

Abstract
The progression of ESCRT (Endosomal Sorting Complexes Required for Transport) pathways, which mediate numerous cellular membrane fission events, is driven by the enzyme Vps4. Understanding of Vps4 mechanism is, therefore, of fundamental importance in its own right and, moreover, it is highly relevant to the understanding of many related AAA+ ATPases that function in multiple facets of cell biology. Vps4 unfolds its ESCRT-III protein substrates by translocating them through its central hexameric pore, thereby driving membrane fission and recycling of ESCRT-III subunits. This mini-review focuses on recent advances in Vps4 structure and mechanism, including ideas about how Vps4 translocates and unfolds ESCRT-III subunits. Related AAA+ ATPases that share structural features with Vps4 and likely utilize an equivalent mechanism are also discussed.
Keywords: AAA proteins, cryo-electron microscopy, endosomal sorting, protein disassembly, protein structure
Introduction
Endosomal Sorting Complex Required for Transport (ESCRT) pathways drive membrane fission in numerous cellular processes, including cytokinesis, multivesicular body formation, plasma membrane repair, neuron pruning, exovesicle shedding, nuclear pore complex quality control, nuclear envelope reassembly, nuclear envelope repair after rupture, unconventional protein secretion, endolysosomal repair, and virus budding [1–8]. These pathways progress when upstream ESCRT factors trigger assembly of ESCRT-III filaments at sites that are destined for membrane fission. In turn, the ESCRT-III filaments recruit Vps4, whose activity subsequently drives both membrane fission and recycling of ESCRT-III subunits to their soluble state. The mechanism of membrane fission is not entirely clear, but an attractive possibility is that formation of the ESCRT-III filaments stabilizes a metastable conformation of the lipid bilayer, and the subsequent removal of ESCRT-III introduces stress that resolves by membrane fission [2,6]. Regardless, it is well established that Vps4 is essential for this process in multiple ESCRT pathways. Thus, understanding of Vps4 structure and mechanism has been a high priority since the discovery of its importance in the ESCRT pathway that buds vesicles into multivesicular bodies [9].
Domain structure of Vps4
The large family of AAA+ ATPases has been classified into multiple clades [10], including the meiotic clade that includes Vps4 and the microtubule-severing enzymes spastin and katanin [11]. Like many other AAA+ ATPases, Vps4 is active as a hexameric assembly that uses the energy of ATP hydrolysis to drive protein translocation through the central pore of the hexamer, thereby unfolding the substrate protein [12–14]. Also like many other AAA+ ATPases, Vps4 possesses an N-terminal domain that is connected to the ATPase cassette by a flexible linker (Figure 1). The N-terminal domain of Vps4 is a three-helix MIT (Microtubule Interacting and Trafficking) domain that binds to MIM (MIT Interacting Motif) sequences located in the C-terminal regions of ESCRT-III subunits [15–18]. These MIM sequences are inherently flexible and accessible in the ESCRT-III filament, and provide the initial interaction that recruits Vps4 for ESCRT-III processing and membrane fission [19–21].
Figure 1. Domain structure of Vps4.

Top, linear sequence of eukaryotic Vps4 with domains and motifs indicated. Bottom, composite structure of Vps4 (center) with separately determined MIT [18] and ATPase cassette [22,36] structures connected by a flexible linker. The MIT domain binds the MIM sequence of ESCRT-III substrates (left) to recruit Vps4 to ESCRT-III filaments [17]. The β-domain binds the dimeric VSL domain of the Vta1 cofactor [23] (right). PDB codes of the separately determined domain and complex structures are indicated.
The Vps4 ATPase cassette resembles that of other AAA+ ATPases, and comprises a large and small ATPase domain (Figure 1). The large domain contains the Walker A and B motifs that drive ATP binding and hydrolysis, respectively, as well as pore loops that engage substrate in the central pore of the hexamer [22]. The small ATPase domain, which follows the large ATPase domain, comprises four helices, and is followed by a C-terminal helix that packs against the large domain. The β-domain emanates from an internal loop of the small ATPase domain to mediate interactions with the Vta1/LIP5 cofactor protein [22,23]. Vta1/LIP5 binds and stabilizes the assembled Vps4 through its dimeric, C-terminal VSL (Vta1–SBP1–LIP5) domain [24] and also displays flexibly connected N-terminal MIT domains that may further enhance association with ESCRT-III filaments [24–26].
Structure of the active Vps4 complex
For many years, the primary goal of Vps4 structural studies has been to visualize its active, assembled conformation. Early studies established that Vps4 can exist in unassembled (monomeric/dimeric) or assembled states, and that the assembled state is associated with sites of membrane fission and performs ATP binding and hydrolysis [9]. The actual assembly state was the subject of considerable uncertainty, with confusion further worsened by three early EM structures that showed double-ring 12-mer and 14-mer structures that were in conflict with each other [27–29]. It is now apparent that each of those structures was misleading because subsequent biochemical studies have established that the active wild-type complex is, in fact, a hexamer that binds substrates in its central pore [30,31]. An X-ray crystal structure of a Vps4 hexamer [32] now also seems to have been misleading because it displays a high degree of symmetry, whereas three independently determined cryo-EM structures that were reported in 2017 are in good agreement with each other and seem to represent the active, more asymmetric conformation [33–35].
Despite the similarity of the three recently reported Vps4 cryo-EM structures [33–35], they have given rise to quite different mechanistic interpretations. Our preferred model is based on a cryo-EM structure of the yeast Saccharomyces cerevisiae Vps4 ATPase cassette in complex with an ESCRT-III-derived peptide, the Vta1 VSL domain, and ADP·BeFx (Figure 2). The primary advantage of this structure, which was initially determined at 4.3 Å resolution [33] and later reported at 3.2 Å resolution [36], is that it includes a bound ESCRT-III peptide, which defines the substrate-binding groove and constrains mechanistic models.
Figure 2. Structure of the active Vps4 hexamer complex.

Left, top view of the Vps4 complex cryo-EM structure [36]. Middle, side view with the ESCRT-III peptide vertical. Right, zoomed-in side and top views of the ESCRT-III peptide and pore loop 1 residues from each of the six Vps4 subunits. The red-blue (A–E) subunits form a helix that spirals around the ESCRT-III peptide, while the F subunit is displaced from the helical axis and does not contact the ESCRT-III peptide.
Substrate peptide binds a helical array of Vps4 subunits
The cryo-EM structures reveal that the Vps4 hexamer comprises a helical assembly of five subunits (subunits A–E) and a sixth subunit (F) that, as discussed below, appears to be transitioning between the ends of the five-subunit helix. The five-subunit helix is right-handed with a translation of ∼6.3 Å and a rotation of 60° between subunits. This matches the symmetry of the bound ESCRT-III peptide, which adopts a β-strand conformation that spirals tightly around the helix axis to bind in a groove formed by the five helical Vps4 subunits [36]. The concordance in symmetry between enzyme and bound substrate has important mechanistic implications because it means that successive substrate dipeptides bind in the same manner to successive Vps4 subunits, or more accurately, to successive interfaces between Vps4 subunits. Thus, the side chain of the first residue of each dipeptide binds into a ‘class I' binding pocket, while the side chain of the second residue of each dipeptide binds into a ‘class II' binding pocket, and these pockets are repeated at each of the four interfaces between the five helical Vps4 subunits (Figure 3) .
Figure 3. Binding of ESCRT-III by Vps4.

Successive ESCRT-III dipeptides are shown bound at the interfaces between the helical Vps4 subunits in equivalent orientations. The odd-numbered (1, 3, 5, 7) residues of the bound ESCRT-III octapeptide bind to class I pockets between the pore loop 1 tryptophan (W) residues of successive Vps4 subunits. The even-numbered (2, 4, 6, 8) residues bind to class II pockets between the pore loop 1 methionine (M) residues of successive Vps4 subunits.
Class I pockets are primarily formed by the tryptophan residue from pore loop 1 of adjacent Vps4 subunits, with the preceding lysine residue also contributing. The side chain of the first residue of the substrate dipeptide packs between the two tryptophans of adjacent subunits, which form a hydrophobic notch that appears capable of accommodating a wide variety of amino acid side chains. The binding of hydrophilic side chains is presumably accommodated by the highly solvated nature of the Vps4 pore, which will allow water molecules to satisfy the hydrogen-bonding needs of the ESCRT-III residues. The same principles are evident in the architecture of the class II binding sites that bind the second residue of each substrate dipeptide. In this case, the pore loop 1 methionine residues of adjacent Vps4 subunits form a hydrophobic notch, while pore loop 2 residues flank the binding pocket. Once again, the structure shows that a wide variety of amino acids can be accommodated, with the hydrogen-bonding groups of hydrophilic side chains presumably satisfied by interaction with water molecules from the solvent-exposed pore. Thus, Vps4 presents a helical array of equivalent dipeptide-binding sites that can accommodate the diverse amino acid residues of the peptide that is bound in the structure. This indicates that binding will be largely sequence-independent, as expected for translocation of the variable sequences of the ESCRT-III protein substrates.
Model of substrate translocation
The geometry of substrate binding suggests a ‘conveyer-belt’ model of substrate translocation in which the Vps4 hexamer propagates along the ESCRT-III substrate. In this model, Vps4 ‘walks' along the substrate through the action of individual Vps4 subunits peeling off the trailing end of the five-subunit helix (subunit E), passing through the ‘transitioning' conformation (subunit F), and joining the growing end of the helix (subunit A) to bind the next dipeptide in the ESCRT-III sequence (Figure 4). Consistent with this model, the large ATPase domain of subunit F is displaced from the helix axis, disengaged from the ESCRT-III peptide, and makes only minor contacts with the large domains of neighboring Vps4 subunits. Subunit F is maintained within the Vps4 assembly primarily through more peripheral contacts of the small ATPase domains. Advancement of the Vps4 hexamer along the extended ESCRT-III polypeptide is equivalent to conveying the ESCRT-III through the Vps4 hexamer pore, and will result in unfolding and disassembly of ESCRT-III. An attractive feature of the model is that each Vps4 subunit only needs to adopt one conformation as it and its associated ESCRT-III dipeptide pass through the pore from the A-subunit position to the E-subunit position. Thus, major conformational changes required of Vps4 are limited to those associated with transitioning from the E-subunit position at one end of the helix, through the F-subunit position and on to the A-subunit position at the other end of the helix.
Figure 4. Model of Vps4 translocation mechanism.
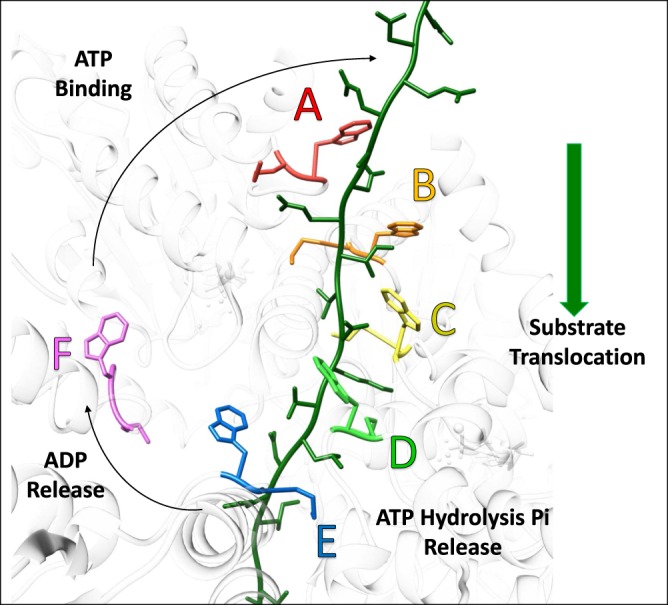
Vps4 cryo-EM structure (white) with the pore loop 1 tryptophan and methionine residues that make the major contribution to binding shown in colors. ESCRT-III peptide (green) with additional residues modeled in extended conformations at the N and C termini. Vps4 subunits A–E bind ESCRT-III while the transitioning F subunit is disengaged. Vps4 subunit interfaces in the helical arrangement of A–E subunits are stabilized by ATP. Hydrolysis and phosphate release in the last (D–E) interface promotes transition from the E to the F conformation with opening of the interface to allow ADP release. Subsequent ATP binding allows the transitioning subunit to pack against the subunit at the top of Vps4 helix (A) and bind the next ESCRT-III dipeptide.
The Vps4 structure was determined in complex with the non-hydrolyzable nucleotide analog ADP·BeFx, which can resemble ATP, the hydrolysis transition state, ADP with dissociated phosphate, or ADP without phosphate [37]. The resolution of the cryo-EM map is not sufficient to be definitive, but it appears to indicate that ADP·BeFx (analogous to ATP) is bound at the A–B, B–C, and C–D subunit interfaces, while ADP is bound at the D–E and E–F interfaces [36]. The nucleotide state of the transitioning subunit F is not apparent due to the low local resolution in this region of the model, but the structure suggests that this site is open to allow nucleotide exchange, which may mean that ATP binding is favored in the cellular context where ATP concentration is typically 10-fold higher than ADP concentration. The E–F interface is also quite open. The most intriguing observation on nucleotide coordination is the apparent binding of ADP at the D–E interface, whereas the very similar A–B, B–C, and C–D interfaces all appear to bind ATP (ADP·BeFx). Despite a high level of similarity to the A–B, B–C, and C–D interfaces, there are some differences at the D–E interface that may reflect the distinct nucleotide states, including a relative ∼1 Å shift in the arginine finger residues of subunit E that co-ordinate the β- and γ-phosphates of ATP (ADP·BeFx).
These observations suggest that ATP is hydrolyzed at the D–E interface, which presumably destabilizes this interaction and promotes transition to the more open EF conformation. Why hydrolysis might preferentially occur at the D–E interface rather than at the similar A–B, B–C, and C–D interfaces is not apparent, but presumably results from conformational changes that result from the more open E–F interface and propagate to the active site at the D–E interface. This model explains why substrate translocation proceeds in the direction indicated in Figure 4. At the other end of the helix, we envision that the open F–A interface allows binding of ATP, which will promote transition to the AB conformation. In this manner, the Vps4 helix will grow at the A-end of the helix as ATP binds and co-ordinately shrink at the E-end of the helix as ATP is hydrolyzed. The direction of translocation indicated by this model is consistent with the proteasome, which has a related structure and is known to translocate substrates in the equivalent direction into the proteasome's proteolytic chamber [38].
Comparison with other AAA+ ATPases
Several recently reported structures of other AAA+ ATPases overlap with the structure of Vps4, which suggests that they may all utilize an equivalent mechanism of substrate translocation. In particular, multiple structures have now been reported with substrate bound in the central pore and with a superimposable helix of subunits [38–44] (Figure 5), which strongly suggests that common mechanisms of nucleotide binding/hydrolysis and substrate binding/translocation will be utilized. Moreover, several AAA+ ATPase structures that have been determined in the absence of engaged substrate also adopt closely similar structures [34,35,45]. In some cases, including katanin [45] and another structure of Vps4 [34], an alternative mechanism of substrate disassembly was proposed. Nevertheless, we favor the conveyor-belt model outlined above because it is consistent with the binding of substrate peptides seen in Vps4 and other AAA+ ATPases, including YME1 [41], HSP104 [40], VAT [39], TRIP13 [42], ClpB [43], and NSF [44]. It should be noted that multiple structures of hexameric AAA+ ATPases that probably translocate protein substrates have been reported in distinctly different conformations, including p97 [46] and ClpX [47]. We favor the model that the mechanism of AAA+ ATPases requires some inherent flexibility, and that these alternative structures are favored in the biochemical/crystallographic environment of sample preparation, but that the active translocating state corresponds to the structure observed for Vps4 and other AAA+ ATPases with bound substrate.
Figure 5. Superposition of Vps4 and other AAA+ ATPase structures.
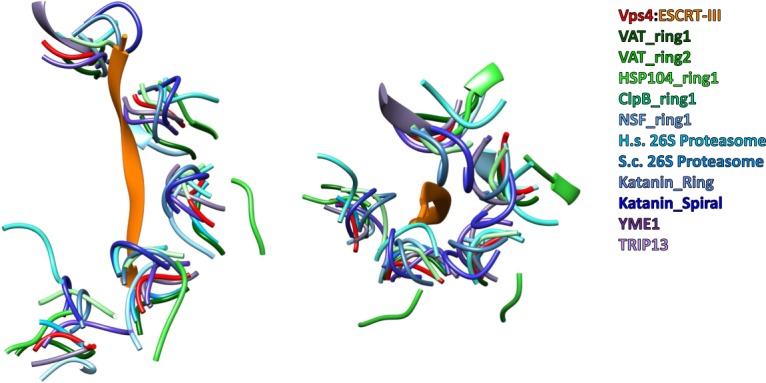
Side and top views of the Vps4 pore loop 1 residues and bound ESCRT-III peptide. The pore loop 1 residues of other AAA+ ATPases are shown following overlap on the ATPase large domain. The structures are listed on the figure and are cited in the text.
Of the AAA+ ATPases structures reported thus far, the mitochondrial quality control protein, YME1, merits discussion. The structure of inactive YME1, in which the catalytic Walker B glutamate was mutated to glutamine, was determined by cryo-EM in the presence of ATP and found to include peptides that fortuitously copurified after expression in E. coli and bind in the same manner as the ESCRT-III peptide binds to Vps4 [41]. The YME1 and Vps4 structures are remarkably similar, but ATP is bound to all four of the subunit interfaces of the five-subunit YME1 helix, whereas the last of these positions appears to be occupied by ADP in the case of Vps4 (D–E interface; above). This led to a subtle difference in mechanistic emphasis, with ATP hydrolysis proposed to occur as the last helical subunit YME1 (subunit E in Vps4 nomenclature) transitions to the F conformation, while the Vps4 model is more consistent with ATP hydrolysis occurring before the closed D–E interface opens and the E subunit transitions to the F conformation. This distinction cannot be resolved from the currently available structures of an inactive mutant in the presence of ATP (YME1) and a wild-type protein in the presence of ADP·BeFx (Vps4). Interestingly, a recent report of structures of active proteasome with engaged substrate indicates that ATP hydrolysis occurs at the interface corresponding to D–E of Vps4 [38].
Vps4 appears to show the ESCRT-III peptide binding in an orientation that is consistent with translocation from the C-terminal to N-terminal direction. This is consistent with biochemical studies that show progressive unfolding toward the N-terminus [14], and with the biological function of engaging the C-terminal residues of ESCRT-III before unfolding the structured N-terminal regions and thereby disassembling ESCRT-III filaments [21]. In contrast, other AAA+ ATPase complexes have been modeled with substrates bound N to C, C to N, or a mixture of both orientations, which is consistent with the view that different AAA+ ATPases can translocate substrates in either or both directions [48]. These observations indicate important questions for future studies, including: (i) Are the peptide orientations assigned for Vps4 and the other AAA+ ATPase complex structures correct? This is pertinent because the resolution attained in the various cryo-EM structures is generally marginal for the assignment of orientation for an isolated peptide. (ii) Is the same orientation also adopted when other peptide sequences bind to Vps4? (iii) Is the assigned orientation for Vps4 favored by a main chain hydrogen bond, as proposed [36], and (iv) what are the factors that dictate substrate-binding orientation for the various AAA+ ATPases?
AAA+ ATPases that act on protein substrates have been proposed to utilize mechanisms that are concerted [49], stochastic [50] or sequential [51]. Although the model that we favor for Vps4 is sequential, the extent to which variations may occur is unclear, especially given that ClpX, which has long served as a model for AAA+ ATPase studies, can accommodate inactive subunits [50] and operate with uneven step sizes [52]. Interestingly, the structures of related AAA+ ATPases that perform work on nucleic acid substrates also appear to utilize mechanisms that are sequential. In particular, the E1 helicase, which translocates DNA, shows five subunits forming a right-handed helix, the sixth subunit appearing to transition from one end of the helix to the other, and with a strand of DNA bound to the pore [53]. An analogous configuration occurs for the Rho RNA helicase, which has a different orientation of ATPase domains but nevertheless shows helical symmetry for five of the ATPase subunits and bound RNA substrate [37].
Future questions
Although considerable progress has been made, numerous important questions about AAA+ ATPase mechanisms remain. These include the timing of ATP hydrolysis, the orientation of substrate binding and translocation, the extent to which substrate specificity is defined by binding preferences in the translocation pore, and whether or not the translocation pore can accommodate more than one substrate polypeptide chain at a time [54–56]. While we believe that structures of Vps4 and other AAA+ ATPase complexes mimic translocating states, the possibility that the Vps4 structure corresponds to the initiation of translocation should be considered because the bound peptide corresponds to a relatively tight-binding segment of an ESCRT-III subunit [31]. On this note, and echoing studies of the proteasome [57], it will be of interest to determine the relative affinities of distinct amino acid sequences, and the impact of binding affinity upon substrate engagement and translocation. More generally, the extent to which other AAA+ ATPases follow the sequential, conveyer-belt mechanism proposed here is unclear, especially in light of the 6-fold rotationally symmetric structures of p97 [46] and CDC48 [58]. Finally, as noted above, our model is at odds with alternative interpretations of independent Vps4 and katanin structures. Resolving these questions will be important priorities for future studies.
Abbreviations
- AAA+
ATPases associated with diverse cellular activities
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- cryo-EM
electron cryo-microscopy
- EM
electron microscopy
- ESCRT
Endosomal Sorting Complexes Required for Transport
- MIM
MIT Interacting Motif
- MIT
Microtubule Interacting and Trafficking
- Vps4
vacuolar protein sorting-associated protein 4
Funding
Work on Vps4 in the Hill laboratory is funded by the National Institutes of Health P50GM082545.
Competing Interests
The Authors declare that there are no competing interests associated with the manuscript.
References
- 1.McCullough J., Colf L.A. and Sundquist W.I. (2013) Membrane fission reactions of the mammalian ESCRT pathway. Annu. Rev. Biochem. 82, 663–692 10.1146/annurev-biochem-072909-101058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schöneberg J., Lee I.H., Iwasa J.H. and Hurley J.H. (2017) Reverse-topology membrane scission by the ESCRT proteins. Nat. Rev. Mol. Cell Biol. 18, 5–17 10.1038/nrm.2016.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanson P.I. and Cashikar A. (2012) Multivesicular body morphogenesis. Annu. Rev. Cell Dev. Biol. 28, 337–362 10.1146/annurev-cellbio-092910-154152 [DOI] [PubMed] [Google Scholar]
- 4.Radulovic M. and Stenmark H. (2018) ESCRTs in membrane sealing. Biochem. Soc. Trans. 46, 773–778 10.1042/BST20170435 [DOI] [PubMed] [Google Scholar]
- 5.Henne W.M., Stenmark H. and Emr S.D. (2013) Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb. Perspect. Biol. 5, a016766 10.1101/cshperspect.a016766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McCullough J., Frost A. and Sundquist W.I. (2018) Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu. Rev. Cell Dev. Biol. 34, 85–109 10.1146/annurev-cellbio-100616-060600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christ L., Raiborg C., Wenzel E.M., Campsteijn C. and Stenmark H. (2017) Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 42, 42–56 10.1016/j.tibs.2016.08.016 [DOI] [PubMed] [Google Scholar]
- 8.Skowyra M.L., Schlesinger P.H., Naismith T.V. and Hanson P.I. (2018) Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360, eaar5078 10.1126/science.aar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babst M., Wendland B., Estepa E.J. and Emr S.D. (1998) The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17, 2982–2993 10.1093/emboj/17.11.2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erzberger J.P. and Berger J.M. (2006) Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 35, 93–114 10.1146/annurev.biophys.35.040405.101933 [DOI] [PubMed] [Google Scholar]
- 11.Monroe N. and Hill C.P. (2016) Meiotic clade AAA ATPases: protein polymer disassembly machines. J. Mol. Biol. 428, 1897–1911 10.1016/j.jmb.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nyquist K. and Martin A. (2014) Marching to the beat of the ring: polypeptide translocation by AAA+ proteases. Trends Biochem. Sci. 39, 53–60 10.1016/j.tibs.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olivares A.O., Baker T.A. and Sauer R.T. (2016) Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 14, 33–44 10.1038/nrmicro.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang B., Stjepanovic G., Shen Q., Martin A. and Hurley J.H. (2015) Vps4 disassembles an ESCRT-III filament by global unfolding and processive translocation. Nat. Struct. Mol. Biol. 22, 492–498 10.1038/nsmb.3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kieffer C., Skalicky J.J., Morita E., De Domenico I., Ward D.M., Kaplan J. et al. (2008) Two distinct modes of ESCRT-III recognition are required for VPS4 functions in lysosomal protein targeting and HIV-1 budding. Dev. Cell 15, 62–73 10.1016/j.devcel.2008.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 16.Stuchell-Brereton M.D., Skalicky J.J., Kieffer C., Karren M.A., Ghaffarian S. and Sundquist W.I. (2007) ESCRT-III recognition by VPS4 ATPases. Nature 449, 740–744 10.1038/nature06172 [DOI] [PubMed] [Google Scholar]
- 17.Obita T., Saksena S., Ghazi-Tabatabai S., Gill D.J., Perisic O., Emr S.D. et al. (2007) Structural basis for selective recognition of ESCRT-III by the AAA ATPase Vps4. Nature 449, 735–739 10.1038/nature06171 [DOI] [PubMed] [Google Scholar]
- 18.Scott A., Gaspar J., Stuchell-Brereton M.D., Alam S.L., Skalicky J.J. and Sundquist W.I. (2005) Structure and ESCRT-III protein interactions of the MIT domain of human VPS4A. Proc. Natl Acad. Sci. U.S.A. 102, 13813–8 10.1073/pnas.0502165102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bajorek M., Schubert H.L., McCullough J., Langelier C., Eckert D.M., Stubblefield W.M.B. et al. (2009) Structural basis for ESCRT-III protein autoinhibition. Nat. Struct. Mol. Biol. 16, 754–762 10.1038/nsmb.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muzioł T., Pineda-Molina E., Ravelli R.B., Zamborlini A., Usami Y., Göttlinger H. et al. (2006) Structural basis for budding by the ESCRT-III factor CHMP3. Dev. Cell 10, 821–830 10.1016/j.devcel.2006.03.013 [DOI] [PubMed] [Google Scholar]
- 21.McCullough J., Clippinger A.K., Talledge N., Skowyra M.L., Saunders M.G., Naismith T.V. et al. (2015) Structure and membrane remodeling activity of ESCRT-III helical polymers. Science 350, 1548–1551 10.1126/science.aad8305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott A., Chung H.-Y., Gonciarz-Swiatek M., Hill G.C., Whitby F.G., Gaspar J. et al. (2005) Structural and mechanistic studies of VPS4 proteins. EMBO J. 24, 3658–3669 10.1038/sj.emboj.7600818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang D. and Hurley J.H. (2010) Structural role of the Vps4-Vta1 interface in ESCRT-III recycling. Structure 18, 976–984 10.1016/j.str.2010.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao J., Xia H., Zhou J., Azmi I.F., Davies B.A., Katzmann D.J. et al. (2008) Structural basis of Vta1 function in the multivesicular body sorting pathway. Dev. Cell 14, 37–49 10.1016/j.devcel.2007.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skalicky J.J., Arii J., Wenzel D.M., Stubblefield W.-M.B., Katsuyama A., Uter N.T. et al. (2012) Interactions of the human LIP5 regulatory protein with endosomal sorting complexes required for transport. J. Biol. Chem. 287, 43910–43926 10.1074/jbc.M112.417899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z., Vild C., Ju J., Zhang X., Liu J., Shen J. et al. (2012) Structural basis of molecular recognition between ESCRT-III-like protein Vps60 and AAA-ATPase regulator Vta1 in the multivesicular body pathway. J. Biol. Chem. 287, 43899–43908 10.1074/jbc.M112.390724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu Z., Gonciarz M.D., Sundquist W.I., Hill C.P. and Jensen G.J. (2008) Cryo-EM structure of dodecameric Vps4p and its 2:1 complex with Vta1p. J. Mol. Biol. 377, 364–377 10.1016/j.jmb.2008.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Landsberg M.J., Vajjhala P.R., Rothnagel R., Munn A.L. and Hankamer B. (2009) Three-dimensional structure of AAA ATPase Vps4: advancing structural insights into the mechanisms of endosomal sorting and enveloped virus budding. Structure 17, 427–437 10.1016/j.str.2008.12.020 [DOI] [PubMed] [Google Scholar]
- 29.Hartmann C., Chami M., Zachariae U., de Groot B.L., Engel A. and Grütter M.G. (2008) Vacuolar protein sorting: two different functional states of the AAA-ATPase Vps4p. J. Mol. Biol. 377, 352–363 10.1016/j.jmb.2008.01.010 [DOI] [PubMed] [Google Scholar]
- 30.Monroe N., Han H., Gonciarz M.D., Eckert D.M., Karren M.A., Whitby F.G. et al. (2014) The oligomeric state of the active Vps4 AAA ATPase. J. Mol. Biol. 426, 510–525 10.1016/j.jmb.2013.09.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han H., Monroe N., Votteler J., Shakya B., Sundquist W.I. and Hill C.P. (2015) Binding of substrates to the central pore of the Vps4 ATPase is autoinhibited by the Microtubule Interacting and Trafficking (MIT) domain and activated by MIT Interacting Motifs (MIMs). J. Biol. Chem. 290, 13490–13499 10.1074/jbc.M115.642355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caillat C., Macheboeuf P., Wu Y., McCarthy A.A., Boeri-Erba E., Effantin G. et al. (2015) Asymmetric ring structure of Vps4 required for ESCRT-III disassembly. Nat. Commun. 6, 8781 10.1038/ncomms9781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Monroe N., Han H., Shen P.S., Sundquist W.I. and Hill C.P. (2017) Structural basis of protein translocation by the Vps4-Vta1 AAA ATPase. eLife 6, e24487 10.7554/eLife.24487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Su M., Guo E.Z., Ding X., Li Y., Tarrasch J.T., Brooks C.L. et al. (2017) Mechanism of Vps4 hexamer function revealed by cryo-EM. Sci. Adv. 3, e1700325 10.1126/sciadv.1700325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun S., Li L., Yang F., Wang X., Fan F., Yang M. et al. (2017) Cryo-EM structures of the ATP-bound Vps4E233Q hexamer and its complex with Vta1 at near-atomic resolution. Nat. Commun. 8, 16064 10.1038/ncomms16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han H., Monroe N., Sundquist W.I., Shen P.S. and Hill C.P. (2017) The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. eLife 6, e31324 10.7554/eLife.31324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomsen N.D. and Berger J.M. (2009) Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell 139, 523–534 10.1016/j.cell.2009.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de la Peña A.H., Goodall E.A., Gates S.N., Lander G.C. and Martin A. (2018) Substrate-engaged 26S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science 362, eaav0725 10.1126/science.aav0725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ripstein Z.A., Huang R., Augustyniak R., Kay L.E. and Rubinstein J.L. (2017) Structure of a AAA+ unfoldase in the process of unfolding substrate. eLife 6, e25754 10.7554/eLife.25754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gates S.N., Yokom A.L., Lin J., Jackrel M.E., Rizo A.N., Kendsersky N.M. et al. (2017) Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 357, 273–279 10.1126/science.aan1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puchades C., Rampello A.J., Shin M., Giuliano C.J., Wiseman R.L., Glynn S.E. et al. (2017) Structure of the mitochondrial inner membrane AAA+ protease YME1 gives insight into substrate processing. Science 358, eaao0464 10.1126/science.aao0464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alfieri C., Chang L. and Barford D. (2018) Mechanism for remodelling of the cell cycle checkpoint protein MAD2 by the ATPase TRIP13. Nature 559, 274–278 10.1038/s41586-018-0281-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deville C., Carroni M., Franke K.B., Topf M., Bukau B., Mogk A. et al. (2017) Structural pathway of regulated substrate transfer and threading through an Hsp100 disaggregase. Sci. Adv. 3, e1701726 10.1126/sciadv.1701726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.White K.I., Zhao M., Choi U.B., Pfuetzner R.A. and Brunger A.T. (2018) Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife 7, e38888 10.7554/eLife.38888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zehr E., Szyk A., Piszczek G., Szczesna E., Zuo X. and Roll-Mecak A. (2017) Katanin spiral and ring structures shed light on power stroke for microtubule severing. Nat. Struct. Mol. Biol. 24, 717–725 10.1038/nsmb.3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Banerjee S., Bartesaghi A., Merk A., Rao P., Bulfer S.L., Yan Y. et al. (2016) 2.3 å resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 351, 871–875 10.1126/science.aad7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glynn S.E., Martin A., Nager A.R., Baker T.A. and Sauer R.T. (2009) Structures of asymmetric ClpX hexamers reveal nucleotide-dependent motions in a AAA+ protein-unfolding machine. Cell 139, 744–756 10.1016/j.cell.2009.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Augustyniak R. and Kay L.E. (2018) Cotranslocational processing of the protein substrate calmodulin by an AAA+ unfoldase occurs via unfolding and refolding intermediates. Proc. Natl Acad. Sci. U.S.A. 115, E4786–E4795 10.1073/pnas.1721811115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gai D., Zhao R., Li D., Finkielstein C.V. and Chen X.S. (2004) Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119, 47–60 10.1016/j.cell.2004.09.017 [DOI] [PubMed] [Google Scholar]
- 50.Martin A., Baker T.A. and Sauer R.T. (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437, 1115–1120 10.1038/nature04031 [DOI] [PubMed] [Google Scholar]
- 51.Bar-Nun S. and Glickman M.H. (2012) Proteasomal AAA-ATPases: structure and function. Biochim. Biophys. Acta 1823, 67–82 10.1016/j.bbamcr.2011.07.009 [DOI] [PubMed] [Google Scholar]
- 52.Rodriguez-Aliaga P., Ramirez L., Kim F., Bustamante C. and Martin A. (2016) Substrate-translocating loops regulate mechanochemical coupling and power production in AAA+ protease ClpXP. Nat. Struct. Mol. Biol. 23, 974–981 10.1038/nsmb.3298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enemark E.J. and Joshua-Tor L. (2006) Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442, 270–275 10.1038/nature04943 [DOI] [PubMed] [Google Scholar]
- 54.Bodnar N.O. and Rapoport T.A. (2017) Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell 169, 722–735.e9 10.1016/j.cell.2017.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee C., Prakash S. and Matouschek A. (2002) Concurrent translocation of multiple polypeptide chains through the proteasomal degradation channel. J. Biol. Chem. 277, 34760–5 10.1074/jbc.M204750200 [DOI] [PubMed] [Google Scholar]
- 56.Burton R.E., Siddiqui S.M., Kim Y.I., Baker T.A. and Sauer R.T. (2001) Effects of protein stability and structure on substrate processing by the ClpXP unfolding and degradation machine. EMBO J. 20, 3092–3100 10.1093/emboj/20.12.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu H., Singh Gautam A.K., Wilmington S.R., Wylie D., Martinez-Fonts K., Kago G. et al. (2016) Conserved sequence preferences contribute to substrate recognition by the proteasome. J. Biol. Chem. 291, 14526–14539 10.1074/jbc.M116.727578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bodnar N.O., Kim K.H., Ji Z., Wales T.E., Svetlov V., Nudler E. et al. (2018) Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1-Npl4. Nat. Struct. Mol. Biol. 25, 616–622 10.1038/s41594-018-0085-x [DOI] [PMC free article] [PubMed] [Google Scholar]