

Summary
Parrots are one of the most distinct and intriguing group of birds, with highly expanded brains [1], highly developed cognitive [2] and vocal communication skills [3], and a long lifespan compared to other similar-sized birds [4]. Yet the genetic basis of these traits remains largely unidentified. To address this question, we have generated a high-coverage, annotated assembly of the genome of the Blue-fronted Amazon (Amazona aestiva), and carried out extensive comparative analyses with 30 other avian species, including 4 additional parrots. We identified several genomic features unique to parrots, including parrot-specific novel genes and parrot-specific modifications to coding and regulatory sequences of existing genes. We also discovered genomic features under strong selection in parrots and other long-lived birds, including genes previously associated with lifespan determination as well as several hundred new candidate genes. These genes support a range of cellular functions, including telomerase activity, DNA damage repair, control of cell proliferation, cancer, immunity, and anti-oxidative mechanisms. We also identified brain-expressed, parrot-specific paralogs with known functions in neural development or vocal learning brain circuits. Intriguingly, parrot-specific changes in conserved regulatory sequences were overwhelmingly associated with genes that are linked to cognitive abilities and have undergone similar selection in the human lineage, suggesting convergent evolution. These findings bring novel insights into the genetics and evolution of longevity and cognition, as well as provide novel targets for exploring the mechanistic basis of these traits.
eTOC blurb
The genetic basis for the complex traits that characterize parrots, including extreme longevity and advanced cognition, remain unknown. Wirthlin et al. present the genome of the Blue-fronted Amazon, Amazona aestiva. Comparisons with other birds and humans reveal genomic similarities suggesting convergent mechanisms in the evolution of these traits.
Results and Discussion
Parrots (order Psittaciformes) possess several attributes that make them excellent models for understanding the evolution of complex traits. Like humans, parrots as a group have (i) large brains relative to body size [1], (ii) a high density of neurons in the forebrain [5], (iii) advanced cognitive abilities including object permanence and tool use [6], (iv) complex social organization [7], (v) vocalizations learned through cultural transmission using specialized brain circuits [3], (vi) cooperative problem solving [8], (vii) extended developmental periods and rearing [9], and (viii) exceptional longevity, especially given their high metabolism [4]. The genetic basis for characteristic parrot traits remains unknown, and no attempt has yet been made to identify genomic features unique to parrots. Genome assemblies are available for the Puerto Rican Parrot, Scarlet Macaw, Rosy-faced lovebird, Budgerigar, and Kea, representing a broad sampling of extant parrot diversity [10] (Figure 1). However, with the exception of the budgerigar, these genomes have been described in low coverage and/or low contiguity (STAR Methods: Genome Sequencing).
Figure 1. Phylogenetic Relationships and Longevity of Parrots, including Amazona aestiva, in Relation to Other Birds.
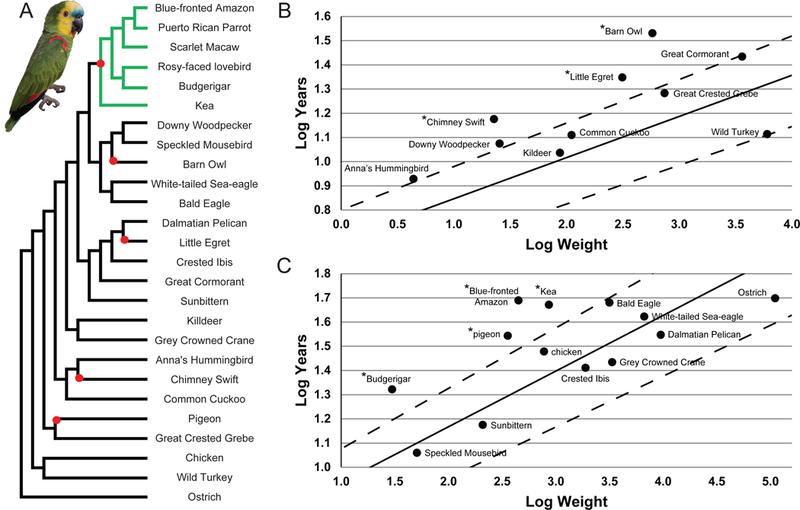
(A) This schematic phylogeny, derived from previously published studies [10, 11], depicts the relationship between parrots and all other birds analyzed in the longevity analyses. Red nodes indicate the occurrence of high longevity, and were used as branches to test for nonneutral sequence evolution in long-lived birds against a null model using a likelihood ratio test. The photo depicts Moises, the adult male Blue-fronted Amazon whose genome was sequenced for this study. (B - C) The expected lifespan of the 23 bird species analyzed for genomic signatures of longevity in plotted as a solid line, based on calculations derived from Prinzinger [12] that describe the relationship between mass (log weight) and longevity (log years) for wild (B) and captive (C) birds (see STAR Methods: Longevity). Species falling outside of the dashed lines deviate significantly from expected lifespan based on mass, and are indicated with a “*”. See also Table S1.
To address these questions, we generated a high-coverage (120.6x) genome assembly for the Blue-fronted Amazon (Amazona aestiva, Linnaeus, 1758). Emblematic of the Brazilian avifauna, this highly vocal parrot species is exceptionally long-lived, even among parrots, with confirmed cases of birds living up to 66 years in captivity, and anecdotal reports of 90+ years [13]. The resultant assembly (Aaes1) contains 3,232 scaffolds with a scaffold N50 size of 1.09 Mb and a contig N50 size of 27.8 kb, surpassing all previous parrot genome assemblies in sequence contiguity (STAR Methods: Genome Sequencing). Genome annotation revealed the A. aestiva to be comparable to other avian genomes in terms of coding genes, miRNAs, and repetitive content [11]. We also provide a set of 3,516 microsatellites with primers that will benefit future conservation, population genetics, and phylogeographic efforts (STAR Methods: Annotation).
A genome-wide comparison of A. aestiva and 4 other parrots with multiple non-parrot outgroups identified a substantial amount of genomic material unique to parrots (STAR Methods: Genome Sequencing). This included a set of novel, parrot-specific genes, resulting from paralogous gene duplications, gene family expansions from successive rounds of duplication, or de novo genes with no known paralog in other species (Table 1; STAR Methods: Novel Genes). There were also numerous non-coding scaffolds likely representing intergenic regions. In limited cases, the mechanism of duplication could be attributed to chromosomal rearrangements (Figure S1). This finding is consistent with our previous findings of gene duplication at chromosomal breakpoints in birds [14].
Table 1.
Novel genes in parrot genomes
| A: Novel genes conserved in parrots (single duplication event) | ||
| Parent gene ortholog | Gene function summary | Budgerigar brain expression |
| ARPC4-TTLL3L | cytoskeletal structural integrity | NO |
| CEP83 | structural integrity of cilia | YES |
| DESI2a,b | post-translational modification | YES |
| PLXNC1 | axon guidance | YES |
| PRKAR2A | cyclic AMP-dependent signaling | YES |
| PSMD6a,b | protease | YES |
| RSPH3 | cyclic AMP-dependent signaling | NO |
| SIDT1 | intercellular RNA transfer | NO |
| SLC9A5 | Na-H exchanger | NO |
| TAGAP | t-cell Rho-gap signaling | NO |
| TAXIBP3 | signal transduction | YES |
| ZNF541a | transcription factor | NO |
| B: Gene families expanded in parrots (multiple duplication events) | ||
| Parent gene ortholog | Gene function summary | |
| AMY1 | dietary starch digestion | |
| GPI-anchored protein 58 | unknown | |
| LRRC37A | innate immunity | |
| MROH7 | unknown | |
| MXRA7 | matrix remodeling | |
| TAGAP | t-cell Rho-gap signaling | |
| TTLL3 | cytoskeletal structural integrity | |
| SUN2 | DNA repair | |
| Viral/retrotransposon related | endogenous retroviral | |
| C: Novel de novo parrot genes | ||
| 136 novel gene predictions | 79 (58%) with predicted protein domains | 17 (12.5%) expressed in budgerigar brain |
| D: Novel de novo Blue-fronted Amazon genes | ||
| 34 novel gene predictions | 21 (62%) with predicted protein domains | |
Summary of novel genes unique to parrot genomes resulting from gene duplication (A), gene family expansion (B), de novo gene origination (C); novel gene predictions unique to the Blue-fronted Amazon (D). Additional details and full list of novel genes are presented in STAR Methods: Novel Genes.
Avian ortholog cannot be unambiguously identified.
Gene duplication in conjunction with chromosomal breakpoint event (see Figure S1).
Insights into longevity from parrots and other long-lived birds
To gain insight into the genetic underpinnings of longevity, we analyzed 23 bird species with reliable longevity data and available genome assemblies for genomic signatures of selection in long-lived birds. We divided species into high-longevity (HL; maximum recorded longevity at least 20% higher than predicted by body mass [12, 15]) and regular-longevity groups (RL; maximum recorded longevity corresponding to or lower than that predicted by body mass) (Figure 1A; Table S1), using estimated corrections for wild vs. captive birds (Figure 1B-C; STAR Methods: Longevity). Besides parrots, HL birds included several other notably long-lived species (Rock Dove, Chimney Swift, Little Egret, and Barn Owl). Importantly, high longevity is most parsimoniously interpreted as having evolved independently in these avian groups, as clearly seen when examining the distribution of this trait mapped onto the currently accepted avian phylogeny (Figure 1A).
We initially searched for the largest possible set of orthologs unequivocally present in all 23 species with high-quality longevity, resulting in 4,132 single gene ortholog clusters (SGOs; gene sets containing one copy from all birds analyzed). We utilized a phylogeny-based likelihood ratio test to test for significant group differences in the ratio of nonsynonymous to synonymous nucleotide substitutions (dn/ds, or ω) indicative of evolutionary selective pressure [16]. We obtained evidence of differential rates of evolution between HL and RL birds in 344 genes (8%) (Figure 2A; STAR Methods: Longevity). Of these, 281 genes had greater ω in the HL group, indicative of more relaxed sequence constraint (favoring variation leading to new functions), whereas the remaining 63 had lower ω in HL birds, suggesting higher stabilizing selection in these genes. Separately, we also detected evidence of positive selection at 90 amino acid sites in 31 proteins, 39 of these within known functional domains (Data S1).
Figure 2. Genes Under Positive Selection in Long-lived Birds.
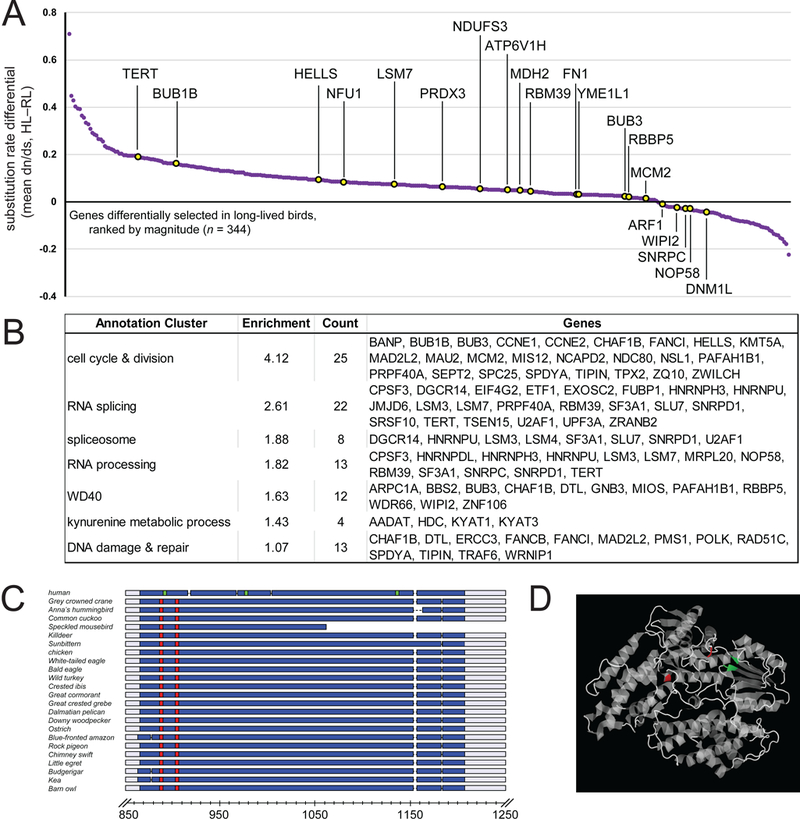
(A) Plot of the strength of selective pressure for the set of genes with significantly different nonsynonymous to synonymous substitution ratios (dn/ds, or ω) between high-longevity birds (HL) and regular-longevity birds (RL, baseline). Genes with known lifespan effect in model systems (from GenAge [15]) are highlighted in yellow; candidate longevity-influencing genes with no previously reported lifespan data are highlighted in purple. (B) Table of the major gene functional categories enriched in genes with differential selection in HL birds. (C) Comparative multiple sequence alignment of TERT proteins across avian species and human, with sites under positive selection in long-lived birds indicated in red and metal binding sites in green, all occurring within the reverse transcriptase domain, in blue. Scales indicate amino acid position within alignments. (D) Crystal structure of TERT reverse transcriptase catalytic subunit. Positively selected residues are in red, which are in close physical proximity to metal binding sites critical for catalytic activity, in green. PDB accession number: TERT, 3DU6. See also Table S2, Data S1.
A subset (n = 20; 6%) of the 344 genes under selection in HL birds have been experimentally shown to impact lifespan in other model organisms (GenAge [15]) (Figure 2A). Besides validating our analyses, this observation supports an association of these genes with life-span determination in birds as well. The remaining genes (n = 324; 94%) had never previously been associated with lifespan determination in any organism to our knowledge, representing promising novel candidates for involvement in longevity (STAR Methods: Longevity). Functional annotation clustering of all genes under selection in long-lived birds revealed highly significant enrichments in annotations related to cell division, cell cycle regulation, and RNA binding/processing, and weaker but significant enrichments in DNA damage and repair, mitochondrial function, and oxidative metabolism (Figure 2B; Table S2).
Within the GenAge subset, the strongest evidence of selective pressure was for TERT (telomerase reverse transcriptase), a key component of the telomerase complex that confers protection against cell senescence [15]. TERT also showed two positively selected sites (Figure 2C; Data S1) within the reverse transcriptase domain in long-lived birds, in proximity to sites critical for catalytic function (Figure 2D). These observations suggest that changes in the TERT catalytic activity may represent a fundamental longevity-promoting mechanism, and substantiate findings that telomerase activity is altered in long-lived birds [17]. Among the genes not previously reported in GenAge, a large subset showed involvement in DNA damage and repair, including POLK, which allows for the DNA replication machinery to bypass sites of DNA lesion, and ERCC3, a helicase that repairs nucleotide excisions (Figure 2B).
While protective against cell senescence, a risk of high TERT activity is an increased rate of uncontrolled proliferation and tumor formation [18]. Highlighting the importance of balancing these processes, we found that several genes under selective pressure in long-lived birds (BUB1B, BUB3, KIF4A, KIF1BP, CCNE1) have been linked to the control cell proliferation and tumor proliferation. Specific mechanisms include regulation of the spindle assembly during cell division by mitotic checkpoint proteins, control of chromosome integrity during mitosis by microtubule-based motor protein, and control of cell cycling and tumor suppression mechanisms (Figure 2B; Table S2). Based on these findings, we suggest that the balanced coevolution of pathways for telomerase activity and cell cycle regulation may represent a mechanism for preventing increased rates of cancer in the evolution of increased longevity in birds.
Further highlighting DNA repair and control of cell proliferation as important pathways in the evolution of parrots, several duplicated genes in parrots also regulate genomic stability and cell senescence. These include: PSMD6, which encodes a 26S proteasome subunit that colocalizes with DNA damage foci in cells that have suffered genotoxic damage, helping to ensure that these cells are targeted for senescence, and DESI2, an apoptosis regulator and part of the early response to DNA damage (see Figure S1A,B for depiction of gene duplication and supportive evidence of gene function).
Parrots and other HL birds also exhibited selection in SOD1 and SOD3, genes essential for protection from oxidative damage stress [19]. We found that SOD3 exhibits high stabilizing selection in HL birds relative to RL birds (STAR Methods: Longevity). Several positively selected sites in SOD1 were within the predicted Cu-Zn binding domain (Data S1), likely modulating metal binding affinity and enzymatic activity [19]. The ‘free radical hypothesis of aging,’ which postulates that increased longevity in birds depends on protection against oxidative damage by free radicals, has been challenged by a failure to detect tissue differences in indicators of oxidative stress when comparing similar sized long- vs. short-lived birds [20]. Our finding of convergent selection in these genes as well as in several others involved in oxidative stress and/or mitochondrial functions in independent avian lineages supports the importance of anti-oxidative protection in the evolution of avian longevity [21]. Further investigation of the specific role of superoxide dismutase in long-lived birds could serve to reconcile the contrasting findings in the previous literature. Lastly, we note a large and significant gene subset related to RNA splicing and processing (Figure 2B; Table S2), which for the most part was not previously reported in GenAge, although a causal relation to life-span determination is unclear.
Overall, our findings suggest that changes in telomerase, DNA repair, cell cycle progression, RNA splicing and processing, and oxidative stress pathways may be critical for heightened longevity in birds. They also provide candidate genes and amino acid sites for future experimental interrogation that could lead to advances in our understanding of lifespan extension. Although genomic studies of individual species with high longevity have been performed previously [16, 22], this represents, to our knowledge, the largest comparative genomic analysis of long-lived vertebrates to date. Because these genes have independently experienced high rates of sequence substitution across multiple long-lived lineages, they could reflect fundamental mechanisms associated with the evolution of heightened longevity.
Insights into cognition from parrot genomes
Supporting our initial hypothesis that novel genes in parrots might relate to brain function and cognition, the ancestral parent gene paralogs of several parrot novel genes have known involvement in neuronal development, physiology, and behavior (Table 1). Most prominently, PLXNC1 was uniquely duplicated in parrots among the bird species examined (Figure S1C). PLXNC1, a regulator of axonal outgrowth, is one of a distinct set of genes with shared differential expression in the specialized vocal learning motor cortical areas of humans, parrots, hummingbirds, and songbirds [23]. This convergent molecular specialization in unrelated vocal learning groups is consistent with the hypothesis that regulation of cortical projections within vocal-motor circuits may be critical to the evolution of vocal learning systems [23]. Determining redundant versus complementary or modulatory roles for the duplicate PLXNC1s represents an important target for gene manipulation studies.
The parent paralogs of several other parrot-specific duplicated genes (CEP83, SLC9A5, RSPH3, LRRC37A; Table 1) are involved in critical aspects of neuronal cell structure and integrity, including regulation of actin cytoskeleton, microtubule sliding, filopodial extension, and the structure of cilia and dendritic spines, disruptions of which can lead to cognitive impairment [24–27]. LRRC37A, in particular, is a member of a large gene family with involvement in the immune system and in nervous system development, and which is greatly expanded in primates [26]. In humans, LRRC37 is broadly expressed, with enrichment in the cerebellum and thymus [26]. Our evidence of a duplication in parrots points to a convergent expansion of this gene family in both primates and parrots, suggesting a possible link with the increased cognitive capacities of these two unrelated taxa.
We further identified a set of parrot de novo novel genes (present in parrot genomes with no paralogs in other species) with evidence of brain expression (n = 17, Table 1C; STAR Methods: Novel Genes). Although all contain open reading frames, roughly half (n = 9; 53%) contain no known functional domains. Whether these expressed transcripts are actually translated or represent regulatory non-coding RNAs, and whether they have been integrated into functional pathways or represent non-functional ‘proto-genes’ [28], are questions that will require further analysis. However, given the precedent in songbirds, where some de novo genes have been shown to be specifically expressed in brain structures devoted to vocal learning behavior [14], parrot de novo genes represent promising candidates for regulating brain regions involved in vocal learning, cognition, and other parrot lineage traits.
Noncoding genomic regions can contribute to trait evolution through changes in gene regulatory elements that cannot be detected by analysis of coding sequences only. To determine whether parrots possess lineage-specific variation in cis-regulatory sequences that could contribute to brain function and cognition, we investigated ultra-conserved noncoding elements (UCNEs; noncoding sequences highly conserved throughout vertebrate phylogeny), which are often associated with enhancers that play critical, conserved roles in development [29]. We found a suite of UCNEs with significant sequence divergence in parrot genomes relative to other vertebrates (Table 2; STAR Methods: UCNEs). Remarkably, of 11 protein-coding genes associated with parrot-divergent UCNEs, 10 (91%) are involved in various aspects of brain function, including forebrain patterning, neuronal subtype differentiation, and adult neurogenesis [30–38] (Table 2). Supporting their activity in the brain, we found budgerigar brain transcriptomic evidence of expression for 9 of these genes (Table 2). The majority of these (n = 7) showed similar evolutionary selection in the human lineage, as evidenced by association with conserved noncoding elements divergent in humans relative to great apes and other mammals [39, 40] (Table 2, Figure S2).
Table 2.
Genes associated with ultra-conserved noncoding elements divergent in parrots
| Gene | haCNSs associated with gene |
HARs associated with gene |
Associations with brain function and cognitive disorders | Brain- expressed in budgerigar |
|---|---|---|---|---|
| AUTS2 | HACNS174, HACNS369 | HAR31 | forebrain development, autism-related [30] | YES |
| BCL11A | HACNS101 | HAR180 | neuron projection development, intellectual disability-related [37] | YES |
| EBF3 | cortical development, intellectual disability-related [36] | NO | ||
| ERBB4 | HACNS183, HACNS505, HACNS977 |
HAR84 | forebrain patterning, schizophrenia-related [31] | YES |
| ESRRG | HAR177 | schizophrenia-related [38] | YES | |
| NPAS3 | HACNS96, HACNS221, HACNS490, HACNS553, HACNS658 |
HAR21, HAR89, HAR96, HAR173, HAR189, HAR202 |
adult neurogenesis, schizophrenia- and intellectual disability-related [35] | YES |
| PROX1 | HACNS878 | forebrain neuron differentiation, adult neurogenesis [34] | YES | |
| SOX6 | neuronal differentiation [33] | YES | ||
| SP8 | forebrain patterning [32] | YES | ||
| ZC3H3 | HACNS71 | no known brain function | YES | |
| ZNF608 | HACNS214, HACNS852 | no known brain function | YES |
Genes associated with ultra-conserved noncoding elements (UCNEs) showing accelerated evolution in the Blue-fronted Amazon, budgerigar, and Kea genomes. The majority of these genes are also associated with human accelerated conserved noncoding sequences (haCNS [39]) and/or human accelerated regions (HAR [40]). See also Figure S2.
Among the 11 genes with regulatory regions containing parrot-divergent UCNEs, a subset is associated with diseases that disrupt cognitive function in humans (Table 2). Noteworthy is AUTS2, whose mutations in humans are associated with a range of cognitive disabilities including autism, intellectual impairment, developmental delay, and language deficits [30]. AUTS2 has also received attention as a gene that may have been critical to the evolution of human cognitive abilities [30]. Also noteworthy are NPAS3, in which breakpoint translocations result in schizophrenia and intellectual disability [35]; and BCL11A, a regulator of axonal branching and outgrowth in developing neurons, in which deletions result in brain malformation and intellectual disability [37]. Two further genes, ERBB4 and ESRRG, have known associations with cognitive disorders, with variants established as risk factors for schizophrenia [31, 38]. These results suggest that humans and parrots may have undergone convergent selection in the regulatory regions of a crucial set of genes related to brain development and cognition.
Conclusions
Parrot genomes are distinguished by the presence of domain-specific modifications in existing gene cohorts, novel genes, and variations in noncoding sequences thought to regulate expression. The discovery of a distinct gene set under evolutionary selection in long-lived birds provides independent support for genes previously associated with longevity in non-vertebrate model systems, and identifies a large suite of genes with no previous association with longevity, representing promising targets for further experimental interrogation. Parrot lineage-specific changes in genes and regulatory regions associated with the brain represent candidate mechanisms for the evolution of the larger brains and more advanced cognitive abilities of parrots, with intriguing parallels to evolutionary mechanisms thought to have facilitated the emergence of these traits in humans. These findings support parrots, which outperform even great apes in several measurements of intelligence [2], as an excellent experimental model for uncovering the genetic basis of higher cognition. Finally, as Blue-fronted Amazon populations have declined in recent decades, owing to drastic reduction in natural habitat due to urban and agricultural expansions and illegal trading, its sequenced genome should be a valuable tool in ongoing conservation efforts.
STAR Methods Text
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Claudio V. Mello (melloc@ohsu.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The Blue-fronted Amazon, Amazona aestiva, individual whose genome was sequenced is alive as of the writing of this paper. The individual is an adult male, born in captivity in 2003 in the Vale Verde Ecological Park in Betim, Minas Gerais, Brazil, under register number FVVF132, and tissue voucher B04212 deposited at Centro de Coleções Taxonômicas of Universidade Federal de Minas Gerais. His parents were obtained in southern Brazil, where subspecies A. aestiva xanthopterix is commonly found. Blood for whole genome sequencing was collected in 2013 through brachial venipuncture according to protocol 202/2007, approved by by CEUA-UFMG (Comissão de Ética no Uso de Animais - Ethics Committee for Animal Use in Research at the Federal University of Minas Gerais).
METHOD DETAILS
Genome Sequencing
About 600 μl of whole blood for genomic DNA was collected in heparinized capillary tubes, promptly mixed with EDTA 50 mM 1:1 (v/v), and kept in ice until DNA extraction. DNA was extracted using DNeasy Blood & Tissue Kit (Qiagen), according to manufacturer’s instructions; 10 μl of the blood/EDTA solution was used as starting input material. DNA integrity was checked by electrophoresis in 0.8% agarose gel and quantified by spectrophotometry using a Nanodrop 2000C.
The 1.126 gigabase genome of the Blue-fronted Amazon was sequenced using high-coverage Illumina and 454 technologies to 117.59x and 3.1x coverage, respectively. Four Illumina libraries were generated for sequencing: two paired-end with an insert size of 300 bp; and two mate-paired libraries, with estimated insert sizes of ~1.4 kb and ~4 kb. Fourteen single-end 454 libraries, with a mean read length of 493 bp, were also sequenced. The Illumina paired-end sequencing data was filtered for low read quality and adapter trimmed with SeqPrep (https://github.com/jstjohn/SeqPrep). Mate pair libraries were adapter trimmed and identified using Illumina’s NxTrim (https://github.com/sequencing/NxTrim). Reads were corrected by most frequent kmers by Quake (http://www.cbcb.umd.edu/software/quake/). The initial genome assembly was performed using all four Illumina libraries with AllPaths-LG with all filtering and correction parameters selected (http://software.broadinstitute.org/allpaths-lg/blog/?page_id=12). The n = 14 454 libraries were incorporated by means of intra-scaffold gap filling with GapFiller (http://www.baseclear.com/bioinformatics-tools/). An additional round of gap-filling was performed using the Illumina 5 kb library with GapFiller. We also explored other approaches of combining the different libraries using different assemblers, such as ABySS (http://www.bcgsc.ca/platform/bioinfo/software/abyss) and Meraculous (https://jgi.doe.gov/data-and-tools/meraculous/), but determined the current approach produced the best result based on various metrics of contiguity and completeness. The resultant assembly (Aaes1) contains 3,232 scaffolds with a scaffold N50 size of 1.09 Mb and a contig N50 size of 27.8 kb (Table S3). Using BLASTn (https://blast.ncbi.nlm.nih.gov/Blast.cgi), Blue-fronted Amazon scaffolds were mapped to the genomes of chicken and zebra finch for additional comparative genomic analysis (Figure S3).
Genome Annotation
We generated in silico gene predictions through a combination of reference-guided and ab initio procedures. Exonerate (https://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate), a tool developed to predict genes by sequence alignment using proteins or transcripts from related organisms to the genome of interest, was implemented for reference-guided gene prediction, using as reference set the Ensembl 74 protein models from chicken (Gallus gallus, galGal4). This set of reference genome protein models was aligned to the Blue-fronted Amazon scaffold assembly using Exonerate protein2genome with a minimum 40% identity. A gene feature format file (.gff) containing gene predictions was generated from the Exonerate alignments. To generate gene predictions in the Blue-fronted Amazon scaffold assembly based on gene structural features, we used AUGUSTUS (http://bioinf.uni-greifswald.de/augustus/), an ab initio gene predictor that learns to identify genes based on structural similarity to genes provided in a training set. The AUGUSTUS-generated gene predictions were built using known gene models from chicken as a training set (provided by the AUGUSTUS package), which were corroborated with the alignment-based predictions produced by Exonerate (converted from .gff to .hints format for use in AUGUSTUS). Predictions were guided by, but not constrained to, the homology evidence generated by Exonerate, allowing for the prediction of novel genes unidentifiable through reference-guided approaches. Predictions in repetitive regions were minimized by using a softmasked version of the genome for gene prediction and the --softmasking parameter of AUGUSTUS. We excluded within AUGUSTUS gene predictions with in-frame stop codons, returning only complete gene models with start and stop codons. We used SABIA (http://www.sabia.lncc.br), an automated annotation pipeline, to annotate all gene predictions with an open reading frame (ORF), taking into account BLAST similarity to sequences in various databases, including the KEGG orthology (KO, http://www.genome.ad.jp/kegg) and Cluster of Orthologous Groups (COG, http://www.ncbi.nlm.nih.gov/COG/) databases, the presence of predicted protein motifs using InterPro (http://www.ebi.ac.uk/interpro/), and Gene Ontology (GO, http://geneontology.org/).
We performed blastp searches to identify orthologs of Blue-fronted Amazon gene predictions among the complete protein sets of chicken, zebra finch, budgerigar, and kea. Using the Blue-fronted Amazon predicted proteins as query, we considered as orthologs BLAST alignments which corresponded to best hits with an e-value < 1e-10, and both query and subject coverage > 60%. Using JVenn (http://bioinfo.genotoul.fr/jvenn/example.html) we clustered genes from the other four birds that were the best hit for the same Blue-fronted Amazon gene prediction.
The ab initio and reference-based gene prediction methodologies used to annotate the Blue-fronted Amazon genome (Exonerate and AUGUSTUS) yielded a combined total of 16,200 gene predictions. 12,839 of these represented open reading frames (ORFs) containing both start and stop codons, which were validated and annotated using SABIA. BLAST analysis against chicken, zebra finch, budgerigar, and kea assemblies identified 11,094 presumed orthologs, with 7,692 present in all 5 genomes (Complete list of gene predictions with orthology information in Data S1A).
In order to find microRNA (miRNA) precursors orthologous to those found in chicken and zebra finch we used mirDeep (https://github.com/rajewsky-lab/mirdeep2) and RNAfold (https://www.tbi.univie.ac.at/RNA/RNAfold.1.html). 1,230 mature microRNAs from chicken and zebra finch were retrieved from the miRBase database (http://www.mirbase.org/) and used as queries against the Blue-fronted Amazon genome with mirDeep, using default parameters. The miRNA genome hits were retrieved and the secondary structure solved with RNAfold. Solved secondary structures were filtered to include minimum free energy <10 kcal/mol, no more than three gaps of 3 to 4 nt in length, two of 5 nt, or one of length 6 to 8 nt. The resulting sequences were considered to be putative microRNA precursors. We predicted 86 miRNA precursors, belonging to 70 different miRNA families (Data S1B).
We conducted a survey of the repetitive content of the Blue-fronted Amazon genome for sequences falling into two distinct categories: transposable elements (TEs) and tandem repeats (i.e. microsatellites). TEs were identified through a combination of de novo, homology-based, and structure-based prediction methodologies. The homology-based approach involved searching the Blue-fronted Amazon genome for expansions of known TEs using publicly available databases, as follows. We used RepeatMasker (http://www.repeatmasker.org/) to identify elements similar to other known TEs in Repbase 19.06, using - species=aves. RepeatMasker was also used for estimating the amount of small RNAs, satellite DNA, low complexity regions, and simple repeats. To identify and classify parrot-specific repetitive elements, we employed RECON and RepeatScout, two de novo repeat finding packages invoked from within the RepeatModeler package, using default parameters (http://www.repeatmasker.org/). To predict candidate miniature inverted-repeat transposable elements (MITEs), we used MITE-hunter (http://target.iplantcollaborative.org/mite_hunter.html) with default parameters. The sequence alignments obtained by MITE-hunter were manually checked and edited in Jalview v2 (http://www.jalview.org/). All consensus output from RepeatModeler and MITE-Hunter was used to build a library in RepeatMasker, which was run again to find additional examples of these elements in the Blue-fronted Amazon genome. In order to accurately determine the number of TE copies found, including reconstruction of full-length copies, we applied the ‘One code to find them all’ perl tool (http://doua.prabi.fr/software/one-code-to-find-them-all) to parse the .out files obtained from the RepeatMasker analyses. The final results obtained from this analysis for each of the three tools (RepeatMasker, RepeatModeler and MITE-Hunter) were combined, removing redundancies. Finally, we identified TE units, which consisted of genomic regions containing contiguous TE predictions obtained by all three tools, which could be complete, incomplete, or nested TEs. These were classified as retrotransposons (Class I TEs) or DNA transposons (Class II TEs). TE units identified by more than one tool, but that did not fit within these categories, were designated unclassified TEs.
The combined TE count identified by three prediction methods (homology-based RepBase searches in RepeatMasker and de novo searches in RepeatModeler and MITE-hunter) totaled 267,799, representing 138.2 Mb or 12.23% of the Blue-fronted Amazon genome assembly (Table S4). Inclusion of small RNAs, satellites, low complexity regions, and simple repeats raise this total to 13%. The majority of TE predictions were categorized as long interspersed nuclear elements (LINEs; 7.14% of the total genome assembly), followed by unclassified TEs (2.38%), miniature inverted-repeat transposable elements (MITEs; 2.06%), long terminal repeats (LTRs; 0.64%), short interspersed nuclear elements (SINEs; 0.01%), and other DNA transposon families (0.02%).
Selection of microsatellite markers and primers was performed using QDD v3.1 (http://net.imbe.fr/~emeglecz/qdd.html) with default parameters, which filtered candidate markers based on similarity to known transposable elements in order to increase the genotyping success rate. We first identified 3,754 candidate microsatellite loci excluding 238 loci that overlapped with subsequently identified TEs resulted in a final count of 3,516 microsatellite loci in 1,040 different scaffolds, containing from 5 to 29 repeats. Of these, 2,775 were found to represent dinucleotide microsatellite loci, followed by smaller proportions of trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide loci, in this order. For each locus we also identified primers designed to generate amplicons varying between 90 and 300 bp for use in population genetics studies (Data S1C).
Longevity
Protein and CDS sequences from 22 birds in addition to Blue-fronted Amazon (Table S1) were retrieved from the GigaScience repository associated with the Avian Phylogenomics Consortium [11]. These species reflect a wide range of phylogeny [11] and were selected based on the availability of annotated genomes and reliable maximum longevity data, either from wild or controlled captive conditions, which were obtained from the AnAge database [15]. We note that lifespan data meeting the threshold for inclusion in the study were not available in the AnAge database for any songbirds with an available genome assembly, including zebra finch.
These birds were divided into two groups: regular-longevity (RL, when maximum observed longevity was lower than or comparable to maximum predicted longevity based on body mass) and high-longevity (HL, when maximum observed longevity was at least 20% higher than maximum predicted longevity, see Table S1). To infer expected longevity, we treated separately birds where longevity data derived from captive versus wild birds. We used the formulas Ac = 5.07 ± 1.63xW0.23±0.02 (where Ac is age in years for captive birds and W is weight in grams) and Aw = 4.75 ± 1.55xW0.17±0.01 (where Aw is age in years for wild birds) as formulated by Prinzinger [12]. Adult weight was extracted from AnAge [15].
The complete set of protein sequences from all 23 bird species analyzed was used in a blastp search and clustered with orthoMCL (http://orthomcl.org/orthomcl/) generating 22,380 clusters. Of these, 4,132 represented single-gene ortholog clusters (SGOs), with no more than one copy in each of the 23 bird species. In order to infer the level of selective pressure acting on longevity gene sets, phylogenetically-aware CDS alignments were generated by PRANK (http://wasabiapp.org/software/prank/), rooted with a tree obtained from the Avian Phylogenomics data [11]. To assess ratios of nonsynonymous (Ka) to synonymous (Ks) substitution rates we implemented branch-sites test of selection using codeml from the PAML (Phylogenetic Analysis by Maximum Likelihood) package (http://abacus.gene.ucl.ac.uk/software/paml.html), rooted with the same avian tree as before. For all models, codeml was run with the following parameters: runmode = 0, Codonfreq = 2, kappa = 3, omega = 0.2, fix_alpha = 1. For the null model control file (no selection in HL birds relative to RL birds) we specified model = 0, whereas for the alternative model (differential selection in HL birds relative to RL birds) we specified model = 2. To identify genes under selection in HL birds, the likelihood values of the two models were compared using a likelihood ratio test (LRT, p-value < 0.01, degrees of freedom = 1), and differential Ka/Ks (ω) values of HL versus RL birds were obtained to identify the direction of selection. Proteins predicted to be under significantly differential selective pressure were manually inspected and suspect protein predictions representing partial or artifactual gene models were removed from subsequent analyses. The complete list of genes under significant selection in HL birds relative to RL birds, ranked by differential ω (HL-RL), is presented in Data S1D. In general, Ka/Ks means were small, indicating strong purifying selection acting on these genes (62.5% and 82.6% ≤ 0.2 for HL and RL, respectively). Genes with divergent selection in long-lived birds were grouped into gene ontology (GO) functional annotation clusters using DAVID 6.8 (https://david.ncifcrf.gov/) (Table S2).
The Ka/Ks analyses considered all codon sites in protein-coding sequences. As a complementary test of differential selective pressure at specific sites, SGOs were analyzed with a site model implemented in codeml (http://abacus.gene.ucl.ac.uk/software/paml.html), comparing the likelihood values of a neutral (runmode = 0, model = 0, and NSites = 1) against a positive selection model (runmode = 0, model = 0, and NSites = 2) with LRT (p-value < 0.01, degrees of freedom = 2). Phylogenetic trees were constructed with PHYML (http://www.atgc-montpellier.fr/phyml/). The posterior probabilities of positively selected sites were estimated with Bayes Empirical Bayes (BEB) with α = 0.05. InterProScan 5 (http://www.ebi.ac.uk/interpro/interproscan.html) was used to annotate positively selected sites that correspond to residues located within discrete functional domains (Data S1E).
For the protein structural analysis of the catalytic domain of TERT, a model derived from Tribolium castaneum was used (Protein Data Bank ID: 3DU6). A multiple alignment was performed with PRANK to determine the residues corresponding to those with evidence of positive selection in the avian high longevity group. The Jmol visualizer (www.jmol.org/) was used to load and manipulate the PDB files, highlighting the position of selected residues.
Novel Genes
We sought to identify novel genes common to all parrots – and present in no other species – that arose from parent genes through gene duplication events or de novo through other mechanisms, and which could explain some aspects of psittacine biology. We searched for parrot novel genes using two complementary but separate strategies. The first strategy, based on sequence identity, was to manually examine the set of 1,822 Blue-fronted Amazon gene predictions for which no KEGG orthology group could be assigned during gene annotation. The second, based on gene synteny, was to employ a custom ‘locus-based’ strategy to identify gene predictions in the Blue-fronted Amazon genome that did not overlap with alignments of orthologous genes from chicken and zebra finch. For the second approach, we initially identified orthologous loci in the Blue-fronted Amazon genome by aligning, in independent analyses, the complete sets of chicken and zebra finch models from Ensembl 80 using BLAT (http://hgdownload.soe.ucsc.edu/admin/exe/), optimizing parameters to maximize model alignment while minimizing artifactual mappings (stepSize=3, maxNtSize=200000, repMatch=2253, minScore=100, minIdentity=0), and retaining only the best alignment of each query using pslReps (parameters: noIntrons, singleHit, minAli=0). Chicken and zebra finch were chosen as background species as they represent the non-parrot avian species with the most complete genomes and best-supported gene annotations, as well as representing the major Neoaves and Galloanseriforme lineages of birds (parrots belong to Neoaves). Blue-fronted Amazon gene predictions that significantly overlapped with the orthologous loci in these two other species were removed using BedOPS (https://github.com/bedops/bedops); the remaining predictions were considered candidate novel genes. The combined novel gene predictions from both approaches and subtracting species were filtered to remove predictions likely to represent artifactual models or retroviral elements (BLAT score <100, score/span ratio ≥0.7). Using this ‘locus-based’ approach, we identified 2,416 gene predictions in the Blue-fronted Amazon that were non-overlapping with non-parrot ortholog alignments.
After filtering to exclude small, low-scoring predictions and partial or compacted predictions likely to represent alignment artifacts, we performed extensive multi-species alignments and manual synteny verification to validate or exclude novel gene predictions (i.e. shared synteny in Blue-fronted Amazon, budgerigar, and kea; and present in no other avian or non-avian genomes). We first performed a blastp alignment of all novel gene predictions against the NCBI non-redundant protein sequence database, excluding predictions that returned significant hits in non-parrot species. This database represents over 68 million unique protein sequence predictions submitted to GenBank, including over 60 avian species to date (Genbank release 208). As a further exclusionary step, those predictions that were not excluded through the BLAST alignment were aligned to two distantly related parrot species (budgerigar, Kea) and outgroup species (zebra finch, Peregrine Falcon, chicken, American alligator, Anole lizard, and human) using BLAT with sensitized parameters optimized for these cross-species alignments, in order to confirm the presence of orthologous novel genes in all parrots and absence in non-parrots. This exhaustive validation step has been previously shown to be necessary for accurate novel gene prediction and exclusion of orthologs that may be partially sequenced, highly divergent, or unpredicted and thus not represented in NCBI’s sequence databases [14].
We note that synteny verification is a critical factor in maximizing the effectiveness of orthology determination, largely because BLAT or BLAST alignments, even when performed mutually and recursively, often cannot discriminate across paralogous genes and/or closely related members of the same gene family. Synteny verification is also particularly useful in situations (e.g. due to genomic gaps) when only a fragment of a gene is present and shows similar partial cross alignments to queries from multiple members of the same gene family. As previously discussed [14], this step is performed manually, by verifying across multiple species the flanking genes of search alignment hits for a given query. A gene is considered novel (de novo) or a novel paralog within a given species (or clade) when it is present in that species (or group) but fails to align at that same syntenic context in any other species (or clade); conversely, a gene is only considered the correct ortholog if there is evidence of alignment to the same syntenic context in other species and outgroups. Failure to utilize synteny criteria results in the frequent misidentification of orthologs, which confounds the correct classification of novel genes in a given species. As we took a conservative stance in excluding models where shared gene synteny in parrots could not be confirmed, ours is likely an underrepresentation of the true set of parrot-specific genes.
To further validate these predictions, we performed a brain transcriptome analysis to determine whether there is any evidence of expression of parrot novel genes. Given the unavailability of high-quality brain tissue from the Blue-fronted Amazon, a species protected by CITES (Convention on the Trade of Endangered Species), the expression of conserved parrot novel genes was assessed using the complete set of brain transcriptome data available from budgerigar.
The total brain transcriptomes of 3 male budgerigars were retrieved from NCBI (SRA Accession: SRR029329–30) and GigaDB (sample ID: GK0K2XF01). Trimmomatic (http://www.usadellab.org/cms/?page=trimmomatic) was used for adapter trimming as well as trimming of low-quality sequence. Only reads with length >30 bp were retained. An index of the budgerigar genome was built using bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml), and budgerigar transcriptome reads were mapped onto the budgerigar genome using tophat2 with the default recommended parameters (https://ccb.jhu.edu/software/tophat/index.shtml). Mapped reads were selected using SAMtools (http://www.htslib.org/). All non-uniquely mapped reads were removed, so that expression among closely related genes could be distinguished. Evidence of gene expression was assessed in terms of counts of reads wholly or partially mapped to exons of budgerigar gene models, or overlapping with such reads.
In order to investigate potential gene function, parrot novel gene predictions were subsequently characterized in terms of their predicted protein domains. Protein sequences were annotated using InterProScan 5 (http://www.ebi.ac.uk/interpro/interproscan.html), retrieving domain information from multiple publicly available databases including PROSITE, Pfam, PRINTS, ProDom, SMART, and PANTHER. Sequences were further annotated through searches against NCBI’s conserved domains database (https://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml) and blastp alignments to NCBI’s GenBank sequence database (release 208, https://www.ncbi.nlm.nih.gov/genbank/) to identify potential related genes in other species that could provide clues to gene family membership.
We identified several categories of high-confidence novel protein-coding genes that could be found exclusively in parrot genomes. A small subset of novel genes (n = 12; Table 1A, Data S1F) were single paralogs of an orthologous parent gene. For two of the genes, PSMD6 and DESI2, the duplicated copies are located at sites of syntenic disruption that are indicative of chromosomal rearrangements, suggesting their origination resulted from a breakpoint duplication event (Figure S1A and B). The fact that both copies in parrots share partial synteny with the parent gene in non-parrot genomes precludes a clear ortholog / duplicate distinction. It was also not possible to determine if the duplications resulted from intra- or inter-chromosomal rearrangements, as the Blue-fronted Amazon and budgerigar scaffolds have not yet been assembled into chromosomes. For the rest of the genes in this subset (Table 1A), we could unambiguously identify the parent gene ortholog that gave rise to the parrot-specific gene duplication by establishing conserved synteny of an ortholog in parrot and non-parrot genomes, and the presence of a novel paralog at a syntenic location unique to parrot genomes. None of these novel paralogs could be found in any non-parrot genomes, including the genomes of songbirds. In some cases the duplication was partial, as only one or few domains present in the parent gene could be seen in the duplicated copy, for example, PLXNC1L compared to PLXNC1 (Figure S1C)
A further subset of novel genes represented expanded gene families (n = 10 family expansions; Table 1B, Data S1G): these are genes that have undergone extensive duplication at multiple syntenic locations in parrot genomes, whereas only a single copy can be readily identified in non-parrot genomes. For six of these gene family expansions, we were able to infer function, discussed in the paper, based on predicted conserved domains and cross-alignments to NCBI sequence databases. Other gene expansions had no predicted conserved protein domains or significant BLAST alignments that could provide clues as to molecular function. A distinct subset were found to contain viral / retrotransposon-like elements, suggesting they largely represent retrotransposon-mediated expansions. Since these predictions are not present in non-parrot genomes, they seem to represent parrot specific viral-related expansions.
In addition to genes with clear gene family relationships, we have also discovered a large set (n = 136; Table 1C, Data S1H) of novel gene predictions unique to parrots that appear to represent de novo novel genes without a parent ortholog in any species. RNA-seq data confirm brain expression for a subset of these predictions (Data S1H1; n = 17, 12%), supporting their validity but also indicating a link to brain function. Half of this brain-expressed set (n = 8) had either predicted protein domains (e.g., transmembrane domain or signal peptide suggestive of membrane localization or secreted peptide, respectively) and/or partial BLAST hits to genes of known function (mostly enzymes) in other organisms, suggesting relations to those genes or functions. The other half (n = 9) did not have predicted domains or detectable BLAST hits to other genes. They likely do not represent false positive predictions, given their high conservation across parrot genomes. However, even though they had a predicted ORF, they might not be expressed as proteins and thus could represent long-noncoding RNAs, or even ‘proto-genes’ [28]. Growing evidence points to prominent roles of noncoding RNAs in gene expression regulation, and the expression of ‘pre-functional’ long-noncoding RNAs has also been suggested as a prerequisite for de novo novel gene evolution [28]. Distinguishing among the possibilities above will require further analyses of these brain-expressed novel transcripts, which would include assessment of expression at the protein level and/or examining possible transcriptional regulatory targets of noncoding RNAs.
Similarly, among novel predictions that lacked brain expression evidence, one subset had either predicted protein domains and/or partial BLAST hits to genes in other organisms (Data S1H2; n = 27, 20%), suggesting relations to other genes of known function. Additional subsets with no brain expression or predicted domains had either only very low-scoring BLAST hits (Data S1H3; n = 45, 33%), or no hits to non-parrot species (Data S1H4; n = 47, 35%), and thus their function and family relatedness are unclear. As with brain-expressed novel genes, these predictions may represent non-coding RNAs, but the lack of expression suggests that some could also be pseudogenes. The majority (73%) of these de novo novel gene variants showed substantial conservation across parrot species (>70% cross-species BLAT alignment), indicating that they likely represent real genetic sequences unique to parrots.
A distinct set (n = 34; Table 1D, Data S1I) of novel gene predictions from the Blue-fronted Amazon could not be found in other species and thus may represent unique features of the Blue-fronted Amazon genome. This set included both single gene paralogs as well as gene family expansions. The majority of these contain predicted protein domains (n = 21, 62%) Some of these genes were not detected in budgerigar only, and could represent novel psittacine genes that were lost in budgerigar only, or alternatively they might be absent in budgerigar genome only due to sequence gaps in the current assembly of the latter. We were unable to distinguish these possibilities, due to the fragmentary nature of the current Kea, Scarlet Macaw, and Puerto Rican Parrot assemblies, which precludes the syntenic analysis necessary for accurate orthology determination.
Ultra-Conserved Noncoding Elements
4,351 UCNE consensus sequences from chicken were retrieved from UCNEbase (http://ccg.vital-it.ch/UCNEbase/; [29]). Orthologous UCNEs were identified in Blue-fronted Amazon, as well as several parrot and non-parrot avian (outgroup) species where high-coverage genomes were available (zebra finch and chicken from Ensembl 80; Budgerigar, Kea, Golden-collared Manakin, Peregrine Falcon, Downy Woodpecker, Crested Ibis, Hoatzin, Pigeon, and Domestic duck from the GigaScience repository associated with the Avian Phylogenomics Consortium [11]), by aligning the chicken UCNE set to each genome using BLAST 2.2.31 with parameters -blastn -dust no -evalue .01 -max_target_seqs 1 - max_hsps 1. As UCNEs are defined as non-coding regions >200bp with ≥95% sequence identity between human and chicken, we considered UCNEs that exhibited a ratio of mismatches to total sequence length of 5% or more to be divergent. In order to identify UCNEs specifically divergent in the parrot lineage, we implemented the phylogenetic ANOVA in the R package GEIGER v 2.0.6 (https://cran.r-project.org/package=geiger), using the species tree from the avian phylogenomics consortium [11] and 1,000 in silico simulations. We thus identified 20 UCNEs that were significantly more divergent in parrots relative to other birds (phylogenetic ANOVA, α < 0.05, Table S5). Syntenic gene position of each UCNE was checked manually; UCNEs where orthology across species could not be confirmed by shared synteny were removed from subsequent analyses.
QUANTIFICATION AND STATISTICAL ANALYSIS
To identify genes under selection in HL birds relative to RL birds, null versus alternate models of selection were compared using a likelihood ratio test (LRT, p-value < 0.01, degrees of freedom = 1). To identify specific codon sites under selection in HL birds, a LRT was used to compare the likelihood values of a neutral agains a positive selection model (p-value < 0.01, degrees of freedom = 2; posterior probabilities of positively selected sites were estimated with Bayes Empirical Bayes with α = 0.05). After identifying a set of UCNEs putatively divergent in parrots (ratio of mismatches 5% or more relative to chicken consensus sequence) we performed additional testing to confirm significant divergence in 3 parrots relative to 9 avian outgroup species using a phylogenetic ANOVA (α < 0.05), using the species tree from the avian phylogenomics consortium [11] and 1,000 in silico simulations. All statistical tests were carried out in R (https://cran.r-project.org/).
Supplementary Material
Highlights.
The Blue-fronted Amazon Amazona aestiva and other parrots share unique novel genes
Convergent selection in long-lived birds suggests new lifespan-influencing genes
Parrot genomes share genetic changes related to genes critical for brain function
Similar changes in parrot and human genomes suggest convergent evolution of cognition
Acknowledgments
This work was supported through the Brazilian Avian Genome Consortium (SISBIO-Aves project, coordinated by MPCS, ATRV and CVM), funded by CNPq/FAPESPA. Further funding support that contributed to this project was provided by FAPERJ to FP (E-26/202.810/2015) and HHMI to EDJ. We also acknowledge that the comparative methods for novel genes analysis were developed with support from the NIH/NIDCD (R21 DC014432) to CVM. We would like to thank biologist Paulo Augusto Ribeiro Machado for providing access to the A. aestiva specimen (FVVF132).
Author Contributions
CVM, FP, ATRV, MT, MSF, MPCS, and AA conceived the project
CVM and MPCS were co-coordinators of the consortium (SISBIO-AVES) that provided funding and resources for the project, ATRV, FRS, CYM, EDJ, MSF and AA were participating members of the consortium
AVC, FRS, and MT performed sample identification, tissue collection, and DNA extraction, with contributions from AA, PNS, MSF, and CYM
JH and EDJ coordinated the genome sequencing
FP, ATRV, and LP conducted the genome assembly projects, with contributions from NCBL, AERS, MS, and EDJ
NC performed the repetitive element analyses
NCBL, FP, RLMG, and LP carried out the gene prediction and orthology annotation
MW and CVM performed the novel gene analyses
MW and NCBL carried out the brain transcriptome analyses
GM performed the miRNA analyses
MW, RLMG, and CVM performed the longevity analyses
MW performed the UCNE analyses
MW, CVM, FP, and EDJ wrote the manuscript, with contributions from RLMG, NCBL, AERS, NC, AA, FRS, and CYM.
All authors read and approved the final manuscript.
Footnotes
Declaration of Interests
The authors declare no competing interests
DATA AND SOFTWARE AVAILABILITY
The genome assembly, associated raw sequence data, and gene predictions for the Amazona aestiva have been deposited in GenBank, EMBL-EBI, and DDBJ under the accession ID LMAW00000000. The version described in this paper is version LMAW01000000.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iwaniuk AN, Dean KM, and Nelson JE (2005). Interspecific Allometry of the Brain and Brain Regions in Parrots (Psittaciformes): Comparisons with Other Birds and Primates. Brain, Behavior and Evolution 65, 40–59. [DOI] [PubMed] [Google Scholar]
- 2.Emery NJ (2006). Cognitive ornithology: the evolution of avian intelligence. Philosophical Transactions of the Royal Society of London B: Biological Sciences 361, 23–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brauth SE, Heaton JT, Shea SD, Durand SE, and Hall WS (1997). Functional Anatomy of Forebrain Vocal Control Pathways in the Budgerigar (Melopsittacus undulatus). Annals of the New York Academy of Sciences 807, 368–385. [DOI] [PubMed] [Google Scholar]
- 4.Munshi-South J, and Wilkinson GS (2006). Diet influences life span in parrots (Psittaciformes). The Auk 123, 108–118. [Google Scholar]
- 5.Olkowicz S, Kocourek M, Lučan RK, Porteš M, Fitch WT, Herculano-Houzel S, and Němec P (2016). Birds have primate-like numbers of neurons in the forebrain. Proceedings of the National Academy of Sciences 113, 7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emery NJ, and Clayton NS (2005). Evolution of the avian brain and intelligence. Current Biology 15, R946–R950. [DOI] [PubMed] [Google Scholar]
- 7.Hobson EA, Avery ML, and Wright TF (2014). The socioecology of Monk Parakeets: Insights into parrot social complexity. The Auk 131, 756–775. [Google Scholar]
- 8.Péron F, Rat-Fischer L, Lalot M, Nagle L, and Bovet D (2011). Cooperative problem solving in African grey parrots (Psittacus erithacus). Animal Cognition 14, 545–553. [DOI] [PubMed] [Google Scholar]
- 9.Iwaniuk AN, and Nelson JE (2003). Developmental differences are correlated with relative brain size in birds: a comparative analysis. Canadian Journal of Zoology 81, 1913–1928. [Google Scholar]
- 10.Wright TF, Schirtzinger EE, Matsumoto T, Eberhard JR, Graves GR, Sanchez JJ, Capelli S, Müller H, Scharpegge J, Chambers GK, et al. (2008). A Multilocus Molecular Phylogeny of the Parrots (Psittaciformes): Support for a Gondwanan Origin during the Cretaceous. Molecular Biology and Evolution 25, 2141–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW, et al. (2014). Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346, 1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prinzinger R (1993). Life span in birds and the ageing theory of absolute metabolic scope. Comparative Biochemistry and Physiology Part A: Physiology 105, 609–615. [Google Scholar]
- 13.Young AM, Hobson EA, Lackey LB, and Wright TF (2012). Survival on the ark: life-history trends in captive parrots. Animal Conservation 15, 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirthlin M, Lovell PV, Jarvis ED, and Mello CV (2014). Comparative genomics reveals molecular features unique to the songbird lineage. BMC Genomics 15, 1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tacutu R, Craig T, Budovsky A, Wuttke D, Lehmann G, Taranukha D, Costa J, Fraifeld VE, and de Magalhães JP (2013). Human Ageing Genomic Resources: Integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Research 41, D1027–D1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keane M, Semeiks J, Webb Andrew E., Li Yang I., Quesada V, Craig T, Madsen Lone B., van Dam S, Brawand D, Marques Patricia I., et al. (2015). Insights into the Evolution of Longevity from the Bowhead Whale Genome. Cell Reports 10, 112–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haussmann MF, Winkler DW, Huntington CE, Nisbet ICT, and Vleck CM (2004). Telomerase Expression Is Differentially Regulated in Birds of Differing Life Span. Annals of the New York Academy of Sciences 1019, 186–190. [DOI] [PubMed] [Google Scholar]
- 18.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, and Shay JW (1994). Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- 19.Zelko IN, Mariani TJ, and Folz RJ (2002). Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radical Biology and Medicine 33, 337–349. [DOI] [PubMed] [Google Scholar]
- 20.Montgomery MK, Buttemer WA, and Hulbert AJ (2012). Does the oxidative stress theory of aging explain longevity differences in birds? II. Antioxidant systems and oxidative damage. Experimental Gerontology 47, 211–222. [DOI] [PubMed] [Google Scholar]
- 21.Lambert AJ, Boysen HM, Buckingham JA, Yang T, Podlutsky A, Austad SN, Kunz TH, Buffenstein R, and Brand MD (2007). Low rates of hydrogen peroxide production by isolated heart mitochondria associate with long maximum lifespan in vertebrate homeotherms. Aging Cell 6, 607–618. [DOI] [PubMed] [Google Scholar]
- 22.Keane M, Craig T, Alföldi J, Berlin AM, Johnson J, Seluanov A, Gorbunova V, Di Palma F, Lindblad-Toh K, Church GM, et al. (2014). The Naked Mole Rat Genome Resource: facilitating analyses of cancer and longevity-related adaptations. Bioinformatics 30, 3558–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pfenning AR, Hara E, Whitney O, Rivas MV, Wang R, Roulhac PL, Howard JT, Wirthlin M, Lovell PV, Ganapathy G, et al. (2014). Convergent transcriptional specializations in the brains of humans and song-learning birds. Science 346, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Failler M, Gee Heon Y., Krug P, Joo K, Halbritter J, Belkacem L, Filhol E, Porath Jonathan D., Braun Daniela A., Schueler M, et al. (2014). Mutations of CEP83 Cause Infantile Nephronophthisis and Intellectual Disability. The American Journal of Human Genetics 94, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diering GH, Mills F, Bamji SX, and Numata M (2011). Regulation of dendritic spine growth through activity-dependent recruitment of the brain-enriched Na+/H+ exchanger NHE5. Molecular Biology of the Cell 22, 2246–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giannuzzi G, Siswara P, Malig M, Marques-Bonet T, Program NCS, Mullikin JC, Ventura M, and Eichler EE (2013). Evolutionary dynamism of the primate LRRC37 gene family. Genome Research 23, 46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu X, Yan R, Song L, Lu X, Chen S, and Zhao S (2014). Subcellular localization and function of mouse radial spoke protein 3 in mammalian cells and central nervous system. Journal of Molecular Histology 45, 723–732. [DOI] [PubMed] [Google Scholar]
- 28.Carvunis A-R, Rolland T, Wapinski I, Calderwood MA, Yildirim MA, Simonis N, Charloteaux B, Hidalgo CA, Barbette J, Santhanam B, et al. (2012). Proto-genes and de novo gene birth. Nature 487, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dimitrieva S, and Bucher P (2013). UCNEbase—a database of ultraconserved non-coding elements and genomic regulatory blocks. Nucleic Acids Research 41, D101–D109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oksenberg N, Stevison L, Wall JD, and Ahituv N (2013). Function and Regulation of AUTS2, a Gene Implicated in Autism and Human Evolution. PLoS Genet 9, e1003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaaro-Peled H, Hayashi-Takagi A, Seshadri S, Kamiya A, Brandon NJ, and Sawa A (2009). Neurodevelopmental mechanisms of schizophrenia: understanding disturbed postnatal brain maturation through neuregulin-1-ErbB4 and DISC1. Trends in Neurosciences 32, 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zembrzycki A, Griesel G, Stoykova A, and Mansouri A (2007). Genetic interplay between the transcription factors Sp8 and Emx2 in the patterning of the forebrain. Neural Development 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Azim E, Jabaudon D, Fame RM, and Macklis JD (2009). SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat Neurosci 12, 1238–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karalay Ö, Doberauer K, Vadodaria KC, Knobloch M, Berti L, Miquelajauregui A, Schwark M, Jagasia R, Taketo MM, Tarabykin V, et al. (2011). Prospero-related homeobox1 gene (Prox1) is regulated by canonical Wnt signaling and has a stage-specific role in adult hippocampal neurogenesis. Proceedings of the National Academy of Sciences 108, 5807–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michaelson JJ, Shin M-K, Koh J-Y, Brueggeman L, Zhang A, Katzman A, McDaniel L, Fang M, Pufall M, and Pieper AA (2017). Neuronal PAS Domain Proteins 1 and 3 Are Master Regulators of Neuropsychiatric Risk Genes. Biological Psychiatry 82, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sleven H, Welsh SJ, Yu J, Churchill MEA, Wright CF, Henderson A, Horvath R, Rankin J, Vogt J, Magee A, et al. (2017). De Novo Mutations in EBF3 Cause a Neurodevelopmental Syndrome. The American Journal of Human Genetics 100, 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balci TB, Sawyer SL, Davila J, Humphreys P, and Dyment DA (2015). Brain malformations in a patient with deletion 2p 16.1: A refinement of the phenotype to BCL11A. European Journal of Medical Genetics 58, 351–354. [DOI] [PubMed] [Google Scholar]
- 38.Terwisscha van Scheltinga AF, Bakker SC, van Haren NEM, Derks EM, Buizer-Voskamp JE, Boos HBM, Cahn W, Hulshoff Pol HE, Ripke S, Ophoff RA, et al. (2013). Genetic Schizophrenia Risk Variants Jointly Modulate Total Brain and White Matter Volume. Biological Psychiatry 73, 525–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prabhakar S, Noonan JP, Pääbo S, and Rubin EM (2006). Accelerated Evolution of Conserved Noncoding Sequences in Humans. Science 314, 786–786. [DOI] [PubMed] [Google Scholar]
- 40.Pollard KS, Salama SR, King B, Kern AD, Dreszer T, Katzman S, Siepel A, Pedersen JS, Bejerano G, Baertsch R, et al. (2006). Forces Shaping the Fastest Evolving Regions in the Human Genome. PLoS Genet 2, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.