

Abstract
Tumors bearing homologous recombination deficiency are extremely sensitive to DNA double strand breaks induced by several chemotherapeutic agents. ATM gene, encoding a protein involved in DNA damage response, is frequently mutated in colorectal cancer (CRC), but its potential role as predictive and prognostic biomarker has not been fully investigated. We carried out a multicenter effort aimed at defining the prognostic impact of ATM mutational status in metastatic CRC (mCRC) patients. Mutational profiles were obtained by means of next-generation sequencing. Overall, 35 out of 227 samples (15%) carried an ATM mutation. At a median follow-up of 56.6 months, patients with ATM mutated tumors showed a significantly longer median overall survival (OS) versus ATM wild-type ones (64.9 vs 34.8 months; HR, 0.50; 95% CI, 0.29–0.85; P = 0.01). In the multivariable model, ATM mutations confirmed the association with longer OS (HR, 0.57; 95% CI, 0.33–0.98; P = 0.04). The prognostic impact of ATM mutations was independent from TP53 mutational status and primary tumor location. High heterogeneity score for ATM mutations, possibly reflecting the loss of wild-type allele, was associated with excellent prognosis. In conclusion, we showed that ATM mutations are independently associated with longer OS in patients with mCRC.
Introduction
Significant advances in the implementation of biomarkers in the clinical practice have been achieved in metastatic colorectal cancer (mCRC), even if only few of them (such as RAS and BRAF mutational status or microsatellite instability [MSI]) are endowed with clinical relevance. Furthermore, despite the advances achieved in understanding the molecular bases of resistance to EGFR targeting agents1–3, there is still a lack of biomarkers able to predict sensitivity/resistance to chemotherapy, which remains the cornerstone of treatment for most patients.
Cancer cells may gain the potential for uncontrolled growth by escaping functional cell-cycle checkpoints. By doing so, they simultaneously become vulnerable to both endogenous (e.g. oncogenic-driven replication stress) and exogenous (e.g. DNA-damaging agents) genotoxic insults4. Tumors with homologous recombination deficiency are extremely sensitive to cross-linking agents such as platinum salts, or topoisomerase inhibitors. This mechanism has substantial implications in the clinical practice, specifically concerning the management of those tumors bearing deleterious BRCA1-2 mutations (e.g. BRCA-mutated breast and ovarian cancer)5,6.
Ataxia-Telangiectasia Mutated (ATM) is a gene member of the highly conserved PI3K-related kinases, on which cells rely for orchestrating the DNA damage response (DDR) for both DNA repair and cell-cycle checkpoint activation. Specifically, ATM is recruited upon DNA double strand breaks (DSBs) and is involved in DNA repair via both BRCA1-driven homologous recombination and non-homologous end-joining pathways, as well as in the G1/S cellular checkpoint activation through its major targets p53 and CHK27.
Germline and somatic mutations involving homologous recombination related genes, including ATM, are predicted to confer an enhanced platinum sensitivity8. Specifically, ATM deficient tumors display a higher sensitivity to DNA DSB-inducing treatments9 and loss of function mutations affecting the ATM gene could confer a vulnerability to DNA-damaging agents, especially in combination with p53 deficiency10–13. Because of the consistent prevalence of ATM mutations in CRC (7% in non-hypermutated cases)14 and their potential crucial role as biomarker of chemosensitivity to platinum salts and topoisomerase inhibitors, ATM mutations would therefore characterize mCRC patients with a more favourable outcome, at least when eligible for combination chemotherapy. Moving from this background, we performed a translational study aimed at assessing the prognostic relevance of ATM mutational status in mCRC patients.
Materials and Methods
Patients population
We retrieved pre-treatment tumor tissue blocks of initially unresectable mCRC patients treated at two Italian Institutions (Fondazione IRCCS Istituto Nazionale dei Tumori di Milano and Azienda Ospedaliero-Universitaria Pisana). Clinical, pathological and molecular characteristics at the time of diagnosis of metastatic disease were collected, including age, gender, Eastern Cooperative Oncology Group (ECOG) Performance Status (PS), primary tumor location (right- vs left-sided), primary tumor resection (yes vs no), time-to-metastases (synchronous vs metachronous), number of metastatic sites (1 vs >1), RAS and BRAF mutational status, and MSI status. All included patients received at least one treatment line with doublet or triplet regimens with or without monoclonal antibodies according to standard clinical practice. The study was approved by the Fondazione IRCCS Istituto Nazionale dei Tumori di Milano Institutional Review Board (study ID: INT 117/15) and conducted according to the ethical principles for medical research involving human subjects adopted in the Declaration of Helsinki. All patients signed a written informed consent.
Next-generation sequencing analysis
We centrally collected formalin-fixed paraffin-embedded archival tumor tissue blocks. Next-generation sequencing (NGS) data were obtained through the Ion AmpliSeq Cancer Hotspot Panel v2 (Life Technologies) with the Ion-Torrent™ Personal Genome Machine platform (Life Technologies), as previously described15,16 and detailed in Supplementary Methods (see Supplementary Information). ATM and TP53 mutational status was obtained, and RAS and BRAF mutational status was centrally confirmed. Heterogeneity score (HS) of ATM mutations was calculated as previously described by Normanno et al.17. Briefly, the mutant allelic frequency was normalized for the neoplastic cell content, and the HS was calculated by multiplying by 2 the frequency of mutant alleles in neoplastic cells as somatic mutations usually involve only one allele.
Statistical analysis
Chi-square test or fisher exact test were used, as appropriate, to evaluate the association between ATM mutational status and the other relevant clinical and pathological patients’ characteristics. Overall survival (OS) was calculated as the time from diagnosis of metastatic disease to the death from any cause. Since chemotherapy sensitivity putatively caused by ATM mutations may be boosted by the concomitant presence of TP53 mutations10 or primary tumor sidedness due to enrichment of mesenchymal and stem-like subtypes in right-sided tumors18 we also evaluated the prognostic impact of combined ATM and TP53 mutational status assessment as well as the prognostic impact of combined ATM mutational status and primary tumor location. The Kaplan-Meier method and the Cox proportional-hazards model were used for survival analyses. Hazard ratios (HRs) together with 95% confidence intervals (CI) were provided. Statistical significance threshold was set to a two-tailed 0.05 value. R software (version 3.5.0) and RStudio software (version 1.1.453) were used for Statistical analyses.
Results
Clinical, pathological and molecular features of ATM mutated mCRC
As detailed in the Consort diagram (Supplementary Fig. S1 in Supplementary Information), the final study population included a total of 227 patients, of whom 35 (15%) had ATM mutated tumors and 192 (85%) ATM wild-type tumors. TP53 mutations were found in a total of 148 (65%) of samples, of whom 24 (69%) in the ATM mutated subgroup and 124 (65%) in ATM wild-type one (P = 0.65). Table 1 shows patients’ demographics and disease characteristics, overall and according to ATM mutational status. Of note, ATM mutations were not significantly associated with specific clinical and molecular features. The exposure to specific agents approved for mCRC and the number of treatment lines received are summarized in Supplementary Table S1 (see Supplementary Information). Table 2 illustrates the specific mutations found in ATM gene and concomitant “trunk” mutations affecting TP53, KRAS, NRAS, BRAF and APC, with relative HS. The median HS for ATM mutations was 116 (IQR, 51–197).
Table 1.
Patients’ and disease characteristics, overall and according to ATM mutational status.
| Characteristics | Total (N = 227) N (%) |
ATM mut (N = 35) N (%) |
ATM wt (N = 192) N (%) |
P* | |
|---|---|---|---|---|---|
| Age (years) | <65 ≥65 |
147 (65) 80 (35) |
25 (71) 10 (29) |
122 (64) 70 (36) |
0.40 |
| Gender | Male Female |
93 (41) 134 (59) |
17 (49) 18 (51) |
76 (40) 116 (60) |
0.32 |
| ECOG PS | 0 1–2 NA |
197 (92) 18 (8) 12 |
34 (97) 1(3) 0 |
163 (91) 17 (9) 12 |
0.20 |
| Primary tumor location | Left-sided Right-sided |
159 (70) 68 (30) |
27 (77) 8 (23) |
132 (69) 60 (31) |
0.32 |
| Primary tumor resection | Yes No |
192 (85) 35 (15) |
31 (89) 4 (11) |
161 (84) 31 (16) |
0.48 |
| Synchronous mets | No Yes |
67 (30) 160 (70) |
13 (37) 22 (63) |
54 (28) 138 (72) |
0.28 |
| Metastatic sites (N) | 1> 1 |
135 (59) 92 (41) |
24 (69) 11 (31) |
111 (58) 81 (42) |
0.23 |
| All-RAS status | Wild-type Mutated |
127 (56) 100 (44) |
20 (57) 15 (43) |
107 (56) 85 (44) |
0.88 |
| BRAF status | Wild-type Mutated |
214 (94) 13 (6) |
33 (94) 2 (6) |
181 (94) 11 (6) |
0.99 |
| MSI status | MSS MSI NA |
188 (94) 13 (6) 26 |
26 (87) 4 (13) 5 |
162 (95) 9 (5) 21 |
0.11 |
*Chi-square test or Fisher exact test, as appropriate.
Abbreviations. ECOG PS: Eastern Cooperative Oncology Group Performance Status. MSI: microsatellite instability. MSS: microsatellite stability. Mut: mutated. Wt: wild-type.
Table 2.
Specific mutations found in ATM gene with concomitant “trunk” mutations (affecting TP53, KRAS, NRAS, BRAF and APC) with relative heterogeneity score.
| ID | MSI status | ATM | TP53 | KRAS | NRAS | BRAF | APC | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | HS | Mutation | HS | Mutation | HS | Mutation | HS | Mutation | HS | Mutation | HS | ||
| 1 | MSI | K610T | 60 | R248Q | 70 | G12V | 70 | — | — | — | — | E1464VfsTer8 | 44 |
| 2 | NA | E1325Stop | 94 | R175C | 180 | — | — | — | — | — | — | — | — |
| 3 | MSS | D2870H | 52 | — | — | G12S | 114 | — | — | — | — | R1450Stop | 106 |
| 4 | MSS | R3047Stop | 88 | — | — | G12D | 58 | — | — | — | — | R1450Stop | 46 |
| 5 | MSI | R337C | 32 | — | — | G13D | 36 | — | — | — | — | I1307K | 156 |
| 6 | MSS | P3050L | 254 | P278S | 173 | Q61H | 120 | — | — | — | — | — | — |
| 7 | MSI | P604S | 132 | R196Stop | 194 | G12V | 140 | — | — | — | — | E1286Stop | 198 |
| 8 | MSS | R337C | 30 | I254S | 200 | G12V | 140 | — | — | — | — | R1450Stop | 64 |
| 9 | MSS | A1309T | 136 | S215R | 192 | — | — | — | — | — | — | S1346Stop | 72 |
| 10 | MSS | V410A | 230 | — | — | — | — | — | — | — | — | — | — |
| 11 | MSS | R337H | 34 | — | — | — | — | — | — | — | I1311MfsTer10 | 200 | |
| 12 | NA | R2443Q | 184 | R273C | 290 | — | — | — | — | — | — | E1353FfsTer20 | 284 |
| 13 | MSS | R337C | 114 | — | — | A146T | 212 | — | — | — | — | T1438HfsTer35 | 106 |
| 14 | MSS | E1704D | 240 | — | — | — | — | — | — | — | — | E1379Stop | 710 |
| 15 | MSS | Q2729H | 128 | — | — | A146T | 104 | — | — | — | — | T1556NfsTer3 | 76 |
| 16 | MSS | V410A | 200 | — | — | — | — | — | — | — | — | E1309DfsTer4 | 158 |
| 17 | MSS | R337H | 46 | R249G | 42 | G12V | 46 | — | — | — | — | — | — |
| 18 | MSS | P604S | 194 | R273H | 306 | — | — | — | — | — | — | Q12894Stop | 320 |
| 19 | MSS | R2691H | 52 | C238Y | 122 | — | — | — | — | — | — | E1317Q | 158 |
| 20 | MSS | R337C | 50 | — | — | G12V | 96 | — | — | — | — | H1349QfsTer4 | 166 |
| 21 | MSS | S333F | 40 | G266E | 760 | — | — | — | — | — | — | — | — |
| 22 | MSS | L1939V | 44 | R282W | 268 | — | — | — | — | — | — | — | — |
| 23 | MSS | V410A | 142 | I251S | 98 | — | — | — | — | — | — | E1547Stop | 86 |
| 24 | MSS | V410A | 314 | Y205H | 207 | — | — | — | — | — | — | — | — |
| 25 | MSS | R337H | 108 | R175H | 196 | G13D | 190 | — | — | — | — | — | — |
| 26 | MSS | S333F | 290 | R273C | 140 | — | — | — | — | — | — | — | — |
| 27 | MSS | S1691R | 204 | R175H | 113 | — | — | — | — | — | — | — | — |
| 28 | MSS | splice site 184_185 + K1992T | 212 + 102 | V73fs*50 | 90 | — | — | — | — | V600E | 350 | — | — |
| 29 | MSI | F1928fs*9 | 206 | R27H + R17H | 340 | — | — | — | — | V600E | 468 | — | — |
| 30 | MSS | F858L | 300 | V274F | 66 | — | — | — | — | — | — | — | — |
| 31 | MSS | R337H | 20 | — | — | A146T | 140 | — | — | — | — | — | — |
| 32 | MSS | R2912G | 116 | R2912G | 400 | — | — | G12S | 120 | — | — | — | — |
| 33 | MSS | G2695V | 46 | G245S | 60 | — | — | — | — | — | — | R1450Stop | 50 |
| 34 | MSS | V410A | 132 | R273C | 108 | G12V | — | — | — | — | — | R1450Stop | 62 |
| 35 | MSS | F858L | 146 | R282W | 202 | — | — | — | — | — | — | Q1291Stop | 84 |
Abbreviations. HS: heterogeneity score. MSI: microsatellite instability. MSS: microsatellite stability.
Prognostic role of ATM mutations in mCRC patients
At a median follow-up of 56.6 months (95% CI, 46.3–62.1), patients with ATM mutated tumors showed a significantly longer median OS than patients with ATM wild-type tumors (64.9 versus 34.8 months; HR, 0.50; 95% CI, 0.29–0.85; P = 0.01) (Fig. 1). In the multivariable model (Table 3), including other covariates significantly associated with OS, the presence of ATM mutations confirmed its association with improved OS (HR, 0.57; 95% CI, 0.33–0.98; P = 0.04), along with left-sided primary tumor location (P = 0.005), primary tumor resection (P = 0.003), metachronous metastases (P = 0.005) and the presence of a single site of metastasis (P = 0.03).
Figure 1.
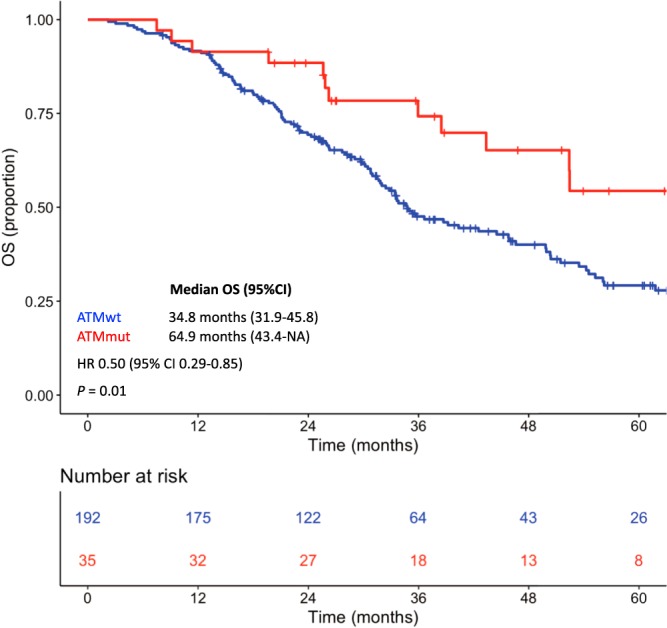
Kaplan-Meier curves for overall survival according to ATM mutational status. Red line indicates patients with ATM mutated tumors, blue line indicates patients with ATM wild-type tumors.
Table 3.
Univariate and multivariate analyses for overall survival.
| Characteristics | Univariate analyses | Multivariable model | |||
|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | ||
| Age (years) | ≥65 vs <65 | 1.60 (1.10–2.20) | 0.009 | — | 0.25 |
| Gender | Female vs Male | — | 0.24 | — | — |
| ECOG PS | 1–2 vs 0 | — | 0.15 | — | — |
| Primary tumor location | Right vs Left | 2.00 (1.40–2.80) | <0.001 | 1.70 (1.17–2.46) | 0.005 |
| Primary tumor resection | No vs Yes | 1.90 (1.20–2.90) | 0.005 | 1.62 (1.04–2.54) | 0.03 |
| Synchronous mets | Yes vs No | 1.80 (1.20–2.60) | 0.003 | 1.76 (1.19–2.61) | 0.005 |
| Metastatic sites (N) | >1 vs 1 | 1.70 (1.20–2.40) | 0.002 | 1.47 (1.03–2.08) | 0.03 |
| All-RAS status | Mut vs wt | — | 0.06 | — | — |
| BRAF status | Mut vs wt | 2.10 (1.10–4.00) | 0.03 | — | 0.09 |
| MSI status | MSI vs MSS | — | 0.16 | — | — |
| ATM status | Mut vs wt | 0.50 (0.29–0.85) | 0.01 | 0.57 (0.33–0.98) | 0.04 |
Abbreviations. ECOG PS: Eastern Cooperative Oncology Group Performance Status. Mets: metastases. MSI: microsatellite instability. MSS: microsatellite stable. Mut: mutated. Wt: wild-type.
Among patients with ATM mutated tumors, an HS ≥ 100 for ATM mutations was associated with a longer median OS compared with an HS < 100 (70.1 versus 38.5 months; HR, 0.28; 95% CI 0.09–0.85; P = 0.02) (Fig. 2). Therefore, when using patients with wild-type ATM as reference, the HR for patients with ATM mutated tumors and HS < 100 was 0.91 (95% CI, 0.44–1.86; P = 0.79), whereas it relevantly decreased for patients with ATM mutated tumors and HS ≥ 100 (HR, 0.57; 95% CI, 0.39–0.84; P = 0.004).
Figure 2.
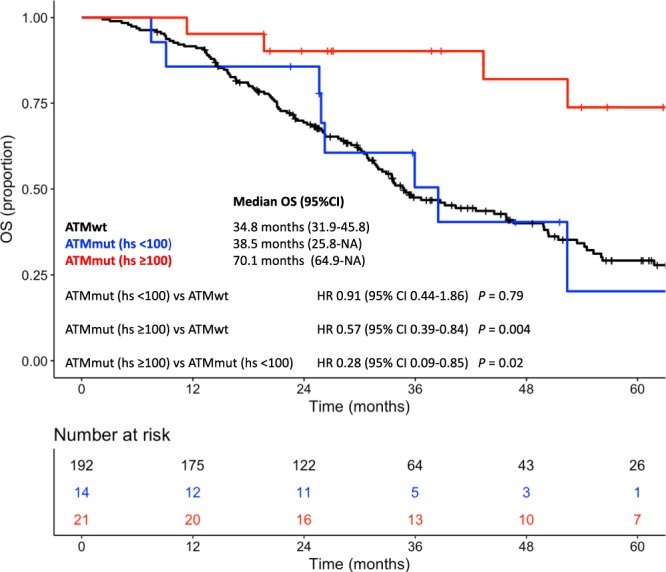
Kaplan-Meier curves for overall survival according to ATM mutational status and ATM mutational heterogeneity score. Red line indicates patients with ATM mutated tumors and ATM HS ≥ 100, blue line indicates patients with ATM mutated tumors and ATM HS < 100, black line indicated patients with ATM wild-type tumors. Abbreviations: HS: heterogeneity score.
Of note, no prognostic significance was observed for TP53 mutational status (P = 0.79), and the prognostic impact of ATM mutations was completely independent from the concomitant presence of TP53 mutations (Supplementary Fig. S2 in Supplementary Information) or primary tumor sidedness (Supplementary Fig. S3 in Supplementary Information).
Finally, since we performed a massively parallel sequencing of multiple cancer-related genes, we assessed the prognostic value of the top mutated genes (i.e. those found mutated in at least 5% of samples: ATM, KRAS, BRAF, NRAS, APC, PIK3CA, SMAD4, FBXW7 and MET) and applied the Benjamini–Hochberg procedure in order to decrease the false discovery rate, demonstrating that the P-value for ATM mutational status remained significant (P = 0.04) (Supplementary Table S2 in Supplementary Information).
Discussion
Given the crucial role of ATM activity in orchestrating the DDR, relevant phenotypic spillover is awaited upon its loss. However, as a result of both biological complexity of the DDR network and heterogeneity across different studies, no conclusive clinical data are available on ATM prognostic and/or predictive impact.
In early stage CRC, low ATM expression has been previously associated with worse outcomes. In a series of 330 early CRCs, the presence of ATM expression detected by immunohistochemistry (IHC) was associated with disease-free survival and OS benefit when considering patients who underwent adjuvant treatments (N = 33)19. Similar results have been confirmed by a subgroup analysis of the VICTOR trial, which included stage II/III CRC patients undergoing adjuvant fluoropyrimidine-based chemotherapy20. Regarding the metastatic setting, a recent monocentric study showed that ATM deficiency (as primarily assessed by IHC) may be associated with improved OS following oxaliplatin-based first-line treatment, but not irinotecan-based one21. Discrepancy in available evidences might be related to the different prognostic role of ATM loss of function according to disease stage, similarly to what reported for MSI22, and the confounding effects derived from the heterogeneity of available regimens and treatment sequences used for metastatic disease.
This is the larger available study assessing the role of ATM mutations as prognostic biomarker in mCRC. Here, the presence of ATM mutations was independently associated with improved OS (adjusted HR, 0.57; 95% CI, 0.33–0.98; P = 0.04). These results suggest that ATM mutations might identify a biologically distinct disease with a survival advantage in the metastatic setting linked, at least in part, to an increased chemosensitivity. Intriguingly, patients with ATM mutations and an HS ≥ 100, showed the best outcomes in terms of OS. As previously described by Normanno et al.17, HS virtually corresponds to the fraction of neoplastic cells bearing a specific mutation. Specifically, an HS > 100 might reflect the loss of the wild-type allele. HS might help identifying tumors with a “functional knock-out” of ATM that lose their ability of properly coping with DNA damage. Therefore ATM HS should be taken into account by future studies and potentially correlated with functional data.
From a preclinical point of view, p53 is one of the most characterized ATM targets, required for G1/S cell arrest and apoptosis. Conceptually, drugs inducing high amount of DNA damage in S phase in cells with both DNA repair and G1/S–G2/M checkpoint deficiency (such as those bearing both ATM and TP53 mutations) are likely to induce a mitotic catastrophe-mediated cell death23. However, we did not find any clinically relevant interaction between ATM and TP53 mutational status in impacting on OS (Supplementary Fig. S2 in Supplementary Information). It must be pointed out that, even if ATM or CHK2 suppression preferentially sensitizes p53 deficient tumors to genotoxic drugs, a chemosensitivity status driven by ATM deficiency might occur independently from TP5324.
In addition, an enrichment of ATM mutations is expected in mCRC patients with right-sided tumors18, MSI-high14 or CMS1 ones25. In our study, the prognostic impact of ATM mutational status was independent from primary tumor location status (Table 3 and Supplementary Fig. S3 in Supplementary Information), even if the low number of patients with ATM mutations and right-sided mCRC highlights the need of larger datasets to specifically assess the impact of DDR alterations according to primary tumor location or disease subtypes.
Our study has some limitations. For instance, despite the strong rationale making ATM mutational status a candidate biomarker of response to oxaliplatin and/or irinotecan26–29, we have not considered response rate or progression-free survival because of the heterogeneity of treatment regimens as per standard practice (fluoropyrimidine monotherapy, doublet or triplet chemotherapy regimens associated or not with anti-VEGF or anti-EGFR). Of course, an integrated assessment of both protein expression and mutational status would be necessary for identifying all tumors with clinically relevant ATM loss of function. In fact, other mechanisms might account for ATM reduced activity, such as low expression due to promoter methylation30. Indeed, a comprehensive assessment of the DDR network on a proteomic scale is expected to reach the best accuracy for predicting chemosensitivity.
Beyond being a sole biomarker of chemosensitivity, ATM mutations might predict response to DDR-targeting agents paralleling recent achievements in other clinical settings, such as castration-resistant prostate cancer (CRPC). Indeed, in the TOPARP-A phase II trial CRPC patients bearing alterations in homologous recombination repair genes displayed a high response rate to the PARP inhibitor olaparib (including 4 out of 5 patients with tumors bearing ATM mutations)31. Similar therapeutic approach would be backed by a strong preclinical rationale also in CRC32. The reader is referred to Choi et al.33 for reviewing potential synthetic lethality strategies (e.g PARP1 or ATR inhibitors) in ATM deficient tumors.
In conclusion, our study suggests that ATM mutations with high HS might characterize a subset of mCRC at better prognosis. From this background, further investigations are needed to cover crucial unresolved issues such as the assessment of functional relevance of specific ATM mutations and their predictive role upon specific DNA damaging and/or DDR-targeting agents. Indeed, synthetic lethality strategies might be preferentially used in ATM deficient tumors, while ATM proficient tumors might be sensitized to conventional therapies by ATM inhibitors34. Thus, ATM mutational status could enter the clinical decision-taking process in parallel with the development of specific targeted strategies.
Supplementary information
Author Contributions
Study conception and design: G.R., G.F., D.R., and fp (Filippo Pagani), F.P. (Filippo Pietrantonio). Acquisition of data: G.R., D.R., f.p., A.R., and f.P. (Federica Perrone), E.T., A.B., G.P., F.M., M.N., S.C., M.A., S.S., B.B., G.Z., and M.M. Analysis and interpretation of data: G.R., G.F., D.R., f.p., F.P., C.C., G.P., and A.R. Drafting of manuscript: G.R., G.F., D.R., f.p., F.P., A.R., F.M., M.N., S.C., and M.A. Critical revision: F.P., C.C., A.F., G.P., M.D.B., and F.D.B.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Giovanni Randon and Giovanni Fucà contributed equally.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-019-39525-3.
References
- 1.Cremolini C, et al. Negative hyperselection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: the PRESSING case-control study. Ann. Oncol. 2017;28:3009–3014. doi: 10.1093/annonc/mdx546. [DOI] [PubMed] [Google Scholar]
- 2.Van Emburgh BO, et al. Acquired RAS or EGFR mutations and duration of response to EGFR blockade in colorectal cancer. Nat. Commun. 2016;7:13665. doi: 10.1038/ncomms13665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pietrantonio F, et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin. Cancer Res. 2017;23:2414–2422. doi: 10.1158/1078-0432.CCR-16-1863. [DOI] [PubMed] [Google Scholar]
- 4.Gavande NS, et al. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016;160:65–83. doi: 10.1016/j.pharmthera.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrski T, et al. Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1- positive metastatic breast cancer. Breast Cancer Res. 2012;14:R110. doi: 10.1186/bcr3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrski T, et al. Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res. Treat. 2014;147:401–405. doi: 10.1007/s10549-014-3100-x. [DOI] [PubMed] [Google Scholar]
- 7.Shiloh Y, Yael Z. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell. Biol. 2013;14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- 8.Pennington KP, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014;20:764–75. doi: 10.1158/1078-0432.CCR-13-2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tribius S, Pidel A, Casper D. ATM protein expression correlates with radioresistance in primary glioblastoma cells in culture. Int. J. Radiat. Oncol. Biol. Phys. 2001;50:511–523. doi: 10.1016/S0360-3016(01)01489-4. [DOI] [PubMed] [Google Scholar]
- 10.Jiang H. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev. 2009;223:1895–1909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biddlestone-Thorpe L, et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013;19:3189–200. doi: 10.1158/1078-0432.CCR-12-3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pietrantonio F, et al. TP53 mutations in advanced colorectal cancer: the dark side of the moon. Oncology. 2014;86:289–294. doi: 10.1159/000360088. [DOI] [PubMed] [Google Scholar]
- 13.Cremona CA, Behrens A. ATM signaling and cancer. Oncogene. 2014;33:3351–3360. doi: 10.1038/onc.2013.275. [DOI] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pietrantonio F, et al. Biomarkers of primary resistance to Trastuzumab in HER2-positive metastatic gastric cancer patients: the AMNESIA case-control study. Clin. Cancer Res. 2018;24:1082–1089. doi: 10.1158/1078-0432.CCR-17-2781. [DOI] [PubMed] [Google Scholar]
- 16.Pietrantonio F, et al. Perioperative Triplet Chemotherapy and Cetuximab in Patients With RAS Wild Type High Recurrence Risk or Borderline Resectable Colorectal Cancer Liver Metastases. Clin. Colorectal Cancer. 2017;16:191–198. doi: 10.1016/j.clcc.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 17.Normanno N, et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann. Oncol. 2015;26:1710–1714. doi: 10.1093/annonc/mdv176. [DOI] [PubMed] [Google Scholar]
- 18.Salem ME, et al. Comparative molecular analyses of left-sided colon, right-sided colon, and rectal cancers. Oncotarget. 2017;8:86356–86368. doi: 10.18632/oncotarget.21169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grabsch H, et al. Expression of DNA double-strand break repair proteins ATM and BRCA1 predicts survival in colorectal cancer. Clin. Cancer Res. 2006;12:1494–1500. doi: 10.1158/1078-0432.CCR-05-2105. [DOI] [PubMed] [Google Scholar]
- 20.Beggs AD, et al. Loss of expression of the double strand break repair protein ATM is associated with worse prognosis in colorectal cancer and loss of Ku70 expression is associated with CIN. Oncotarget. 2013;3:1348–1355. doi: 10.18632/oncotarget.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sundar R, et al. Ataxia Telangiectasia Mutated Protein Loss and Benefit From Oxaliplatin-based Chemotherapy in Colorectal Cancer. Clin. Colorectal Cancer. 2018;18:10. doi: 10.1016/j.clcc.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Copija A, Waniczek D, Witkoś A, Walkiewicz K, Nowakowska-Zajdel E. Clinical Significance and Prognostic Relevance of Microsatellite Instability in Sporadic Colorectal Cancer Patients. Int. J. Mol. Sci. 2017;18:107. doi: 10.3390/ijms18010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol. Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- 24.Batey, M. A. et al. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 12, 959-967 (2013). [DOI] [PMC free article] [PubMed]
- 25.Dienstmann R, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer. 2017;17:79–92. doi: 10.1038/nrc.2016.126. [DOI] [PubMed] [Google Scholar]
- 26.Hickson I, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto K, et al. Kinase-dead ATM protein is highly oncogenic and can be preferentially targeted by Topo-isomerase I inhibitors. Elife. 2016;15:5. doi: 10.7554/eLife.14709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blackford AN, Jackson SP. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 29.Kass EM, et al. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. USA. 2013;110:5564–5569. doi: 10.1073/pnas.1216824110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai AH, et al. Promoter hypermethylation of tumor-related genes in the progression of colorectal neoplasia. Int. J. Cancer. 2004;112:846–853. doi: 10.1002/ijc.20485. [DOI] [PubMed] [Google Scholar]
- 31.Mateo J, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C, et al. ATM-Deficient Colorectal Cancer Cells Are Sensitive to the PARP Inhibitor Olaparib. Transl. Oncol. 2017;10:190–196. doi: 10.1016/j.tranon.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: therapeutic implications. Mol. Cancer Ther. 2016;15:1781–1791. doi: 10.1158/1535-7163.MCT-15-0945. [DOI] [PubMed] [Google Scholar]
- 34.Winkler J, Hofman K, Chen S. Novel targets for ATM deficient malignancies. Mol. Cell. Oncol. 2014;1:e29905. doi: 10.4161/mco.29905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.