

Analyses of fragrance compounds in a rose segregating population show that the biosynthesis of 2-phenylethanol is linked to the expression of one allele of phenylacetaldehyde synthase gene in petals.
Abstract
Floral scent is one of the most important characters in horticultural plants. Roses (Rosa spp.) have been cultivated for their scent since antiquity. However, probably by selecting for cultivars with long vase life, breeders have lost the fragrant character in many modern roses, especially the ones bred for the cut flower market. The genetic inheritance of scent characters has remained elusive so far. In-depth knowledge of this quantitative trait is thus very much needed to breed more fragrant commercial cultivars. Furthermore, rose hybrids harbor a composite genomic structure, which complexifies quantitative trait studies. To understand rose scent inheritance, we characterized a segregating population from two diploid cultivars, Rosa × hybrida cv H190 and Rosa wichurana, which have contrasting scent profiles. Several quantitative trait loci for the major volatile compounds in this progeny were identified. One among these loci contributing to the production of 2-phenylethanol, responsible for the characteristic odor of rose, was found to be colocalized with a candidate gene belonging to the 2-phenylethanol biosynthesis pathway: the PHENYLACETALDEHYDE SYNTHASE gene RhPAAS. An in-depth allele-specific expression analysis in the progeny demonstrated that only one allele was highly expressed and was responsible for the production of 2-phenylethanol. Unexpectedly, its expression was found to start early during flower development, before the production of the volatile 2-phenylethanol, leading to the accumulation of glycosylated compounds in petals.
Roses (Rosa spp.) are among the most important ornamental plants and have been used since antiquity for their beauty and scent. Hundreds of volatile molecules, belonging to different biosynthetic pathways, have been isolated from rose petals. The combination of these molecules makes up the characteristic rose scent bouquet. Monoterpene alcohols such as geraniol and phenylpropanoid-related compounds such as 2-phenylethanol (2PE) have been shown to contribute greatly to the typical rose scent. Many studies have been performed to investigate the various ways in which these major compounds are produced in petals. The biosynthetic pathway leading to 2PE production has been previously studied in detail (Fig. 1), and the key enzymes of the pathway have been identified: PHENYLACETALDEHYDE SYNTHASE (RhPAAS; Kaminaga et al., 2006; Sakai et al., 2007; Farhi et al., 2010) and PHENYLACETALDEHYDE REDUCTASE (PAR; Chen et al., 2011). Recently, an alternative pathway, which is seasonally induced in summer, has been identified in roses for the production of 2PE. This pathway uses AROMATIC AMINO ACID AMINOTRANSFERASE (Hirata et al., 2012) and PHENYLPYRUVATE DECARBOXYLASE (Hirata et al., 2016) to produce 2-phenylacetaldehyde (Fig. 1, dashed arrows). Monoterpene biosynthesis in rose has recently been deciphered. Roses, unlike other plants, use a nudix hydrolase to synthetize geraniol (Magnard et al., 2015). The biosynthetic pathway of 3,5-dimethoxytoluene (DMT) production, which is responsible for the tea scent of some cultivars, involves O-methyltransferases (Scalliet et al., 2006, 2008). These studies have generally shown that the genes are specifically expressed in petals when the scent is emitted and that the amount of volatile compounds is linked to the transcriptional level of the genes involved in the pathway (Farhi et al., 2010).
Figure 1.

The 2PE biosynthetic pathway in roses. AAAT, AROMATIC AMINO ACID AMINOTRANSFERASE (R. hybrida cv Yves Piaget, RyAAAT3 gene; Hirata et al., 2012); AAT, ACETYL-COENZYME A:GERANIOL/CITRONELLOL ACETYL TRANSFERASE (R. hybrida cv Fragrant Cloud, RhAAT1 gene; Shalit et al., 2003); Glu, β-GLUCOSIDASE (R. hybrida cv Hoh-Jun, β-glucosidase enzyme; Sakai et al., 2008); PAAS, PHENYLACETALDEHYDE SYNTHASE (R. hybrida cv Fragrant Cloud, RhPAAS gene; Kaminaga et al., 2006; R. hybrida cv Hoh-Jun, AADC gene; Sakai et al., 2007); PAR, PHENYLACETALDEHYDE REDUCTASE (R. x hybrida cv Hoh-Jun, PAR enzyme; Sakai et al., 2007; R. damascena, PAR gene; Chen et al., 2011); PPDC, PHENYLPYRUVATE DECARBOXYLASE (R. x hybrida cv Yves Piaget, RyPPDC gene; Hirata et al., 2016); ?, putative glycosyltransferase, not yet characterized. Dashed arrows indicate an alternative pathway (Hirata et al., 2012).
Despite these efforts, knowledge of rose fragrance biosynthesis is still incomplete and cannot yet be used to assist breeders in selecting scented roses. One of the reasons is that scent inheritance during crossings has seldom been evaluated in rose. Indeed, most studies have characterized the heritability of a wide range of other floral characteristics, such as resistance to pathogens, vigor, recurrence in flowering, inflorescence architecture, number of petals, and flower color (Linde et al., 2006; Yan et al., 2007; Kawamura et al., 2011; Henz et al., 2015; Roman et al., 2015; Gitonga et al., 2016; Bourke et al., 2018; François et al., 2018). Although genetic dissection of fruit aroma and flower scent in plants is difficult due to the nature and the number of compounds involved, such genetic studies, including quantitative trait locus (QTL) and genome-wide association studies, have been performed in some crop plants such as tomato (Solanum lycopersicum), for which powerful genetic resources exist (Rambla et al., 2017; Tieman et al., 2017). QTL approaches have also been applied to petunia (Petunia spp.) to study the evolution of floral scent in relationship to pollination syndrome (Klahre et al., 2011; Amrad et al., 2016). In Rosaceae, research efforts have been focused on the improvement of fruit flavor characters. Several studies have addressed the identification of QTLs for volatile compounds in apple (Malus domestica; Zini et al., 2005; Dunemann et al., 2012; Costa et al., 2013; Vogt et al., 2013; Souleyre et al., 2014; Yauk et al., 2015, 2017), raspberry (Rubus spp.; Paterson et al., 2013), and strawberry (Fragaria spp.; Zorrilla-Fontanesi et al., 2012). In strawberry and raspberry, the authors observed some genetic regions (clusters of QTLs) controlling several volatile compounds, suggesting either tightly linked multiple loci or single loci with pleiotropic effects, which could be coding for master regulators such as transcription factors or microRNAs. Genes likely responsible for some of the identified QTLs were sometimes assigned. In strawberry, the O-methyltransferase gene FaOMT was identified as the locus controlling natural variation in mesifurane content (Zorrilla-Fontanesi et al., 2012). In apple, candidate genes (coding for O-methyltransferases and alcohol acyltransferases), involved in the production of the phenylpropene estragole, were characterized (Yauk et al., 2017). In rose, the first published analysis of scent in a segregating population was performed on a crossing between tetraploid roses (Cherri-Martin et al., 2007). This analysis showed that a large proportion of descendants lacked volatile compounds of good fragrance value, so that this positive feature may have been lost in such a crossing. Another study of genetic determinism of rose compounds was conducted on diploid roses with a Rosa multiflora genetic background (Spiller et al., 2010). These authors resolved the patterns of inheritance for several important volatile compounds and were able to map six QTLs influencing volatile content. For example, they found two loci influencing the amount of 2PE. However, no functional relationships to known candidate genes in the 2PE pathway were found in that study.
The aim of this study was to identify genomic regions controlling scent compound production in rose petals. We addressed the genetic determinism of volatile compound production over 3 years in a diploid F1 progeny. We focused on the identification of the candidate gene responsible for the segregation of 2PE content in the population. Analyses of the alleles associated with the production of 2PE and their expression during flower development were conducted.
RESULTS
Identification of Major Scent Loci
To study the inheritance of the production of scent in rose flowers, we performed gas chromatography (GC)-flame ionization detection and GC-mass spectrometry (MS) to analyze the volatile compounds in 79 individuals of the HW F1 progeny (named HW progeny; Crespel et al., 2002; Hibrand-Saint Oyant et al., 2008) during 3 successive years. To facilitate compound collection, we used a solvent solid/liquid extraction of petals in full-bloom flowers, which allowed us to analyze the free internal pool of petal volatile compounds. Thirty-nine compounds were recovered, including some nonvolatile long-chain hydrocarbons (Supplemental List). For further studies, the 16 most abundant volatile compounds were chosen. Compound measurements were made each year in the progeny and in the parents (Table 1). Striking differences in scent characteristics between the two parents were observed. The female parent, an R. x hybrida dihaploid cv H190 (H190), predominantly produced DMT and E-2-hexenal, while the male parent, an R. x wichurana hybrid (Rw), produced 2PE and E,E-farnesol at high concentrations. These results were consistent over all 3 years. On average, Rw produced 57.8 times more 2PE than H190. Extracts of both parents contained minor (i.e. less than 0.5 µg g−1 fresh weight) compounds: 2-phenylacetaldehyde, 2-phenylethyl acetate, E-β-farnesene, farnesal, germacrene D, geraniol, geranyl acetate, and benzyl alcohol (Table 1). Some compounds were only detected in one parent, such as DMT and 2-phenylacetaldehyde in H190 and Z-3-hexenol, 2-phenylethyl acetate, farnesyl acetate, and geranyl acetate in Rw.
Table 1. Mean and median values and range of volatile compounds (µg g−1 fresh weight, means of two to four replicates) analyzed by GC-flame ionization detection and GC-MS during three successive years (Y1, Y2, and Y3) in petals of Rw and H190 parental lines and in HW progeny.
| Volatile Compound | Y1 | Y2 | Y3 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HW Range | HW Mean ± SD | HW Median | H190 | Rw | HW Range | HW Mean ± SD | HW Median | H190 | Rw | HW Range | HW Mean ± SD | HW Median | H190 | Rw | |
| E-2-Hexenal | 0.3–32.9 | 5.3 ± 0.7 | 3.7 | 8.3 | 0.4 | 0.7–37.0 | 8.1 ± 1.2 | 5.0 | 5.7 | 1.7 | 0.4–31.1 | 5.8 ± 0.8 | 4.0 | 14.9 | 0.8 |
| Z-3-Hexenol | 0.0–0.9 | 0.0 ± 0.0 | 0.0 | 0.0 | 1.4 | 0.0–14.8 | 2.1 ± 0.5 | 0.0 | 0.0 | 3.3 | 0.0–17.8 | 1.3 ± 0.4 | 0.0 | 0.0 | 1.9 |
| 2PE | 0.0–350.5 | 65.5 ± 10.5 | 1.4 | 2.1 | 97.1 | 0.0–716.9 | 105.2 ± 23.8 | 0.9 | 1.2 | 183.9 | 0.0–487.9 | 73.1 ± 13.2 | 1.9 | 3.2 | 94.5 |
| 2-Phenylacetaldehyde | 0–7.3 | 0.7 ± 0.2 | 0.0 | 0.2 | 0.0 | 0.0–14.4 | 1.2 ± 0.5 | 0.0 | 0.0 | 0.0 | 0.0–21.6 | 1.4 ± 0.5 | 0.0 | 0.0 | 0.0 |
| 2-Phenylethyl acetate | 0.0–40.9 | 0.9 ± 0.5 | 0.0 | 0.0 | 0.5 | 0.0–2.5 | 0.2 ± 0.1 | 0.0 | 0.0 | 0.4 | 0.0–1.5 | 0.2 ± 0.0 | 0.0 | 0.0 | 0.2 |
| 3,5-Dimethoxytoluene | 0.0–14.0 | 4.2 ± 0.4 | 3.5 | 25.9 | 0.0 | 0.2–33.0 | 6.3 ± 0.8 | 4.9 | 6.6 | 0.0 | 0.0–45.4 | 10.8 ± 1.2 | 8.2 | 14.9 | 0.0 |
| Benzyl alcohol | 0.0–1.1 | 0.3 ± 0.0 | 0.2 | 0.2 | 0.3 | 0.0–1.6 | 0.2 ± 0.1 | 0.0 | 0.0 | 0.4 | 0.0–0.8 | 0.1 ± 0.0 | 0.0 | 0.0 | 0.2 |
| Geraniol | 0.0–6.9 | 0.9 ± 0.2 | 0.4 | 0.1 | 0.2 | 0.0–2.8 | 0.3 ± 0.1 | 0.0 | 0.0 | 0.1 | 0.0–3.4 | 0.2 ± 0.1 | 0.0 | 0.0 | 0.0 |
| Geranyl acetate | 0.0–6.4 | 0.2 ± 0.1 | 0.0 | 0.0 | 0.3 | 0.0–1.9 | 0.1 ± 0.0 | 0.0 | 0.0 | 0.0 | 0.0–3.3 | 0.1 ± 0.1 | 0.0 | 0.0 | 0.0 |
| Germacrene D | 0.0–6.7 | 0.5 ± 0.1 | 0.1 | 0.0 | 0.2 | 0.0–1.0 | 0.1 ± 0.0 | 0.0 | 0.0 | 0.0 | 0.0–8.6 | 0.2 ± 0.1 | 0.0 | 0.0 | 0.0 |
| E-β-Farnesene | 0.0–4.2 | 0.7 ± 0.1 | 0.3 | 0.0 | 0.2 | 0.0–0.6 | 0.0 ± 0.0 | 0.0 | 0.0 | 0.0 | 0.0–1.9 | 0.1 ± 0.0 | 0.0 | 0.0 | 0.0 |
| E,E-Farnesol | 0.0–42.5 | 8.7 ± 1.3 | 1.8 | 1.1 | 19.0 | 0.0–25.0 | 6.1 ± 0.7 | 5.6 | 0.6 | 14.6 | 1.1–20.3 | 5.9 ± 0.5 | 5.3 | 2.4 | 12.6 |
| Farnesal | 0.0–1.2 | 0.2 ± 0.0 | 0.0 | 0.1 | 0.4 | 0.0–0.6 | 0.0 ± 0.0 | 0.0 | 0.0 | 0.0 | 0.0–1.3 | 0.3 ± 0.0 | 0.3 | 0.0 | 0.2 |
| Farnesyl acetate | 0.0–38.1 | 1.5 ± 0.6 | 0.4 | 0.0 | 4.9 | 0.0–1.7 | 0.0 ± 0.0 | 0.0 | 0.0 | 0.2 | 0.0–4.5 | 0.1 ± 0.1 | 0.0 | 0.0 | 0.9 |
| Dihydro-β-ionone | 0.0–3.2 | 0.5 ± 0.1 | 0.2 | 1.2 | 0.3 | 0.0–4.5 | 0.6 ± 0.2 | 0.0 | 1.9 | 0.0 | 0.0–4.6 | 0.7 ± 0.1 | 0.3 | 4.8 | 0.0 |
| Dihydro-β-ionol | 0.0–3.0 | 0.5 ± 0.1 | 0.4 | 0.7 | 0.6 | 0.0–6.4 | 1.0 ± 0.2 | 0.0 | 1.3 | 0.5 | 0.0–9.6 | 1.1 ± 0.2 | 0.4 | 2.1 | 0.7 |
Correlations between the different scent compounds could indicate a common biosynthetic pathway. Therefore, we analyzed such correlations for each year and for the 3 years together (denoted by Y1, Y2, and Y3). The pairwise correlation for each volatile was analyzed against every other volatile, and the Spearman correlation coefficients were calculated. Some correlations were observed in more than 1 year (Supplemental Fig. S1). Volatile phenylpropanoid-related compounds and benzenoid compounds (2PE, 2-phenylacetaldehyde, 2-phenylethyl acetate, and benzyl alcohol) were positively correlated. The two apocarotenoid compounds dihydro-β-ionone and dihydro-β-ionol were also coregulated. The amounts of E-2-hexenal and Z-3-hexenol were negatively correlated. Other more unexpected pairwise positive correlations, such as the one implicating geraniol and E,E-farnesol, were also found in Y1 and Y2.
For most of the compounds, we observed a significant variance between genotypes and between years (Supplemental Table S1). Furthermore, as all the variances between technical replicates were weak or nonsignificant (except for farnesal) compared with the variances between genotypes and between years, we decided to average technical replicates and performed analyses on the means of these technical replicates. The heritability ranged from 28.4% to 96.12% (Supplemental Table S1). The highest heritability estimates were obtained for geranyl acetate (96.12%) and dihydro-β-ionol (87.92%). The smallest values of heritability were observed for benzyl alcohol, farnesal, and E-β-farnesene, 28.4%, 41.12%, and 45.13%, respectively.
Data obtained from each experiment (three experiments corresponding to 3 different years) were first used to perform a comprehensive QTL analysis for each parental map. For this analysis, we used the male and female genetic maps previously developed (Hibrand-Saint Oyant et al., 2008; Remay et al., 2009). Due to nonnormality distribution for most of the metabolites, the raw data were analyzed first by the nonparametric Kruskal-Wallis rank-sum test (KW). Interval mapping analysis (IM) was then performed with a step size of 1 centimorgan (cM) to find regions with potential QTL effects (i.e. where the logarithm of odds [LOD] score was greater than the threshold). The percentage explained by the QTL (r2) was also presented. With KW, a total of 112 significant associations were detected for the 3 years (Supplemental Table S2). We assumed that associations detected in the same chromosomal region for the same volatile compound in different years were identical, and 77 QTLs were finally considered. Among them, 24 (31.2%) were stable over 2 or 3 years. Using IM, a total of 34 significant associations were obtained, resulting in 24 QTLs (Fig. 2; Table 2). Among them, eight (33.3%) were stable over 2 or 3 years. Moreover, some clusters of QTLs were detected in the same chromosomal regions (Fig. 2). All 24 QTLs detected with IM were also detected with KW (Supplemental Table S2). Data from the 3 years were then analyzed together by KW (Supplemental Table S3). Most of the 24 common QTLs detected in a single year by KW and IM were also detected when the data were averaged. Some QTLs were only detected when the data were averaged. Only the 24 QTLs detected by KW and IM are described below.
Figure 2.

Locations of QTLs associated with scent compounds as determined by IM, and the major gene controlling 2PE production, RhPAAS, as candidate gene. The genetic linkage maps were constructed for both parents (female parent R. x hybrida cv H190, A1–A7, and male parent R. wichurana, B1–B7). QTLs associated with compounds are located to the right of the chromosomes, with color bars showing one-LOD confidence intervals.
Table 2. Summary of QTLs for scent compounds detected with IM in the HW progeny.
The QTLs were detected in the male parent (Rw) and in the female parent (H190). Dash (–) inidcates no close MM.
| QTL | Compound and Yeara | QTL Characteristics | |||||
|---|---|---|---|---|---|---|---|
| PTb | LGc | LOD Scored | Positione | MMf | r2g | ||
| 1 | E-2-Hexenal Y1 | 2.5 | A4 | 4.8 | 16.2 | RoCol2 | 25.1 |
| E-2-Hexenal Y2 | 2.9 | A4 | 6.1 | 17.5 | – | 41.0 | |
| E-2-Hexenal Y3 | 2.7 | A4 | 4.6 | 17.5 | – | 28.0 | |
| 2 | E-2-Hexenal Y1 | 2.6 | B5 | 2.7 | 75.5 | H22F01 | 15.0 |
| 3 | Z-3-Hexenol Y2 | 2.7 | A4 | 5.9 | 16.5 | – | 40.0 |
| Z-3-Hexenol Y3 | 2.5 | A4 | 4.4 | 16.5 | – | 27.2 | |
| 4 | 2PE Y1 | 2.5 | B6 | 16.8 | 61.3 | RhPAAS | 63.3 |
| 2PE Y2 | 3.1 | B6 | 7.7 | 64.6 | Rw61F2 | 48.8 | |
| 2PE Y3 | 2.7 | B6 | 13.1 | 61.3 | RhPAAS | 62.6 | |
| 5 | 2-Phenylacetaldehyde Y1 | 2.6 | B6 | 6.3 | 67.5 | – | 31.8 |
| 6 | 2-Phenylethyl acetate Y2 | 2.6 | B6 | 3.6 | 65.5 | – | 27.1 |
| 2-Phenylethyl acetate Y3 | 2.6 | B6 | 6.1 | 60.0 | – | 36.7 | |
| 7 | DMT Y3 | 2.6 | A3 | 3.3 | 34.6 | Fro9 | 21.3 |
| 8 | DMT Y1 | 2.7 | A5 | 3.1 | 8.0 | – | 17.9 |
| 9 | DMT Y1 | 2.7 | B5 | 4.2 | 16.0 | – | 23.4 |
| 10 | Benzyl alcohol Y1 | 2.8 | B5 | 5.4 | 16.0 | – | 29.0 |
| 11 | Benzyl alcohol Y2 | 3.0 | B6 | 4.0 | 64.6 | Rw61F2 | 29.5 |
| Benzyl alcohol Y3 | 2.6 | B6 | 5.4 | 58.9 | – | 33.3 | |
| 12 | Geraniol Y1 | 2.6 | B5 | 2.7 | 27.3 | EIM3.05 | 15.1 |
| 13 | Geraniol Y1 | 2.6 | B7 | 4.4 | 19.8 | Rw5G14 | 23.6 |
| Geraniol Y2 | 2.8 | B7 | 3.2 | 22.8 | – | 24.8 | |
| 14 | Geranyl acetate Y1 | 2.4 | B2 | 3.8 | 19.0 | H09B01f | 19.1 |
| 15 | Germacrene D Y1 | 2.5 | B5 | 3.5 | 28.3 | – | 18.8 |
| 16 | E-β-Farnesene Y1 | 2.6 | B7 | 6.6 | 19.8 | Rw5G14 | 33.4 |
| 17 | Farnesal Y3 | 2.8 | A1 | 3.2 | 45.1 | RMS070 | 20.6 |
| 18 | Farnesal Y1 | 2.6 | B7 | 6.2 | 19.8 | Rw5G14 | 31.8 |
| 19 | E,E-Farnesol Y2 | 2.6 | B2 | 3.1 | 64.0 | – | 25.3 |
| 20 | E,E-Farnesol Y1 | 2.8 | B7 | 11.0 | 19.7 | – | 49.2 |
| E,E-Farnesol Y2 | 2.6 | B7 | 5.7 | 18.8 | RwIF9 | 39.3 | |
| 21 | E,E-Farnesol Y3 | 2.7 | B7 | 3.4 | 54.2 | RoAP1a | 21.5 |
| 22 | Dihydro-β-ionone Y1 | 2.6 | A4 | 2.8 | 27.7 | – | 19.2 |
| 23 | Dihydro-β-ionone Y1 | 2.6 | B6 | 4.1 | 41.8 | – | 22.7 |
| 24 | Dihydro-β-ionol Y2 | 2.5 | A5 | 3.0 | 31.8 | RhAB38 | 22.7 |
| Dihydro-β-ionol Y3 | 2.5 | A5 | 3.6 | 31.8 | RhAB38 | 23.0 | |
Name of the scent compound and year (Y).
The threshold of the LOD score was defined by a permutation test (PT).
LGs are as follows: A, female map; B, male map.
QTLs with a LOD higher than the threshold LOD were considered.
Position on the linkage group (cM).
Closest molecular marker (MM) associated.
Percentage of explanation r2.
QTLs for fatty acid derivatives (E-2-hexenal Y1-Y2-Y3 and Z-3-hexenol Y2-Y3) were identified in the same region of the female map on Linkage Group (LG) 4, with a LOD score up to 6.1 for E-2-hexenal in Y2. These QTLs explained a large part of the variability observed for these compounds, between 25.1% and 41% (Table 2). A QTL for E-2-hexenal was also located on LG5 (B5) in Y1. It is noteworthy that these two compounds were negatively correlated (Supplemental Fig. S1).
On LG6 (B6), QTLs for phenylpropanoid-related compounds (2PE, 2-phenylacetaldehyde, and 2-phenylethyl acetate) were detected in the same region. Some LOD scores were up to 16.8 (e.g. 2PE in Y1). The detected QTL for 2PE explained more than 60% of the phenotypic variance, indicating a strong effect of this locus in the control of the variation of the trait. On the same locus, a QTL for benzyl alcohol was also detected in Y2 and Y3. These four compounds showed a strong positive correlation (Supplemental Fig. S1). For benzyl alcohol, a QTL was also detected on LG5 (B5) in Y1.
On LG5 (A5 and B5), one QTL for DMT was detected in Y1 that explains 17.9% and 23.4% of variability, respectively. Another QTL for this compound was located in Y3 on LG3 (A3).
A cluster of QTLs was detected on the male LG7 (B7): one QTL for geraniol (Y1 and Y2), one QTL explaining up to 49.2% of the E,E-farnesol content in Y1 and Y2 (K value < 0.0001), and two QTLs for two other related compounds (E-β-farnesene in Y1 and farnesal in Y1). Another QTL for E,E-farnesol was present on LG7 (B7) but on a different chromosomal region and another on LG2 (B2). One QTL for geraniol was localized on LG5 (B5). A QTL for geranyl acetate was observed on LG2 (B2) and one for farnesal was observed on LG1 (A1). One QTL in the middle of LG5 (B5) was detected for germacrene D.
A QTL explaining 19.2% of the variability of dihydro-β-ionone production was located on LG4 (A4) in Y1 and one on LG6 (B6) in Y1. On LG5 (A5), one QTL for dihydro-β-ionol detected in Y2 and Y3 explains around 23% of the variability.
Altogether, with data from each year analyzed separately or together, we identified 84 associations accounting for the heritability of all 16 major scent compounds. QTL clusters identified as affecting rose scent may suggest common control points in biosynthetic pathways of the different scent compounds or close loci controlling the synthesis of these compounds.
Identification of RhPAAS as a Major Gene Linked to the Production of 2PE
In order to identify the genetic inheritance of rose scent traits, one major compound was of particular interest and was further investigated. For 2PE, we observed a large difference between the mean and median values (Table 1). Analyses of 2PE mean contents over the 3 years showed that the progeny could be divided into two groups: group 1 with mean 2PE contents up to 2.2 µg g−1 fresh weight and group 2 with values from 48.3 to 498.3 µg g−1 fresh weight (Fig. 3). The female parent H190 produced low 2PE contents (between 1.2 and 3.2 µg g−1 fresh weight), while the male parent Rw produced high amounts of 2PE (between 94.5 and 183.9 µg g−1 fresh weight). A Pearson’s χ2 analysis was performed to test for Mendelian segregation and showed no differences between observed and expected ratios for a segregation of 1:1 (P > 0.05; Supplemental Table S4). This qualitative variation suggested a monogenic inheritance of this trait, which prompted us to adopt a candidate gene approach.
Figure 3.

2PE content in petals of Rw male parent (blue) and H190 female parent (red) and in HW progeny. Descendants were ordered according to their 2PE content. Group 1 descendants have up to 2.2 µg g−1 fresh weight (FW) 2PE; group 2 descendants have from 48.3 to 498.3 µg g−1 fresh weight. Values are means of three replicates, obtained during 3 successive years.
The biosynthesis of rose 2PE through the classical pathway described involves two steps, implicating two enzymes: RhPAAS (Kaminaga et al., 2006) and PAR (Chen et al., 2011; Fig. 1). The PAR gene was mapped on LG1 by Spiller et al. (2010) and localized on LG5 in the rose genome (Raymond et al., 2018). It thus cannot correspond to the QTL for 2PE content that was found on LG6. From the results of Kaminaga et al. (2006), we designed primers to isolate the RhPAAS gene in H190 and Rw roses (parents of the HW population). Three different alleles of the putative RhPAAS gene were isolated by PCR from genomic DNA. A 1,527-bp open reading frame without introns was isolated, and 22 single-nucleotide polymorphism (SNP) sites were identified in the parental genotypes (Supplemental Fig. S2). All three alleles shared 99% identity with the previously published RhPAAS sequence (Kaminaga et al., 2006). A comparison of the encoded 508 amino acid proteins and the published sequences from other rose cultivars confirmed the presence of a conserved Phe residue proposed to be responsible for the aldehyde synthase activity (Supplemental Fig. S3; Torrens-Spence et al., 2013).
A single-strand conformation polymorphism (SSCP) marker allowed us to map the RhPAAS gene on the B6 LG (male map; Figs. 2 and 4) with a segregation ratio of 1:1 (44 versus 46). As the major QTL for 2PE was also localized on the same LG, the RhPAAS gene could be the locus of interest for this QTL (Fig. 2). Moreover, the SSCP electrophoresis separated RhPAAS PCR products into three clear bands: one for H190 (a3 allele) and two for Rw (a1 and a2 alleles; Fig. 4). All individuals from the HW population that produced high amounts of 2PE (group 2) had the a1 allele, whereas individuals producing small amounts of or no 2PE (group 1) did not, suggesting that allelic variation in RhPAAS could account for phenotypic variation in the HW progeny.
Figure 4.

SSCP analysis of the RhPAAS gene in HW progeny. The profiles obtained for both parents (female H190 and male Rw) and several members of the progeny are shown. 2PE contents are shown (−, less than 3 µg g−1 fresh weight; +, greater than 45 µg g−1 fresh weight). a1, a2, and a3 are alleles of the RhPAAS gene present in the HW progeny.
The amino acid sequences of the three alleles harbored eight variations, which could influence the functionality of the corresponding proteins (Supplemental Fig. S3). To test this hypothesis, we overexpressed the proteins produced by the three alleles by expressing their cDNAs in Nicotiana benthamiana and in the previously described somatic embryogenic callus derived from Rosa chinensis cv Old Blush (Vergne et al., 2010). For the transient transformation of N. benthamiana, leaves were infiltrated with Agrobacterium tumefaciens harboring 35S::RhPAAS constructs. Stable transgenic embryogenic rose calli were recovered by using the same construct. Thirty and 15 independent transgenic events were recovered in N. benthamiana and rose calli, respectively. For these experiments, plants transformed with the control p19 plasmid alone (N. benthamiana leaves) and untransformed rose callus explants were used as controls. Analyses of volatile compounds from transformed N. benthamiana leaves and rose calli showed that all three alleles were functional and able to induce the production of similar levels of 2PE (Fig. 5). The absence of 2-phenylacetaldehyde in transformed extracts suggested that this molecule was immediately converted in 2PE by an endogenous enzyme that played the role of the phenylacetaldehyde reductase (Chen et al., 2011).
Figure 5.

Overexpression of RhPAAS alleles in planta. A, Stable expression in R. chinensis cv Old Blush calli. Transformation with A. tumefaciens harboring the pK7WGF2 binary vector was performed according to Vergne et al. (2010). Nontransformed plants (NT) were used as controls, and volatile compounds were extracted by hexane from calli and analyzed by GC-MS. B, Transient expression in N. benthamiana. A. tumefaciens harboring the pK7WGF2 binary vector or the viral suppressor p19 into the pBIN61 were mixed and coinfiltrated into the adaxial side of young leaves. Control infiltrations were performed with A. tumefaciens carrying the viral suppressor p19. Four days after injection, volatile compounds were extracted by hexane and analyzed by GC-MS. Error bars indicate se obtained from 30 (N. benthamiana leaves) and 15 (rose calli) independent biological replicates. Means with different letters (a and b) are significantly different (Dunn’s test, P < 0.05). FW, Fresh weight.
Differential Expression of the Three RhPAAS Alleles in the HW Population
As the three proteins encoded by the three RhPAAS alleles had full capacity to synthesize 2PE, the causal link between the capacity to produce 2PE and the presence of a1 allele was not a defect in one of these proteins. Differences in the expression of the three alleles in rose petals could account for variations in 2PE biosynthesis. As a first step to determine whether the RhPAAS gene was indeed expressed and to further characterize its expression pattern in the HW progeny, RhPAAS transcript accumulation was assessed during flower development by reverse transcription quantitative PCR (RT-qPCR) in the parents and in four selected individuals of the HW progeny with a1a3 (two individuals, HW24 and HW530) and a2a3 (two individuals, HW68 and HW149) genotypes. Non-allele-specific primers were used to measure the transcript pool abundance of RhPAAS in petals, the flower organs that produce the most scent. Petals were harvested from flower stages 2 to 5, and free internal pools of petal volatile compounds were collected as previously described (Bergougnoux et al., 2007). Supplemental Table S5 shows 2PE and 2-phenylacetaldehyde contents in the six individuals. During flower development, 2-phenylacetaldehyde pools followed the same trends as 2PE internal pools, the latter being generally much higher. Production of both compounds was higher at late developmental stages. Our analysis showed high expression of RhPAAS in the individuals producing 2PE, Rw parental line, HW24 and HW530 selected individuals (Fig. 6). In contrast, the expression was barely detectable in the lines not producing or weakly producing 2PE, H190 parental line, HW68 and HW149 selected individuals (Fig. 6).
Figure 6.

2PE content analyzed by GC-MS (blue bars) and expression level of RhPAAS during flower development analyzed by RT-qPCR (orange lines) in the parents of the HW progeny, H190 (A) and Rw (B), and in four individuals of the progeny, HW24 (C), HW68 (D), HW149 (E), and HW530 (F). Flower developmental stages were defined by Bergougnoux et al. (2007). Transcript levels of RhPAAS were normalized to α-tubulin and EF1-α according to Dubois et al. (2011). Gene expression values are relative to the expression of Rw at flower stage 2, for which the value was set to 1. Error bars indicate se obtained from three independent biological replicates. Means with different letters (a, b, and c) are significantly different (Dunn’s test, P < 0.05). FW, Fresh weight; a.u., arbitrary units.
Moreover, we used SNPs to elucidate allelic contribution to the RhPAAS expression. High-resolution melting (HRM) technology (Liew et al., 2004; McKinney et al., 2010) was used to specifically estimate allele expression in cDNAs obtained from petals at stage 2. HRM allows allele-specific detection of heterozygotes, since the PCR amplicons containing an SNP produce distinctive HRM profiles. Assuming a correct allele discrimination and using sequenced plasmids as control templates (Supplemental Fig. S4), we analyzed the allelic dosage through a comparison of cDNAs from four selected individuals (Rw, H190, HW149, and HW530). We used varying ratios of mixed plasmids as PCR templates, each containing one of the three-allele full-length cDNAs (Fig. 7; Supplemental Fig. S4). Two different SNPs were used (Supplemental Fig. S2). SNP1 was able to discriminate allele a3 from a1 or a2 and SNP2 could discriminate allele a2 from a1 or a3. Used on the cDNAs, this approach confirmed the monomorphic allelic state of a3 in the dihaploid H190 parental line, as seen in the melting curve profile (Fig. 7B; Table 3). In the a1a2 and a1a3 genotypes of the respective Rw parent and HW530 individual, only allele a1 was amplified, as the melting curve profile corresponded to the a1 control amplicon (100% of the a1 plasmid DNA; Figure 7, A and C). This suggests that in the a1a2 and a1a3 genotypes, both alleles a2 and a3 were subjected to exclusion. In the a2a3 HW149 progeny, although RhPAAS was barely expressed (Fig. 6), we could estimate that a2 accounted for 75% and a3 for 25% of the remaining gene expression (Fig. 7D; Table 3). In conclusion, the presence of the a1 allele was correlated with high RhPAAS expression, while the other two alleles were correlated with a very weak one that did not lead to significant 2PE production. These findings are in agreement with the data obtained in the SSCP analysis.
Figure 7.

HRM expression profiling of RhPAAS. Normalized and temperature-shifted difference plots are shown at left using a control plasmid as reference template for PCR (*). A and B, Parental genotypes. C and D, HW progeny. Two HRM primer pairs were separately used to distinguish allele-specific expression in cDNA obtained from petals at stage 2. SNP1 was used to differentiate the a3 amplicon from the a1/a2 amplicon in the a3 H190 (B, orange lines) and the a1a3 HW530 (C, green lines) backgrounds. SNP2 was used to specifically discriminate the a2 amplicon from a1/a3 in the a1a2 Rw (A, red lines) and the a2a3 HW149 (D, blue lines) backgrounds. Plasmid DNA controls were used either as pure solutions (A–C) or mixed in specific ratios to estimate allelic composition in the cDNA mixture (D; Supplemental Fig. S4). The estimated calling shown at right of expressed RhPAAS alleles was obtained using the genotype-calling tool of the Rotor-Gene Q Series software with a confidence of greater than 95%.
Table 3. Expression of RhPAAS alleles a1, a2, and a3 in the parental lines and selected individuals of the HW progeny and 2PE production.
| Line | 2PE Production | Genotype | Allele Expression (%) | ||
|---|---|---|---|---|---|
| a1 | a2 | a3 | |||
| Rw | + | a1a2 | 100 | 0 | – |
| H190 | − | a3a3 | – | – | 100 |
| HW149 | − | a2a3 | – | 75 | 25 |
| HW530 | + | a1a3 | 100 | 0 | – |
Spatiotemporal Expression of RhPAAS
We observed an unexpected early expression of the gene compared with the stage of volatile 2PE production (Fig. 6). In HW individuals producing high amounts of 2PE, HW24 and HW530, RhPAAS expression was barely detectable at stages 4 and 5, which are the scent-production stages. In recombinant lines that produce low amounts or no 2PE, such as HW68 and HW149, the expression of RhPAAS was very weak, but slightly higher at stage 4. It is well known that 2PE and 1-phenylethanol can be conjugated to glycosides to be stored in the vacuoles in soluble forms (Watanabe et al., 2001; Zhou et al., 2014). To know if such forms were present in early stages of flower development in our population and explain the timing of RhPAAS expression, we analyzed the internal free and conjugated 2PE contents in the same samples. After incubation with a glycosidase solution, free internal volatile forms could be recovered (Fig. 8). Free internal volatile 2PE contents increased during flower development, whereas glycosylated form contents decreased, the highest being in flower buds. Moreover, 2PE free forms were present in lower quantities, irrespective of stage. These results show that 2PE is produced in the early stages of flower development through the activity of the RhPAAS enzyme and is then transformed into glycosylated soluble forms. The soluble forms are finally released at later stages. To know if this pattern of expression was specific to the a1 allele, RhPAAS expression was analyzed throughout development in cv The Mac Cartney rose (Supplemental Fig. S5A). Expression was also analyzed at two stages of development (S2 and S4) in five different rose cultivars (Supplemental Fig. S5B). Early expression of the RhPAAS gene was confirmed in all the tested cultivars. Partial sequencing of alleles in these different rose cultivars showed high percentage identities with a1, a2, and a3 alleles, although none of them were identical (Supplemental Fig. S6).
Figure 8.

Evolution of free and glycosylated forms of 2PE in R. wichurana petals during flower development. Flower developmental stages were defined by Bergougnoux et al. (2007). Error bars indicate se obtained from three independent biological replicates. Means with different letters (a, b, and c) are significantly different (Dunn’s test, P < 0.05). FW, Fresh weight.
DISCUSSION
Scent QTLs
We performed a genetic analysis of rose scent segregation using a diploid cross from parents with contrasted scent compounds. Our study provides a comprehensive map of QTL clusters identified as affecting rose scent. QTLs were found for all main 16 volatile compounds. Thirty percent of these QTLs were stable over 2 or 3 years, whereas some were found only once (Table 2; Supplemental Tables S2 and S3). Volatile organic compound analyses to identify QTLs have not always been performed over several successive years. In a previous study on strawberry, 50% of the detected QTLs were stable over at least 2 years (Zorrilla-Fontanesi et al., 2012), whereas only about 30% were stable in our study. In Citrus reticulata, the percentage of stable QTLs was even less (12.14%; Yu et al., 2017). Similarly, in tomato, a large percentage of metabolites also showed variation over years (Bauchet et al., 2017). These variations might be due to differences in environmental growth conditions. Indeed, in most studies, including ours, plants were cultivated in the field under noncontrolled conditions. It was shown in raspberry that the cultivation method (open field or under tunnel) and the season greatly influenced the volatile contents and the associated QTLs (Paterson et al., 2013). As was done in apple (Costa et al., 2013) and in rose (Bourke et al., 2018), the validation of the major QTLs that we found could be done by cultivating the population in different locations or by using other segregating populations or genetic association panels. The number of QTLs detected in other studies was generally higher, probably because we focused on major volatile compounds emitted by rose cultivars and also because the two cultivars that were used are moderately scented. The availability of highly scented diploid populations limits the power of such approaches for rose. Moreover, in our study, we focused on the internal pools of volatile compounds. Analyzing the emitted volatile compounds could lead to the discovery of other QTLs, possibly related to the emission, storage, or conjugation processes.
Localizing QTLs can be used to find putative candidate genes underlying these QTLs. For example, in tomato, using both the genome sequence and transcriptomic data, Bennewitz et al. (2018) were able to propose several candidate genes for two of their identified QTLs involved in glandular trichome shapes. Although less powerful genetic tools are available in rose, putative candidate genes can sometimes be proposed using recently published genome sequences. Terpenes are very important compounds for rose scent. For example, germacrene D is a sesquiterpene frequently encountered in rose petals (Guterman et al., 2002). We found one QTL for this compound on LG5, which could correspond to GERMACRENE D SYNTHASE, the terpene synthase responsible for the biosynthesis of this compound. Very recently, a GERMACRENE D SYNTHASE gene was annotated on chromosome 5 of the assembled rose genome (Raymond et al., 2018), thus corroborating our observation. Spiller et al. (2010) were able to identify other loci in rose that influence terpene volatile contents in an F1 progeny. One QTL for geraniol content was located on LG1. For geranyl acetate, they found a very complex inheritance pattern with four possible loci. An acetyltransferase, RhAAT1, was proposed as a candidate gene corresponding to one of the loci on LG2 (Shalit et al., 2003). Our study also identified a QTL for geranyl acetate in the same chromosomal region, possibly related to the presence of the same RhAAT1 gene. Although generally present in small quantities, apocarotenoids, which are derived from carotenoid degradation, have low odor thresholds and may have a great impact on the odor of flowers such as roses (Huang et al., 2009a). In our progeny, two major apocarotenoids were produced: dihydro-β-ionone and dihydro-β-ionol. It is likely that they are derived from one another because their amounts are highly correlated in the progeny. We found three QTLs present in 1 or 2 years: two for dihydro-β-ionone on LG4 and LG6 and one for dihydro-β-ionol on LG5. In raspberry fruit, apocarotenoids were found to be the most abundant volatile compounds, and overlapping QTLs for multiple carotenoid compounds were found (Paterson et al., 2013). Enzymes involved in the biosynthesis of carotenoid derivatives are carotenoid cleavage dioxygenases (CCDs), the genes encoding which are present in multiple copies in the rose genome. For example, CCD1 has been mapped on LG1 (Spiller et al., 2010), and we found one QTL on LG1 using KW (Supplemental Table S2). CCD4, which is localized on LG4 (Raymond et al., 2018), could be a candidate for the QTL on the same chromosome. However, its biological role in the rose flower is still unclear (Huang et al., 2009b).
For other important scent compounds such as geraniol, no obvious gene candidate could be proposed for the QTLs we found. Geraniol production in both parents and most individuals of the progeny was very small, below 1 µg g−1 fresh weight. Some individuals in the progeny produced higher amounts, up to 6.9 µg g−1 fresh weight, indicating a transgressive segregation. Two QTLs were found, one on LG5 and one on LG7. It has recently been shown that the first step of geraniol biosynthesis in rose involved a nudix hydrolase, whose gene was localized on LG2 (Magnard et al., 2015). Therefore, the identified QTLs do not colocalize with this enzyme. Another step of the pathway or a regulatory protein could correspond to such QTLs. It is noteworthy that QTLs for geraniol, E,E-farnesol, farnesal, and E-β-farnesene were all clustered in the same chromosomic region of LG7. This could indicate that these compounds share a common biosynthetic step and/or a common regulatory gene. In tomato, it was also shown that QTLs for aroma volatile compounds derived from the same metabolic pathway were often colocalized (Saliba-Colombani et al., 2001). In rose, more transcriptomic data sets of roses with contrasting scent profiles and ultra-dense genetic maps are needed to fully exploit the QTLs that we discovered.
Involvement of RhPAAS in 2PE Production
2PE is one of the key compounds responsible for the rose odor. A cluster of QTLs was found on LG6 for 2PE as well as other phenylpropanoid-related compounds that share the same biosynthetic pathway (Fig. 1). The locus contributing to the production of 2PE was found to colocalize with one of the genes belonging to this pathway, RhPAAS. The corresponding enzyme catalyzes the first step in 2PE biosynthesis, having amine oxidase as well as decarboxylase activity (Kaminaga et al., 2006). We were able to show the existence of three alleles of this gene in the HW progeny. As shown by overexpression in rose calli and N. benthamiana leaves, the three alleles encoded perfectly functional RhPAAS isoforms. However, only genotypes having at least one copy of a1 emitted 2PE in significant amounts. The weak accumulation of a3 and a2 allele transcripts certainly explains the very low production of 2PE in H190 and in the descendants with an a2a3 genotype. To summarize, a1, whose transcripts are strongly accumulated, is the only allele responsible for the production of 2PE in high amounts and therefore can be considered as a dominant allele for 2PE production.
In parallel to its high expression, our data also showed an unexpected early expression of the a1 RhPAAS allele during flower development. The a1 allele transcripts were accumulated at high levels just before and during the opening of the flower and were almost no longer accumulated at later stages of flowering. Although it has been reported that the expression of RhPAAS peaked 1 to 2 d before maximum scent emission (Farhi et al., 2010), to the best of our knowledge, the significance of the shift in expression of RhPAAS with respect to the emission of 2PE, which takes place at the blooming stage, has not been addressed thus far. Pioneering research on Clarkia breweri and Antirrhinum majus has shown that scent production is often regulated at the transcription level (Schnepp and Dudareva, 2006), and this seems to be a general feature of plant specialized metabolism (Colinas and Goossens, 2018). In rose, for instance, the pathway leading to the biosynthesis of DMT, partly responsible for the tea scent, has been described (Lavid et al., 2002; Scalliet et al., 2006, 2008). O-Methyltransferases, the enzymes that are responsible for DMT synthesis, are specifically expressed in petals of roses emitting DMT, and their expression peaks when the emission of DMT is at its maximum (Scalliet et al., 2006). However, such changes in timing of transcript accumulation between different alleles (heterochronic allelic variation) have already been shown in plants. One allele, responsible for the increase in fruit size of cultivated tomatoes, was shown to harbor such a heterochronic mutation (Frary et al., 2000; Cong et al., 2002). Our study shows that such a regulation mechanism might exist for scent-biosynthesis genes and that alleles with precocious expression might have been selected, at least in the rose cultivars that we have studied. This could have major implications for the manipulation of floral scent, which could be focused on early stages of flower development. In rose, the early expression of RhPAAS probably coincides with the storage of 2PE in a glycosylated form in early stages of floral development, just after its biosynthesis. At later stages, enzymatic hydrolysis would release the compound in volatile form (Oka et al., 1999; Watanabe et al., 2001). Indeed, a β-glucosidase was partially purified from rose petals by Sakai et al. (2008). However, no gene corresponding to this enzyme has been characterized yet. In many plants, aroma compounds exist both in volatile free forms and in glycosylated forms, usually stored in vacuoles. For instance, in grape (Vitis vinifera), nonvolatile glycosides account for a large fraction of berries (Li et al., 2017). In tea (Camellia sinensis) leaves, scent compounds typically accumulate as water-soluble glycoside forms, and specific glycosyltransferases are responsible for the conjugation of various scent volatile compounds, such as geraniol, to β-primeveroside (Ohgami et al., 2015). Interestingly, a glycosyltransferase was identified as a major candidate gene for the variation of phenylacetaldehyde and 2PE contents in apple fruits (Bauchet et al., 2017). In tea leaves, two glycosyltransferases responsible for the biosynthesis of aroma β-primeverosides were characterized. It was shown that the glycosides were stored in young leaves and they were supposed to play a role in defense against herbivores, after their release upon injury. In flowers, the potential role of these storage compounds is less clear, as volatile compounds such as 2PE or geraniol are supposed to attract pollinators. Furthermore, it has been shown that 2PE attracts herbivore insects such as flower flies in the field (Imai et al., 1998). However, in case of an attack of other herbivore insects on young petals in the bud, the release of these volatile compounds could possibly play the same role of chemical defense against herbivores.
Two other QTLs for the production of 2PE have been previously described in a progeny derived from R. multiflora (Spiller et al., 2010). A major QTL on LG5 accounted for 60% of the variability of the trait. Using KW, we were also able to detect for 2 years a QTL for 2PE on the same LG (Supplemental Table S2). Although no sequence was provided, the authors indirectly linked this QTL to an EST whose corresponding protein was homologous to the AMINO ACID DECARBOXYLASE RhAADC isolated by Sakai et al. (2007). A minor QTL on LG2 accounted for 28% of the variability and was tentatively related to paralogs of the RhAAT1 ALCOHOL ACETYLTRANSFERASE gene (Shalit et al., 2003). RhAADC is an amino acid decarboxylase, isolated from R. x hybrida cv Hoh-Jun, with 99.8% similarity to RhPAAS reported by Kaminaga et al. (2006). The segregation patterns also differed from one another in the two populations. Fifty percent of the descendants produced significant amounts of 2PE in the HW progeny, whereas in the population studied by Spiller et al. (2010), the quantity of 2PE was nearly normally distributed. This difference is probably at the origin of the different QTLs explaining the production of this molecule. These results suggest that there are several genes encoding phenylacetaldehyde synthases on different chromosomes in rose, which could be responsible 2PE production in different segregating populations. Indeed, examination of the rose genome shows that there are multiple proteins with putative Tyr decarboxylase activities. RhPAAS, which corresponds to the QTL on LG6 and is responsible for the capacity to synthetize 2PE in our progeny, is the only one that was functionally characterized. The functions of other proteins present in the rose genome need to be studied. Moreover, it is not uncommon that QTLs for volatile compounds show a low degree of overlapping in studies on different populations in one species, as the genetic background of the parents can be different. In tomato, such lack of redundancy was attributed to environmental or methodological factors and to specific genetic variations (Rambla et al., 2017).
The gene corresponding to rose PAR, which converts phenylacetaldehyde to 2PE (Chen et al., 2011), has been localized on LG1 in the integrative genetic map (Spiller et al., 2010), while the gene is localized on LG5 in the rose genome (Raymond et al., 2018), and thus its activity does not appear to be a major limiting factor in 2PE biosynthesis in our progeny. Indeed, the expression of the rose RhPAAS gene in N. benthamiana leaves led to the production of high amounts of 2PE, but no 2-phenylacetaldehyde, suggesting that the reduction step may be supported by nonspecific endogenous reductases. An alternative pathway has been identified for the biosynthesis of 2PE in rose (Hirata et al., 2012; Fig. 1). It has been proposed that this pathway is used by the plant only in hot conditions (Hirata et al., 2016). Our plants were grown in the field and harvested during spring and summer. Our results showed that the RhPAAS gene was the limiting step for the production of 2PE in our progeny. Moreover, none of the genes of the alternative pathway were localized on chromosome 6. However, it would be interesting to see if the alternative pathway is turned on in the progeny under certain conditions and if it can account for some of the variations we saw in the amount of 2PE produced.
CONCLUSION
In this study, we identified a set of QTLs associated with major volatile compounds of rose. One of these QTLs was found to colocalize with a gene involved in the pathway for 2PE production, namely, RhPAAS, the expression of which was responsible for the capacity of descendants to produce 2PE. For the other QTLs, the identification of underlying genes and their functional characterization would be of great interest to elucidate the regulation of the levels of volatile scent compounds in rose petals. The rose genome, which has recently been published (Hibrand Saint-Oyant et al., 2018; Raymond et al., 2018), in combination with transcriptome data sets (Dubois et al., 2012; Han et al., 2017) and ultra-dense genetic maps (Bourke et al., 2017), will help assign candidate genes to the QTLs we have identified.
Moreover, these results would need to be validated in crosses involving other rose cultivars and species. Such validation will help to better understand the genetics of rose scent and to develop molecular markers for key alleles involved in volatile biosynthesis. In addition to QTL studies, association studies, such as the ones that have already been performed on color (Schulz et al., 2016), will have to be applied on scent traits. Modern rose cultivars are tetraploids, and understanding the inheritance of desired traits is expected to be complicated (Grover et al., 2012). Finally, we observed transgressive segregations for all the analyzed compounds, with some individuals showing very high concentrations for desired scent molecules. This suggests that there are valuable genetic resources available for breeding, with the help of scent markers combined to markers for other desirable traits.
MATERIALS AND METHODS
Plant Material and Scent Analyses
The Rosa mapping progeny HW, as previously described (Crespel et al., 2002; Hibrand-Saint Oyant et al., 2008), consists of a full-sib family of 91 hybrids derived from a cross between a dihaploid rose, H190, obtained from haploidization of the tetraploid Rosa x hybrida cv Zambra (Meynet et al., 1994) and a hybrid of a diploid Rosa wichurana, Rw, originating from Jardin de Bagatelle (Paris, France). The population was planted in one copy in the field (Institut National de la Recherche Agronomique, Angers, France). R. x hybrida cv Alister Stella Gray, Rosa × damascena cv bifera, R. x damascena cv Kazanlik, R. x hybrida cv The Mac Cartney rose, R. x hybrida cv Papa Meilland, and Rosa rugosa were all cultivated outside at the University of Saint-Etienne. For the HW progeny, samples were collected during 3 successive years from 2007 to 2009 from most of the hybrids, when flowers were available. All flowers were harvested at 10 am at stage 4 of flower development, as previously described (Bergougnoux et al., 2007). Each year, for each individual of the HW progeny, petals from different flowers of the same individual were harvested (from four to six independent flowers depending on the number of petals per flower). The petals were mixed, and two to four technical replicates were prepared by placing 1 g of petals into a glass vial for volatile compound extraction. Compounds were extracted after 24 h at 4°C with 2 mL of hexane containing 5 mg L−1 (±)-camphor (148075; Merck) as the internal standard. Fragrance volatile compounds were analyzed as previously described (Bergougnoux et al., 2007). The same protocol was used to analyze volatile compounds produced in Nicotiana benthamiana leaves and rose calli. For the analysis of 2PE glycosides, 20% (w/v) of Rw petals were crushed in citrate-phosphate buffer (pH 4.5) and centrifuged twice at 12,000g during 5 min. The final supernatant volume was measured (1 v) and separated into two vials (0.5 v each). In each vial, 0.5 v of the citrate-phosphate buffer was added, but in one vial, it contained the appropriate concentration of Rapidase (AR 2000; DSM) to obtain a final concentration of 20 mg mL−1. Rapidase contained a mix of glycosidases, among other enzymes, from Aspergillus niger. After overnight incubation at 37°C, 0.25 v of hexane containing (±)-camphor was added, mixed vigorously by hand, and incubated 10 min at room temperature. The supernatant was then centrifuged twice at 10,000g during 10 min and analyzed by GC-MS.
Statistical and QTL Analyses
Data were analyzed using R software to determine the variance components for genotypic effect (σG), year interaction (σGY), and the variance between replicated samples from the same genotype (σGR). Broad-sense heritability (h2) based on genotypic mean values averaged across years was calculated as follows (Holland et al., 2003):
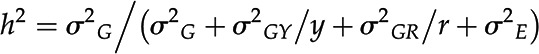 |
where y is the number of replication years and r is the number of replication plants per genotype.
JoinMap 4.0 (Van Ooijen, 2006) was used in the construction of the linkage map. Details of the map construction are given by Hibrand-Saint Oyant et al. (2008) and Remay et al. (2009). According to the pseudo-test cross strategy (Grattapaglia and Sederoff, 1994), parental maps were separately constructed using uniparental and common biparental markers. The homologous parental linkage groups having common biparental markers were then combined, and integrated maps were constructed based on mean recombination frequencies and combined LOD scores (Van Ooijen, 2006). QTL analysis was carried out using MapQTL 5.0 (Van Ooijen, 2004). A LOD threshold at which a QTL was declared significant was determined according to a genome-wide error rate of 0.05 over 1,000 permutations (Churchill and Doerge, 1994). The Shapiro-Wilk test (Shapiro and Wilk, 1965) was applied to test normality of trait distributions. For those volatile compounds deviating from normality, several transformations (Box-Cox, Log10, and Log2) were tested without success. Because of nonnormality for most of the metabolites, the data were thus analyzed first by the nonparametric KW. A significance level of P = 0.01 was used as a threshold. IM was then performed with a step size of 1 cM to find regions with potential QTL effects (i.e. where the LOD score was greater than the threshold). The percentage explained by the QTL (r2) was also presented. Other statistical analyses (normality tests, Spearman correlation tests, χ2 tests, and Dunn’s tests) were performed with XLSTAT software.
RhPAAS Isolation and Mapping on HW Populations
A forward primer for the 22 first nucleotides and two different reverse primers for the 3′ untranslated transcribed region of the published RhPAAS rose sequence (Kaminaga et al., 2006) were used for PCR amplification of the rose RhPAAS sequences from petal cDNAs and genomic DNA from the HW progeny. Forward (5′-TGTTGGAATCAACACGGAGA-3′) and reverse (5′-TCCATTCTCACAAGCCCTTC-3′) primers were used to amplify a 597-bp cDNA fragment from six different rose cultivars. Sequences were cloned in the Stratagene StrataClone Blunt PCR cloning vector according to the manufacturer’s instructions. Sequence alignments were performed using ClustalW and Geneious software. RhPAAS was localized on the genetic map with SSCP markers. Forward (5′-GAAAACATAGTCATGGATTGG-3′) and reverse (5′-CCGTGTTGATTCCAACAATTT-3′) primers were defined to amplify a 247-bp genomic DNA fragment. PCR was carried out in 15 µL with GoTaq Flexi DNA Polymerase according to the manufacturer’s recommendations (Promega) under the following conditions: 94°C for 3 min, 40 cycles consisting of 60°C for 1 min and 72°C for 2 min, and a final elongation at 72°C for 7 min. The SSCP marker was detected as described by Remay et al. (2009).
Overexpression of RhPAAS in N. benthamiana Leaves and Rose Calli
The RhPAAS coding sequence was amplified and cloned into the Gateway vector pENTR/D-TOPO (Invitrogen) and then subcloned in the binary expression vector pk7WGF2 via Gateway LR recombinant reaction between attL and attR sites for the expression in Rosa chinensis cv Old Blush callus (Vergne et al., 2010). pK7WGF2 was the Ti plasmid used for the expression of the gene encoding the N-terminal GFP fusions by CaMV35S (Karimi et al., 2002), to allow monitoring of the transformation state of the explants. Positive clones were transformed into the Agrobacterium tumefaciens EHA105 strain (Hood et al., 1993). Agrobacteria cultures were used to infiltrate leaves of N. benthamiana as previously described (Batoko et al., 2000). Four days after infiltration, 1 g of infected leaf sectors was kept in hexane and analyzed for volatile composition as described. Rose embryogenic calli were transformed as previously described (Vergne et al., 2010), except that no transgenic rose plants were regenerated and the transformed calli were used directly for GC-MS analyses.
RNA Preparation and Gene Expression Analyses
Total RNA preparation and cDNA synthesis were performed as previously described (Dubois et al., 2011). In short, total RNA was isolated from petals of Rw, H190, four hybrids (HW24, HW68, HW149, and HW530) that were chosen based on their contrasting levels of 2PE, and six rose cultivars (R. x hybrida cv Alister Stella Gray, R. x damascena cv bifera, R. x damascena cv Kazanlik, R. x hybrida cv The Mac Cartney rose, R. x hybrida cv Papa Meilland, and R. rugosa) using the NucleoSpin RNA Plant kit (Macherey-Nagel) according to the manufacturer’s instructions. After a DNase treatment performed with the Turbo DNA-free kit (Ambion), cDNA was obtained with the Revert Aid M-MulV reverse transcriptase kit (Fermentas) at 42°C for 1 h using an oligo(dT; T11VN) primer with 1 µg of RNA. RT-qPCR was performed with the FastStart Universal SYBR Green Master (Rox; Roche Diagnostics) using the StepOnePlus real-time PCR system (Applied Biosystems). Reactions were run in duplicate and quantified against a relative standard curve prepared for a serially diluted stock cDNA containing the target sequence. Data collection and analysis were performed using the StepOne Software v2.1 (Applied Biosystems). Relative quantification of candidate genes was performed using rose orthologs of α-TUBULIN (GenBank accession no. AF394915) and EF1-α (GenBank accession no. BI978089) as calibrators according to Vandesompele et al. (2002). Geometric means of the arbitrary units of the calibrator’s transcripts were used to normalize the relative amount of candidate gene transcripts. Primers specific to RhPAAS cDNAs were used for expression analysis by RT-qPCR (5′-CGTGTGGGTTCATGTGGAT-3′ and 5′-CCGAAATTCTGGACAAATGC-3′).
HRM Assay and Data Analysis
HRM assays were performed using the MeltDoctor HRM Master Mix (Applied Biosystems) according to the manufacturer’s instructions. In short, primers were designed using Primer-3-Plus so as to discriminate SNP1 (G-to-A variant discriminating a3 allele from a1 and a2) and SNP2 (G-to-A variant discriminating a2 allele from a1 and a3). The primers used for SNP1 were 5′-TGAACCCATCTCAACCATCC-3′ and 5′-AGCAGTGCTAGCAGTTGAAGAG-3′, and those used for SNP2 were 5′-CGTAGAGGGCGCAAATTCTT-3′ and 5′-GGATTCGTCGACAGTGAACTTG-3′ (Supplemental Fig. S2). Relative standard curves describing PCR efficiencies for each primer pair were generated for each amplicon according to Larionov et al. (2005). HRM-qPCR amplifications were performed using 10 µL of the MeltDoctor HRM Master Mix in a 20-µL reaction volume containing 5 µm of each primer and either 10 ng µL−1 plasmid DNA or 1 pg µL−1 cDNA obtained as described earlier. PCR was performed using the Qiagen thermocycler in a 72-well rotor with the following thermal profile: 95°C for 10 min, followed by amplification for 40 cycles consisting of 95°C for 15 s and 60°C for 1 min. Amplicon dissociation was immediately started by a melting step in the same real-time PCR machine, with 0.1°C temperature increments from 60°C to 85°C. Normalization, melt calibration, and data analysis were all performed with the Rotor-Gene Q Series Software (version 2.0.2). Differences in allelic expression were estimated by using serial dilutions of plasmid DNA. The shape of the melting peaks and melting temperature values of PCR products were compared with the known dilution mixes of plasmids containing each allele (a1, a2, and a3; Chateigner-Boutin and Small, 2007). Triplicate technical repeats were performed with a minimum of two biological replicates.
Accession Numbers
Sequences used in this article can be found in the GenBank database under the following accession numbers: RhPAAS a1, MH553438; RhPAAS a2, MH553439; and RhPAAS a3, MH553440.
Supplemental Data
The following supplemental data are available.
Supplemental Figure S1. Correlation map of the quantity of volatile scent compounds found in the rose progeny.
Supplemental Figure S2. Nucleotide sequence alignment of the alleles of RhPAAS.
Supplemental Figure S3. Amino acid sequence alignment of the alleles of RhPAAS.
Supplemental Figure S4. HRM analysis of amplicons from plasmid DNA control mixtures.
Supplemental Figure S5. Expression level of RhPAAS during flower development analyzed by RT-qPCR in different rose cultivars.
Supplemental Figure S6. Partial nucleotide sequence alignment of the alleles of RhPAAS from different rose cultivars.
Supplemental Table S1. Estimates of broad sense heritability and different variance components.
Supplemental Table S2. Summary of QTLs for scent compounds detected with nonparametric KW in the HW progeny. The 3 year data were analyzed separately.
Supplemental Table S3. Summary of QTLs for scent compounds detected with nonparametric KW in the HW progeny. Mean values corresponding to the 3 years were considered for the scent contents.
Supplemental Table S4. Pearson’s χ2 test of 2PE contents in the HW progeny.
Supplemental Table S5. 2PE and 2-phenylacetaldehyde contents analyzed by GC-MS in the parents of the HW progeny, H190 and Rw, and in four individuals of the progeny, HW24, HW68, HW149, and HW530.
Supplemental List. Complete list of 39 compounds extracted from rose petals.
Acknowledgments
We thank Nicolas Boyer, Loic Sarrabère, Isabelle Desbouchages, Priscilla Villand, and Alexis Lacroix for providing plant material. We thank the experimental unit HORTI for field plant management and the platform ANAN (Muriel Bahut) from the Structure Fédérative de Recherche Qualité et Santé du végétal for nucleic acid analysis. We thank the platform Analyse Génétique et Cellulaire of the Structure Fédérative de Recherche BioSciences Lyon (UMS3444/US8) for HRM and qPCR analyses. We thank Frédéric Hache from the English department of Saint-Etienne University for text revisions. We also thank Florence Gros, Saretta Paramita, Jérome Chameau, Sandrine Pierre, and Gilles Michel for technical assistance.
Footnotes
This work was supported by funding from the Région Rhône-Alpes, Centre National de la Recherche Scientifique, and INRA, France. Additional support was provided by CNRS GDR MediatEC (3658) and ANR Rosascent.
References
- Amrad A, Moser M, Mandel T, de Vries M, Schuurink RC, Freitas L, Kuhlemeier C (2016) Gain and loss of floral scent production through changes in structural genes during pollinator-mediated speciation. Curr Biol 26: 3303–3312 [DOI] [PubMed] [Google Scholar]
- Batoko H, Zheng HQ, Hawes C, Moore I (2000) A rab1 GTPase is required for transport between the endoplasmic reticulum and Golgi apparatus and for normal Golgi movement in plants. Plant Cell 12: 2201–2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauchet G, Grenier S, Samson N, Segura V, Kende A, Beekwilder J, Cankar K, Gallois JL, Gricourt J, Bonnet J, et al. (2017) Identification of major loci and genomic regions controlling acid and volatile content in tomato fruit: Implications for flavor improvement. New Phytol 215: 624–641 [DOI] [PubMed] [Google Scholar]
- Bennewitz S, Bergau N, Tissier A (2018) QTL mapping of the shape of type VI glandular trichomes in tomato. Front Plant Sci 9: 1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergougnoux V, Caissard JC, Jullien F, Magnard JL, Scalliet G, Cock JM, Hugueney P, Baudino S (2007) Both the adaxial and abaxial epidermal layers of the rose petal emit volatile scent compounds. Planta 226: 853–866 [DOI] [PubMed] [Google Scholar]
- Bourke PM, Arens P, Voorrips RE, Esselink GD, Koning-Boucoiran CF, Van’t Westende WP, Santos Leonardo T, Wissink P, Zheng C, van Geest G, et al. (2017) Partial preferential chromosome pairing is genotype dependent in tetraploid rose. Plant J 90: 330–343 [DOI] [PubMed] [Google Scholar]
- Bourke PM, Gitonga VW, Voorrips RE, Visser RGF, Krens FA, Maliepaard C (2018) Multi-environment QTL analysis of plant and flower morphological traits in tetraploid rose. Theor Appl Genet 131: 2055–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chateigner-Boutin AL, Small I (2007) A rapid high-throughput method for the detection and quantification of RNA editing based on high-resolution melting of amplicons. Nucleic Acids Res 35: e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XM, Kobayashi H, Sakai M, Hirata H, Asai T, Ohnishi T, Baldermann S, Watanabe N (2011) Functional characterization of rose phenylacetaldehyde reductase (PAR), an enzyme involved in the biosynthesis of the scent compound 2-phenylethanol. J Plant Physiol 168: 88–95 [DOI] [PubMed] [Google Scholar]
- Cherri-Martin M, Jullien F, Heizmann P, Baudino S (2007) Fragrance heritability in hybrid tea roses. Sci Hortic (Amsterdam) 113: 177–181 [Google Scholar]
- Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138: 963–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colinas M, Goossens A (2018) Combinatorial transcriptional control of plant specialized metabolism. Trends Plant Sci 23: 324–336 [DOI] [PubMed] [Google Scholar]
- Cong B, Liu J, Tanksley SD (2002) Natural alleles at a tomato fruit size quantitative trait locus differ by heterochronic regulatory mutations. Proc Natl Acad Sci USA 99: 13606–13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa F, Cappellin L, Zini E, Patocchi A, Kellerhals M, Komjanc M, Gessler C, Biasioli F (2013) QTL validation and stability for volatile organic compounds (VOCs) in apple. Plant Sci 211: 1–7 [DOI] [PubMed] [Google Scholar]
- Crespel L, Chirollet M, Durel E, Zhang D, Meynet J, Gudin S (2002) Mapping of qualitative and quantitative phenotypic traits in Rosa using AFLP markers. Theor Appl Genet 105: 1207–1214 [DOI] [PubMed] [Google Scholar]
- Dubois A, Remay A, Raymond O, Balzergue S, Chauvet A, Maene M, Pécrix Y, Yang SH, Jeauffre J, Thouroude T, et al. (2011) Genomic approach to study floral development genes in Rosa sp. PLoS ONE 6: e28455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois A, Carrere S, Raymond O, Pouvreau B, Cottret L, Roccia A, Onesto JP, Sakr S, Atanassova R, Baudino S, et al. (2012) Transcriptome database resource and gene expression atlas for the rose. BMC Genomics 13: 638–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunemann F, Ulrich D, Malysheva-Otto L, Weber WE, Longhi S, Velasco R, Costa F (2012) Functional allelic diversity of the apple alcohol acyl-transferase gene MdAAT1 associated with fruit ester volatile contents in apple cultivars. Mol Breed 29: 609–625 [Google Scholar]
- Farhi M, Lavie O, Masci T, Hendel-Rahmanim K, Weiss D, Abeliovich H, Vainstein A (2010) Identification of rose phenylacetaldehyde synthase by functional complementation in yeast. Plant Mol Biol 72: 235–245 [DOI] [PubMed] [Google Scholar]
- François L, Verdenaud M, Fu X, Ruleman D, Dubois A, Vandenbussche M, Bendahmane A, Raymond O, Just J, Bendahmane M (2018) A miR172 target-deficient AP2-like gene correlates with the double flower phenotype in roses. Sci Rep 8: 12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frary A, Nesbitt TC, Grandillo S, Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert KB, Tanksley SD (2000) fw2.2: A quantitative trait locus key to the evolution of tomato fruit size. Science 289: 85–88 [DOI] [PubMed] [Google Scholar]
- Gitonga VW, Stolker R, Koning-Boucoiran CFS, Aelaei M, Visser RGF, Maliepaard C, Krens FA (2016) Inheritance and QTL analysis of the determinants of flower color in tetraploid cut roses. Mol Breed 36: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grattapaglia D, Sederoff R (1994) Genetic linkage maps of Eucalyptus grandis and Eucalyptus urophylla using a pseudo-testcross: Mapping strategy and RAPD markers. Genetics 137: 1121–1137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grover CE, Gallagher JP, Szadkowski EP, Yoo MJ, Flagel LE, Wendel JF (2012) Homoeolog expression bias and expression level dominance in allopolyploids. New Phytol 196: 966–971 [DOI] [PubMed] [Google Scholar]
- Guterman I, Shalit M, Menda N, Piestun D, Dafny-Yelin M, Shalev G, Bar E, Davydov O, Ovadis M, Emanuel M, et al. (2002) Rose scent: Genomics approach to discovering novel floral fragrance-related genes. Plant Cell 14: 2325–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Wan H, Cheng T, Wang J, Yang W, Pan H, Zhang Q (2017) Comparative RNA-seq analysis of transcriptome dynamics during petal development in Rosa chinensis. Sci Rep 7: 43382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henz A, Debener T, Linde M (2015) Identification of major stable QTLs for flower color in roses. Mol Breed 35: 190 [Google Scholar]
- Hibrand Saint-Oyant L, Ruttink T, Hamama L, Kirov I, Lakhwani D, Zhou NN, Bourke PM, Daccord N, Leus L, Schulz D, et al. (2018) A high-quality genome sequence of Rosa chinensis to elucidate ornamental traits. Nat Plants 4: 473–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibrand-Saint Oyant L, Crespel L, Rajapakse S, Zhang L, Foucher F (2008) Genetic linkage maps of rose constructed with new microsatellite markers and locating QTL controlling flowering traits. Tree Genet Genomes 4: 11–23 [Google Scholar]
- Hirata H, Ohnishi T, Ishida H, Tomida K, Sakai M, Hara M, Watanabe N (2012) Functional characterization of aromatic amino acid aminotransferase involved in 2-phenylethanol biosynthesis in isolated rose petal protoplasts. J Plant Physiol 169: 444–451 [DOI] [PubMed] [Google Scholar]
- Hirata H, Ohnishi T, Tomida K, Ishida H, Kanda M, Sakai M, Yoshimura J, Suzuki H, Ishikawa T, Dohra H, et al. (2016) Seasonal induction of alternative principal pathway for rose flower scent. Sci Rep 6: 20234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland JB, Nyquist WE, Cervantes-Martinez CT (2003) Estimating and interpreting heritability for plant breeding: An update. Plant Breed Rev 22: 9–112 [Google Scholar]
- Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2: 208–218 [Google Scholar]
- Huang FC, Horváth G, Molnár P, Turcsi E, Deli J, Schrader J, Sandmann G, Schmidt H, Schwab W (2009a) Substrate promiscuity of RdCCD1, a carotenoid cleavage oxygenase from Rosa damascena. Phytochemistry 70: 457–464 [DOI] [PubMed] [Google Scholar]
- Huang FC, Molnár P, Schwab W (2009b) Cloning and functional characterization of carotenoid cleavage dioxygenase 4 genes. J Exp Bot 60: 3011–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Maekawa M, Tsuchiya S, Fujimori T (1998) Field attraction of Hoplia communis to 2-phenylethanol, a major volatile component from host flowers, Rosa spp. J Chem Ecol 24: 1491–1497 [Google Scholar]
- Kaminaga Y, Schnepp J, Peel G, Kish CM, Ben-Nissan G, Weiss D, Orlova I, Lavie O, Rhodes D, Wood K, et al. (2006) Plant phenylacetaldehyde synthase is a bifunctional homotetrameric enzyme that catalyzes phenylalanine decarboxylation and oxidation. J Biol Chem 281: 23357–23366 [DOI] [PubMed] [Google Scholar]
- Karimi M, Inzé D, Depicker A (2002) GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7: 193–195 [DOI] [PubMed] [Google Scholar]
- Kawamura K, Hibrand-Saint Oyant L, Crespel L, Thouroude T, Lalanne D, Foucher F (2011) Quantitative trait loci for flowering time and inflorescence architecture in rose. Theor Appl Genet 122: 661–675 [DOI] [PubMed] [Google Scholar]
- Klahre U, Gurba A, Hermann K, Saxenhofer M, Bossolini E, Guerin PM, Kuhlemeier C (2011) Pollinator choice in Petunia depends on two major genetic loci for floral scent production. Curr Biol 21: 730–739 [DOI] [PubMed] [Google Scholar]
- Larionov A, Krause A, Miller W (2005) A standard curve based method for relative real time PCR data processing. BMC Bioinformatics 6: 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavid N, Wang J, Shalit M, Guterman I, Bar E, Beuerle T, Menda N, Shafir S, Zamir D, Adam Z, et al. (2002) O-Methyltransferases involved in the biosynthesis of volatile phenolic derivatives in rose petals. Plant Physiol 129: 1899–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Wen YQ, Meng N, Qian X, Pan QH (2017) Monoterpenyl glycosyltransferases differentially contribute to production of monoterpenyl glycosides in two aromatic Vitis vinifera varieties. Front Plant Sci 8: 1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew M, Pryor R, Palais R, Meadows C, Erali M, Lyon E, Wittwer C (2004) Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clin Chem 50: 1156–1164 [DOI] [PubMed] [Google Scholar]
- Linde M, Hattendorf A, Kaufmann H, Debener T (2006) Powdery mildew resistance in roses: QTL mapping in different environments using selective genotyping. Theor Appl Genet 113: 1081–1092 [DOI] [PubMed] [Google Scholar]
- Magnard JL, Roccia A, Caissard JC, Vergne P, Sun P, Hecquet R, Dubois A, Hibrand-Saint Oyant L, Jullien F, Nicolè F, et al. (2015) Plant volatiles: Biosynthesis of monoterpene scent compounds in roses. Science 349: 81–83 [DOI] [PubMed] [Google Scholar]
- McKinney JT, Nay LM, De Koeyer D, Reed GH, Wall M, Palais RA, Jarret RL, Wittwer CT (2010) Mutation scanning and genotyping in plants by high-resolution DNA melting. In Meksem K and Kahl G, eds, The Handbook of Plant Mutation Screening: Mining of Natural and Induced Alleles. Wiley, Weinheim, Germany, pp 149–165 [Google Scholar]
- Meynet J, Barrade R, Duclos A, Siadous R (1994) Dihaploid plants of roses (Rosa × hybrida, cv ‘Sonia’) obtained by parthenogenesis induced using irradiated pollen and in vitro culture of immature seeds. Agronomie 14: 169–175 [Google Scholar]
- Ohgami S, Ono E, Horikawa M, Murata J, Totsuka K, Toyonaga H, Ohba Y, Dohra H, Asai T, Matsui K, et al. (2015) Volatile glycosylation in tea plants: Sequential glycosylations for the biosynthesis of aroma β-primeverosides are catalyzed by two Camellia sinensis glycosyltransferases. Plant Physiol 168: 464–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka N, Ohishi H, Hatano T, Hornberger M, Sakata K, Watanabe N (1999) Aroma evolution during flower opening in Rosa damascena Mill. Z Naturforsch 54c: 889–895 [Google Scholar]
- Paterson A, Kassim A, McCallum S, Woodhead M, Smith K, Zait D, Graham J (2013) Environmental and seasonal influences on red raspberry flavour volatiles and identification of quantitative trait loci (QTL) and candidate genes. Theor Appl Genet 126: 33–48 [DOI] [PubMed] [Google Scholar]
- Rambla JL, Medina A, Fernández-Del-Carmen A, Barrantes W, Grandillo S, Cammareri M, López-Casado G, Rodrigo G, Alonso A, García-Martínez S, et al. (2017) Identification, introgression, and validation of fruit volatile QTLs from a red-fruited wild tomato species. J Exp Bot 68: 429–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond O, Gouzy J, Just J, Badouin H, Verdenaud M, Lemainque A, Vergne P, Moja S, Choisne N, Pont C, et al. (2018) The Rosa genome provides new insights into the domestication of modern roses. Nat Genet 50: 772–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remay A, Lalanne D, Thouroude T, Le Couviour F, Hibrand-Saint Oyant L, Foucher F (2009) A survey of flowering genes reveals the role of gibberellins in floral control in rose. Theor Appl Genet 119: 767–781 [DOI] [PubMed] [Google Scholar]
- Roman H, Rapicault M, Miclot AS, Larenaudie M, Kawamura K, Thouroude T, Chastellier A, Lemarquand A, Dupuis F, Foucher F, et al. (2015) Genetic analysis of the flowering date and number of petals in rose. Tree Genet Genomes 11: 85 [Google Scholar]
- Sakai M, Hirata H, Sayama H, Sekiguchi K, Itano H, Asai T, Dohra H, Hara M, Watanabe N (2007) Production of 2-phenylethanol in roses as the dominant floral scent compound from L-phenylalanine by two key enzymes, a PLP-dependent decarboxylase and a phenylacetaldehyde reductase. Biosci Biotechnol Biochem 71: 2408–2419 [DOI] [PubMed] [Google Scholar]
- Sakai M, Tomita S, Hirata H, Asai T, Dohra H, Hara M, Watanabe N (2008) Purification and characterization of β-glucosidase involved in the emission of 2-phenylethanol from rose flowers. Biosci Biotechnol Biochem 72: 219–221 [DOI] [PubMed] [Google Scholar]
- Saliba-Colombani V, Causse M, Langlois D, Philouze J, Buret M (2001) Genetic analysis of organoleptic quality in fresh market tomato. 1. Mapping QTLs for physical and chemical traits. Theor Appl Genet 102: 259–272 [Google Scholar]
- Scalliet G, Lionnet C, Le Bechec M, Dutron L, Magnard JL, Baudino S, Bergougnoux V, Jullien F, Chambrier P, Vergne P, et al. (2006) Role of petal-specific orcinol O-methyltransferases in the evolution of rose scent. Plant Physiol 140: 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalliet G, Piola F, Douady CJ, Réty S, Raymond O, Baudino S, Bordji K, Bendahmane M, Dumas C, Cock JM, et al. (2008) Scent evolution in Chinese roses. Proc Natl Acad Sci USA 105: 5927–5932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp J, Dudareva N (2006) Floral scent: Biosynthesis, regulation and genetic modifications. In Ainsworth C, ed, Annual Plant Reviews 20: Flowering and Its Manipulation. Blackwell Publishing, Oxford, UK, pp 240–257 [Google Scholar]
- Schulz DF, Schott RT, Voorrips RE, Smulders MJM, Linde M, Debener T (2016) Genome-wide association analysis of the anthocyanin and carotenoid contents of rose petals. Front Plant Sci 7: 1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalit M, Guterman I, Volpin H, Bar E, Tamari T, Menda N, Adam Z, Zamir D, Vainstein A, Weiss D, et al. (2003) Volatile ester formation in roses: Identification of an acetyl-coenzyme A. Geraniol/Citronellol acetyltransferase in developing rose petals. Plant Physiol 131: 1868–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro SS, Wilk MB (1965) An analysis of variance test for normality (complete samples). Biometrika 52: 591–611 [Google Scholar]
- Souleyre EJ, Chagné D, Chen X, Tomes S, Turner RM, Wang MY, Maddumage R, Hunt MB, Winz RA, Wiedow C, et al. (2014) The AAT1 locus is critical for the biosynthesis of esters contributing to ‘ripe apple’ flavour in ‘Royal Gala’ and ‘Granny Smith’ apples. Plant J 78: 903–915 [DOI] [PubMed] [Google Scholar]
- Spiller M, Berger RG, Debener T (2010) Genetic dissection of scent metabolic profiles in diploid rose populations. Theor Appl Genet 120: 1461–1471 [DOI] [PubMed] [Google Scholar]
- Tieman D, Zhu G, Resende MFR Jr, Lin T, Nguyen C, Bies D, Rambla JL, Beltran KSO, Taylor M, Zhang B, et al. (2017) A chemical genetic roadmap to improved tomato flavor. Science 355: 391–394 [DOI] [PubMed] [Google Scholar]
- Torrens-Spence MP, Liu P, Ding H, Harich K, Gillaspy G, Li J (2013) Biochemical evaluation of the decarboxylation and decarboxylation-deamination activities of plant aromatic amino acid decarboxylases. J Biol Chem 288: 2376–2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F (2002) Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3: H0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ooijen JW. (2004) MapQTL 5.0 Software for the Mapping of Quantitative Trait Loci in Experimental Populations. Plant Research International, Wageningen, The Netherlands [Google Scholar]
- Van Ooijen JW. (2006) JoinMap 4.0 Software for the Calculation of Genetic Linkage Maps in Experimental Populations. Plant Research International, Wageningen, The Netherlands [Google Scholar]
- Vergne P, Maene M, Gabant G, Chauvet A, Debener T, Bendahmane M (2010) Somatic embryogenesis and transformation of the diploid Rosa chinensis cv Old Blush. Plant Cell Tissue Organ Cult 100: 73–81 [Google Scholar]
- Vogt J, Schiller D, Ulrich D, Schwab W, Dunemann F (2013) Identification of lipoxygenase (LOX) genes putatively involved in fruit flavour formation in apple (Malus × domestica). Tree Genet Genomes 9: 1493–1511 [Google Scholar]
- Watanabe S, Hashimoto I, Hayashi K, Yagi K, Asai T, Knapp H, Straubinger M, Winterhalter P, Watanabe N (2001) Isolation and identification of 2-phenylethyl disaccharide glycosides and mono glycosides from rose flowers, and their potential role in scent formation. Biosci Biotechnol Biochem 65: 442–445 [DOI] [PubMed] [Google Scholar]
- Yan Z, Visser PB, Hendriks T, Prins TW, Stam P, Dolstra O (2007) QTL analysis of variation for vigour in rose. Euphytica 154: 53–62 [Google Scholar]
- Yauk YK, Chagné D, Tomes S, Matich AJ, Wang MY, Chen X, Maddumage R, Hunt MB, Rowan DD, Atkinson RG (2015) The O-methyltransferase gene MdoOMT1 is required for biosynthesis of methylated phenylpropenes in ripe apple fruit. Plant J 82: 937–950 [DOI] [PubMed] [Google Scholar]
- Yauk YK, Souleyre EJF, Matich AJ, Chen X, Wang MY, Plunkett B, Dare AP, Espley RV, Tomes S, Chagné D, et al. (2017) Alcohol acyl transferase 1 links two distinct volatile pathways that produce esters and phenylpropenes in apple fruit. Plant J 91: 292–305 [DOI] [PubMed] [Google Scholar]
- Yu Y, Bai J, Chen C, Plotto A, Yu Q, Baldwin EA, Gmitter FG Jr (2017) Identification of QTLs controlling aroma volatiles using a ‘Fortune’ × ‘Murcott’ (Citrus reticulata) population. BMC Genomics 18: 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Dong F, Kunimasa A, Zhang Y, Cheng S, Lu J, Zhang L, Murata A, Mayer F, Fleischmann P, et al. (2014) Occurrence of glycosidically conjugated 1-phenylethanol and its hydrolase β-primeverosidase in tea (Camellia sinensis) flowers. J Agric Food Chem 62: 8042–8050 [DOI] [PubMed] [Google Scholar]
- Zini E, Biasioli F, Gasperi F, Mott D, Aprea E, Mark TD, Patocchi A, Gessler C, Komjanc M (2005) QTL mapping of volatile compounds in ripe apples detected by proton transfer reaction-mass spectrometry. Euphytica 145: 269–279 [Google Scholar]
- Zorrilla-Fontanesi Y, Rambla JL, Cabeza A, Medina JJ, Sánchez-Sevilla JF, Valpuesta V, Botella MA, Granell A, Amaya I (2012) Genetic analysis of strawberry fruit aroma and identification of O-methyltransferase FaOMT as the locus controlling natural variation in mesifurane content. Plant Physiol 159: 851–870 [DOI] [PMC free article] [PubMed] [Google Scholar]