | −13.2 | 234 | AT dinucleotide |
| PQA11_3 | −8.7 | 212 | AT dinucleotide |
| 1F4_3 | −8.9 | 268 | AT dinucleotide |
| 2G9_5 | −13.1 | 104 | AT dinucleotide |
| 2G9_3 | −10.2 | 194 | AT dinucleotide |
| 33XC3_5c | −13.1 | 118 | AT dinucleotide |
| 33XC3_3c | −9 | 30 | Inverted repeat |
| 3C3_5 | −6.8 | 342 | Inverted repeat |
| 3C3_3 | −9 | 261 | Inverted repeat |
| R2B2_5 | −7.3 | 2 | Inverted repeat |
| R2B2_3 | −10.7 | 95 | AT dinucleotide |
| 1B2ch10_5 | −8.4 | 0 | AT dinucleotide |
| 1B2ch10_3 | −8.3 | 123 | AT dinucleotide |
| 1B2ch12_5c | −8 | 2 | AT dinucleotide |
| 1B2ch12_3c | −7.9 | 171 | AT dinucleotide |
| Mean of all | −8.6 ± 2.0 | 132.6 ± 106.3 | NA |
NA, not applicable. _5, upstream breakpoint. _3, downstream breakpoint.
aTrack-hairpin distance was calculated to the nearest stably predicted hairpin. Distances of 0: the A/T track breakpoint is participating in hairpin formation.
bSequences derived from PCR across breakpoints.
cUtilize A/T dinucleotides as the breakpoint rather than A/T tracks.
In all cases, multiple stable hairpins were detected in close proximity to the shared breakpoints (see Figure 1, where multiple peaks reach or fall below the dotted line); it was not clear which structure was contributing to CNV formation, the closest or the most stable hairpin. We therefore used this data to investigate whether there was a critical A/T track-hairpin distance; we determined the mean ΔG at each base pair traveling away from the A/T tracks for the five shared breakpoints. When we compared this profile with that from random A/T tracks across the genome that do not participate in CNV formation (see ‘Materials and Methods’ section for details about these sequences were chosen), we detected a ΔG minima for the shared breakpoints at a distance of ∼80 and ∼360 bp (Figure 2A, P < 0.05 for both). This analysis provided evidence that stable hairpins within very close proximity, <400 bp, to the breakpoint A/T track likely contributed to CNV formation.
Figure 2.

Mean-free energy profiles highlight a critical distance for stable hairpins. The mean ΔG of folding in close proximity to shared (A) and all (B) CNV breakpoints is plotted. This was done by setting the A/T track breakpoint at a distance of 0 bp and calculating the mean ΔG for each window of 50 bp as the sequence is shifted by 1 bp (A: purple line and B: blue line). As a comparator, the mean ΔG profile of 36 randomly chosen A/T tracks not associated with CNV formation (20–40 bp in length) was plotted (green line, see characteristics in ‘Materials and Methods’ section). Mean values with 95% confidence interval are shown. Shared breakpoints are DSM1C/F_3, CladoA/C_5, F7/3B6_5, 1F4/1E3_5 and 3B6/1E3_3 (see Table 2)
We extended our analysis to the remainder of the high-quality CNV breakpoints identified in the above analysis (Supplementary Table S1). Although less pronounced than with the shared breakpoints, the mean ΔG profile for all CNV breakpoints indicated that the most stable structure is within ∼400 bp (Figure 2B). Minima were identified at similar distances from the breakpoints and were significantly stronger than random A/T tracks (P < 0.05). In line with this result, stably predicted hairpins were found in very close proximity to all CNV breakpoints (mean hairpin distance of 133 ± 106 bp, mean ΔG of −8.6 kCal/mol). Overall, 42% of breakpoints had a highly stable structure within 100 bp of the A/T track breakpoint, 60% within 150 bp distance and all but one within 400 bp (Table 2). These proximal structures were frequently composed of inverted repeats or AT dinucleotide repeats (Table 2).
As has been noted before, the majority of CNV breakpoints occurred at very long A/T tracks (>20 bp, Supplementary Table S4). There were a few exceptions; AT dinucleotide repeats sat at both junctions for 33XC3 and 1B2 ch10 and an imperfect A/T track was found on the 3′ end of the 1F4 clone (88% pure T’s).
CNV breakpoint features are enriched in intergenic regions
We noted previously that CNV breakpoints are more often found in intergenic than genic regions (8). To explore this further, we expanded our analysis across these two regions of the P. falciparum genome. Specifically, we investigated (i) the quantity and length of A/T tracks, (ii) the propensity for DNA hairpin formation, as measured by ΔG of folding and (iii) the distance relationship between these two features. First, when compared to expected numbers, long A/T tracks >9 bp were highly enriched across the genome (Supplementary Figure S3, P < 0.01 for A/T tracks > 9 bp). When comparing genic to intergenic regions of the genome, we found about twice as many long A/T tracks in intergenic sequences than genic (42 026 in intergenic versus 19 408 in genic, Table 3, P < 0.001). A more striking difference was observed if the quantity of very long A/T tracks, >20 bp, were compared (∼4-fold increase: 9509 in intergenic regions and 2410 in genic, Table 3, P < 0.001). Second, we predicted a greater number of stable structures (ΔG <−5.8 kCal/mol) in intergenic compared to genic regions (37 439 intergenic and 23 442 genic, Table 3, P < 0.05) and an increase in the mean hairpin strength of these stable hairpins (−7.56 kCal/mol for intergenic compared to −7.23 kCal/mol for genic, P < 0.01). Finally, we found that the distance between A/T tracks and hairpins differed greatly between genic and intergenic regions. The mean A/T track-hairpin distance when considering long A/T tracks was 99 bp in intergenic regions and 277 bp in genic regions (Table 3, P < 10−13). This trend was conserved when considering very long A/T tracks (mean of 104 bp distance in intergenic and 163 bp in genic, P < 10−6).
Table 3.
Quantitation of A/T track frequency, hairpin frequency and distance relationships across the genome
| A/T tracks > 9 bp | A/T tracks > 20 bp | Stable hairpin minimaa | Mean distance (bp): A/T tracks > 9bp | Mean distance (bp): A/T tracks > 20 bp | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genic | Intergenic | Genic | Intergenic | Genic | Intergenic | Genic | Intergenic | Genic | Intergenic | |
| Chr. 1 | 434 | 1223 | 70 | 250 | 566 | 958 | 269 | 103.0 | 145.3 | 101.3 |
| Chr. 2 | 742 | 1629 | 93 | 371 | 915 | 1484 | 254.1 | 99.6 | 160.6 | 115.4 |
| Chr. 3 | 962 | 1824 | 126 | 401 | 1117 | 1568 | 249.5 | 98.8 | 143.8 | 105.9 |
| Chr. 4 | 1040 | 2005 | 106 | 460 | 1270 | 1734 | 318.8 | 107.3 | 195.5 | 110.9 |
| Chr. 5 | 1085 | 2404 | 131 | 576 | 1307 | 2085 | 287.1 | 92.5 | 143.0 | 101.9 |
| Chr. 6 | 1155 | 2365 | 143 | 526 | 1447 | 2449 | 299.8 | 92.1 | 193.5 | 95.9 |
| Chr. 7 | 1292 | 2350 | 153 | 532 | 1558 | 2037 | 302.6 | 102.7 | 190.9 | 105.8 |
| Chr. 8 | 1274 | 2722 | 146 | 631 | 1562 | 2466 | 274.6 | 99.4 | 173.6 | 105.8 |
| Chr. 9 | 1286 | 2977 | 167 | 675 | 1543 | 2604 | 234.3 | 103.3 | 120.3 | 100.3 |
| Chr. 10 | 1336 | 3160 | 175 | 710 | 1657 | 3009 | 266.0 | 99.4 | 152.2 | 101.7 |
| Chr. 11 | 1746 | 3821 | 219 | 858 | 2060 | 3477 | 273.9 | 95.3 | 170.1 | 100.5 |
| Chr. 12 | 1935 | 4169 | 239 | 989 | 2273 | 3655 | 272.1 | 97.7 | 151.1 | 98.2 |
| Chr. 13 | 2336 | 5321 | 283 | 1197 | 2870 | 4690 | 280.7 | 102.9 | 177.6 | 102.7 |
| Chr. 14 | 2785 | 6056 | 359 | 1333 | 3297 | 5223 | 282.6 | 100.9 | 175.7 | 106.2 |
| Total | 19 408 | 42 026 | 2410 | 9509 | 23 442 | 37 439 | 277.3 | 99.2 | 163.8 | 103.8 |
aStable hairpin minima were determined by identifying the most stably predicted structure, most negative ΔG. If contiguous windows had the same minimum, the windows were combined into the same structure feature for calculations. Distances between A/T tracks and stable hairpin minimum were calculated from the edge of A/T tracks to the edge of stable hairpin minima.
By visualizing these distributions on a whole genome scale, the disparities between the two genomic regions and the close A/T track-hairpin association in intergenic regions are emphasized (Figure 3A and B, Kolmogorov–Smirnov test, P < 10−15). Due to the characteristics of these features that are associated with observed CNV breakpoints, we propose that there is an optimal range for A/T track lengths (∼20–40 bp) and track-hairpin distances (<400 bp) (yellow highlight in Figure 3A and B). We defined genome positions with these characteristics as CNV ‘trigger sites’: those locations that are competent to generate CNVs. Using these parameters, there are 9130 intergenic and 2222 genic trigger sites across the P. falciparum genome which corresponds to 19.0% of intergenic and 9.6% of genic A/T tracks.
Figure 3.

Stable hairpins near long A/T tracks are overrepresented in the Plasmodium falciparum genome. The distribution of absolute distances between long A/T tracks (>9 bp) and the nearest stable hairpin (<−5.8 kCal/mol) for genic sequences (A) and intergenic sequences (B) in the 3d7 genome. The yellow highlight indicates the critical ranges noted in our analysis: A/T tracks between 20 and 40 bp in length (the range detected in our analysis of CNV breakpoints, see Supplementary Table S4) and distance of <400 bp (the distance limit for the most highly stable structures identified in mean profiles, Figure 2). All plots exclude absolute distance values >4000bp (few data points fell beyond this distance).
Identifying DNA repair pathways utilized in CNV formation
The above analysis was performed using parent sequence prior to CNV formation (pre-CNV, Figure 4A). In order to pinpoint which repair pathways may be acting in this process, we also studied the sequence from resistant clones after CNV formation (post-CNV, Figure 4B). This was accomplished by comparing pre- and post-CNV sequences from two sources, when available: PCR sequence of the A/T track breakpoint (for two DSM1 resistant clones) and split-reads from breakpoint alignment sequences (for another 14 clones). We found that the post-CNV A/T track lengths were 16.6 ± 19.0% shorter than the pre-CNV lengths (Supplementary Table S4, P < 0.01). Despite the almost ubiquitous shortening of the breakpoint A/T track, hairpin predictions using post-CNV sequence from DSM1 resistant clones yielded a pattern similar to that of pre-CNV sequence due to a general lack of mutations surrounding the A/T tracks (Figure 4C; Supplementary Figure S4B and D). In two exceptions (of seven post-CNV breakpoints analyzed), a novel stable hairpin was generated (Figure 4D; Supplementary Figure S4A and C), indicating sequence changes following CNV generation. Analysis of deep sequencing reads at these locations further confirmed these findings (unpublished data). These two different patterns suggest the action of multiple repair pathways in CNV generation (see ‘Discussion’ section).
Figure 4.

Post-CNV sequences indicate two models of repair. (A and B) Steps leading to the generation of a novel junction after CNV formation. (A) Upstream (5′, red chevron) and downstream (3′, blue chevron) sequences in the parent clone undergo recombination (yellow bolt), amplifying the genome surrounding the target gene (gray bar with star). Sequence outside of the amplified region is indicated in yellow. (B) Following recombination, a tandem duplication with two copies of the target gene and a novel junction at the upstream and downstream sequence (arrow) is formed. Sequence outside of the amplicon is conserved (yellow). (C and D) Use of hairpin prediction at the novel junction to identify signatures of repair pathways. (C) Hairpin prediction pattern is conserved at the novel junction, indicating action of MMEJ (red/blue: predicted error-free repair, black: observed sequence, plot shown for DSM1 resistant D clone, see Supplementary Figure S4 for DSM1 resistant F clone). Repair via MMEJ occurs through resection, A/T track exposure, and annealing of two complementary genomic locations. The method of repair does not affect upstream and downstream sequence but may remove nucleotides from the A/T track. (D) Hairpin prediction pattern is altered at the junction/novel downstream hairpins and mismatched locations indicate action of microhomology-mediated break induced replication (MMBIR, red/blue: predicted error-free repair, black: observed sequence, plot shown for DSM1 resistant C clone, see Supplementary Figure S4 for DSM1 resistant E clone). Repair via MMBIR uses error prone replication that induces mutations around the A/T track to resolve a stalled replication fork (arrow). This likely occurs through A/T track invasion at another genomic location for CNV generation and resolution.
DISCUSSION
CNVs are an established contributor to clinical antimalarial resistance (3,13,17,24,31,61,62). From conservative estimates on wild parasite populations, as much as 6% of P. falciparum genes are encompassed within CNVs (13). It is important to note that this estimate is distinct from laboratory selections because it quantifies stable CNVs that persist following purifying selection in the mosquito or human parasite stages. Recent laboratory selections have shown that CNVs are as frequently observed as non-synonymous SNPs within in vitro selected P. falciparum clones (43). However, CNVs affect more to total base pairs and are distributed across all chromosomes (13,43). This broad distribution is somewhat unique. CNVs are often biased to certain chromosomes in organisms as diverse as rice (63), rats (64), cattle (65) and humans (66). However, organisms that show vast phenotypic diversity and high-selective pressures appear to have a broader CNV distribution such as dogs (67) and mice (68).
Here, we took a novel approach to dissect CNV generation across the genome of the protozoan parasite; we performed in-depth bioinformatic analysis of sequences found at known CNV breakpoints across all chromosomes. In doing so, we gained an understanding of DNA features and molecular pathways that can trigger CNV formation. We and others have postulated that CNV formation is the initial step in P. falciparum that leads to the accumulation of high level, stable, resistance-conferring SNPs (8,69). This hypothesis is consistent with the role of CNVs as an adaptation strategy that is broadly relevant to the parasite as well as other organisms (13,69–72).
Shared CNV breakpoints reveal a model of CNV formation
High-quality deep sequencing of parasites from several controlled laboratory selections provided a unique opportunity to study CNV formation in the P. falciparum genome (Supplementary Table S1). Three characteristics facilitated these studies: (i) the availability of sequence from parent clones (prior to selection or pre-CNV) allowed for analysis of the native genome architecture at the position of the future CNV breakpoint, (ii) sequence from resistant clones (post-CNV) allowed for mechanistic studies on the pathways that enacted the change and (iii) breakpoints that occurred more than once in independent selections (or ‘shared’ breakpoints) allowed us to identify features that likely contribute to CNV formation.
Overall, five shared breakpoints were detected in our analysis; due to their occurrence, we speculated that there was an additional CNV signal beyond the almost ubiquitous A/T track present at these locations. Indeed, secondary structure predictions identified extremely stable hairpins in close proximity to these shared breakpoints (Figures 1, 2 and Table 2). The specific hairpins identified in this analysis were more stable than 99.8% of hairpins predicted across the genome (∼23.5 million structures overall) or the top 8% of stable hairpins (∼61 000 structures with ΔG of <−5.8 kCal/mol in total). This finding increased our confidence that hairpins within close proximity to the breakpoint A/T track were of importance. Structure predictions on the remaining unique CNV breakpoints displayed a similar profile with a mean ΔG in the top 12% of stably predicted hairpins across the genome.
DNA hairpins and other secondary structures have been implicated in mechanisms of immune evasion by P. falciparum (41,51,73). Additionally, such structures are known to cause problems during DNA replication in other organisms: they result in higher levels of replication fork collapse and DNA breakage (40,74) and hairpin-binding proteins can stimulate recombination at these sites (75–77). When repaired erroneously, these events can lead to the formation of CNVs (50,51,74,78).
In light of these previous studies and our results, we propose a model of CNV generation (Figure 5): DNA hairpins in close proximity to long A/T tracks throughout the P. falciparum genome have the propensity to create DSBs by replication fork collapse (Step 1A) or cleavage by hairpin-binding proteins (Step 1B). These DSBs are subsequently repaired in a non-faithful manner to create CNVs (Step 2). Resulting amplifications are initially rare throughout P. falciparum populations but then undergo selection to remove deleterious CNVs and promote the maintenance of beneficial CNVs (Step 3).
Figure 5.

Model of CNV development and selection in Plasmodium falciparum. In Step 1, DNA hairpins trigger double strand breaks throughout the P. falciparum genome presumably by either halting replication fork progression (Step 1A) or recognition by hairpin-binding proteins (Step 1B). In Step 2, long A/T tracks (gray circles) within 400 bp of the double strand break are utilized as microhomology for error-prone repair pathways to generate CNVs (blue, red and green bars). CNV breakpoints (vertical dotted lines) are generated semi-randomly across the genome but more stable hairpins are more likely to generate recurrent breakpoints (purple dotted line). De novo CNVs can either contain beneficial genes (gray bar with star) or those unrelated to the selection. New CNVs are generated frequently and could randomly occur throughout the highly repetitive P. falciparum genome (green bar), but may increase under selective pressure (see Discussion). In Step 3, selection (i.e. drug or fitness effects) enriches for beneficial CNVs (blue and red parasites) and purges deleterious CNVs (green parasite) from the population.
CNV trigger sites are enriched within intergenic regions
We detected elevated numbers of long A/T tracks (>9 bp) and stable hairpins (>−5.8 kCal/mol) in intergenic regions when compared to genic regions of the P. falciparum genome (Table 2). Furthermore, we identified a closer track-hairpin relationship in intergenic regions (Table 3 and Figure 3) and a corresponding enrichment in trigger sites (defined as A/T tracks between 20 and 40 bp in length within 400 bp of a stable hairpin, which occurs for 19.0% of intergenic A/T tracks). These data indicate that there may be a selective benefit of their association in non-coding regions of the genome. We hypothesize that one such benefit includes increased CNV generation and thus, increased adaptability especially in the face of antimalarial selection. In support of this hypothesis, the presence of CNV trigger sites across the genome poises every potential drug target for amplification (Figures 3 and 5). It is interesting to speculate that characteristics of CNV trigger sites could contribute to the observation that some clones develop resistance in vitro more readily than others (8,79). This would be the first time that DNA sequence itself, as opposed to the regulation of specific repair proteins (62,80), has been implicated in the ability of P. falciparum to develop resistance.
Potential DNA repair mechanisms leading to CNV formation in Plasmodium falciparum
Through the analysis of post-CNV sequences, we detect evidence for two DNA repair pathways acting in the generation of P. falciparum CNVs: microhomology-mediated end joining (MMEJ, (81–83)) and microhomology-mediated break-induced repair (MMBIR, (74,84)). The ubiquitous shortening of long A/T tracks after CNV generation as well as several single nucleotide insertions after repair implicates MMEJ, which can cause deletions with and without small insertions (clones D and F, Figure 4C, Supplementary Figure S4 and Supplementary Table S2). Alternatively, the presence of short repeat expansions points to MMBIR, which has not been characterized in P. falciparum (clones C and E, Figure 4D). Nucleotide addition is a common consequence of fork slippage during replication-mediated repair processes (74,82,84,85). Fork slippage is also a hallmark of an alternate and possibly unique pathway to P. falciparum, synthesis-dependent MMEJ, which appears to be a mixture of MMEJ and MMBIR (82).
One major influence on the use of microhomology-mediated pathways (MMEJ and MMBIR) versus homologous recombination is the distance of DNA resection which is the distance from DNA lesion to homologous sequence used for repair. For example, short-range resection biases repair toward microhomology-mediated pathways and extensive resection biases repair toward homologous recombination (86,87). Furthermore, when excluding homologous recombination, short resection distances of <50 bp are more likely to lead to MMEJ as a means of repair and longer distances <250 bp are more likely enacted by MMBIR (88). Our CNV ‘trigger site’ model suggests an important role for the A/T track-hairpin distance (Figure 5); we speculate that the span of sequence between each component could reflect the resection distance for either of these two repair pathways. Given the proposed 400 bp distance limit (Figure 3), there are 9130 intergenic and 2222 genic trigger sites capable of being utilized by these pathways (Table 3 and Figure 3). Although our study only assessed amplifications, repair of DSB breaks at these sites can lead to deletions as well but further investigation is required to understand the mechanisms involved in the generation of deletions as well as how they contribute to the adaptability of the parasite.
Homologous recombination is highly active in the parasite (16,22,82,83); what then leads to the use of these error-prone pathways for repair? We propose that antimalarial treatment, which causes metabolic stress, skews repair toward MMEJ and MMBIR in P. falciparum. Microhomology-mediated pathways in other organisms have been shown to exhibit increased activity when cells are under stress (89–91). For example, under normal conditions in mammalian cells, RAD51 inhibits MMBIR activity and facilitates the use of homologous recombination for DSB repair (92). However, RAD51 is downregulated during hypoxic stress in tumors, dNTP depletion, and the starvation response in Escherichia coli and cancer as well as during replication stress in humans (90,92–96). Future studies on the levels of key repair proteins will be required to see if this is the case in P. falciparum.
Overall, we propose that a close A/T track-hairpin relationship in the P. falciparum genome leads to the utilization of error-prone microhomology-mediated pathways. These events lead to enhanced generation of CNVs and adaptability of this parasite under selective pressure. Further investigation of these mechanisms may identify DNA repair pathways that can be targeted to limit parasite adaptability.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Cory Wheeler, Ali Guler and members of Jennifer Guler, Bill Petri and Barbara Mann's labs for their valuable input and advice on the study design, statistical approaches, and critical evaluation of the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Health [R01GM101192 to Y.H.W.]; University of Virginia, Start-up Funds (to J.L.G.); Infectious Disease, Cell and Molecular Biology and Biomedical Data Sciences Training Grants [T32AI007046 to A.H., T32GM008136-32 to M.C., T32LM012416 to B.A.]. Funding for open access charge: University of Virginia, Start-up Funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. Carter R., Mendis K.N.. Evolutionary and historical aspects of the burden of malaria. Clin. Microbiol. Rev. 2002; 15:564–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Corey V.C., Lukens A.K., Istvan E.S., Lee M.C.S., Franco V., Magistrado P., Coburn-Flynn O., Sakata-Kato T., Fuchs O., Gnädig N.F. et al. A broad analysis of resistance development in the malaria parasite. Nat.Commun. 2016; 7:11901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Foote S.J., Thompson J.K., Cowman A.F., Kemp D.J.. Amplification of the multidrug resistance gene in some chloroquine-resistant isolates of P. falciparum. Cell. 1989; 57:921–930. [DOI] [PubMed] [Google Scholar]
- 4. Heinberg A., Siu E., Stern C., Lawrence E.A., Ferdig M.T., Deitsch K.W., Kirkman L.A.. Direct evidence for the adaptive role of copy number variation on antifolate susceptibility in Plasmodium falciparum. Mol. Microbiol. 2013; 88:702–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lynch M., Conery J.S.. The evolutionary fate and consequences of duplicate genes. Science. 2000; 290:1151–1155. [DOI] [PubMed] [Google Scholar]
- 6. Kondrashov F.A., Rogozin I.B., Wolf Y.I., Koonin E.V.. Selection in the evolution of gene duplications. Genome Biol. 2002; 3:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kondrashov F.A. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proceedings of the Royal Society B. 2012; 1749:5048–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guler J.L., Freeman D.L., Ahyong V., Patrapuvich R., White J., Gujjar R., Phillips M.A., DeRisi J., Rathod P.K.. Asexual populations of the human malaria parasite, Plasmodium falciparum, use a two-step genomic strategy to acquire accurate, beneficial DNA amplifications. PLoS Pathog. 2013; 9:e1003375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rottmann M., McNamara C., Yeung B.K., Lee M.C., Zou B., Russell B., Seitz P., Plouffe D.M., Dharia N.V., Tan J. et al. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010; 329:1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phillips M.A., Lotharius J., Marsh K., White J., Dayan A., White K.L., Njoroge J.W., El Mazouni F., Lao Y., Kokkonda S. et al. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci. Transl. Med. 2015; 7:296ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thaithong S., Ranford-Cartwright L.C., Siripoon N., Harnyuttanakorn P., Kanchanakhan N.S., Seugorn A., Rungsihirunrat K., Cravo P.V., Beale G.H.. Plasmodium falciparum: gene mutations and amplification of dihydrofolate reductase genes in parasites grown in vitro in presence of pyrimethamine. Exp. Parasitol. 2001; 98:59–70. [DOI] [PubMed] [Google Scholar]
- 12. Triglia T., Foote S.J., Kemp D.J., Cowman A.F.. Amplification of the multidrug resistance gene pfmdr1 in Plasmodium falciparum has arisen as multiple independent events. Mol. Cell. Biol. 1991; 11:5244–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheeseman I.H., Gomez-Escobar N., Carret C.K., Ivens A., Stewart L.B., Tetteh K.K.A., Conway D.J.. Gene copy number variation throughout the Plasmodium falciparum genome. BMC Genomics. 2009; 10:353–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sidhu A.B.S., Uhlemann A.-C., Valderramos S.G., Valderramos J.-C., Krishna S., Fidock D.A.. Decreasing pfmdr1 copy number in plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J. Infect. Dis. 2006; 194:528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kidgell C. A systematic map of genetic variation in plasmodium falciparum. PLoS Pathog. 2006; 2:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bopp S.E.R., Manary M.J., Bright A.T., Johnston G.L., Dharia N.V., Luna F.L., McCormack S., Plouffe D., McNamara C.W., Walker J.R. et al. Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLos Genet. 2013; 9:e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nair S., Miller B., Barends M., Jaidee A., Patel J., Mayxay M., Newton P., Nosten F., Ferdig M.T., Anderson T.J.. Adaptive copy number evolution in malaria parasites. PLos Genet. 2008; 4:e1000243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ribacke U., Mok B.W., Wirta V., Normark J., Lundeberg J., Kironde F., Egwang T.G., Nilsson P., Wahlgren M.. Genome wide gene amplifications and deletions in Plasmodium falciparum. Mol. Biochem. Parasitol. 2007; 155:33–44. [DOI] [PubMed] [Google Scholar]
- 19. Nair S., Nkhoma S., Nosten F., Mayxay M., French N., Whitworth J., Anderson T.. Genetic changes during laboratory propagation: copy number at the recitulocyte-binding protein 1 locus of Plasmodium falciparum. Mol. Biochem. Parasitol. 2010; 172:145–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Banyal H.S., Inselburg J.. Plasmodium falciparum: induction, selection, and characterization of pyrimethamine-resistant mutants. Exp. Parasitol. 1986; 62:61–70. [DOI] [PubMed] [Google Scholar]
- 21. Cowman A.F., Galatis D., Thompson J.K.. Selection for mefloquine resistance in Plasmodium falciparum is linked to amplification of the pfmdr1 gene and cross-resistance to halofantrine and quinine. PNAS. 1994; 91:1143–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crabb B.S., Cowman A.F.. Characterization of promoters and stable transfection by homologous and nonhomologous recombination in Plasmodium falciparum. PNAS. 1996; 93:7289–7294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Price R.N., Uhlemann A.-C., Brockman A., McGready R., Ashley E., Phaipun L., Patel R., Laing K., Looareesuwan S., White N.J. et al. Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004; 364:438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dharia N.V., Sidhu A.B., Cassera M.B., Westenberger S.J., Bopp S.E., Eastman R.T., Plouffe D., Batalov S., Park D.J., Volkman S.K. et al. Use of high-density tiling microarrays to identify mutations globally and elucidate mechanisms of drug resistance in Plasmodium falciparum. Genome Biol. 2009; 10:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Singh A., Rosenthal P.J.. Selection of cysteine protease inhibitor-resistant malaria parasites is accompanied by amplification of falcipain genes and alteration in inhibitor transport. J. Biol. Chem. 2004; 279:35236–35241. [DOI] [PubMed] [Google Scholar]
- 26. Cheeseman I.H., Miller B., Tan J.C., Tan A., Nair S., Nkhoma S.C., De Donato M., Rodulfo H., Dondorp A., Branch O.H. et al. Population structure shapes copy number variation in malaria parasites. Mol. Biol. Evol. 2015; 33:603–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Auburn S., Serre D., Pearson R.D., Amato R., Sriprawat K., To S., Handayuni I., Suwanarusk R., Russell B., Drury E. et al. Genomic analysis reveals a common breakpoint in amplifications of the plasmodium vivax multidrug resistance 1 locus in Thailand. J. Infect. Dis. 2016; 214:1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Menard D., Chan E.R., Benedet C., Ratsimbasoa A., Kim S., Chim P., Do C., Witkowski B., Durand R., Thellier M. et al. Whole genome sequencing of field isolates reveals a common duplication of the duffy binding protein gene in malagasy plasmodium vivax strains. PLoS Negl. Trop. Dis. 2013; 7:e2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gunalan K., Lo E., Hostetler J.B., Yewhalaw D., Mu J., Neafsey D.E., Yan G., Miller L.H.. Role of Plasmodium vivax Duffy-binding protein 1 in invasion of Duffy-null Africans. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:6271–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Samarakoon U., Gonzales J.M., Patel J.J., Tan A., Checkley L., Ferdig M.T.. The landscape of inherited and de novo copy number variants in a plasmodium falciparum genetic cross. BMC Genomics. 2011; 12:457–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nair S., Nash D., Sudimack D., Jaidee A., Barends M., Uhlemann A.C., Krishna S., Nosten F., Anderson T.J.. Recurrent gene amplification and soft selective sweeps during evolution of multidrug resistance in malaria parasites. Mol. Biol. Evol. 2007; 24:562–573. [DOI] [PubMed] [Google Scholar]
- 32. Zhang H., Freudenreich C.H.. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell. 2007; 27:367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shah S.N., Opresko P.L., Meng X., Lee M.Y., Eckert K.A.. DNA structure and the Werner protein modulate human DNA polymerase delta-dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2010; 38:1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burrow A.A., Marullo A., Holder L.R., Wang Y.H.. Secondary structure formation and DNA instability at fragile site FRA16B. Nucleic Acids Res. 2010; 38:2865–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walsh E., Wang X., Lee M.Y., Eckert K.A.. Mechanism of replicative DNA polymerase delta pausing and a potential role for DNA polymerase kappa in common fragile site replication. J. Mol. Biol. 2013; 425:232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zheng G.X., Kochel T., Hoepfner R.W., Timmons S.E., Sinden R.R.. Torsionally tuned cruciform and Z-DNA probes for measuring unrestrained supercoiling at specific sites in DNA of living cells. J. Mol. Biol. 1991; 221:107–122. [DOI] [PubMed] [Google Scholar]
- 37. Cromie G.A., Millar C.B., Schmidt K.H., Leach D.R.. Palindromes as substrates for multiple pathways of recombination in Escherichia coli. Genetics. 2000; 154:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rogers F.A., Tiwari M.K.. Triplex-induced DNA damage response. Yale J. Biol. Med. 2013; 86:471–478. [PMC free article] [PubMed] [Google Scholar]
- 39. van Kregten M., Tijsterman M.. The repair of G-quadruplex-induced DNA damage. Exp. Cell Res. 2014; 329:178–183. [DOI] [PubMed] [Google Scholar]
- 40. Mirkin E.V., Mirkin S.M.. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 2007; 71:13–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stanton A., Harris L.M., Graham G., Merrick C.J.. Recombination events among virulence genes in malaria parasites are associated with G-quadruplex-forming DNA motifs. BMC Genomics. 2016; 17:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Herman J.D., Rice D.P., Ribacke U., Silterra J., Deik A.A., Moss E.L., Broadbent K.M., Neafsey D.E., Desai M.M., Clish C.B. et al. A genomic and evolutionary approach reveals non-genetic drug resistance in malaria. Genome Biol. 2014; 15:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cowell A.N., Istvan E.S., Lukens A.K., Gomez-Lorenzo M.G., Vanaerschot M., Sakata-Kato T., Flannery E.L., Magistrado P., Owen E., Abraham M. et al. Mapping the malaria parasite druggable genome by using in vitro evolution and chemogenomics. Science. 2018; 359:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okonechnikov K., Conesa A., García-Alcalde F.. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 2016; 32:292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chiang C., Layer R.M., Faust G.G., Lindberg M.R., Rose D.B., Garrison E.P., Marth G.T., Quinlan A.R., Hall I.M.. SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat. Methods. 2015; 12:966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Abyzov A., Urban A.E., Snyder M., Gerstein M.. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011; 21:974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Layer R.M., Chiang C., Quinlan A.R., Hall I.M.. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol. 2014; 15:R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thorvaldsdottir H., Robinson J.T., Mesirov J.P.. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 2013; 14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dillon L.W., Pierce L.C.T., Ng M.C.Y., Wang Y.-H.. Role of DNA secondary structures in fragile site breakage along human chromosome 10. Hum. Mol. Genet. 2013; 22:1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sander A.F., Lavstsen T., Rask T.S., Lisby M., Salanti A., Fordyce S.L., Jespersen J.S., Carter R., Deitsch K.W., Theander T.G. et al. DNA secondary structures are associated with recombination in major Plasmodium falciparum variable surface antigen gene families. Nucleic Acids Res. 2014; 42:2270–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Balakrishnan L., Bambara R.A.. Okazaki fragment metabolism. Cold Spring Harb. Perspect. Biol. 2013; 5:a010173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gruber A.R., Lorenz R., Bernhart S.H., Neuböck R., Hofacker I.L.. The Vienna RNA websuite. Nucleic Acids Res. 2008; 36:W70–W74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rice P., Longden I., Bleasby A.. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000; 16:276–277. [DOI] [PubMed] [Google Scholar]
- 55. Thys R.G., Lehman C.E., Pierce L.C.T., Wang Y.-H.. DNA secondary structure at chromosomal fragile sites in human disease. Curr. Genomics. 2015; 16:60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dechering K.J., Cuelenaere K., Konings R.N., Leunissen J.A.. Distinct frequency-distributions of homopolymeric DNA tracts in different genomes. Nucleic Acids Res. 1998; 26:4056–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2009; NY: Springer-Verlag. [Google Scholar]
- 59. R Development Core team R Foundation for Statistical Computing. R: A language and environment for statistical computing. 2016; Vienna. [Google Scholar]
- 60. Manary M.J., Singhakul S.S., Flannery E.L., Bopp S.E., Corey V.C., Bright A.T., McNamara C.W., Walker J.R., Winzeler E.A.. Identification of pathogen genomic variants through an integrated pipeline. BMC Bioinformatics. 2014; 15:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brown T., Smith L.S., Oo E.K., Shawng K., Lee T.J., Sullivan D., Beyrer C., Richards A.K.. Molecular surveillance for drug-resistant Plasmodium falciparum in clinical and subclinical populations from three border regions of Burma/Myanmar: cross-sectional data and a systematic review of resistance studies. Malar. J. 2012; 11:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lee A.H., Fidock D.A.. Evidence of a mild mutator phenotype in Cambodian Plasmodium falciparum Malaria Parasites. PLoS One. 2016; 11:e0154166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu P., Wang C.H., Xu Q., Feng Y., Yuan X.P., Yu H.Y., Wang Y.P., Tang S.X., Wei X.H.. Genome-wide copy number variations in Oryza sativa L. BMC Genomics. 2013; 14:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Guryev V., Saar K., Adamovic T., Verheul M., van Heesch S.A., Cook S., Pravenec M., Aitman T., Jacob H., Shull J.D. et al. Distribution and functional impact of DNA copy number variation in the rat. Nat. Genet. 2008; 40:538–545. [DOI] [PubMed] [Google Scholar]
- 65. Fadista J., Thomsen B., Holm L.E., Bendixen C.. Copy number variation in the bovine genome. BMC Genomics. 2010; 11:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zarrei M., MacDonald J.R., Merico D., Scherer S.W.. A copy number variation map of the human genome. Nat. Rev. Genet. 2015; 16:172–183. [DOI] [PubMed] [Google Scholar]
- 67. Nicholas T.J., Baker C., Eichler E.E., Akey J.M.. A high-resolution integrated map of copy number polymorphisms within and between breeds of the modern domesticated dog. BMC Genomics. 2011; 12:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Locke M.E., Milojevic M., Eitutis S.T., Patel N., Wishart A.E., Daley M., Hill K.A.. Genomic copy number variation in Mus musculus. BMC Genomics. 2015; 16:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Anderson T.J., Patel J., Ferdig M.T.. Gene copy number and malaria biology. Trends Parasitol. 2009; 25:336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hendrickson H., Slechta E.S., Bergthorsson U., Andersson D.I., Roth J.R.. Amplification-mutagenesis: evidence that “directed” adaptive mutation and general hypermutability result from growth with a selected gene amplification. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:2164–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Roth J.R., Andersson D.I.. Amplification-mutagenesis–how growth under selection contributes to the origin of genetic diversity and explains the phenomenon of adaptive mutation. Res. Microbiol. 2004; 155:342–351. [DOI] [PubMed] [Google Scholar]
- 72. Elde N.C., Child S.J., Eickbush M.T., Kitzman J.O., Rogers K.S., Shendure J., Geballe A.P., Malik H.S.. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell. 2012; 150:831–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Harris L.M., Monsell K.R., Noulin F., Famodimu M.T., Smargiasso N., Damblon C., Horrocks P., Merrick C.J.. G-quadruplex DNA motifs in the Malaria Parasite Plasmodium falciparum and their potential as Novel Antimalarial Drug Targets. Antimicrob. Agents Chemother. 2018; 62:e01828-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hastings P.J., Ira G., Lupski J.R.. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLos Genet. 2009; 5:e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ma Y., Pannicke U., Schwarz K., Lieber M.R.. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002; 108:781–794. [DOI] [PubMed] [Google Scholar]
- 76. Chiruvella K.K., Rajaei N., Jonna V.R., Hofer A., Astrom S.U.. Biochemical characterization of Kat1: a domesticated hAT-transposase that induces DNA hairpin formation and MAT-switching. Sci. Rep. 2016; 6:21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brázda V., Laister R.C., Jagelská E.B., Arrowsmith C.. Cruciform structures are a common DNA feature important for regulating biological processes. BMC Mol. Biol. 2011; 12:33–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Carvalho C.M., Pehlivan D., Ramocki M.B., Fang P., Alleva B., Franco L.M., Belmont J.W., Hastings P.J., Lupski J.R.. Replicative mechanisms for CNV formation are error prone. Nat. Genet. 2013; 45:1319–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rathod P.K., McErlean T., Lee P.-C.. Variations in frequencies of drug resistance in Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:9389–9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gupta D.K., Patra A.T., Zhu L., Gupta A.P., Bozdech Z.. DNA damage regulation and its role in drug-related phenotypes in the malaria parasites. Sci. Rep. 2016; 6:23603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Ottaviani D., LeCain M., Sheer D.. The role of microhomology in genomic structural variation. Trends Genet. 2014; 30:85–94. [DOI] [PubMed] [Google Scholar]
- 82. Kirkman L.A., Lawrence E.A., Deitsch K.W.. Malaria parasites utilize both homologous recombination and alternative end joining pathways to maintain genome integrity. Nucleic Acids Res. 2014; 42:370–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Lee A.H., Symington L.S., Fidock D.A.. DNA repair mechanisms and their biological roles in the malaria parasite plasmodium falciparum. Microbiol. Mol. Biol. Rev. 2014; 78:469–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang F., Khajavi M., Connolly A.M., Towne C.F., Batish S.D., Lupski J.R.. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat. Genet. 2009; 41:849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Verdin H., D’Haene B., Beysen D., Novikova Y., Menten B., Sante T., Lapunzina P., Nevado J., Carvalho C.M.B., Lupski J.R. et al. Microhomology-mediated mechanisms underlie non-recurrent disease-causing microdeletions of the FOXL2 gene or its regulatory domain. PLos Genet. 2013; 9:e1003358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Symington L.S., Gautier J.. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011; 45:247–271. [DOI] [PubMed] [Google Scholar]
- 87. Mimitou E., LS S.. DNA end resection: many nucleases make light work. DNA Repair (Amst.). 2009; 8:983–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chung W.-H., Zhu Z., Papusha A., Malkova A., Ira G.. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLos Genet. 2010; 6:e1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Galhardo R.S., Hastings P.J., Rosenberg S.M.. Mutation as a stress response and the regulation of evolvability. Crit. Rev. Biochem. Mol. Biol. 2007; 42:399–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Scanlon S.E., Glazer P.M.. Multifaceted control of DNA repair pathways by the hypoxic tumor microenvironment. DNA Repair (Amst.). 2015; 32:180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Arlt M.F., Mulle J.G., Schaibley V.M., Ragland R.L., Durkin S.G., Warren S.T., Glover T.W.. Replication stress induces Genome-wide copy number changes in human cells that resemble polymorphic and pathogenic variants. Am. J. Hum. Genet. 2009; 84:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bindra R.S., Schaffer P.J., Meng A., Woo J., Maseide K., Roth M.E., Lizardi P., Hedley D.W., Bristow R.G., Glazer P.M.. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol. Cell. Biol. 2004; 24:8504–8518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bristow R.G., Hill R.P.. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer. 2008; 8:180–192. [DOI] [PubMed] [Google Scholar]
- 94. Slack A., Thornton P.C., Magner D.B., Rosenberg S.M., Hastings P.J.. On the mechanism of gene amplification induced under stress in Escherichia coli. PLos Genet. 2006; 2:e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Mannava S., Moparthy K.C., Wheeler L.J., Natarajan V., Zucker S.N., Fink E.E., Im M., Flanagan S., Burhans W.C., Zeitouni N.C. et al. Depletion of deoxyribonucleotide pools is an endogenous source of DNA damage in cells undergoing Oncogene-Induced senescence. Am. J. Pathol. 2013; 182:142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bhattacharya S., Srinivasan K., Abdisalaam S., Su F., Raj P., Dozmorov I., Mishra R., Wakeland E.K., Ghose S., Mukherjee S. et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017; 45:4590–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.