

Abstract
The endoplasmic reticulum (ER), as a multifunctional organelle, plays crucial roles in lipid biosynthesis and calcium homeostasis as well as the synthesis and folding of secretory and membrane proteins. Therefore, it is of high importance to maintain ER homeostasis and to adapt ER function and morphology to cellular needs. Here, we show that signal peptide peptidase (SPP) modulates the ER shape through degradation of morphogenic proteins. Elevating SPP activity induces rapid rearrangement of the ER and formation of dynamic ER clusters. Inhibition of SPP activity rescues the phenotype without the need for new protein synthesis, and this rescue depends on a pre-existing pool of proteins in the Golgi. With the help of organelle proteomics, we identified certain membrane proteins to be diminished upon SPP expression and further show that the observed morphology changes depend on SPP-mediated cleavage of ER morphogenic proteins, including the SNARE protein syntaxin-18. Thus, we suggest that SPP-mediated protein abundance control by a regulatory branch of ER-associated degradation (ERAD-R) has a role in shaping the early secretory pathway.
Keywords: intramembrane proteolysis, endoplasmic reticulum-associated protein degradation (ERAD), organelle, proteolysis, cellular regulation, organelle morphology, substrate identification, tail-anchored protein
Introduction
Eukaryotic cells are highly compartmentalized with various membrane-enclosed organelles that fulfill different important functions. Key determinants of this complex membrane system are organelle-specific proteins. Because cells face varying environmental conditions, the secretory pathway constantly has to respond to changing needs. A dynamic protein homeostasis (proteostasis) network is capable of targeting specific proteins for degradation and thereby maintaining and adapting organelle function. Whereas regulated endocytosis selectively targets proteins in the late secretory pathway for lysosomal degradation (1), endoplasmic reticulum (ER)3 proteins are extracted in an ATP-dependent process via the ER-associated degradation (ERAD) pathway for proteasomal turnover (2). Specificity in these proteostasis systems is commonly provided by E3 ubiquitin ligases, which label proteins for degradation by ubiquitin attachment. This ubiquitination process is reversible, as deubiquitinating enzymes can revert this post-translational modification, thereby increasing fidelity and dynamics of the selection process (3, 4). Therefore, tight regulation is crucial, and different strategies have evolved to maintain protein homeostasis. An evolutionarily conserved regulatory branch of ERAD (ERAD-R), which is centered around the intramembrane protease signal peptide peptidase (SPP) and the E3 ubiquitin ligases TRC8 and MARCH6, initiates membrane protein turnover by a direct proteolytic cut in transmembrane (TM) segments (5–8). As for classical protein dislocation, fragments released from the membrane by SPP are efficiently degraded by the proteasome (5, 6). Related mechanisms, including intramembrane proteases and ATP-dependent extraction mechanisms, exist in other cellular organelles (9).
Another key determinant ensuring maintenance and dynamics of cellular organelles is regulated vesicular trafficking (10). Within the secretory pathway, this is mainly achieved by controlled vesicle budding from one compartment followed by intracellular transport and coordinated fusion of the transport vesicle with the target organelle. Major players in this process that ensure selectivity of the fusion step are soluble NSF attachment protein receptor (SNARE) proteins. They form a large protein family of over 60 members, each found in a specific set of cellular compartments (11). The hallmark of SNAREs is a conserved cytoplasmic heptad repeat sequence that forms coiled-coil structures followed by a TM anchor close to the C terminus, a special class of so-called tail-anchored (TA) proteins that is post-translationally targeted to the ER (12). Although the so-called trans-SNARE complex between vesicle and target membrane induces specificity and lowers the energy barrier of membrane fusion (11), there is growing evidence that certain SNAREs also play a role in organelle morphology control, including cell cycle–dependent ER rearrangement, homotypic membrane fusion, and organization of the ER network (13–16).
The ER not only serves as the entry point of newly synthesized proteins, it also constitutes the largest part of the endomembrane system and provides the physical boundary for the nuclear compartment. The physiological function of the peripheral ER is as diverse as protein folding, quality control, intracellular signaling, and lipid biosynthesis (17). To meet these different tasks, the ER is divided in several morphologically distinct subdomains such as rough and smooth ER, consisting of different structures like tubules and sheets (17, 18). Therefore, control of its morphology and subdomain structure is a particular challenge. It has been found that relative amounts of different ER domains vary significantly depending on the cell type, and distorted ER morphology is linked to several physiological disorders (19, 20). Mechanisms that determine the ER morphology can be divided into three classes. The first class of proteins, which are associated to or inserted into the lipid bilayer such as reticulons, mediate curvature and shaping of the membrane itself (21–23). The second class mediates dynamics and continuity of ER membranes by an atlastin-mediated homotypic fusion process (24–26). This process is constitutively active but thought to be controlled by several factors, including the SNARE protein syntaxin-18 (STX18) (15, 16, 27). The third group of proteins, including CLIMP63, STIM1, and REEP1, links the ER with N-terminal cytoplasmic domains to the microtubule cytoskeleton and thus helps to maintain ER architecture (28–31). During mitosis, CLIMP63 phosphorylation triggers loss of ER–microtubule interaction, a process that is thought to enable segregation between mother and daughter cells (31). In somatic cells, the SNARE protein syntaxin-5 (STX5) has been shown to regulate CLIMP63-mediated ER–microtubule contact points (14, 31). Likewise, in neuronal cells, phosphorylation of CLIMP63 reduces ER–microtubule interaction, resulting in local zones of higher ER complexity, which leads to polarized secretion required for formation of new dendritic branching points (32). The emerging picture is that both ER-resident SNARE proteins STX5 and STX18 tune morphology of the peripheral ER in addition to their key function in membrane fusion. However, little is known of how the abundance and activity of these TA proteins are regulated. Here, we show by a gain-of-function assay that the intramembrane protease SPP targets, among other substrates, the SNARE protein STX18 for degradation.
Results
Organelle proteomics identifies STX18 as an endogenous SPP substrate
To identify potential endogenous substrates for an SPP-dependent ERAD pathway, we followed a stable isotope labeling with amino acids in cell culture (SILAC) approach combined with quantitative organelle proteomics. By mining publicly available RNA expression databases, we observed that SPP (referred to as HM13, its gene name) is transcriptionally up-regulated in several cancer tissues compared with corresponding healthy tissues, including liver hepatocellular carcinoma (Fig. S1A) (33). Consistent with this, two recent reports showed an increase of SPP expression in glioblastoma, lung, and breast cancer cells (34, 35). Therefore, we asked what the consequence of uncoupled SPP activity may be on the cellular level. HEK293T cells transfected with empty vector, SPP WT, or the catalytic mutant SPP-DA (36) labeled with light, heavy, and medium amino acids, respectively, were analyzed for changes in their membrane proteome (Fig. 1A). Differential proteomics of this SPP gain-of-function assay combined with isolation of membrane proteins by subcellular fractionation and high-salt and sodium carbonate extraction identified 1,686 proteins of which 1,531 were quantified in both biological replicates (Table S1). Of the 910 proteins with a membrane-related gene ontology annotation, 31 proteins were strongly reduced compared with control cells (cutoff, 40% reduction) upon 20-fold SPP overexpression, whereas expression of the catalytic SPP mutant did not have a significant effect (Fig. 1B and Table S1). HO1 and cytochrome B5A (CYB5A), two TA proteins that previously had been identified as SPP substrates (5), scored among the top hits (Fig. 1B), demonstrating the power of our approach. Interestingly, we identified depletion of four additional TA proteins by >40%, whereas 58 TA proteins were identified without a pronounced difference in the SPP WT–overexpressing cells (Table S2). Of those, several TA proteins such as metabolic enzyme sterol-4-α-carboxylate 3-dehydrogenase and sarcolemmal membrane–associated protein reside in the ER. Taken together with the predicted preference of SPP for type II–oriented TM segments (36), colocalization and our proteomics data suggest that ectopically expressed SPP triggers turnover of a specific set of membrane proteins. To test this prediction, we picked several TA proteins, including the ER–Golgi cycling SNARE STX18 (11), which was one of the highly reduced proteins in our proteomics list (Fig. 1B), and tested whether coexpression with SPP accelerates their degradation in cycloheximide chase experiments (Fig. 1C). Coexpression of SPP with FLAG-tagged STX18 destabilized the full-length form, leading to generation of a faster migrating cleavage fragment, whereas SPP-DA had no effect compared with control cells (Fig. 1C). To study the fate of cleaved STX18, we performed cycloheximide chase experiments in the presence or absence of proteasome inhibition. Addition of the proteasome inhibitor epoxomicin during chase stabilized the cleavage fragment (Fig. 1D), indicating that the cleaved protein is degraded by the proteasome. Consistent with a proteolysis-triggered degradation route, cellular fractionation showed that the majority of the cleaved STX18 is released into the cytosol (Fig. S1B) and thereby is available for proteasomal turnover. Coexpression of WT or catalytic mutant SPP-DA with another ER-resident SNARE, STX5, did not have any significant effect on the degradation rate, showing similar stability as in control cells (Fig. 1C). These results confirm that SPP shows specificity for certain substrates and that STX18 fulfils the requirements for efficient turnover by the SPP-dependent ERAD pathway. Consistent with this, coimmunoprecipitation experiments with ectopically expressed HA-tagged SPP-DA, which previously had been shown to act as a dominant-negative substrate-trapping mutant (6, 37), showed a pronounced physical interaction with endogenous STX18 (>10% of total amount; Fig. 1E). Notably, we observed a significant but lower recovery of STX18 by immunoprecipitation of ectopically expressed SPP WT, suggesting that upon binding to the enzyme intramembrane proteolysis is a multistep process and a subpopulation interacts with SPP at a precleavage step as has been suggested previously (6). In a related manner, we also observed a strong physical interaction of SPP with endogenous STX5 but only a modest increase of interaction with the SPP-DA substrate-trapping mutant (Fig. 1E). In contrast, the structural protein CLIMP63 was not copurified with either SPP form (WT or catalytic mutant) (Fig. 1E), demonstrating that the substrate-trapping approach recovers only selected membrane proteins. Despite that, cleavage efficiency significantly varies between different client proteins, indicating that SPP samples a wide range of clients but efficiently cleaves only those with cognate substrate features. To test this idea, we performed cycloheximide chase experiments in HEK293T cells stably expressing WT or the catalytic mutant form of SPP, comparing endogenous STX18 and STX5 degradation rates (Fig. S1, C and D). Induction of SPP WT but not the catalytic mutant led to slightly accelerated degradation of endogenous STX18 and enhanced generation of the N-terminal cleavage fragment (Fig. S1C), whereas STX5 was stable under these conditions and not significantly affected by SPP expression (Fig. S1D). This corroborates that endogenous STX18 is a bona fide SPP substrate. Next, we asked whether STX18 is a substrate also for endogenous SPP. STX18 is a relatively long-lived protein (Fig. S1C, mock condition); thus, blocking its abundance control machinery is expected to lead to only minor changes. Accordingly, steady-state analysis of endogenous STX18 levels revealed a small but statistically significant increase of 10% upon treatment with the SPP inhibitor (Z-LL)2-ketone for 16 h compared with vehicle-treated cells (Fig. 1F and Fig. S1E). Consistent with this low-level turnover of STX18, cycloheximide chase experiments in the presence of the proteasome inhibitor epoxomicin revealed traces of an endogenous N-terminal STX18 cleavage fragment over time (Fig. 1G). Addition of (Z-LL)2-ketone completely blocked generation of this fragment, demonstrating that STX18 is cleaved by endogenous SPP, generating this N-terminal cleavage fragment. Altogether, these results indicate that in tissue culture cells STX18 is an endogenous substrate of the SPP-dependent ERAD machinery; albeit as a functional native protein only a small fraction is targeted to this degradation pathway under steady-state conditions.
Figure 1.

Protease “degradomics” identifies SNAREs as SPP substrates. A, experimental outline of the SILAC-based MS analysis of high-salt, EDTA-washed, sodium carbonate–extracted membrane proteins. B, graphical representation of proteomics results showing -fold change in membrane proteins upon SPP WT expression from two replicates (mean heavy/light (H/L) ratio). Selected proteins are highlighted in red. C, cycloheximide (CHX) chase experiments showing turnover of ectopically expressed FLAG-tagged STX18 and STX5 long form upon coexpression of SPP WT and catalytic mutant SPP-DA. Actin is used as a loading control. Quantifications are shown on the right. Error bars represent S.E. (n = 3). Arrows represent full-length (black) and cleaved (white) forms of the proteins. D, cycloheximide chase experiments in the presence of 2 μm epoxomicin or vehicle control (DMSO) shows that the N-terminal cleavage fragment of STX18 (white arrow) is degraded by the proteasome. Actin is used as a loading control. Quantifications are shown on the right. Error bars represent S.E. (n = 3). E, coimmunoprecipitation assays with ectopically expressed SPP-HA and endogenous STX18, STX5, and CLIMP63. The long form and short form of STX5 and monomer and SDS-stable dimer forms of SPP are indicated. The asterisk represents antibody heavy chain. IP, immunoprecipitation. F, treatment of HEK293T cells with 50 μm (Z-LL)2-ketone and the analysis of endogenous STX18 by Western blotting (wb). The bar graph shows quantifications of the steady-state level of STX18. Error bars represent S.E. (n = 6). Significant chance is indicated (**, p < 0.01, Student's t test). See Fig. S1E for data on biological replicates. G, cycloheximide chase of endogenous STX18 in the presence of 5 μm epoxomicin and the absence or presence of 50 μm (Z-LL)2-ketone, showing N-terminal cleavage fragment accumulation upon proteasome inhibition.
Uncoupled SPP activity induces rapid rearrangement of the ER network
Next, we asked whether SPP-triggered degradation of ER-resident bioactive factors such as STX18 has an impact on HEK293T cell physiology. To test a potential effect of SPP-mediated SNARE abundance on ER to Golgi trafficking, we used metabolic labeling in HEK293T cells transfected with the type I plasma membrane protein CD44 together with SPP or empty vector and followed its trafficking by chase. We did not observe any significant difference in the trafficking rate of CD44 through the secretory pathway upon SPP expression (Fig. S2A). In addition, using deglycosylation enzymes EndoH and PNGaseF to distinguish between ER- and Golgi-modified N-linked glycans, we observed similar CD44 glycosylation patterns between control and SPP-overexpressing cells (Fig. S2B). Of note, we observed increased steady-state levels of the early secretory pathway form of CD44 upon SPP coexpression (Fig. S2B); however, the half-life of this form was not significantly affected during metabolic chase (Fig. S2A), suggesting an accumulated effect upon longer expression of the secretory pathway proteins. Consistent with this, we did not detect any signs of unfolded protein response induction in SPP-overexpressing cells (6). However, when we analyzed ER morphology in SPP-overexpressing cells, about 80% of the transfected cells showed a striking accumulation of the ER marker RFP-KDEL into large clusters (Fig. 2A). In contrast, expressing the catalytically inactive mutant SPP-DA or transfection of the empty plasmid had no significant effect on ER morphology (Fig. 2A). Because overexpression of GFP-tagged membrane proteins may lead to membrane alterations such as organized smooth ER (OSER) (38), we used a construct expressing untagged SPP in combination with the soluble ER luminal marker protein RFP-KDEL (39) for our microscopy analysis. To follow SPP expression in live cells, we used a plasmid with a GFP under the control of an internal ribosomal entry site (IRES). We observed the cluster formation starting from 10-fold SPP overexpression; however, it required about 25-fold overexpression to reach the high penetrance of 80–90% of cells showing the phenotype (Fig. S2C). Time-lapse microscopy revealed that these ER clusters are relatively dynamic and frequently changing shape and size (Fig. S2D). OSERs induced by high-level overexpression of GFP-Sec61β (38) are immobile structures that give a characteristic drop of the ER marker signal in the middle of the structure (Fig. S2E, upper panel), whereas SPP-induced ER clusters show a homogenous distribution (Fig. S2E, lower panel). After taking confocal image series of the cells, three-dimensional (3D) reconstructions of the ER were rendered, showing the varying dimensions and locations of the clusters within the ER network (Fig. 2B and Movie S1). These SPP-induced clusters are not specific to HEK293T cells as similar structures were observed in COS7, HeLa, and Huh7 cells upon ectopic SPP expression (Fig. S2F). Importantly, expression of the related enzyme SPPL3 (40, 41) as well as the ER rhomboid protease RHBDL4 (42) did not cause any significant ER morphology changes (Fig. S2G). Taken together, these results show that the observed ER cluster phenotype is a specific consequence of uncoupled SPP activity. Consistent with this, the SPP inhibitor (Z-LL)2-ketone and a chemically distinct, cross-reacting γ-secretase inhibitor, L685,458 (43), efficiently suppressed the ER phenotype (Fig. 2C). These results suggest that the ER clusters are caused by proteolytic cleavage of an endogenous ER protein. Intriguingly, time-lapse microscopy revealed that rescue upon SPP inhibitor treatment occurs in only a few minutes. To capture a rescue event of a whole ER, we performed 3D confocal imaging over time. By displaying single confocal sections as well as 3D reconstructions over time, the rapid disassembly of all ER clusters and the reappearance of the reticular ER network can clearly be seen (Fig. 2D and Movie S2). This suggests that uncoupled SPP activity affects a regulatory function that controls ER morphology instead of depleting a major structural component. Consistent with this idea, we observed that SPP inhibitor treatment in the presence of cycloheximide did not affect kinetics of the ER restructuring (Fig. 2D, lower panel), indicating that the rapid rescue relies on a pool of a bioactive molecule that is not susceptible to SPP activity and that can refill the ER pool under SPP inhibition without any need for new protein synthesis. Furthermore, the time-lapse series showed that the ER clusters indeed seem to consist of stacked membranes that upon inhibitor treatment start to resolve and finally appear in a typical network-like shape (Fig. 2D and Movie S2). Taken together, these findings indicate that membranes within the ER clusters are dynamic, loosely associated, and not terminally aggregated.
Figure 2.

Uncoupling SPP activity leads to local zones of high ER complexity. A, fluorescence microscopy of HEK293T cells ectopically expressing empty vector (with IRES-GFP), SPP WT, or SPP-DA together with RFP-KDEL, showing the ER cluster phenotype. Quantification of cells with ER morphology change is shown below. Error bars represent S.D. (n = 3; 50 cells were analyzed per condition). Scale bars, 5 μm. B, 3D reconstruction from a confocal z-stack of the ER as in A. ER clusters and reticular ER (*) of a region next to the cell's nucleus are shown. C, live cell imaging of HEK293T cells ectopically expressing SPP WT together with RFP-KDEL and treated with DMSO control or SPP inhibitors. Quantification of cells with ER morphology change is shown below. Error bars represent S.D. (n = 3; 50 cells were analyzed per condition). Scale bars, 5 μm. D, time-lapse microscopy of HEK293T cells transfected with RFP-KDEL and SPP-IRES-GFP (WT) and treated with 5 μm L685,458 SPP inhibitor alone (upper panel) or together with 100 μg/ml cycloheximide (CHX; lower panel) at time point 0. For GFP-expressing cells, the RFP-KDEL signal is shown. Scale bars, 5 μm.
Next, we aimed to get a better understanding of how these ER clusters are generated and which processes could be affected by SPP to modulate the cluster architecture. To analyze how the ER clusters relate to the overall organization of the cell, we coexpressed ER markers with other organelle and cytoskeleton markers and examined the SPP-induced phenotype by 3D confocal microscopy. Coexpression of SPP with the ER luminal marker RFP-KDEL and the outer mitochondrial membrane protein TOMM20-GFP showed no obvious divergence from the norm (Fig. 3A). Both organelles, even in the presence of large clusters, displayed the typical partial association between the ER and mitochondria (44). We did not observe any mitochondrion wrapped up largely or fully engulfed by the ER clusters (Fig. 3A). Coexpression of SPP with the ER membrane protein GFP-Sec61β and the cytoskeleton component α-tubulin-mCherry revealed that tubulin was not excluded from the regions occupied by the ER clusters (Fig. 3B). Confocal sections of the ER clearly showed the presence of tubulin within the clustered ER structures, indicating the clusters to be rather mesh-like structures than swollen ER tubules, which would exclude any other components (Fig. 3B). In addition, the presence of dividing cells showing both ER clusters and the typical spindle apparatus indicates that ER clustering does not affect segregation of the ER into mother and daughter cells (Fig. 3C). To further analyze the membrane organization in SPP-induced ER clusters, we performed confocal microscopy both with deconvolution and stimulated emission depletion (STED) super-resolution microscopy (45) with live HEK293T cells expressing WT SPP together with the luminal ER marker RFP-KDEL (Fig. 3D). The higher spatial resolution gained with these techniques enabled a more detailed presentation of the ER cluster substructure. Older and very large clusters, which are formed upon SPP expression, showed very dense and hardly resolvable mesh-like structures throughout the cluster (Fig. S3A, upper panel). Younger and often smaller clusters, which are formed shortly after inhibitor washout, displayed tight, but less dense structures that could better be resolved by STED with the ER luminal marker (Fig. S3A, lower panel). STED microscopy also revealed that the membranes of the ER are tightly intertwined with each other and ruled out any swollen (i.e. blown up) ER membranes as alternative explanations for the large ER clusters (Fig. 3D). Consistent with this, correlative light and EM (CLEM) of fixed cells showed that in HEK293T and COS7 cells, SPP-induced ER clusters mainly consist of a highly branched network of irregular ER structures that are loosely stacked in concentric order (Fig. 3E and Fig. S3B). The outermost shell decorated with ribosomes is continuous with rough ER, whereas there are only a few ribosomes inside of the clusters. Inner structures anastomosing between layers establish networks, and their lumen is wider and more densely packed with content (Fig. 3E and Fig. S3B). Similar to the results from confocal microscopy, we observed mitochondria in close vicinity of the ER clusters, but they were never fully embedded (Fig. S3C). Likewise, Golgi stacks were observed in close vicinity of the ER clusters but showed no sign of distortion (Fig. S3D). This result suggests that tight packing of the ER network may affect the access of cytosolic components. To test this idea, we compared RFP-KDEL–stained structures in cells with freshly formed ER clusters after a few minutes of inhibitor washout (Fig. 3F, upper panel) with cells expressing SPP-IRES-GFP for 16 h (Fig. 3F, lower panel). GFP served as a proxy for cytoplasmic content and allowed us to analyze the distribution into cluster regions. Although the overall shape of ER clusters was unchanged, a significantly reduced GFP signal could be observed in the affected regions of cells that had clusters for a prolonged time, suggesting limited access of soluble GFP to cluster-containing regions of the cell (Fig. 3F, lower panel). Apart from this, no significant morphological difference was observed. Taken together with the CLEM experiment of cells expressing SPP for ∼16 h (Fig. 3E and Fig. S3B), these results show that SPP-induced clusters consist of ER zones of high complexity that show usual spatial interaction with mitochondria and the microtubule network but impaired access of other cytosolic components such as translating ribosomes.
Figure 3.

Membrane organization in SPP-induced ER clusters. A, confocal sections of HEK293T cells transfected with WT SPP, RFP-KDEL, and TOMM20-GFP as indicated. Shown are subregions of the cells next to the nuclei (*). Four frames, each from z-stacks at step sizes of 0.3 μm, are displayed. Plot profiles of signal intensities from the yellow lines superimposed on the image at 0.6 μm each are shown next to the z-stacks. The z axis represents the distance of the line, and the y axis represents the arbitrary intensities. Scale bar, 1 μm. B, confocal sections of HEK293T cells transfected with WT SPP, GFP-Sec61β, and α-tubulin-mCherry as in A. Scale bar, 1 μm. C, maximum intensity projection from confocal sections of a mitotic HEK293T cell transfected with WT SPP, GFP-Sec61β, and α-tubulin-mCherry. Scale bar, 1 μm. D, confocal (upper panel) and STED (lower panel) images of a single ER cluster of an HEK293T cell transfected with WT SPP and RFP-KDEL. Four frames, each from a z-stack at step sizes of 0.2 μm, are displayed. Scale bar, 1 μm. E, CLEM of HEK293T cells transfected with RFP-KDEL and SPP-IRES-GFP (WT). Upper left panel, low-power transmission electron micrograph of a cell of interest with fluorescence overlay (upper right panel, fluorescence light microscopy data for RFP and GFP signals). The two boxes depict ER clusters shown at higher resolution in the middle panel. The lower panel shows a closeup of the respective ER clusters. Scale bars, middle panel, 5 μm and lower panel, 1 μm. F, single confocal images from midsections of HEK293T cells transfected with WT SPP, RFP-KDEL, and GFP a few minutes after inhibitor washout (new cluster; upper panel) or 16 h after cluster formation (old cluster; lower panel). Plot profiles of signal intensities from the yellow lines superimposed on each merge image are shown. The z axis represents the distance of the line, and the y axis represents the arbitrary intensities. Scale bars, 10 μm.
Deregulation of SPP detaches the ER from microtubule network
Because we observed the close vicinity of the ER cisterna to microtubules in SPP-induced clusters, we asked how ER–cytoskeleton contact points affect this morphology change. Similar ER clusters have been described when microtubules are depolymerized by nocodazole treatment (46) or upon expression of a dominant-negative mutant of the ER–microtubule linking protein CLIMP63 (31). In contrast, overexpression of WT CLIMP63 leads to an overalignment of the ER with microtubules (31). To test whether the SPP-induced ER phenotype is affected by altered ER–microtubule connection, we coexpressed mCherry-tagged CLIMP63 with SPP-IRES-GFP and quantified the number of SPP-IRES-GFP–expressing cells with ER clusters >1 μm (Fig. 4). Increasing ER–microtubule contact points significantly suppressed the formation of SPP-induced ER clusters (by >30%) (Fig. 4). Conversely, an N-terminal deletion construct (ΔCLIMP63) that is unable to interact with microtubules (31) had no effect on cluster formation (Fig. 4). Because protein kinase C (PKC) phosphorylates two critical serine residues of the CLIMP63 N-terminal domain, leading to ER–microtubule detachment in mitosis (31), we tested whether PKC interferes with SPP-induced ER cluster formation. We followed reformation of ER clusters in SPP-IRES-GFP–expressing cells after SPP inhibitor washout (Fig. 5). Starting from 5 min after inhibitor washout, pronounced ER clusters were observed in control cells (Fig. 5, upper panel). Interestingly, inhibition of PKC activity by chelerythrine chloride significantly slowed down ER cluster formation after SPP inhibitor washout compared with control cells (Fig. 5, lower panel), suggesting that impaired CLIMP63 phosphorylation in somatic cells delays detachment of the ER network from microtubules. Consistent with this, expression of a phosphorylation-deficient CLIMP63 mutant (CLIMP63-3A) reduced cluster formation, whereas phosphomimicking CLIMP63 mutant (CLIMP63-3E) (31) had no effect (Fig. 4). Taken together, these results suggest that uncoupling SPP activity influences ER morphology by suppressing ER–microtubule interaction points. Of note, no significant differences in CLIMP63 steady-state level and stability were observed upon SPP expression as assessed by our proteomics analysis (Fig. 1B and Table S1) and cycloheximide chase experiments (Fig. S4), and we did not detect CLIMP63 interaction in our SPP-DA substrate-trapping experiments (Fig. 1D).
Figure 4.
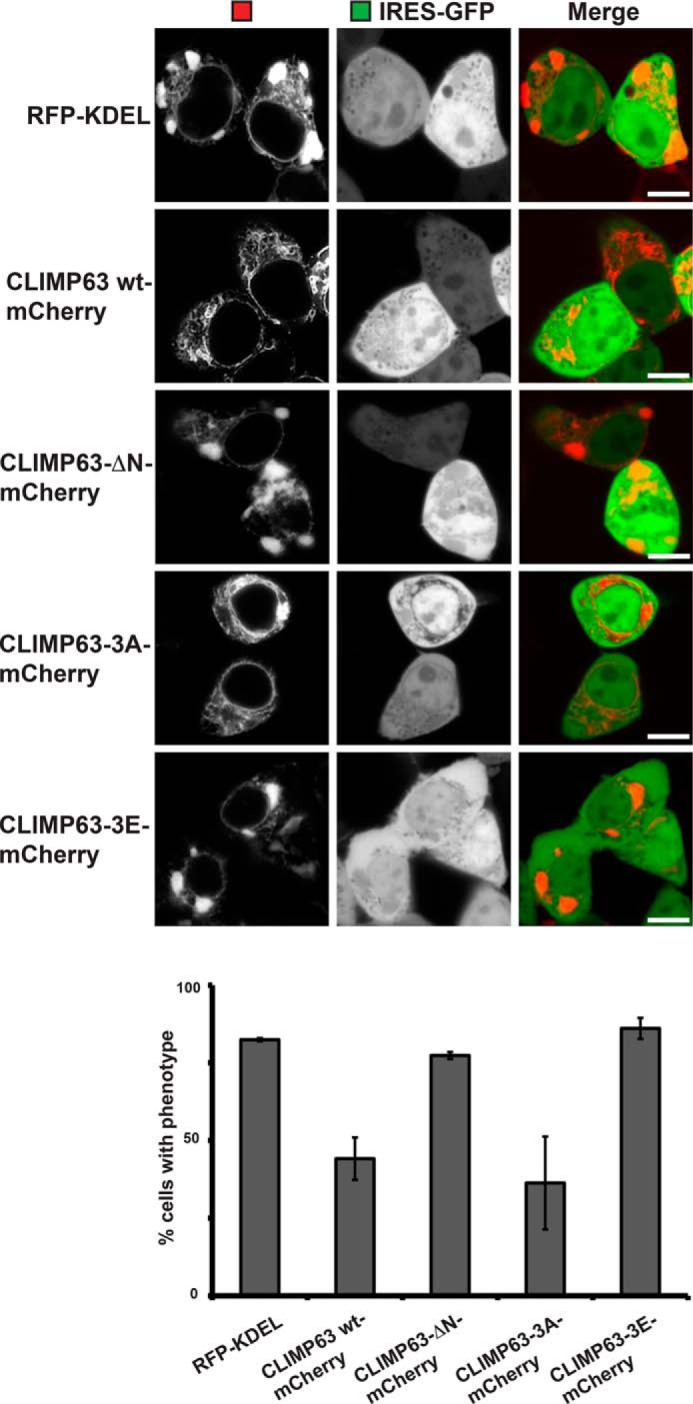
SPP modulates CLIMP63-dependent ER–microtubule contacts. Fluorescence microscopy of HEK293T cells coexpressing SPP-IRES-GFP (WT) and different CLIMP63 constructs with mCherry tag is shown. Quantification of cells with ER morphology change is shown below. Error bars represent S.D. (n = 3; 100 cells were analyzed per condition). Scale bars, 5 μm.
Figure 5.

PKC affects formation of ER clusters. Time-lapse microscopy of HEK293T cells transfected with RFP-KDEL and SPP-IRES-GFP (WT) shows phenotype recovery after SPP inhibitor washout alone (upper panel) or together with PKC inhibitor (lower panel). Scale bars, 5 μm.
Rescue of ER clusters depends on pre-existing pool of STX18 in the Golgi
SPP-triggered ER membrane clusters are readily resolved by treatment with SPP inhibitors. As the rescue occurs in a range of minutes and does not depend on de novo protein synthesis, we asked whether there is a pre-existing pool of a bioactive molecule that can refill the ER pool under SPP inhibition. As the ER is closely associated to the Golgi apparatus and continuous protein transport takes place between both organelles, we asked whether this bioactive protein fraction localizes to the Golgi or ER–Golgi intermediate compartment. SPP inhibitor treatment in the presence of a small-molecule inhibitor of retrograde transport from Golgi to the ER, known as Retro-2 (47), significantly slowed down the rescue by SPP inhibition as ER clusters were still visible after 20 min (Fig. 6A, lower panel). This result suggests that the rapid rescue upon SPP inhibition depends on a second protein pool located at the Golgi apparatus, and SNARE proteins involved in retrograde transport might interfere with SPP-induced ER cluster formation. Consistent with this idea, both STX5 and STX18 have been shown to affect ER morphology (14–16). Therefore, the equilibrium between the ER and Golgi pools of these SNAREs may be crucial for controlling ER morphology. Indeed, ectopic expression of STX18 in inducible stable cell lines significantly reduced the number of cells that show the ER clustering phenotype (Fig. 6B). Consistent with this, treating SPP overexpressing cells with brefeldin A, which leads to redistribution of cis- and medial Golgi components toward the ER (48), dissolved the SPP-induced ER clusters in a similar time range as SPP inhibition (Fig. 6C). Taken together, these results corroborate our model that uncoupling SPP activity has a profound effect on shaping the early secretory pathway. The SNARE STX18 is potentially one of these bioactive substrates, but given the relatively loose substrate specificity of SPP, other regulators of ER morphology may also be affected.
Figure 6.

Modulation of cycling pool of STXs is responsible for ER condensation. A, time-lapse microscopy of HEK293T cells transfected with RFP-KDEL and SPP-IRES-GFP (WT) and treated with 5 μm L685,458 SPP inhibitor alone (upper panel) or together with 30 μm Retro-2 (lower panel) at time point 0. Scale bars, 5 μm. B, formation of SPP-induced ER clusters was suppressed by increased expression of STX18 as shown by fluorescence microscopy of HEK293 T-REx cells and stable HEK293 T-REx-STX18. Quantification of cells with ER clusters >1 μm. Error bars represent S.D. (n = 5; 100 cells were analyzed per condition) was performed, and representative pictures are shown. Significant change between SPP expression in parenteral HEK293 T-REx cells and HEK293 T-REx-STX18 is indicated (**, p < 0.01, Student's t test). Scale bars, 10 μm. C, time-lapse microscopy of HEK293T cells transfected with SPP-IRES-GFP, ER marker RFP-KDEL, and Golgi marker CFP-GalT and treated with 10 μg/ml brefeldin A, showing the rescue of ER morphology phenotype. Scale bars, 5 μm.
Discussion
Maintenance of ER morphology and function are highly interdependent, and multiple mechanisms exist that safeguard key properties such as the organelle proteome, membrane dynamics, and attachment to the cytoskeleton (17). In addition to their canonical role in vesicle trafficking, the two ER-resident SNAREs STX5 and STX18 both play an important role in fine-tuning ER morphology (13–16). In the present study, by a proteomics work flow and subsequent validation in cell-based assays, we identified that SPP cleaves endogenous STX18, leading to an N-terminal cleavage fragment that is released from the membrane and degraded by the proteasome. Because we also uncovered that uncoupled catalytic activity of SPP leads to massive ER rearrangements, we hypothesize that these effects are functionally related. Given the role of SPP in ERAD-mediated abundance control of TA proteins (5), identifying the ER-resident intramembrane protease as a regulator of ER morphogenesis could provide answers to the question of how cells adapt their ER structure in response to varying growth conditions and functional needs.
Uncoupled SPP activity induces highly mobile ER clusters
The most commonly observed type of alteration of the dispersed ER network in tissue culture cells is formation of OSER structures, which involves membrane clustering and can be artificially induced by overexpressing ER integral membrane proteins (38, 49–53). This highly organized ER membrane structure has been linked to pathological conditions such as Emery–Dreifuss disease, torsion dystonia, and Hodgkin's lymphoma (54–56). The commonly accepted model of OSER formation is that the cytosolic domains of ER membrane proteins from opposing membranes interact with each other, leading to the zippering of these membranes in smooth ER structures. It has been shown that especially overexpression of proteins harboring a cytosolic GFP tag with its tendency to dimerize runs the risk to induce structural ER artifacts (38). To this end, in this study, we used primarily untagged SPP expressed from a vector harboring GFP under the control of an IRES to identify SPP-positive cells. All of the previously observed structures, independent of their origin, have a high degree of membrane organization. In contrast, our enhanced light microscopy as well as CLEM analysis of the membrane organization in SPP-induced ER clusters showed no similarity to OSER structures; as in the ER clusters presented here, membranes appear to be randomly raveled and relatively loosely packed. Although the ER network is severely compromised, we observed mitochondria and Golgi stacks in close vicinity to the SPP-induced ER clusters, indicating that regular organelle interaction is maintained. However, prolonged ER cluster occurrence seems to reduce the access of large cytosolic components such as translating ribosomes and shows reduced interaction with microtubules. Despite that, SPP-induced clusters are extremely mobile as they constantly merge and split and change their shape. So far described highly organized ER rearrangements such as OSER structures do not show such dynamic behaviors (38), indicating that the SPP-induced phenotype relies on an alternative mechanism. The physiological relevance of this putative SPP-dependent rheostat of ER morphology remains an important question. Although on a reduced scale, local zones of higher ER complexity have been observed in dendrites of primary neurons and are thought to ensure a spatially defined secretion of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)-type glutamate receptors (32). Recent findings indicate the importance of ER morphology control in highly polarized neurons (57, 58). Hence, it is attractive to speculate that SPP controls the local concentration of ER-modulating proteins and thereby locally tunes secretion dynamics. Interestingly, elevated SPP levels have been shown to facilitate cytokine secretion in tissue culture cells and progression of glioblastoma in mice (34).
How does SPP change ER morphology?
The ER forms an interconnected membrane network within the cell, and, as various as its tasks, the shape and organization can differ. To maintain the diversity in ER morphology, many shaping mechanisms exist (17). We showed that ectopically expressed SPP destabilizes the SNARE protein STX18, which has been implicated in ER morphology control by tuning homotypic fusion (15, 59). Based on the findings of our overexpression system, we hypothesize that also endogenous SPP alters ER morphology by modulating the local concentration of morphogenic ER proteins such as STX18. Linkage to microtubules also impacts ER morphology, and ER phenotypes that are similar to that observed under enhanced SPP activity have been reported in cells where ER–microtubule contact is disturbed (61). For example, silencing of the ER–microtubule linker huntingtin (62) and ectopic expression of the microtubule binding–impaired mutant of CLIMP63 (61) both induce similar changes in ER morphology. Parallels of the phenotype observed in SPP-overexpressing cells and the sensitivity of the SPP inhibitor washout experiment to PKC inhibitor point toward a so far unrecognized role of intramembrane proteolysis in the control of ER–microtubule interaction. In line with this, we found that increasing the amount of ER–microtubule interactions by overexpression of WT CLIMP63 prevented formation of SPP-induced ER clusters. Because we showed that CLIMP63 is not a substrate of SPP and a sheer depletion of an ER structural protein would fail to explain the rapid rescue upon SPP inhibition in the presence of cycloheximide, we conclude that the Golgi harbors a second pool of bioactive molecules that rapidly refills the ER reservoir upon SPP inhibition. However, as several ER proteins have been reported to trigger ER–microtubule contact points (61), the identity of all SPP substrates involved and the exact mechanism of how SPP-mediated ERAD-R impacts the ER morphology remains to be investigated.
SPP-catalyzed cleavage acts as a potent degradation signal
Regulated intramembrane proteolysis is widely used by cells to regulate trafficking and activity of important bioactive molecules (63). However, intramembrane proteolysis is not restricted to limited proteolysis. Recent studies by us and others showed that SPP targets proteins for ERAD, affecting a wide range of physiological processes in the cell (5–7). Because in this context intramembrane proteolysis serves primarily as an abundance control regulator of native proteins, introducing an irreversible post-translational modification that targets proteins for complete destruction by the proteasome, we introduced the term ERAD-R (7). We now extend the role of SPP-mediated ERAD-R to the turnover of the SNARE protein STX18 and processing of a not yet fully defined substrate spectrum that modulates ER morphology. Further investigation into the substrate spectrum of ERAD-R likely will help to explain how SPP affects the dynamics of the ER network and its association with the microtubule network. Another important question is whether SPP-like proteases found in the Golgi (SPPL3) and late secretory pathway (SPPL2a and SPPL2b) (64) also cleave SNARE proteins, potentially introducing a so far unrecognized layer of protein abundance control for this important membrane protein class. In an analogous manner, we recently showed that the ER rhomboid intramembrane protease RHBDL4 tunes ER export of a defined set of cargoes in response to G protein–coupled receptors (65), indicating that the role of intramembrane proteases in controlling secretion dynamics is a more general phenomenon.
Experimental procedures
Plasmids
Constructs based on pcDNA3.1 (Invitrogen) encoding human SPP with a triple HA tag inserted between residue 373 and the C-terminal KKEK ER-retention signal (SPP-HA) and human SPPL3-Myc-KKEK have been described before (6). An open reading frame (ORF) coding human SPP with an S tag (KETAAAKFERQHMDS) inserted between residue 373 and the C-terminal KKEK ER-retention signal was ordered as a gene block (Integrated DNA Technologies) and cloned into pcDNA5/FRT/TO (Invitrogen). SPP-IRES-GFP was cloned into pcDH-IRES-GFP (66). The active-site mutant D265A of SPP was introduced by QuikChange site-directed mutagenesis (Stratagene). STX5 long form (gene block corresponding to UniGene ID Hs. 654602, isoform 1) and STX18 (full-ORF Gateway clone 126176896) were cloned with an N-terminal triple FLAG tag into pcDNA3.1. Human RHBDL4 cloned into pEGFP-N1 and FLAG-tagged human CD44 cloned into pcDNA3.1 were described before (65). Plasmid encoding an ER-targeting signal sequence fused to RFP followed by a KDEL ER-retention signal (RFP-KDEL) (39), human GFP-Sec61β (23), human TOMM20-GFP (66), α-tubulin-mCherry (67), CFP-GalT (68), and human CLIMP63 (32) have been described previously.
Cell lines and transfection
HEK293T cells were grown in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum at 37 °C in 5% CO2. Transient transfections were performed using 25-kDa linear polyethyleneimine (Polysciences) (69). Typically, 1 μg of plasmid encoding substrate candidate and 300 ng of plasmid encoding SPP were used per well of a 6-well plate. Total transfected DNA (2 μg/well) was held constant by the addition of empty plasmid. If not otherwise stated, cells were harvested 24 h after transfection. For inhibition of the proteasome and SPP, 2 μm epoxomicin (Calbiochem) and 50 μm (Z-LL)2-ketone (Calbiochem) were added from stock solutions in dimethyl sulfoxide (DMSO). As a vehicle control, the same amount of DMSO was used. To generate inducible stable cell lines, Flp-In HEK293 T-REx cells were cotransfected with pOG44 (Invitrogen) and either pcDNA5/FRT/TO/SPP-S tag, pcDNA5/FRT/TO/SPP-D265A-S tag, or pcDNA5/FRT/TO/STX18–3xFLAG followed by selection with blasticidin (10 μg/ml) and hygromycin B (100 μg/ml). For microscopy experiments, transfection was done in 8-well Ibidi plates, typically with 50 ng of plasmid encoding SPP and 50 ng of plasmid for ER, Golgi, mitochondrial, or microtubule markers. Total DNA amount was kept at 200 ng by the addition of empty plasmid. If not stated otherwise, cells were fixed with 4% (w/v) paraformaldehyde in PBS or imaged live 16 h after transfection. Inhibitor treatment were done with the following concentrations for the indicated times: 5 μm L685,458 (Calbiochem), 50 μm (Z-LL)2-ketone (Calbiochem), 2 μm epoxomicin (Calbiochem), 100 μg/ml cycloheximide (Sigma-Aldrich), 30 μm Retro-2 (Sigma-Aldrich), 5 μm chelerythrine chloride (Sigma-Aldrich), and 10 μg/ml brefeldin A (Sigma-Aldrich).
Antibodies
The following antibodies were used: mouse monoclonal anti-FLAG (M2; Sigma-Aldrich), mouse monoclonal anti-HA (HA.11; Covance), mouse monoclonal anti-Myc (9B11; New England Biolabs), rabbit polyclonal anti-syntaxin-18 and -syntaxin-5 (Synaptic Systems), mouse monoclonal anti-CLIMP63 (G1/296; Enzo Life Sciences), mouse monoclonal anti-β actin (Sigma-Aldrich), and rabbit polyclonal anti-calnexin (Abcam).
Cellular fractionation and proteomics analysis
For identification of SPP substrates by differential organelle proteomics, we used SILAC. HEK293T cells were grown for five doublings in SILAC medium supplemented with heavy amino acids (l-[13C6,15N4]Arg and l-[13C6,15N2]Lys; Silantes), medium amino acids (l-[13C6]Arg and 4,4,5,5-d4-l-Lys; Silantes), or unlabeled amino acids before transfection. For the SPP gain-of-function assay, per condition (SPP WT, SPP-DA, or mock transfection) 3 × 150-mm culture plates were transiently transfected with polyethyleneimine as described above. At day of harvest, cells were detached by cold PBS-EDTA, and an equal number of cells from each transfection were resuspended in hypotonic buffer (10 mm HEPES-KOH, pH 7.4, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT) containing 10 μg/ml phenylmethylsulfonyl fluoride and Complete EDTA-free protease inhibitor mixture (Roche Applied Science). After 10-min incubation on ice, cells were lysed by passing five times through a 27-gauge needle. Cellular debris and nuclei were discarded after centrifugation at 1,000 × g for 5 min at 4 °C. The supernatant was spun at 100,000 × g for 20 min at 4 °C. The membrane pellet was resuspended in RM buffer (250 mm sucrose, 50 mm HEPES-KOH, pH 7.4, 50 mm KOAc, 2 mm Mg(OAc)2, 1 mm DTT) and extracted by 500 mm KOAc, 50 mm EDTA for 15 min followed by centrifugation through a high-salt sucrose cushion (500 mm sucrose, 50 mm HEPES-KOH, pH 7.4, 500 mm KOAc, 5 mm Mg(OAc)2). The pellet thereof was resuspended in a freshly prepared, ice-cold sodium carbonate solution (100 mm) and transferred onto an alkaline sucrose cushion (125 mm sucrose, 100 mm Na2CO3) followed by centrifugation for 20 min at 100,000 × g at 4 °C. The pellet was resuspended in RM buffer and snap frozen in liquid nitrogen.
For proteomics analysis, protein samples were dissolved in SDS sample buffer (see below) and separated by SDS-PAGE for 1 cm. Each lane was cut in two gel pieces, which were processed as described with minor modifications (70). In brief, after reduction with DTT and alkylation with iodoacetamide, trypsin digestion was done overnight at 37 °C. The reaction was quenched by addition of 20 μl of 0.1% trifluoroacetic acid (TFA; Biosolve, Valkenswaard), and the supernatant was dried in a vacuum concentrator before LC-MS analysis. Nanoflow LC-MS2 analysis was performed with an Ultimate 3000 LC system coupled to an QExactive HF mass spectrometer (Thermo Fischer Scientific). Dried samples were dissolved in 0.1% TFA and loaded on a C18 Acclaim PepMap100 trap-column (Thermo Fisher Scientific) with a flow rate of 30 μl/min 0.1% TFA. Peptides were eluted and separated on an C18 Acclaim PepMap RSLC analytical column (75 μm × 250 mm; Thermo Fisher Scientific) with a flow rate of 300 nl/min in a 120-min gradient of 3% buffer A (0.1% formic acid) to 40% buffer B (0.1% formic acid, acetonitrile). The mass spectrometer was operated in data-dependent acquisition mode, automatically switching between MS and MS2. Collision-induced dissociation MS2 spectra were generated for up to 20 precursors with normalized collision energy of 29%.
Raw files were processed using MaxQuant version 1.5.3.30 (71) for peptide identification and quantification. MS2 spectra were searched against the UniProt human proteome database and the contaminants database by Andromeda search engine with the following parameters: carbamidomethylation of cysteine residues, acetylation of protein N termini, and oxidation of Met were considered as variable modifications, and trypsin as the proteolytic enzyme with up to two missed cleavages was allowed. The maximum false discovery rate for proteins and peptides was 0.01, and a minimum peptide length of 7 amino acids was required. All other parameters were the default parameters of MaxQuant. Quantitative normalized ratios were calculated by MaxQuant and used for further data analysis by Perseus (72).
SDS-PAGE and Western blotting
Proteins were solubilized in SDS sample buffer (50 mm Tris-Cl, pH 6.8, 10 mm EDTA, 5% glycerol, 2% SDS, 0.01% bromphenol blue) containing 5% β-mercaptoethanol. All samples were incubated for 15 min at 65 °C. For deglycosylation, solubilized proteins were treated with EndoH and PNGaseF (New England Biolabs) according to the manufacturer's protocol. For Western blot analysis, proteins were separated by SDS-PAGE using Tris-glycine acrylamide gels and transferred onto polyvinylidene difluoride membrane followed by enhanced chemiluminescence analysis (Pierce) using the LAS-4000 system (Fuji). Data shown are representative of at least three independent experiments. For quantification, we used Multi Gauge (Fuji) or ImageJ/Fiji software and data acquired from the LAS-4000.
Chase experiments
Cycloheximide (100 μg/ml) chase was performed 24 h after transfection of HEK293T cells or after doxycycline induction of HEK293 T-REx stable cell lines. Cell extracts were subjected to SDS-PAGE and Western blot analysis. For pulse-label chase experiments, cells were metabolically labeled for 10 min with 55 μCi/ml [35S]methionine/cysteine protein labeling mixture (PerkinElmer Life Sciences) and chased in cold medium. Radiolabeled proteins were then isolated by immunoprecipitation and analyzed by SDS-PAGE and autoradiography.
Immunoprecipitation
For immunoprecipitation experiments, 2 mm cross-linker dithiobis(succinimidyl propionate) (Pierce) in PBS was added to the cells, incubated for 30 min on ice, and quenched with 20 mm Tris-Cl, pH 7.5, for 15 min at room temperature. Cells were then solubilized with 1% (w/v) CHAPS in IP buffer (50 mm HEPES-KOH, pH 7.4, 150 mm KCl, 2 mm Mg(OAc)2, 10% glycerol, 1 mm EGTA) containing 10 μg/ml phenylmethylsulfonyl fluoride and Complete EDTA-free protease inhibitor mixture. Nonsolubilized proteins were removed by centrifugation. Immunoprecipitation with the indicated antibody was performed with overnight incubation at 4 °C. Immunoprecipitates were washed three times in IP buffer containing 0.1% (w/v) CHAPS and eluted in SDS sample buffer by heating at 65 °C.
Fluorescence microscopy
Confocal microscopy for Figs. 2, 4, 5, 6 and S2 was performed on a Zeiss LSM 780 (Carl Zeiss) and on a Leica TCS SP8 (Leica Microsystems) system for all other confocal images. Lasers used were the 458, 488, and 514 nm lines of an argon laser; a 561 nm diode-pumped solid-state laser (Carl Zeiss); and a white light laser (Leica Microsystems). Plan Apochromat objectives were used for the experiments on the microscopes: 40×/1.3 numerical aperture (NA) oil, 63×/1.4 NA oil, 100×/1.4 NA oil (Carl Zeiss), 100×/1.4 NA oil, and 93×/1.3 glycerol (Leica Microsystems). All pictures were taken with a pinhole setting of 1 airy unit. Zeiss ZEN 2010 and Leica Application Suite (LAS) software were used during imaging. For 3D projections, z-stack image series were taken, and maximum intensity was projected with ImageJ/Fiji. Quantification of cells with ER clusters was performed by a microscopist without knowledge of the actual sample ID to guarantee unbiased data collection.
STED microscopy was performed on a Leica TCS SP8 STED 3X system with a high-contrast Plan Apochromat CS2 93×/1.3 NA glycerol objective (Leica Microsystems). Confocal and STED images were acquired in sequential mode using a white light laser at 561 nm and a STED laser of 660 nm for mCherry imaging. The voxel sizes were 20 nm in x and y (lateral) and 200 nm in z (axial) directions. The pinhole was set to 1 airy unit. Leica's HyD detectors were used for signal detection. All STED images and their corresponding confocal images were deconvolved with Huygens Professional (Scientific Volume Imaging (SVI)).
Correlative light and EM
CLEM studies were performed as described (73). Briefly, COS7 and HEK293T cells were transiently cotransfected with SPP and RFP-KDEL, grown on gridded glass coverslips (175 μm; Cellocate, Eppendorf), fixed in buffered aldehyde (4% paraformaldehyde, 2% glutaraldehyde, 1 mm MgCl2, 1 mm CaCl2, 100 mm cacodylate, pH 7.3), and then prepared for fluorescence light microscopy (LM) (rinsed with cacodylate buffer and mounted in Vectashield). LM image acquisition (Zeiss LSM 780) included low-power views of sites of interest to document the grid position for later relocation. Coverslips were recovered by floating on buffer, cell layers were post-fixed with OsO4 following uranyl en bloc, dehydration, and flat-embedding in epoxide (glycidether, nadic methyl anhydride, and dodecenyl succinic anhydride; Serva). From the cured resin blocks, the glass coverslips were detached under liquid nitrogen. Once the grid imprint became visible on the shiny block face, ultrathin sections were prepared from the site of interest. Transmission EM images (Zeiss EM910/Ditabis image plate scanner) taken at low power allowed relocation of cells of interest and comparison with the LM data, and basic affine image matching using open source computer software (Fiji) allowed correlation of fluorescence signals with cellular ultrastructure.
Author contributions
D. A., N. S. M., and M. K. L. conceptualization; D. A., N. S. M., R. H., H. L., K. R., M. N., and M. K. L. data curation; D. A., N. S. M., and M. K. L. formal analysis; D. A., N. S. M., R. H., H. L., and M. K. L. validation; D. A., N. S. M., R. H., H. L., K. R., M. N., and M. K. L. investigation; D. A., N. S. M., R. H., H. L., K. R., and M. K. L. visualization; D. A., N. S. M., H. L., K. R., and M. K. L. methodology; D. A., N. S. M., and M. K. L. writing-original draft; D. A., H. L., and M. K. L. writing-review and editing; M. K. L. supervision; M. K. L. funding acquisition; M. K. L. project administration.
Supplementary Material
Acknowledgments
We thank Anne-Lore Schlaitz and Sara Suna Yücel for critical reading of the manuscript and Bernd Hessling and Thomas Ruppert (ZMBH, MS facility) for MS analysis.
This work was supported by Deutsche Forschungsgemeinschaft Grants LE 2749/2-1, ZUK 49/2 6.1 DKFZ-ZMBH-Alliance, and INST 35/1485-1 FUGG and a Boehringer Ingelheim Fonds fellowship (to N. S. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4, Tables S1 and S2, and Movies S1 and S2.
- ER
- endoplasmic reticulum
- SPP
- signal peptide peptidase
- ERAD
- ER-associated degradation
- ERAD-R
- regulatory branch of ERAD
- SNARE
- soluble NSF attachment protein receptor
- NSF
- N-ethylmaleimide–sensitive factor
- TM
- transmembrane
- TA
- tail-anchored
- STX
- syntaxin
- SILAC
- stable isotope labeling with amino acids in cell culture
- Z
- benzyloxycarbonyl
- EndoH
- endoglycosidase H
- PNGaseF
- peptide:N-glycosidase F
- RFP
- red fluorescent protein
- OSER
- organized smooth ER
- IRES
- internal ribosomal entry site
- STED
- stimulated emission depletion
- CLEM
- correlative light and EM
- PKC
- protein kinase C
- CFP
- cyan fluorescent protein
- GalT
- galactosyltransferase
- NA
- numerical aperture
- LM
- light microscopy
- HA
- hemagglutinin
- SPP-DA
- SPP-D265A.
References
- 1. Piper R. C., and Lehner P. J. (2011) Endosomal transport via ubiquitination. Trends Cell Biol. 21, 647–655 10.1016/j.tcb.2011.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Christianson J. C., and Ye Y. (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335 10.1038/nsmb.2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Balut C. M., Loch C. M., and Devor D. C. (2011) Role of ubiquitylation and USP8-dependent deubiquitylation in the endocytosis and lysosomal targeting of plasma membrane KCa3.1. FASEB J. 25, 3938–3948 10.1096/fj.11-187005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Z. R., Bonifacino J. S., and Hegde R. S. (2013) Deubiquitinases sharpen substrate discrimination during membrane protein degradation from the ER. Cell 154, 609–622 10.1016/j.cell.2013.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Boname J. M., Bloor S., Wandel M. P., Nathan J. A., Antrobus R., Dingwell K. S., Thurston T. L., Smith D. L., Smith J. C., Randow F., and Lehner P. J. (2014) Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J. Cell Biol. 205, 847–862 10.1083/jcb.201312009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen C. Y., Malchus N. S., Hehn B., Stelzer W., Avci D., Langosch D., and Lemberg M. K. (2014) Signal peptide peptidase functions in ERAD to cleave the unfolded protein response regulator XBP1u. EMBO J. 33, 2492–2506 10.15252/embj.201488208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Avci D., Fuchs S., Schrul B., Fukumori A., Breker M., Frumkin I., Chen C. Y., Biniossek M. L., Kremmer E., Schilling O., Steiner H., Schuldiner M., and Lemberg M. K. (2014) The yeast ER-intramembrane protease Ypf1 refines nutrient sensing by regulating transporter abundance. Mol. Cell 56, 630–640 10.1016/j.molcel.2014.10.012 [DOI] [PubMed] [Google Scholar]
- 8. Stefanovic-Barrett S., Dickson A. S., Burr S. P., Williamson J. C., Lobb I. T., van den Boomen D. J., Lehner P. J., and Nathan J. A. (2018) MARCH6 and TRC8 facilitate the quality control of cytosolic and tail-anchored proteins. EMBO Rep. 19, e45603 10.15252/embr.201745603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Avci D., and Lemberg M. K. (2015) Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol. 25, 611–622 10.1016/j.tcb.2015.07.003 [DOI] [PubMed] [Google Scholar]
- 10. Lippincott-Schwartz J., Roberts T. H., and Hirschberg K. (2000) Secretory protein trafficking and organelle dynamics in living cells. Annu. Rev. Cell Dev. Biol. 16, 557–589 10.1146/annurev.cellbio.16.1.557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y. A., and Scheller R. H. (2001) SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2, 98–106 10.1038/35052017 [DOI] [PubMed] [Google Scholar]
- 12. Hegde R. S., and Keenan R. J. (2011) Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 12, 787–798 10.1038/nrm3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee M., Ko Y. J., Moon Y., Han M., Kim H. W., Lee S. H., Kang K., and Jun Y. (2015) SNAREs support atlastin-mediated homotypic ER fusion in Saccharomyces cerevisiae. J. Cell Biol. 210, 451–470 10.1083/jcb.201501043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Miyazaki K., Wakana Y., Noda C., Arasaki K., Furuno A., and Tagaya M. (2012) Contribution of the long form of syntaxin 5 to the organization of the endoplasmic reticulum. J. Cell Sci. 125, 5658–5666 10.1242/jcs.105304 [DOI] [PubMed] [Google Scholar]
- 15. Kano F., Kondo H., Yamamoto A., Kaneko Y., Uchiyama K., Hosokawa N., Nagata K., and Murata M. (2005) NSF/SNAPs and p97/p47/VCIP135 are sequentially required for cell cycle-dependent reformation of the ER network. Genes Cells 10, 989–999 10.1111/j.1365-2443.2005.00894.x [DOI] [PubMed] [Google Scholar]
- 16. Nakajima K., Hirose H., Taniguchi M., Kurashina H., Arasaki K., Nagahama M., Tani K., Yamamoto A., and Tagaya M. (2004) Involvement of BNIP1 in apoptosis and endoplasmic reticulum membrane fusion. EMBO J. 23, 3216–3226 10.1038/sj.emboj.7600333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shibata Y., Hu J., Kozlov M. M., and Rapoport T. A. (2009) Mechanisms shaping the membranes of cellular organelles. Annu. Rev. Cell Dev. Biol. 25, 329–354 10.1146/annurev.cellbio.042308.113324 [DOI] [PubMed] [Google Scholar]
- 18. Nixon-Abell J., Obara C. J., Weigel A. V., Li D., Legant W. R., Xu C. S., Pasolli H. A., Harvey K., Hess H. F., Betzig E., Blackstone C., and Lippincott-Schwartz J. (2016) Increased spatiotemporal resolution reveals highly dynamic dense tubular matrices in the peripheral ER. Science 354, aaf3928 10.1126/science.aaf3928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baumann O., and Walz B. (2001) Endoplasmic reticulum of animal cells and its organization into structural and functional domains. Int. Rev. Cytol. 205, 149–214 10.1016/S0074-7696(01)05004-5 [DOI] [PubMed] [Google Scholar]
- 20. Westrate L. M., Lee J. E., Prinz W. A., and Voeltz G. K. (2015) Form follows function: the importance of endoplasmic reticulum shape. Annu. Rev. Biochem. 84, 791–811 10.1146/annurev-biochem-072711-163501 [DOI] [PubMed] [Google Scholar]
- 21. Chen S., Novick P., and Ferro-Novick S. (2012) ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell Biol. 14, 707–716 10.1038/ncb2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Craene J. O., Coleman J., Estrada de Martin P., Pypaert M., Anderson S., Yates J. R. 3rd, Ferro-Novick S., and Novick P. (2006) Rtn1p is involved in structuring the cortical endoplasmic reticulum. Mol. Biol. Cell 17, 3009–3020 10.1091/mbc.e06-01-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Voeltz G. K., Prinz W. A., Shibata Y., Rist J. M., and Rapoport T. A. (2006) A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell 124, 573–586 10.1016/j.cell.2005.11.047 [DOI] [PubMed] [Google Scholar]
- 24. Bian X., Klemm R. W., Liu T. Y., Zhang M., Sun S., Sui X., Liu X., Rapoport T. A., and Hu J. (2011) Structures of the atlastin GTPase provide insight into homotypic fusion of endoplasmic reticulum membranes. Proc. Natl. Acad. Sci. U.S.A. 108, 3976–3981 10.1073/pnas.1101643108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hu J., Shibata Y., Zhu P. P., Voss C., Rismanchi N., Prinz W. A., Rapoport T. A., and Blackstone C. (2009) A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 138, 549–561 10.1016/j.cell.2009.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orso G., Pendin D., Liu S., Tosetto J., Moss T. J., Faust J. E., Micaroni M., Egorova A., Martinuzzi A., McNew J. A., and Daga A. (2009) Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature 460, 978–983 10.1038/nature08280 [DOI] [PubMed] [Google Scholar]
- 27. Gillingham A. K., Sinka R., Torres I. L., Lilley K. S., and Munro S. (2014) Toward a comprehensive map of the effectors of rab GTPases. Dev. Cell 31, 358–373 10.1016/j.devcel.2014.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pendin D., McNew J. A., and Daga A. (2011) Balancing ER dynamics: shaping, bending, severing, and mending membranes. Curr. Opin. Cell Biol. 23, 435–442 10.1016/j.ceb.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen S., Novick P., and Ferro-Novick S. (2013) ER structure and function. Curr. Opin. Cell Biol. 25, 428–433 10.1016/j.ceb.2013.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park S. H., and Blackstone C. (2010) Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 11, 515–521 10.1038/embor.2010.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vedrenne C., Klopfenstein D. R., and Hauri H. P. (2005) Phosphorylation controls CLIMP-63-mediated anchoring of the endoplasmic reticulum to microtubules. Mol. Biol. Cell 16, 1928–1937 10.1091/mbc.e04-07-0554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cui-Wang T., Hanus C., Cui T., Helton T., Bourne J., Watson D., Harris K. M., and Ehlers M. D. (2012) Local zones of endoplasmic reticulum complexity confine cargo in neuronal dendrites. Cell 148, 309–321 10.1016/j.cell.2011.11.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang Z., Li C., Kang B., Gao G., Li C., and Zhang Z. (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45, W98–W102 10.1093/nar/gkx247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wei J. W., Cai J. Q., Fang C., Tan Y. L., Huang K., Yang C., Chen Q., Jiang C. L., and Kang C. S. (2017) Signal peptide peptidase, encoded by HM13, contributes to tumor progression by affecting EGFRvIII secretion profiles in glioblastoma. CNS Neurosci. Ther. 23, 257–265 10.1111/cns.12672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hsu F. F., Chou Y. T., Chiang M. T., Li F. A., Yeh C. T., Lee W. H., and Chau L. Y. (2019) Signal peptide peptidase promotes tumor progression via facilitating FKBP8 degradation. Oncogene, in press 10.1038/s41388-018-0539-y [DOI] [PubMed] [Google Scholar]
- 36. Weihofen A., Binns K., Lemberg M. K., Ashman K., and Martoglio B. (2002) Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296, 2215–2218 10.1126/science.1070925 [DOI] [PubMed] [Google Scholar]
- 37. Dev K. K., Chatterjee S., Osinde M., Stauffer D., Morgan H., Kobialko M., Dengler U., Rueeger H., Martoglio B., and Rovelli G. (2006) Signal peptide peptidase dependent cleavage of type II transmembrane substrates releases intracellular and extracellular signals. Eur. J. Pharmacol. 540, 10–17 10.1016/j.ejphar.2006.04.011 [DOI] [PubMed] [Google Scholar]
- 38. Snapp E. L., Hegde R. S., Francolini M., Lombardo F., Colombo S., Pedrazzini E., Borgese N., and Lippincott-Schwartz J. (2003) Formation of stacked ER cisternae by low affinity protein interactions. J. Cell Biol. 163, 257–269 10.1083/jcb.200306020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Snapp E. L., Sharma A., Lippincott-Schwartz J., and Hegde R. S. (2006) Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc. Natl. Acad. Sci. U.S.A. 103, 6536–6541 10.1073/pnas.0510657103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Friedmann E., Lemberg M. K., Weihofen A., Dev K. K., Dengler U., Rovelli G., and Martoglio B. (2004) Consensus analysis of signal peptide peptidase and homologous human aspartic proteases reveals opposite topology of catalytic domains compared with presenilins. J. Biol. Chem. 279, 50790–50798 10.1074/jbc.M407898200 [DOI] [PubMed] [Google Scholar]
- 41. Voss M., Fukumori A., Kuhn P. H., Künzel U., Klier B., Grammer G., Haug-Kröper M., Kremmer E., Lichtenthaler S. F., Steiner H., Schröder B., Haass C., and Fluhrer R. (2012) Foamy virus envelope protein is a substrate for signal peptide peptidase-like 3 (SPPL3). J. Biol. Chem. 287, 43401–43409 10.1074/jbc.M112.371369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fleig L., Bergbold N., Sahasrabudhe P., Geiger B., Kaltak L., and Lemberg M. K. (2012) Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol. Cell 47, 558–569 10.1016/j.molcel.2012.06.008 [DOI] [PubMed] [Google Scholar]
- 43. Weihofen A., Lemberg M. K., Friedmann E., Rueeger H., Schmitz A., Paganetti P., Rovelli G., and Martoglio B. (2003) Targeting presenilin-type aspartic protease signal peptide peptidase with γ-secretase inhibitors. J. Biol. Chem. 278, 16528–16533 10.1074/jbc.M301372200 [DOI] [PubMed] [Google Scholar]
- 44. Simmen T., and Herrera-Cruz M. S. (2018) Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol. 53, 61–69 10.1016/j.ceb.2018.04.014 [DOI] [PubMed] [Google Scholar]
- 45. Hell S. W., and Wichmann J. (1994) Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19, 780–782 10.1364/OL.19.000780 [DOI] [PubMed] [Google Scholar]
- 46. Terasaki M., Chen L. B., and Fujiwara K. (1986) Microtubules and the endoplasmic reticulum are highly interdependent structures. J. Cell Biol. 103, 1557–1568 10.1083/jcb.103.4.1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stechmann B., Bai S. K., Gobbo E., Lopez R., Merer G., Pinchard S., Panigai L., Tenza D., Raposo G., Beaumelle B., Sauvaire D., Gillet D., Johannes L., and Barbier J. (2010) Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 141, 231–242 10.1016/j.cell.2010.01.043 [DOI] [PubMed] [Google Scholar]
- 48. Lippincott-Schwartz J., Yuan L. C., Bonifacino J. S., and Klausner R. D. (1989) Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56, 801–813 10.1016/0092-8674(89)90685-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gong F. C., Giddings T. H., Meehl J. B., Staehelin L. A., and Galbraith D. W. (1996) Z-membranes: artificial organelles for overexpressing recombinant integral membrane proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 2219–2223 10.1073/pnas.93.5.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Anderson R. G., Orci L., Brown M. S., Garcia-Segura L. M., and Goldstein J. L. (1983) Ultrastructural analysis of crystalloid endoplasmic reticulum in UT-1 cells and its disappearance in response to cholesterol. J. Cell Sci. 63, 1–20 [DOI] [PubMed] [Google Scholar]
- 51. Korkhov V. M., and Zuber B. (2009) Direct observation of molecular arrays in the organized smooth endoplasmic reticulum. BMC Cell Biol. 10, 59 10.1186/1471-2121-10-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lenormand C., Spiegelhalter C., Cinquin B., Bardin S., Bausinger H., Angénieux C., Eckly A., Proamer F., Wall D., Lich B., Tourne S., Hanau D., Schwab Y., Salamero J., and de la Salle H. (2013) Birbeck granule-like “organized smooth endoplasmic reticulum” resulting from the expression of a cytoplasmic YFP-tagged langerin. PLoS One 8, e60813 10.1371/journal.pone.0060813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Costantini L. M., Fossati M., Francolini M., and Snapp E. L. (2012) Assessing the tendency of fluorescent proteins to oligomerize under physiologic conditions. Traffic 13, 643–649 10.1111/j.1600-0854.2012.01336.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fidziańska A., Rowińska-Marcinska K., and Hausmanowa-Petrusewicz I. (2004) Coexistence of X-linked recessive Emery-Dreifuss muscular dystrophy with inclusion body myositis-like morphology. Acta Neuropathol. 107, 197–203 10.1007/s00401-003-0794-y [DOI] [PubMed] [Google Scholar]
- 55. Gonzalez-Alegre P., and Paulson H. L. (2004) Aberrant cellular behavior of mutant torsinA implicates nuclear envelope dysfunction in DYT1 dystonia. J. Neurosci. 24, 2593–2601 10.1523/JNEUROSCI.4461-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Parmley R. T., Spicer S. S., and Garvin A. J. (1976) Multilaminar endoplasmic reticulum and abnormal mitosis in Hodgkin tumor cells. Cancer Res. 36, 1717–1724 [PubMed] [Google Scholar]
- 57. Valenzuela J. I., Jaureguiberry-Bravo M., and Couve A. (2011) Neuronal protein trafficking: emerging consequences of endoplasmic reticulum dynamics. Mol. Cell. Neurosci. 48, 269–277 10.1016/j.mcn.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 58. Renvoisé B., and Blackstone C. (2010) Emerging themes of ER organization in the development and maintenance of axons. Curr. Opin. Neurobiol. 20, 531–537 10.1016/j.conb.2010.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Uchiyama K., Jokitalo E., Kano F., Murata M., Zhang X., Canas B., Newman R., Rabouille C., Pappin D., Freemont P., and Kondo H. (2002) VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J. Cell Biol. 159, 855–866 10.1083/jcb.200208112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deleted in proof.
- 61. Vedrenne C., and Hauri H. P. (2006) Morphogenesis of the endoplasmic reticulum: beyond active membrane expansion. Traffic 7, 639–646 10.1111/j.1600-0854.2006.00419.x [DOI] [PubMed] [Google Scholar]
- 62. Omi K., Hachiya N. S., Tokunaga K., and Kaneko K. (2005) siRNA-mediated inhibition of endogenous Huntington disease gene expression induces an aberrant configuration of the ER network in vitro. Biochem. Biophys. Res. Commun. 338, 1229–1235 10.1016/j.bbrc.2005.10.061 [DOI] [PubMed] [Google Scholar]
- 63. Lemberg M. K. (2011) Intramembrane proteolysis in regulated protein trafficking. Traffic 12, 1109–1118 10.1111/j.1600-0854.2011.01219.x [DOI] [PubMed] [Google Scholar]
- 64. Mentrup T., Fluhrer R., and Schröder B. (2017) Latest emerging functions of SPP/SPPL intramembrane proteases. Eur. J. Cell Biol. 96, 372–382 10.1016/j.ejcb.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 65. Wunderle L., Knopf J. D., Kühnle N., Morlé A., Hehn B., Adrain C., Strisovsky K., Freeman M., and Lemberg M. K. (2016) Rhomboid intramembrane protease RHBDL4 triggers ER-export and non-canonical secretion of membrane-anchored TGFα. Sci. Rep. 6, 27342 10.1038/srep27342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Meissner C., Lorenz H., Weihofen A., Selkoe D. J., and Lemberg M. K. (2011) The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 117, 856–867 10.1111/j.1471-4159.2011.07253.x [DOI] [PubMed] [Google Scholar]
- 67. Friedman J. R., Webster B. M., Mastronarde D. N., Verhey K. J., and Voeltz G. K. (2010) ER sliding dynamics and ER-mitochondrial contacts occur on acetylated microtubules. J. Cell Biol. 190, 363–375 10.1083/jcb.200911024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Cole N. B., Smith C. L., Sciaky N., Terasaki M., Edidin M., and Lippincott-Schwartz J. (1996) Diffusional mobility of Golgi proteins in membranes of living cells. Science 273, 797–801 10.1126/science.273.5276.797 [DOI] [PubMed] [Google Scholar]
- 69. Durocher Y., Perret S., and Kamen A. (2002) High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 30, E9 10.1093/nar/30.2.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fecher-Trost C., Wissenbach U., Beck A., Schalkowsky P., Stoerger C., Doerr J., Dembek A., Simon-Thomas M., Weber A., Wollenberg P., Ruppert T., Middendorff R., Maurer H. H., and Flockerzi V. (2013) The in vivo TRPV6 protein starts at a non-AUG triplet, decoded as methionine, upstream of canonical initiation at AUG. J. Biol. Chem. 288, 16629–16644 10.1074/jbc.M113.469726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 10.1038/nbt.1511 [DOI] [PubMed] [Google Scholar]
- 72. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 10.1038/nmeth.3901 [DOI] [PubMed] [Google Scholar]
- 73. Richter K., Reichenzeller M., Görisch S. M., Schmidt U., Scheuermann M. O., Herrmann H., and Lichter P. (2005) Characterization of a nuclear compartment shared by nuclear bodies applying ectopic protein expression and correlative light and electron microscopy. Exp. Cell Res. 303, 128–137 10.1016/j.yexcr.2004.09.022 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.