

Abstract
Background
Pathogenic variants in FBN1 cause autosomal dominant Marfan syndrome but can also be found in patients presenting with apparently isolated features of Marfan syndrome. Moreover, several families with autosomal recessive Marfan syndrome caused by pathogenic variants in FBN1 have been described. The aim of this report was to underline the clinical variability that can be associated with the pathogenic variant c.1453C>T, p.(Arg485Cys) in FBN1.
Methods
We provide the clinical details of two autosomal dominant families with this specific FBN1 variant, which was previously associated with autosomal recessive Marfan syndrome.
Results
Clinical data of 14 individuals carrying this variant from these two families were collected retrospectively. In both families, the diagnosis of autosomal dominant Marfan syndrome was established based on the characteristics of the variant and the phenotype which includes aortic aneurysms and dissections. Of interest, in one of the families, multiple relatives were diagnosed with early onset abdominal aortic aneurysms.
Conclusion
In conclusion, FBN1 variant c.1453C>T, p.(Arg485Cys) is a pathogenic variant that can cause autosomal dominant Marfan syndrome characterized by a high degree of clinical variability and apparently isolated early onset familial abdominal aortic aneurysms.
Keywords: abdominal aortic aneurysm, autosomal dominant inheritance, autosomal recessive inheritance, clinical heterogeneity, FBN1, Marfan syndrome
1. INTRODUCTION
Marfan syndrome (MFS, OMIM #154700) is a multisystem disorder with an estimated prevalence of 1 in 5,000–10,000. MFS is caused by pathogenic variants in FBN1 (OMIM #134797), encoding fibrillin‐1 and is classically characterized by autosomal dominant inheritance (Dietz et al., 1991). However, several MFS families with an apparently autosomal recessive mode of inheritance have been reported (Fried & Krakowsky, 1977; Hilhorst‐Hofstee et al., 2010; Khan, Bolz, & Bergmann, 2014; Vries, Pals, Odink, & Hamel, 2007). A large proportion of pathogenic FBN1 variants causing MFS are missense variants, commonly occurring in EGF‐like domains and involving cysteine residue substitutions with a predicted dominant negative effect (Dietz et al., 1993). MFS is classically characterized by skeletal features, ectopia lentis (EL) and thoracic aortic aneurysms and dissections. The diagnosis is based on the revised Ghent criteria (Loeys et al., 2010). Diagnosing MFS is essential since cardiological surveillance and, when indicated, timely aortic surgery is lifesaving (Cameron et al., 2009). The most feared complication of MFS, aortic dissection, is reported in up to 50% of undiagnosed MFS patients and may be the presenting feature of unrecognized MFS (Ammash, Sundt, & Connolly, 2008). Aortic aneurysms and dissections in MFS are typically located in the aortic root and ascending aorta; however, the descending and abdominal aorta may be involved as well (Engelfriet, Boersma, Tijssen, Bouma, & Mulder, 2006; Loeys et al., 2010; Mariucci et al., 2013; Wolfgarten, Krüger, & Gawenda, 2001). Pathogenic variants in FBN1 may result in classical MFS but have also been reported in families presenting with, for example, apparent isolated thoracic aortic aneurysms and dissections (Wang et al., 2013).
The clinical features of two families with autosomal dominant MFS caused by FBN1 variant c.1453C>T, p.(Arg485Cys) and a high rate of abdominal aneurysms is presented here. Homozygosity for this variant was previously reported to cause autosomal recessive MFS in a consanguineous family (Vries et al., 2007). In addition, this variant was reported in a heterozygous state in one patient in a Taiwanese MFS cohort (Hung et al., 2009). Only limited clinical information was provided in this publication. Our report illustrates the importance of clinical follow‐up in FBN1 mutation carriers, irrespective of previously reported phenotypes associated with that specific variant and suggested mode of inheritance.
2. MATERIAL AND METHODS
We retrospectively collected the clinical data of two families (n = 14 patients) with the heterozygous c.1453C>T variant in FBN1 (NC_000015.9(NM_000138.4):c.1453C>T p.(Arg485Cys)). The families were referred for DNA diagnostics by their clinical geneticists from VU University Medical Center and Leiden University Medical Center, the Netherlands. Informed consent for DNA diagnostics was obtained from all patients. Next‐generation sequencing (NGS) gene panel diagnostics including 13 genes associated with hereditary thoracic aortic disease (ACTA2, COL3A1, FBN1, FBN2, MYH11, MYLK, PLOD1, SLC2A10, SMAD3, TGFBR1, TGFBR2, EFEMP2 and ELN) was performed. Assessment of the study protocol by our ethics committee was not required since only anonymized data collected during regular patient care were used. Both pedigrees have been slightly adapted in order to ensure privacy.
3. RESULTS
Family 1 (Figure 1a indicates the pedigree at initial presentation of the family, Figure 1b indicates the pedigree after several years follow‐up, Table 1.): The proband (III:2) and her daughter (IV:2) were referred for genetic analysis because of the familial occurrence of abdominal aortic aneurysms (AAA) and a type B aortic dissection at older age. Both were diagnosed with an AAA (4.0 cm at the age of 62 years and 5.0 cm at the age of 38 years, respectively). Ophthalmological and physical examination did not reveal any signs of MFS. NGS gene panel diagnostics in IV:2 revealed FBN1 variant c.1453C>T, p.(Arg485Cys) which was confirmed by Sanger sequencing in III:2. This variant substitutes an arginine by a cysteine in a calcium‐binding(cb)‐EGF‐like domain of fibrillin 1. Introduction of a cysteine in a cb‐EGF‐like domain likely affects the formation of disulfide bridges within the domain. This type of alteration is generally considered to be pathogenic (Loeys et al., 2010). However, because of the nonspecific phenotype and the fact that this variant had been reported in a family with autosomal recessive MFS (Vries et al., 2007), the heterozygous variant was initially classified as likely pathogenic. In order to clarify the clinical significance of this variant in heterozygote state, we offered combined clinical and genetic screening to first‐degree relatives of family members with an aneurysm or dissection. During follow‐up, the proband, her daughter, and several other family members carrying the FBN1 variant (III:2, III:4, III:5, IV:1, IV:2) were diagnosed with hallmark cardiovascular features of MFS (Figure 1b, Table 1). Of note, no relatives were diagnosed with significant ocular and/or skeletal involvement. Based on the cosegregation and the associated cardiovascular phenotype during follow‐up, the variant was re‐classified to a dominant pathogenic variant and the diagnosis of MFS was established in this family.
Figure 1.
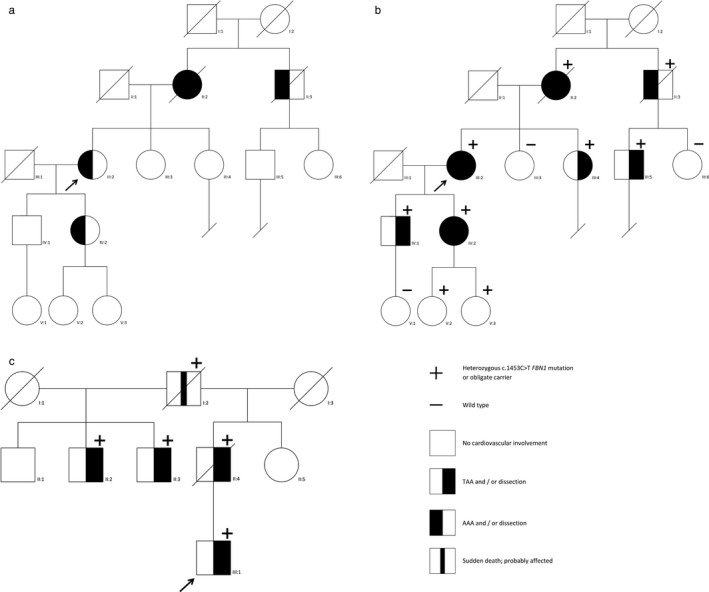
Pedigrees of families 1 and 2. (a) indicates the pedigree at initial presentation of family 1, (b) indicates the pedigree of this family after several years follow‐up. (c) Shows family 2. The proband is indicated with an arrow
Table 1.
Clinical details of families 1 and 2, and the previously published family (De Vries et al). Given the initial uncertainty about the pathogenicity of the variant, cardiologic and/or ophthalmologic evaluation was also performed in several individuals without the variant in family 1 (patient III:3, III:6 and V:1)
| Patient | Genotype | Phenotype | ||
|---|---|---|---|---|
| c.1453C>Ta | Cardiovascular involvement | Ocular involvement | Skeletal involvement, other features | |
| Family 1 | ||||
| II:2 | OC | Type B dissection 63y, rupture AAA 73y | Unknown | Unknown |
| II:3 | OC | Rupture AAA 80y | Unknown | Unknown |
| III:2 | Het | AAA 62y (E.S.), bilateral subclavian aneurysm 66y (E.S.), TAA 69y | None | Elongated facies, malar hypoplasia |
| III:4 | Het | Type A dissection 59y | None | Malar hypoplasia, pectus carinatum, scoliosis |
| III:5 | Het | Type B dissection 58y | None | None |
| IV:1 | Het | TAA 46y (E.S.) | None | Pectus excavatum, pes plani |
| IV:2 | Het | AAA 38y (E.S.), type B dissection 41y | None | None |
| V:2 | Het | None 18y | None | None |
| V:3 | Het | None 14y | None | None |
| III:3 | WT | None 62y | None | None |
| III:6 | WT | None 48y | None | None |
| V:1 | WT | Unknown | NP | Unknown |
| Family 2 | ||||
| I:2 | OC | Sudden death 57y | Unknown | Unknown |
| II:2 | Het | Borderline TAA 51y | Myopia>3 dpt | Span to height ratio >1.05 |
| II:3 | Het | TAA 47y | None | Downslanting palpebral fissures, elbow contractures, pectus carinatum, pes plani |
| II:4 | OC | Type A dissection 42y, died at 59y heart failure | Unknown | Unknown |
| III:1 | Het | Type A dissection 39y, dilatation coronary artery 39y | NP | Downslanting palpebral fissures, scoliosis, pes plani |
| De Vries et al. | ||||
| II:1 | Het | None 43y | None | Span to height ratio >1.05, high palate |
| II:2 | Het | None 43y | None | None |
| II:3 | Het | None 37y | None | Span to height ratio >1.05, high palate |
| II:4 | Het | Aortic root 40 mmb 40y | None | None |
| III:1 | Hom | MVP 13y, distal TAA dissection 20y, TAA 22y (E.S.), died 23y | Bilateral lens subluxation, ptosis | Scoliosis, elbow contractures, pectus excavatum, highly arched palate, facial appearance, pneumothorax |
| III:4 | Hom | None 13y | Bilateral lens subluxation, flat cornea | Highly arched palate, lumbosacral dural ectasia |
AAA: abdominal aortic aneurysm; E.S.: elective surgery; Het: heterozygous; Hom: homozygous; MVP: mitral valve prolapse; NP: opthalmological examination not performed; OC: obligate carrier; TAA: thoracic aortic aneurysm; WT: wild type; y: age in years.
Nomenclature FBN1 variant according to HGVS: NC_000015.9(NM_000138.4):c.1453C>T p.(Arg485Cys).
Considered normal for BSA.
Family 2 (Figure 1c, Table 1): The proband (III:1) was referred to a clinical genetics outpatient clinic at 39 years of age for genetic counseling after a type A aortic dissection and an aneurysm of a coronary artery. Physical examination revealed downslanting palpebral fissures, scoliosis, and pes plani. His father (II:4) was diagnosed with a type A dissection at the age of 42 years. He died at the age of 59 years due to heart failure. The paternal grandfather (I:2) died suddenly at the age of 57 years. The c.1453C>T, p.(Arg485Cys) variant in FBN1 was identified by NGS gene panel diagnostics resulting in the diagnosis MFS. Both the father and the paternal grandfather were obligate carriers, since the paternal half‐brothers (II:2 and II:3) of the father were also found to carry the FBN1 variant. II:2 had an aortic sinus of 4.0 cm and an elongated sinotubular junction at the age of 51 years, whereas II:3 was diagnosed with a thoracic aortic aneurysm of 4.1 cm at the age of 47 years. In addition, both of them had minor signs of MFS at physical and/or ophthalmological examination.
In both families, NGS analysis revealed no other (likely) pathogenic variants or variants of unknown significance.
4. DISCUSSION
In total, we present the phenotypic features of 10 genetically confirmed carriers and four obligate carriers of variant c.1453C>T, p.(Arg485Cys) in FBN1. These data show that this variant—contrary to earlier observations—is a cause of autosomal dominant MFS. In 2007, de Vries et al. reported two cousins with MFS caused by the homozygous c.1453C>T FBN1 variant, while the four heterozygous parents (ages 37–43 years) did not fulfill the original Ghent criteria for MFS at that time (Loeys et al., 2010). This variant has not been identified in large population databases (ExAC, gnomAD, and GoNL) and has, to our knowledge, only been published in one additional patient from a Taiwanese MFS cohort (Hung et al., 2009).
Though MFS is generally characterized by a dominant mode of inheritance, several other MFS families with an apparently autosomal recessive mode of inheritance have been reported in the literature (Fried & Krakowsky, 1977; Hilhorst‐Hofstee et al., 2010; Khan et al., 2014). Prior to the availability of FBN1 analysis, Fried and Krakowsky (1977) already suggested the possibility of an autosomal recessive mode of inheritance in MFS. Hilhorst‐Hofstee et al. (2010) described three MFS patients homozygous for FBN1 variant c.7454A>T, p.(Asp2485Val). In this family, 13 heterozygous relatives were identified, of which only one was diagnosed with MFS based on the original Ghent criteria (Loeys et al., 2010). Khan et al. (2014) reported a 3‐year‐old girl with bilateral lens subluxation and facial features suggestive of MFS carrying FBN1 variant c.7258A>C, p.(Asn2420His) homozygously. Her heterozygous parents were unaffected. In addition, several families with autosomal dominant MFS have been described in which family members carrying either homozygous or compound heterozygous FBN1 variants were more severely affected; however, this was not always the case (Arnaud et al., 2017; Hogue et al., 2013; Karttunen, Raghunath, Lönnqvist, & Peltonen, 1994; VanDijk et al., 2009). Because the c.1453C>T, p.(Arg485Cys) FBN1 families we describe show a clear autosomal dominant pattern of inheritance, the former report of apparently autosomal recessive MFS due to homozygosity of this variant might be due to age‐dependent penetrance and clinical variability. The age at evaluation of the heterozygous parents of the apparently autosomal recessive family varied between 37 and 43 years, and unfortunately, cardiological follow‐up data are not available. The age at diagnosis of aortic aneurysms and/or dissections in the two presented autosomal dominant families ranged from 38 to 80 years. Therefore, the cardiological phenotype in the unaffected carriers of the variant might still develop during further follow‐up. In the literature, a high degree of clinical variability has been reported concerning the age of onset, the severity, and extent of the clinical manifestations. Different genetic mechanisms, including a second pathogenic variant in another gene associated with thoracic aortic aneurysm and a polygenic model involving multiple modifier loci, are suggested to be a cause of this clinical variability in MFS by recent research (Aubart et al., 2018).
The variability of cardiovascular involvement is also illustrated by family 1 in which the apparent early onset familial AAA was the reason for referral. AAA have been reported as a feature in MFS, and in rare cases even as the presenting feature (Ooijen, 1988; Takayama, Miyata, & Nagawa, 2009; Ugwu et al., 2003; Wolfgarten et al., 2001). Family 2 in this report underlines the importance of DNA testing in individuals with a family history of young patients with AAA and the importance of regular imaging of the abdominal aorta in individuals with Marfan syndrome.
5. CONCLUSIONS
This study corroborates the high degree of clinical variability associated with variants in FBN1 and provides novel insights into the pattern of inheritance of FBN1 variant c.1453C>T, p.(Arg485Cys). Moreover, it underlines the importance of clinical follow‐up in heterozygous FBN1 mutation carriers irrespective of the previously suggested mode of inheritance related to a specific variant.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ACKNOWLEDGMENTS
The authors would like to thank Gerard Pals, Marlies Kempers and the families for participation in this study.
Overwater E, Efrat R, Barge‐Schaapveld DQCM, et al. Autosomal dominant Marfan syndrome caused by a previously reported recessive FBN1 variant. Mol Genet Genomic Med. 2019;7:e518 10.1002/mgg3.518
REFERENCES
- Ammash, N. M. , Sundt, T. M. , & Connolly, H. M. (2008). Marfan syndrome‐diagnosis and management. Current Problems in Cardiology, 33(1), 7–39. 10.1016/j.cpcardiol.2007.10.001 [DOI] [PubMed] [Google Scholar]
- Arnaud, P. , Hanna, N. , Aubart, M. , Leheup, B. , Dupuis‐Girod, S. , Naudion, S. , … Boileau, C. (2017). Homozygous and compound heterozygous mutations in the FBN1 gene: Unexpected findings in molecular diagnosis of Marfan syndrome. Journal of Medical Genetics, 54(2), 100–103. [DOI] [PubMed] [Google Scholar]
- Aubart, M. , Gazal, S. , Arnaud, P. , Benarroch, L. , Gross, M.‐S. , Buratti, J. , … Boileau, C. (2018). Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. European Journal of Human Genetics. 10.1038/s41431-018-0164-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron, D. E. , Alejo, D. E. , Patel, N. D. , Nwakanma, L. U. , Weiss, E. S. , Vricella, L. A. , … Gott, V. L. (2009). Aortic root replacement in 372 Marfan patients: Evolution of operative repair over 30 years. The Annals of Thoracic Surgery, 87(5), 1344–1349; discussion 1349–1350. [DOI] [PubMed] [Google Scholar]
- de Vries, B. B. , Pals, G. , Odink, R. , & Hamel, B. C. (2007). Homozygosity for a FBN1 missense mutation: Clinical and molecular evidence for recessive Marfan syndrome. European Journal of Human Genetics, 15(9), 930–935. 10.1038/sj.ejhg.5201865 [DOI] [PubMed] [Google Scholar]
- Dietz, H. C. , Cutting, C. R. , Pyeritz, R. E. , Maslen, C. L. , Sakai, L. Y. , Corson, G. M. , … Francomano, C. A. (1991). Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature, 252(6333), 337–339. [DOI] [PubMed] [Google Scholar]
- Dietz, H. C. , McIntosh, I. , Sakai, L. Y. , Corson, G. M. , Chalberg, S. C. , Pyeritz, R. E. , & Francomano, C. A. (1993). Four novel FBN1 mutations: Significance for mutant transcript level and EGF‐like domain calcium bindin in the pathogenesis of Marfan syndrome. Genomics, 17(2), 468–475. [DOI] [PubMed] [Google Scholar]
- Engelfriet, P. M. , Boersma, E. , Tijssen, J. G. , Bouma, B. J. , & Mulder, B. J. (2006). Beyond the root: Dilatation of the distal aorta in Marfan's syndrome. Heart, 92(9), 1238–1243. 10.1136/hrt.2005.081638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried, K. , & Krakowsky, D. (1977). Probable autosomal recessive Marfan syndrome. Journal of Medical Genetics., 14(5), 359–361. 10.1136/jmg.14.5.359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilhorst‐Hofstee, Y. , Rijlaarsdam, M. E. , Scholte, A. J. , Swart‐van den Berg, M. , Versteegh, M. I. , van derSchoot‐van Velzen, I. , … Pals, G. (2010). The clinical spectrum of missense mutations of the first aspartic acid of cbEGF‐like domains in fibrillin‐1 including a recessive family. Human Mutation, 31(12), E1915–E1927. 10.1002/humu.21372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogue, J. , Lee, C. , Jelin, A. , Strecker, M. N. , Cox, V. A. , & Slavotinek, A. M. (2013). Homozygosity for a FBN1 missense mutation causes a severe Marfan syndrome phenotype. Clinical Genetics, 84(4), 392–393. [DOI] [PubMed] [Google Scholar]
- Hung, C.‐C. , Lin, S.‐Y. , Lee, C.‐N. , Cheng, H.‐Y. , Lin, S.‐P. , Chen, M.‐R. , … Su, Y.‐N. (2009). Mutation spectrum of the fibrillin‐1 (FBN1) gene in Taiwanese patients with Marfan syndrome. Annals of Human Genetics, 73(Pt 6), 559–567. [DOI] [PubMed] [Google Scholar]
- Karttunen, L. , Raghunath, M. , Lönnqvist, L. , & Peltonen, L. (1994). A compound‐heterozygous Marfan patient: Two defective fibrillin alleles result in a lethal phenotype. American Journal of Human Genetics, 55(6), 1083–1091. [PMC free article] [PubMed] [Google Scholar]
- Khan, A. O. , Bolz, H. J. , & Bergmann, C. (2014). Results of fibrillin‐1 gene analysis in children from inbred families with lens subluxation. Journal of American Association for Pediatric Ophthalmology and Strabismus, 18(2), 134–139. 10.1016/j.jaapos.2013.11.012 [DOI] [PubMed] [Google Scholar]
- Loeys, B. L. , Dietz, H. C. , Braverman, A. C. , Callewaert, B. L. , DeBacker, J. , Devereux, R. B. , … DePaepe, A. M. (2010). The revised Ghent nosology for the Marfan syndrome. Journal of Medical Genetics, 47(7), 476–485. 10.1136/jmg.2009.072785 [DOI] [PubMed] [Google Scholar]
- Mariucci, E. M. , Lovato, L. , Rosati, M. , Palena, L. M. , Bonvicini, M. , & Fattori, R. (2013). Dilation of peripheral vessels in Marfan syndrome: Importance of thoracoabdominal MR angiography. International Journal of Cardiology, 167(6), 2928–2931. 10.1016/j.ijcard.2012.08.001 [DOI] [PubMed] [Google Scholar]
- Ooijen, B. v. (1988). Marfan's syndrome and isolated aneurysm of the abdominal aorta. British Heart Journal, 59(1), 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama, T. , Miyata, T. , & Nagawa, H. (2009). True abdominal aortic aneurysm in Marfan syndrome. Journal of Vascular Surgery, 49(5), 1162–1165. 10.1016/j.jvs.2008.12.007 [DOI] [PubMed] [Google Scholar]
- Ugwu, B. T. , Ardill, W. , Yiltok, S. J. , Momoh, J. T. , Lenkop, D. W. , & Uba, F. A. (2003). Marfan's syndrome presenting with abdominal aortic aneurysm: A case for vigilance. West African Journal of Medicine, 22(1), 95–97. [DOI] [PubMed] [Google Scholar]
- VanDijk, F. S. , Hamel, B. C. , Hilhorst‐Hofstee, Y. , Mulder, B. J. M. , Timmermans, J. , Pals, G. , & Cobben, J. M. (2009). Compound‐heterozygous Marfan syndrome. European Journal of Medical Genetics, 52(1), 1–5. 10.1016/j.ejmg.2008.11.004 [DOI] [PubMed] [Google Scholar]
- Wang, W.‐J. , Han, P. , Zheng, J. , Hu, F.‐Y. , Zhu, Y. , Xie, J.‐S. , … Tian, X.‐L. (2013). Exon 47 skipping of fibrillin‐1 leads preferentially to cardiovascular defects in patients with thoracic aortic aneurysms and dissections. Journal of Molecular Medicine (Berlin), 91(1), 37–47. 10.1007/s00109-012-0931-y [DOI] [PubMed] [Google Scholar]
- Wolfgarten, B. , Krüger, I. , & Gawenda, M. (2001). Rare manifestation of abdominal aortic aneurysm and popliteal aneurysm in a patient with Marfan's syndrome: A case report. Vascular Surgery, 35(1), 81–84. 10.1177/153857440103500118 [DOI] [PubMed] [Google Scholar]