

Summary
Many pathogenic intracellular bacteria manipulate the host phago-endosomal system to establish and maintain a permissive niche. The fate and identity of these intracellular compartments is controlled by phosphoinositide lipids. By mechanisms that have remained undefined, a Francisella pathogenicity island-encoded secretion system allows phagosomal escape and replication of bacteria within host cell cytoplasm. Here, we report the discovery that a substrate of this system, OpiA, represents a family of wortmannin-resistant bacterial phosphatidylinositol (PI) 3-kinase enzymes with members found in a wide range of intracellular pathogens, including Rickettsia and Legionella spp. We show that OpiA acts on the Francisella- containing phagosome and promotes bacterial escape into the cytoplasm. Furthermore, we demonstrate that the phenotypic consequences of OpiA inactivation are mitigated by endosomal maturation arrest. Our findings suggest that FramciselL·, and likely other intracellular bacteria, override the finely-tuned dynamics of phagosomal PI(3)P in order to promote intracellular survival and pathogenesis.
eTOC blurb
Bacterial effector proteins can modulate host cell trafficking to establish a replicative niche. Ledvina et al. report the discovery that the Francisella effector protein OpiA is a phosphatidylinositol 3-kinase (PI3K) that acts on Francisella - containing phagosomes, promoting bacterial escape into the cytoplasm. OpiA-like PI3Ks are also found to be broadly distributed.
Graphical Abstract
Introduction
The success of most bacterial pathogens relies on their capacity to actively override host immune defense pathways. Those that replicate intracellularly face the additional requirement of manipulating cellular trafficking to reach and maintain an inhabitable niche (Escoll et al., 2016; Thi et al., 2012). These feats are often accomplished by effector proteins that are delivered directly to host cells by specialized secretion systems, including the well characterized type III and IV secretion systems (T3SS, T4SS) (Backert and Meyer, 2006; Byndloss et al., 2017; Galan and Wolf-Watz, 2006).
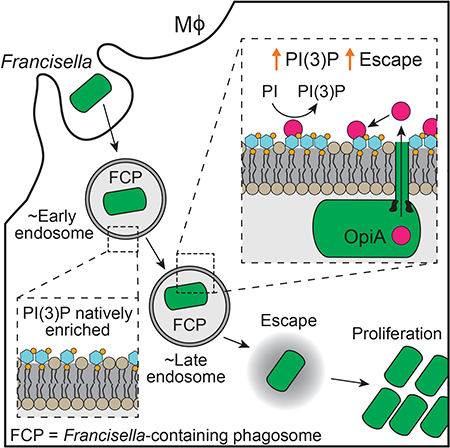
Francisella tularensis is an intracellular Gram-negative pathogen and the etiological agent of tularemia, a zoonotic infection that can be fatal if untreated (Kingry and Petersen, 2014). Subsequent to internalization, this bacterium escapes the Francisella-containing phagosome (FCP) and replicates in the cytoplasm. The molecular mechanisms enabling the intracellular lifestyle of F. tularensis remain unknown. However, studies indicate that a type VI secretion subtype 2 (T6SSii) pathway encoded on the Francisella pathogenicity island (FPI) is required for this bacterium to establish a productive intracellular niche (Barker et al., 2009; Eshraghi et al., 2016; Kadzhaev et al., 2009; Nano and Schmerk, 2007; Nano et al., 2004; Russell et al., 2014). On the basis of phylogenetic analyses, three T6SS subtypes have been defined: T6SSi and T6SSiii are widely distributed in the phyla Proteobacteria and Bacteroidetes, respectively, and are generally involved in interbacterial antagonism via the delivery of toxins to recipient bacterial cells (Hood et al., 2017; Russell et al., 2014). On the contrary, the T6SSii pathway appears to be restricted to Francisella and closely related host-associated bacteria, suggesting that this T6SS subtype evolved to mediate interactions with eukaryotic cells.
To begin to understand how the FPI-encoded T6SSii facilitates intracellular proliferation of F. tularensis, we recently performed a proteome-wide search for its substrates (Eshraghi et al., 2016). This work identified four T6SSii-exported effector proteins and several additional proteins that transit the T6SSii pathway to support core function of the apparatus. A surprising finding to emerge from this work was that two T6SSii effector proteins, OpiA (outside pathogenicity island A) and OpiB, are encoded by loci distal to the FPI in F. tularensis genomes (Eshraghi et al., 2016). Macrophage infection assays demonstrated that these proteins contribute to F. tularensis subspecies novicida (F. novicida) intracellular growth; however, their mechanism of action was not defined.
F. tularensis cells defective in T6SSii activity fail to escape the FCP and instead are shuttled to lysosomes and killed, suggesting that the pathway is important for modulating the trafficking of bacteria within cells (Nano and Schmerk, 2007). Phosphoinositide (PI) molecules are a family of dynamic signaling lipids that define the identity and fate of many eukaryotic vesicular compartments, including those within the phago-endosomal system (De Craene et al., 2017). As such, these molecules are frequently manipulated by intracellular pathogens (Pizarro-Cerda et al., 2015; Weber et al., 2009). For example, Legionella pneumophila utilizes its Dot/Icm T4SS to deliver PI 3-phosphatases and a PI 4-kinase to the cytoplasm of infected phagocytes (Dong et al., 2016; Hsu et al., 2012; Toulabi et al., 2013). The concerted action of these proteins causes the accumulation of PI(4)-phosphate (PI(4)P) on the Legionella containing vacuole (LCV), which leads to the binding of additional Legionella effectors and likely promotes the recruitment of endoplasmic reticulum-derived vesicles (Weber et al., 2006).
It has been over 15 years since genetic screens first implicated the FPI in the pathogenesis of F. tularensis (Gray et al., 2002). Despite this, the molecular mechanisms underlying its requirement for intracellular growth remain elusive. Here, we show that the T6SSii effector protein OpiA belongs to a previously unrecognized bacterial PI 3-kinase (PI3K) family. We provide biochemical and genetic evidence that OpiA enforces high PI(3)-phosphate (PI(3)P) levels on the FCP, leading to delayed maturation of this compartment and efficient escape of bacteria into the cytoplasm.
Results
OpiA family proteins share motifs with eukaryotic phosphoinositide kinases
OpiA is present in the majority of Francisella spp., including all subspecies of F. tularensis (Eshraghi et al., 2016) (Figure 1A). However, database searches failed to reveal characterized proteins with homology to OpiA beyond this group of organisms. To gain insight into the function of OpiA, we therefore turned to a hidden Markov model-based approach, which can identify small sequence elements conserved between functionally related proteins (Finn et al., 2015). This analysis identified a constellation of amino acids that is conserved among all Francisella OpiA orthologs and between a group of otherwise non-homologous bacterial proteins (Figure 1A). We subsequently refer to the latter as OpiA family proteins (OFPs). These residues constitute two consensus motifs, DxHxxN and IDH, separated by 14 amino acids in OpiA. Interestingly, OFPs are enriched in host-associated bacteria, including species belonging to the genera Legionella, Vibrio and Rickettsia (Figures 1B and S1). Moreover, an OFP encoded by Legionella pneumophila, previously designated LegA5, was shown to transit the Dot/Icm T4SS (de Felipe et al., 2008). Although the activity of the protein was not investigated, subsequent high-throughput studies further demonstrated that LegA5 is toxic and membrane-associated when expressed in yeast (Heidtman et al., 2009; Weigele et al., 2017).
Figure 1.

OpiA and related bacterial proteins share conserved motifs with eukaryotic PI kinases.
(A) Overview of conserved sequence motifs found in OFPs. Graphical representations of OpiA orthologous proteins from F. tularensis subsp novicida, tularensis and holarctica, and the Legionella pneumophila OFP LegA5. Conserved residues (black), portions of the gene encode by a separate ORF (blue) and ankyrin repeats in LegA5 (dark grey) are highlighted. Sequence logo indicates conserved catalytic motifs derived from an alignment of OFP sequences from bacterial species representative of the genera shown in panel B (alignment shown in Figure S1).
(B) Bacterial genera found to contain OFPs, grouped by class and phylum and with lifestyle indicated by box colors (eukaryotic-associated, purple; pathogen, grey; other, green).
(C) Alignment depicting the residues shared between OFPs and the indicated human PI3K and PI4K enzymes (Bact, bacterial; Euk, eukaryotic).
Literature searches revealed that the sequence and relative positions of the motifs shared among the OFPs closely resemble elements conserved between a subset of phosphoinositide kinases. Specifically, the catalytic and activation loops of phosphoinositide 3-kinases (PI3Ks) and certain phosphoinositide 4-kinases PI4Ks possess DxHxxN and IDF consensus sequences, respectively, with spacing similar to that found in OFPs (Figure 1C) (Gehrmann and Heilmeyer, 1998). However, outside of these motifs, OFPs bear no clear similarity with these proteins. For instance, OFPs lack lipid binding and protein interaction domains typical of this subset of eukaryotic PI kinases (Sasaki et al., 2009). Together, our sequence analyses suggested that OFPs may represent a previously unrecognized and unique family of bacterial phosphoinositide kinases that act in eukaryotic cells as effector proteins.
OpiA and LegA5 are phosphoinositide 3-kinase enzymes
Despite the divergence between OpiA and eukaryotic PI kinases, we tested whether the effector synthesizes phosphatidylinositol phosphate(s) in vitro. F. novicida OpiA was heterologously expressed, purified, and incubated with PI- or PI(4,5)P2-containing liposomes in the presence of [γ32P]-ATP (Figure S2A). These molecules were chosen based on the substrate preferences of eukaryotic OFP-related lipid kinase enzymes. PI3Ks of this group act on PI (class I-III) or PI(4,5)P (class III) to generate PI(3)P or PI(3,4,5)P3, respectively, whereas PI4Ks of this group (class III) act on PI to generate PI(4)P (Marat and Haucke, 2016). Separation of products by thin layer chromatography revealed a species consistent with the retention factor of PIP generated in the OpiA reaction (Figure 2A). This product was not observed in a similar reaction prepared using OpiA bearing a substitution of the predicted catalytic histidine residue it shares with characterized PI3K and PI4K enzymes (OpiAH261A) (Figures 2A and S2A). In contrast to the class I PI3K p100±, OpiA did not act on PIP2. We next purified the L. pneumophila OFP LegA5, and similarly examined its PI kinase activity. Like OpiA, LegA5 generated PIP from PI and displayed no detectable activity against PIP2 (Figures 2B and S2B).
Figure 2.

OpiA and LegA5 are class III-like phosphatidylinositol 3-kinase enzymes.
(A and B) Autoradiographs of thin layer chromatography (TLC)-separated lipid products produced by incubating the denoted proteins with [γ32P]-ATP and the indicated lipid substrates.
(C) Quantification of PIP species generated from incubating the indicated proteins with solubilized derivatives of PI (left and middle panels) and PIP2 (right), normalized to activity of control proteins for each reaction (dark grey). Data presented as mean values ± s.d. Asterisks represent statistically significant differences (Student’s t-test; ***p ≤ 0.0001).
Our data to this point indicated that OpiA produces PIP; however, it did not identify the position of the phosphate on the inositol moiety. To determine the specific PIP produced by OpiA, we utilized a competitive ELISA strategy that exploits the high selectivity of PI(3)P and PI(4)P binding domains. PI(5)P is produced in vivo by the action of phosphatases and is not produced directly by PI kinases; thus, we did not test for the capacity of OpiA to generate this molecule (Sasaki et al., 2009). The results of these assays indicated that the action of OpiA on PI leads exclusively to the production of PI(3)P (Figure 2C). Moreover, the efficiency of PI(3)P production by OpiA approaches that of p110α, suggesting that this is a physiologically relevant product of the enzyme. This assay additionally confirmed that PIP2 is not a substrate for OpiA.
The capacity of OpiA to phosphorylate PI and not PIP2 places its substrate specificity within PI3Ks belonging to classes II and III (Marat and Haucke, 2016). Class II and III PI3Ks can be further separated by the ability of the former to act on PI(4)P, generating PI(3,4)P2. We were unable to detect activity against PI(4)P by OpiA, indicating that, biochemically, the effector is most reminiscent of eukaryotic class III PI3K enzymes (Figure S2C).
OpiA PI3K activity contributes to F. novicida growth in vivo
In the course of our studies, we noted that OpiA over-expression is toxic to Saccharomyces cerevisiae (Figure 3A). Given the catalytic activity of the enzyme in vitro, we reasoned that this is due to excess PI(3)P production. Consistent with this, OpiAH261A did not inhibit S. cerevisiae growth. To further interrogate the activity of OpiA in vivo, we examined its toxicity in S. cerevisiae vps34Δ. Vps34 is a class III PI3K and is the only enzyme known to generate PI(3)P in S. cerevisiae (Reidick et al., 2017).
Figure 3.

The PI3K activity of OpiA leads to toxicity when expressed in yeast and contributes to F. novicida growth in vivo
(A and B) Growth resulting from serial dilution of the indicated yeast strains carrying plasmids to express the noted OpiA alleles (A) or Mcm1(B) plated on inducing (+) or non-inducing (−) media.
(C-E) Growth at 24hrs of the indicated strains of F. novicida in RAW 264.7 cells, normalized to level of growth by the wild-type strain (100%). Data are shown as the mean ± s.d. Asterisks represent statistically significant differences (Student’s t-test; ***p ≤ 0.0001).
(F) Western blot analysis of OpiA or OpiAH261A in the supernatant (sup) and cellular (cell) fractions of the indicated strains of F. novicida grown in the presence of 5% (w/v) potassium chloride.
(G) Quantification of the bacterial burden in the lungs of mice infected via aerosolization with the indicated strains of F. novicida. Data are shown as the mean ± s.d. and asterisks represent statically significant differences (ANOVA; *p ≤ 0.05, **p ≤ 0.001).
Deletion of this gene, but not the deletion of vps30 (a component of the Vps34 complex that is dispensable for PI(3)P production), significantly suppressed the toxicity of OpiA (Figure 3A). The suppression of the OpiA phenotype by Vps34 inactivation is specific, as the expression of another toxic protein (Mcm1) was similarly inhibitory to both S. cerevisiae wild-type and vps34Δ (Figure 3B) (Espinet et al., 1995).
Motivated by the toxicity of OpiA to yeast, we next asked whether OpiA promotes F. novicida pathogenesis. However, macrophage infection assays showed that inactivating opiA in wild-type F. novicida does not attenuate intracellular proliferation (Figure 3C). In the close relative of F. novicida, L. pneumophila, it is commonplace for the activity of an effector protein to mask that of another in infection models (Ghosh and O’Connor, 2017; Qiu and Luo, 2017). For example, the deletion of genes encoding the effector pairs WipB and LidA, MavP and LidA or WipB and LegC8 leads to intracellular replication defects, while inactivation of any of these effectors individually has no effect on in vivo growth (O’Connor et al., 2012). We reasoned this may explain the lack of a discernable phenotype for ΔopiA in the wild-type F. novicida background. To test this, we performed infection assays with F. novicida strains containing ΔopiA in combination with deletions in genes encoding each of the T6SSii effectors previously identified by our group (Eshraghi et al., 2016). This analysis uncovered a significant growth phenotype attributable to OpiA specifically in a strain lacking the FPI-encoded effector PdpC (Figure 3D). Genetic complementation of this phenotype was achieved by expressing OpiA from a neutral chromosomal site under the control of its native promoter. OpiAH261A failed to restore growth despite expression and secretion at levels similar to the wild-type protein, suggesting that the PI3K activity of OpiA is critical for its role in vivo (Figures 3E and3F).
Next, we used an aerosol delivery-based murine model to determine if the cell culture phenotype of ΔopiA extends to an animal infection (West et al., 2008). As observed in our macrophage infection assays, inactivation of opiA in the wild-type background did not yield a detectable mouse infection phenotype (Figure S3B). Previous studies have demonstrated that inactivation of pdpC attenuates bacterial growth and virulence in mice (Brodmann et al., 2017; Chou et al., 2013). We found that deletion of opiA further diminished the in vivo growth of F. novicida ΔpdpC and led to an additional decrease in morbidity compared to that exhibited by mice infected with F. novicida ΔpdpC (Figures 3G and S3B). In summary, these data show that the PI3K activity of OpiA can play an important role in the pathogenesis of F. novicida.
OpiA generates PI(3)P on endosomes
Our observation that the contribution of OpiA to F. novicida intramacrophage growth is apparent specifically in strains lacking PdpC led us to hypothesize that these two proteins function in a common pathway. An alternative explanation is that the absence of PdpC cripples F. novicida in vivo and non-specifically sensitizes the bacterium to the loss of additional effectors. However, a strain containing deletions of opiB and pdpC does not have an intracellular growth defect beyond that of one lacking only pdpC (Figure S3C). Sequence-based algorithms fail to identify candidate functions for PdpC, although recent experimental data implicate the protein in the escape of F. tularensis from phagosomes (Ozanic et al., 2016). While wild-type cells typically reach the cytoplasm within two hours, cells lacking PdpC remain co-localized with endosomal markers for an extended period and a large proportion are killed in lysosomes. Notably, PI(3)P - the product of OpiA - accumulates on early endosomes and is a key regulator of their trafficking (Marat and Haucke, 2016). These observations lend credence to the hypothesis that PdpC and OpiA promote F. novicida pathogenesis by influencing related cellular processes.
To probe the effects of OpiA on cellular PI(3)P, we utilized a well described PI(3)P fluorescent probe that is based on tandem FYVE domains (2xFYVE-mCherry) (Florey et al., 2015; Gaullier et al., 1998). Confocal microscopy was used to examine the co-localization of this probe with markers of major cellular compartments in control HeLa cells and those transfected with an opiA expression plasmid (Figures 4A, 4B and S4A). Consistent with the known association of PI(3)P with early endosomes, the probe displayed strong co-localization specifically with EEAl-positive vesicles in control cells. OpiA-expressing cells possessed a similar pattern of 2xFYVE-mCherry localization, with the exception that in these cells the probe showed significantly greater co-localization with LAMP 1-positive vesicles, representing late endosomes and lysosomes. These data suggest that OpiA-catalyzed PI(3)P production is limited to sites in the endosomal pathway. However, due to the background of endogenous PI(3)P on early endosomes, they do not distinguish whether OpiA acts on these vesicles or exclusively on membranes derived from later stages of the endosomal pathway.
Figure 4.

OpiA is a wortmannin-insensitive PI3K that generates PI(3)P on endosomes. (A) Pearson correlation coefficients indicating degree of co-localization between PI(3)P probe 2xFYVE-mCherry and markers for the Golgi network (TGN46), endoplasmic reticulum (calnexin), mitochondria (Mt-tar), and peroxisomes (PMP70) in HeLa cells co-transfected with an empty plasmid or a plasmid expressing OpiA, with or without wortmannin (wort) treatment (mitochondria: cells co-transfected to express GFP-labeled mitochondrial targeting sequence (Mt-tar); all other compartments: immunofluorescence with antibodies for indicated proteins). Data are shown as the mean ± SEM and asterisks represent statically significant differences (Student’s t-test; **p ≤ 0.001, ***p ≤ 0.0001). (B) Representative immunofluorescence fluorescent images of HeLa cells expressing 2xFYVE-mCherry, cotransfected with vectors expressing OpiA (or empty vector control) and stained for EEA1 or LAMP1(green), with or without wortmannin treatment. Insets depict 6-fold magnification of boxed regions. Scale bar, 10 μm.
(C) Quantification of the PI3K activity of p110α and OpiA treated with the indicated concentration of wortmannin. Activity normalized to non-wortmannin treated samples (set to 100% relative activity).
(D) Autoradiographs of thin layer chromatography (TLC)-separated lipid products produced by incubating the denoted proteins with [γ32P]-ATP, PI, and 300 nM wortmannin (+) or DMSO (−).
(E) Representative fluorescence images of HeLa cells co-transfected with plasmids expressing GFP-2xFYVE and the indicated genes (or empty vector control) for 24hrs followed by treatment with 800 nM wortmannin or a DMSO control for 40min. Insets represent 6-fold magnification of boxed regions.
(F) Proportion of HeLa cells transfected with the indicated plasmids in which GFP-2xFYVE localized to vesicle-like structures. A minimum of 100 cells over two biological replicates were imaged for quantification and the data are shown as the mean ± s.d. Asterisks represent statistically significant differences (Student’s t-test; ***p ≤ 0.0001).
The potential overlap between the site of OpiA-catalyzed PI(3)P production and the localization of this phosphoinositide in unperturbed cells prompted us to pursue a means to selectively inhibit endogenous PI3K activity. Wortmannin is a fungal-derived natural product that displays potent inhibitory activity against all previously described classes of PI3Ks (Knight et al., 2006). The mechanism of inhibition by wortmannin relies on the formation of a covalent adduct between the molecule and the sidechain of a conserved lysine residue present in the active site of target kinases (Lys802 of p110α) (Wymann et al., 1996). Given the high degree of sequence divergence between OpiA and known PI3K enzymes, we hypothesized that wortmannin may not efficiently inhibit the bacterial protein. In vitro inhibition studies confirmed the capacity of wortmannin to significantly inhibit p110α at concentrations below 10 nM; however, no inhibition of OpiA was observed at wortmannin concentrations exceeding 10 μM (Figures 4C, S4B and S4C). Likewise, a high concentration of wortmannin had no detectable impact on Legionella OFP LegA5 activity (Figure 4D). From these data, we conclude that OpiA, and likely the OFPs more generally, are resistant to inhibition by wortmannin.
The selectivity of wortmannin for endogenous PI3Ks allowed us to specifically define the site(s) of PI(3)P production by OpiA. As previously observed, 2xFYVE-GFP appeared diffusely localized in cells treated with wortmannin (Figures 4E and4F) (Pattni et al., 2001). On the contrary, the probe clearly localized to vesicular structures resembling endosomes in wortmannin-treated cells expressing OpiA. This pattern was not observed in wortmannin-treated cells expressing catalytically inactive OpiA. Co-localization analyses indicated that OpiA-catalyzed PI(3)P is significantly enriched on both early and late endosomal compartments (Figures 4A and4B, + wort). This localization pattern does not simply reflect the distribution of the substrate of OpiA - PI can be found on many cellular membranes (Hammond and Balla, 2015). Therefore, these data strongly suggest that OpiA acts in a site-selective manner in vivo.
OpiA is a selective PI(3)P-binding protein
The apparent selectivity of OpiA for the endosomal pathway suggested that the protein may be specifically recruited to these vesicles. In immunoprecipitation studies we were unable to identify OpiA-binding proteins that could mediate this recruitment. Accordingly, we tested if OpiA-lipid interactions could be responsible. Using a lipid overlay assay, we found that OpiA exhibited selective binding to its product, PI(3)P (Figure 5A). Based on the distribution of PI(3)P in cells, we posited that similar to several early endosomal proteins (e.g. EEA1 and PIKFYVE), binding to this lipid could facilitate the recruitment of OpiA to endosomes (Patki et al., 1998; Sbrissa et al., 2002).
Figure 5.

OpiA binds PI(3)P specifically and with high affinity.
(A) Measurement of the specific lipid binding capacity of purified OpiA-His6, determined by a lipid-overlay assay and ±-His detection of the protein.
(B) Bio-layer interferometry (BLI)-based quantification of the PI(3)P binding capacity of OpiA. The binding kinetics for three concentrations of OpiA with PI(3)P are shown (333nM, green; 111nM, blue; 37nM, purple).
(C) Measurement of the specific lipid binding capacity of purified His6-LegA5, determined by a lipid-overlay assay and α-His detection of the protein.
Using bio-layer interferometry coupled with synthetic streptavidin-conjugated PI(3)P, we determined that the interaction between the lipid and OpiA is likely of sufficient strength (Kd =190 nM) to target the protein in vivo (Figure 5B). The active sites of kinases, like those of other enzymes, can possess some affinity for their products; however, the strength of the OpiA-PI(3)P interaction strongly suggests that binding occurs at a distal site on the protein. Although OpiA lacks canonical PI(3)P binding domains, it is a basic protein (pi =10.0) with short polycationic motifs that are analogous to those previously shown to mediate specific interactions between eukaryotic proteins and phosphoinositide species (Choy et al., 2017). A tight association between OpiA and PI(3)P is in-line with our data demonstrating that OpiA-produced PI(3)P is localized to early endosomes in the presence of the pan PI3K inhibitor wortmannin. Whereas de novo synthesis of PI(3)P is inhibited by wortmannin, during endocytosis the enzyme inositol polyphosphate-4-phosphatase (INPP4A/B) acts on plasma membrane PI(3,4)P2 to generate an initial, small pool of early endosomal PI(3)P (Ivetac et al., 2005; Nigorikawa et al., 2015).
We sought to determine if PI(3)P binding is a common feature of OFPs. Lipid overlay assays indicated that LegA5 exhibits only weak binding to PI(3)P and preferentially associates with phosphatidylserine (Figure 5C). Together, these findings show that OpiA binds PI(3)P in a selective and high-affinity manner. Furthermore, they suggest PI(3)P binding serves as a mechanism for the specific recruitment of OpiA to endosomal membranes.
OpiA contributes to PI(3)P production on Francisella-containing phagosomes
Our data to this point were consistent with a feed-forward model to explain the behavior of OpiA (Figure 6A). Upon delivery via the T6SSii pathway, OpiA binds the FCP via the PI(3)P present on the cytoplasmic face of this early endosome-like vacuole. The effector generates additional PI(3)P, which promotes a positive feedback mechanism for the continued production of PI(3)P on the FCP. This process leads to a departure from the normal localization pattern of PI(3)P on maturing endosomes; OpiA maintains PI(3)P on the FCP even as the vacuole matures to resemble a late endosome.
Figure 6.

OpiA-mediated alterations to phagosomal trafficking contributes to intracellular growth of F. novicida.
(A) Model depicting localization and hypothesized role of OpiA during intracellular replication. OpiA exported by the T6SSii binds to PI(3)P on the cytosolic face of the FCP, and facilitates accumulation of additional PI(3)P through phosphorylation of PI. We speculate that FCP lysis is then promoted by additional effectors secreted by the T6SSii.
(B) Proportion of F. novicida cells that co-localize with the PI(3)P probe mCherry-2xFYVE at the indicate times post infection of primary murine BMDMs. Data represent means ± s.d. of three independent experiments. Asterisks represent statistically significant differences (Two-way ANOVA with Sidak’s correction for multiple comparisons; **p ≤ 0.001).
(C) Representative images acquired at 10 and 40 min post-infection of the experiment described in (B) (PI(3)P, (red); GFP-expressing bacteria, green). Insets represent 2.3-fold magnification of the regions indicated by arrows. Scale bars, 10 μm and 1 μm (insets).
(D) Proportion of F. novicida cells that co-localize with the late endosomal marker LAMP1 at the indicated times post infection of primary murine BMDMs. Colors indicate strains tested as in panel B. Data represent means ± s.d. of three independent experiments. Asterisks represent statistically significant differences (One-way ANOVA with Tukey’s correction for multiple comparisons; *p ≤ 0.05).
(E) Proportion of infected macrophages that contained replicating bacteria (greater than 5 bacteria/cell) at the indicated times post-infection of primary murine BMDMs. Colors indicate strains tested as in panel B. Data represent means ± s.d. of three independent experiments. Asterisks represent statistically significant differences (One-way ANOVA with Tukey’s correction for multiple comparisons; **p ≤ 0.001,*** p ≤ 0.0001).
(F) Representative images acquired at indicated time post-infection of the experiment described in (D) and (E) (PI(3)P, (red); GFP-expressing bacteria, green). Insets represent 3-fold magnification of the regions indicated by arrows. When >5 bacteria present in cytosol, insets are not included. Scale bars, 10 μm and 1 μm (insets).
(G) Intracellular growth (16hrs post-infection) of the indicated F. novicida strains infecting HeLa cells transfected with empty vector or a construct to express Rab7T22N. Data represent means ± s.d. Asterisks indicate statistically greater replication in experimental treatments compared to vector-only controls (Student’s t-test; *p ≤ 0.05).
We interrogated this model in vivo by following the co-localization of F. novicida wild-type versus ΔopiA with PI(3)P during the infection of murine bone marrow-derived primary macrophages (BMDMs). As expected, we observed F. novicida within PI(3)P-containing vacuoles at high frequency (>70%) immediately following phagocytosis (Figure 6B). The frequency of co-localization then decreased over the next 30 minutes, consistent with the known dynamics of phagosome maturation. In macrophages infected with ΔopiA cells, co-localization of PI(3)P with FCPs continued to decrease until reaching baseline by 40 minutes post-infection. In contrast, infection with wild-type F. novicida was associated with prolonged FCP-PI(3)P co-localization, up to 50 minutes post-infection. Inactivation of pdpC had no impact on these dynamics in either the wild-type or ΔopiA backgrounds (Figures 6B and6C). Prior studies show that 40–50 minutes post-infection corresponds to the time during which wild-type Francisella are beginning to escape the FCP (Chong et al., 2008). Therefore, this finding strongly suggests that, as predicted by our model, OpiA activity promotes PI(3)P accumulation on FCPs that have acquired late endosomal characteristics.
Studies suggest that Francisella escape from late endosome-like compartments before beginning replication within the cytoplasm (Chong and Celli, 2010). Based on our data indicating that the product of OpiA is present on this compartment at the approximate time of bacterial exit, we posited that OpiA-generated PI(3)P promotes endosomal escape. We reasoned that retardation of this process could explain why a ΔopzA-dependent in vivo growth defect is specifically observed in the ΔpdpC background, as PdpC is thought to also participate in endosomal escape. To evaluate phagosomal escape, we tracked the co-localization of F. novicida with LAMP1 during initial stages of BMDM infection. This assay confirmed the previously established phenotype of ΔpdpC, and moreover it revealed that a significant OpiA-dependent contribution to escape is observable in the absence of PpdC (Figures 6D and6F). As expected, the escape defect of ΔopiA cells was concomitant with a delay in replication (Figures 6E and6F).
While PI(3)P is required for early phagosome biogenesis, recent reports suggest that prolonging its presence inhibits maturation of these vesicles (Liu et al., 2016; Rapiteanu et al., 2016). We reasoned that if OpiA-catalyzed PI(3)P accumulation promotes bacterial escape by delaying endosomal maturation, then the impact of opiA inactivation should be minimized in cells wherein this process is interrupted. To test this, we examined the effect of arresting endosome maturation on a panel of F. novicida strains. Rab7 is a small GTPase that is required for the progression of early to late endosomes and expression of a dominant negative allele of this protein (Rab7T22N) leads to well characterized defects in endosomal maturation in HeLa cells (Bucci et al., 2000; Guerra and Bucci, 2016). Remarkably, we found that Rab7T22N restores the intracellular proliferation of ΔpdpC ΔopiA to the level of ΔpdpC in HeLa cells (Figure 6G). The effect of endosomal maturation arrest was specific to the ΔopiA genetic background, as neither the growth of ΔdotU nor ΔpdpC strains was enhanced by this allele. In total, these data strongly suggest that OpiA-catalyzed accumulation of PI(3)P on the FCP enhances Francisella intracellular survival by promoting timely escape from late endosomes.
Discussion
We have shown that the FPI-encoded T6SSii pathway substrate OpiA belongs to a previously unreported family of bacterial PI3K enzymes. As might be expected based on the phylogenetic distribution of PI, OFPs are found primarily in bacteria that associate intimately with eukaryotic cells. The Rickettsia and bacteria belonging to the related genus OrieMia are particularly replete with genes encoding OFPs. These bacteria are obligate intracellular parasites and cause a wide range of diseases in human (Renvoise et al., 2011). Like Francisella, the intracellular lifecycle of Rickettsia and Oriernia spp requires their escape from the endosomal pathway into the cytoplasm (Renvoise et al., 2011). Little is understood mechanistically about the factors required for phagosomal escape by Rickettsia. However, it is conceivable that despite their sequence divergence, OpiA and the OFPs of these bacteria function in a conserved manner to support escape.
Our biochemical analyses demonstrate that the L. pneumophila Dot/Icm T4SS effector LegA5 catalyzes the production of PI(3)P. Intracellular growth of L. pneumophila occurs within the Legionella containing vacuole (LCV), a compartment with a PI content that is highly orchestrated by the bacterium. At least four effector proteins delivered by L. pneumophila are direct PI-processing enzymes and an additional three effectors specifically bind PI(3)P (Dong et al., 2016; Finsel et al., 2013; Gaspar and Machner, 2014; Harding et al., 2013; Hsu et al., 2012; Jank et al., 2012; Toulabi et al., 2013). An effector central to the coordinated action of these proteins is LepB, a PI4K that generates PI(3,4)P2 from PI(3)P (Dong et al., 2016). SidF then acts downstream of LepB as a PI 3-phosphatase to generate PI(4)P (Hsu et al., 2012). The latter is the apparent end product of the pathway and plays an important role in the recruitment of other L. pneumophila effectors to the LCV and in the acquisition of ER-derived material necessary to support growth (Weber et al., 2006). Interestingly, the source of PI(3)P that LepB phosphorylates during LCV maturation in unknown (Dong et al., 2016). Work has demonstrated that L. pneumophila actively inhibits endosome fusion with the LCV, thus it is unlikely that the substrate of LepB is produced by a host enzyme (Gaspar and Machner, 2014). It is tempting to speculate that LegA5 is the enzyme responsible for the generation of PI(3)P on the LCV. Our data suggesting that LegA5 preferentially interacts with phosphatidylserine lends additional credence to this possibility. This phospholipid is enriched on the plasma membrane, the structure from which the LCV derives (Yeung et al., 2009). Although phosphatidylserine is depleted from the LCV by six hours post-infection, this is after the accumulation of PI(4)P and hence also subsequent to the proposed action of LegA5 (Hsu et al., 2012; Weber et al., 2006; Yeung et al., 2009).
Our data show that in the absence of PdpC, OpiA contributes to the intracellular growth of F. novicida by promoting bacterial escape into the cytoplasm. Since PdpC and OpiA are both present in the genomes of most Francisella spp., it is likely that each protein independently contributes to the fitness of the bacterium. We posit that the apparent functional redundancy in these proteins represents a limitation in the restrictiveness and relevance of the tissue culture and animal models commonly employed to study Francisella. It is therefore possible that, if examined in additional models, an OpiA phenotype may become apparent. Nevertheless, when taken together with our biochemical and cell biological data, the apparent functional overlap between OpiA and PdpC strongly implicate OpiA in the endosomal phase of cellular infection by F. novicida. Recent reports wherein individual endosomes were tracked during maturation demonstrate that PI(3)P levels are more dynamic than previously appreciated (Lu et al., 2012). Moreover, fine-tuned temporal regulation of PI(3)P levels appears to be important for the progression of early endosomes to lysosomes. For instance, deletions in negative regulators of Vps34 activity, WDR81 and WDR91, markedly delay phagosomal and endosomal maturation (Liu et al., 2016; Rapiteanu et al., 2016). By generating PI(3)P through a positive feedback mechanism, OpiA could override the natural dynamics of PI(3)P, in turn allowing additional time for other factors to mediate bacterial escape. This model is supported by our finding that cells with arrested phagosomal maturation are permissive to replication of F. novicida lacking OpiA.
The opiA genes in F. tularensis subsp tularensis and holarctica are split into two open reading frames and, in the case of F. holarctica, the N-terminal portion of the gene is truncated (Figure 1A). Despite these differences, the proteins encoded at these loci share >97% amino acid identity and critical catalytic residues are preserved. This conservation could indicate that the function of OpiA during infection is conserved across F. tularensis subsp. However, it is also possible that subspecies-specific differences in OpiA sequence have an impact on protein function. If this is the case, OpiA may be a factor that contributes to the variable host ranges of F. tularensis subsp (Kingry and Petersen, 2014). Work to define the role of OpiA during infection by other F. tularensis subsp. is ongoing.
Phosphoinositides are key regulators for a wide range of cell processes, including membrane trafficking, cellular proliferation, and autophagy. These phospholipids act in a spatiotemporal manner, which makes their functional dissection challenging. We demonstrate that OFPs are recalcitrant to inhibition by wortmannin, a potent inhibitor of virtually all known PI3K enzymes. This property of OFP enzymes could afford the opportunity to study the generation of PI(3)P in specific cellular compartments dictated by the unique intracellular biology of the bacterium from which they derive. Therefore, OFPs may provide a tool to study the role of phosphoinositides in eukaryotic cell biology.
Experimental procedures
Key Resources Table
See separate document
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph Mougous (mougous@uw.edu).
Experimental Model and Subject Details
Francisella movicida
F. novicida strains U112 and MFN245 were grown in tryptic soy broth or agar supplemented with 0.1% (w/v) cysteine (TSBC or TSAC). For macrophage infections, F. novicida and all derived strains were grown in Chamberlain’s defined media (CDM). All strains were grown at 37°C while shaking. All strains were stored at −80°C in TSBC supplemented with 40% (v/v) glycerol.
Escherichia coli
E. coli strains DH5α and BL21 Rosetta 2 DE3 were utilized for cloning and expression, respectively. All strains were routinely grown in Lysogeny broth (LB) at 37°C while shaking. All strains were stored at −80°C in LB supplemented with 20% (v/v) glycerol.
Saccharomyces cerevisiae
S. cerevisiae strain BY4742 MATalpha his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 was grown at 30°C in yeast extract peptone dextrose broth or agar (YPD). Mutant strains (vps34Δ and vps30Δ, Yeast Knockout Collection) were grown at 25°C on YPD. Mutations were confirmed by PCR. Strains carrying the inducible expression plasmid pCM190 were grown at 25°C on synthetic drop out media lacking uracil (C-ura) containing 1μg mL−1 doxycycline. All strains were stored at −80°C in YPD supplemented with 20% (v/v) glycerol.
Cell lines
All cell lines were authenticated by microscopic morphologic evaluation. RAW264.7 cells, derived from male mice, and HeLa cells, derived from a human female, were maintained at 37°C in the presence of 5% CO2. All cells were cultured in supplemented Dulbecco’s Modified Eagle’s Medium (DMEM; supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS; Atlanta biologicals), 4.5 g L−1 glucose, 2 mM glutamine, 110 mg L−1 sodium pyruvate, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin, and 0.05 mM beta-mercaptoethanol).
Human embryonic kidney 293T cells (HEK293T/17), derived from a human fetus, were grown in DMEM (4.5 g/l glucose and sodium pyruvate) supplemented with 10% (v/v) heat-inactivated FBS (Gibco, Life Technologies) and 4 mM L-glutamine at 37 °C and 10% CO2.
Murine bone marrow-derived macrophages (BMDMs) were generated from bone marrow cells collected from female 6–12 week-old C57BL/6NHsd mice (Envigo) and differentiated into macrophages for 5 days at 37°C and 10% CO2 in 1g/L glucose DMEM (with L-glutamine, and sodium pyruvate) supplemented with 10% (v/v) FBS (Invitrogen) and 20% (v/v) L-929 mouse fibroblasts-conditioned medium in non tissue culture-treated Petri dishes. After 5 days, loosely adherent BMMs were washed with PBS, harvested by incubation in chilled cation-free PBS supplemented with 1g/l D-glucose on ice for 10 min, resuspended in complete medium and replated in 24-well tissue culture treated plates at a density of 5×104 cell/well. BMMs were further incubated at 37°C under 10% CO2 atmosphere for 72 h, replenishing with complete medium 48 and 24 h before infection.
Mice
Female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME) and were 8–10 weeks of age when enrolled in experiments. Animals were group-housed according to experimental group in HEPA-filtered laminar flow cages with cage enrichment and unrestricted access to sterile food and water in the vivarium of Harborview Medical Center. The vivarium is managed by the University of Washington Department of Comparative Medicine in compliance with all policies and regulations of the Office of Laboratory Animal Welfare of the Public Health Service. The facility is fully accredited by the American Association for Laboratory Animal Care. All experimental procedures were approved in advance by the University of Washington Institutional Animal Care and Use Committee.
Method Details
Construction of Expression Plasmids
Heterologous expression in E. coli employed plasmid pET28b. To enable IPTG-inducible expression of opiA in this plasmid, the open reading frame of the gene was amplified using primers #5 and #6 (see supplemental table 1) from F. novicida genomic DNA and cloned into the NcoI and XhoI sites. The open reading frame of legA5 was amplified from L. pneumophilia subsp pneumophilla genomic DNA using primers #9 and #10 and cloned into the NdeI and XhoI sites of pET28b. The H178A mutation of legA5 was generated using SOE (primers #11 and #12).
The doxycycline repressible plasmid pCM190 was employed for yeast expression experiments (Gari et al., 1997). The opiA reading frame was amplified from F. novicida genomic DNA using primers #13 and #14, and the control gene mcm1 was amplified from S. cerevisiae genomic DNA using primers #15 and #16. Both genes were cloned separately into the NotI and BamHI sites of the plasmid. To generate the OpiA mammalian expression vector, the open reading frame of opiA was cloned in the XhoI and EcoRI sites of pMSCVpuro (Clontech) using primers #17 and #18. The H261A point mutation in OpiA was generated by SOE using primers #7 and #8, and was then cloned into E. coli, yeast and mammalian expression vectors using the same primers employed for the wild-type opiA allele.
Construction of Genetically Modified F. novicida Strains
For complementation or expression of GFP in F. novicida, a miniTn7 system was used to integrate constructs ectopically, downstream of glmS. For complementation of OpiA, the opiA open reading frame plus the intergenic regions upstream (containing the native promoter for the gene) and downstream of the gene were amplified by PCR and cloned into the HindIII and BamHI sites of the mini-Tn7 integration vector pMP749 using primers #1 and #2 (Table S1)(LoVullo et al., 2009). To generate GFP positive bacteria, the super-folder GFP open reading frame was cloned into pMP749. Subsequently, the constitutively active bfr promoter was cloned upstream of sfGFP (Zaide et al., 2011).
To generate strains containing mini-Tn7 insertions, pMP749 constructs were first transformed into the plasmid compatible strain F. novicida MFN245 carrying the helper plasmid pMP720 (LoVullo et al., 2009). Competent cells were prepared by growing overnight cultures of F. novicida, back-diluting 1:100 in 2 mLs TSBC, growing for 3hrs at 37°C with shaking, harvesting by centrifugation and resuspending in 1mL Francisella transformation buffer (per liter; L-arginine. 0.4 g; L-aspartic acid 0.4 g; L-histidine, 0.2 g, DL-methionine, 0.4g; spermine phosphate, 0.04 g; sodium chloride, 15.8 g, calcium chloride, 2.94g; tris(hydroxymethyl) aminomethane 6.05g).10 μg of the appropriate pMP749 vectors were added to prepared competent F. novicida MFN245 (pMP720), and the bacterial suspensions were then grown at 37°C with shaking for 30min. Transformed cells were recovered by adding 2 mLs TSBC and followed by incubation at 37°C with shaking for 3 hrs. Transformants were selected by plating on TSAC + 15 μg mL−1 kanamycin. Colonies were screen by PCR using primers #3 and #4. To move chromosomal insertions from F. novicida MFN245 to F. novicida U112, genomic DNA was prepared from appropriate MFN245 strains and 10 ng was used to transform competent U112, prepared as described above.
To generate deletion strains, previously generated deletion constructions (Eshraghi et al., 2016) were transformed into competent U112 strains as described above. Resulting merodiploids were grown overnight in non-selective TSBC, diluted 1:100 into CDM containing 0.1% p-chlorophenylalanine (w/v) and allowed to grow to stationary phase. These cultures were streaked onto non-selective TSAC and resulting kanamycin sensitive colonies were screened for deletions by colony PCR.
Bioinformatic identification of OpiA Family Proteins
The protein sequence of OpiA was submitted to the EMBL-EBI HMMMER server Jackhmmer (https://www.ebi.ac.uk/Tools/hmmer/search/jackhmmer) for iterative BLAST searches against the Uniprot protein database (Finn et al., 2015). Six iterations or hidden markov model BLASTs were performed. All identified bacterial proteins were then analyzed to determine if they contain the DxHxxN---IDH/F motif. In order to be considered an OFP, the two amino acid constellations had to be less than 25 residues apart; this criterion distinguished between OFPs and homologs of HipA, a protein kinase that contains the DxHxxN sequence but lacks a recognizable IDH/F (Correia et al., 2006).
Protein Expression and Purification
For protein expression, E. coli BL21 cells carrying pET28b expression plasmids were back diluted in 2xYT broth and grown at 37°C until the optical density at 600nM (OD600) was equal to 0.5. Expression was induced by the addition of 1 mM IPTG and cultures were incubated for 18 hours at 18°C with shaking. Cells were harvested post induction by centrifugation at 7,000 x g for 15 min and resuspended in buffer containing 20 mM Tris-HCl pH 7.5, 500 mM NaCl, 5mM imidazole, 1 mM AEBSF, 10 mM leupeptin, 1 mM pepstatin, 1 mM lysozome and 1mU benzonase. Cells were disrupted by sonication and cellular debris was removed by centrifugation at 45,000 x g for 45 min. His-tagged proteins were purified from lysates using a 1 mL HisTrap FF NI-TA cartridge on an AKTA FPLC purification system. Bound proteins were eluted using a linear imidazole gradient from 5 mM to 500 mM. The purity of each protein sample was assessed by SDS-PAGE and Coomassie brilliant blue staining, and fractions with high purity were used in biochemical and lipid binding assays.
Lipid Kinase Assays
Lipid kinase activity was measured using autoradiography to detect transfer of 32P from [γ32P]-ATP to phosphoinositide species in liposomes, separated by thin layer chromatography (TLC). Liposomes were composed of either PI or PI(4,5)P2 and phosphatidylserine (1:3, Promega V1711, V1701). In PI kinase activity and qualitative wortmannin inhibition screens, we employed 10 nM P110α, and 100 nM PIP5K1C, OpiA, OpiAH261A, LegA5 and LegA5H170A. In quantitative wortmannin inhibition assays, 1 nM P110α and 2 nM OpiA were employed. Reactions were started by the addition of ATP (subnanomolar [γ32P]-ATP and 50 nM ATP) and MgCl2 (2 mM). Kinase reactions were incubated at 25°C for 20 or 10min, for qualitative or wortmannin titration experiments respectively, and then terminated by the addition 1M hydrochloric acid and at least 4x volume of a 1:1 solution of methanol and chloroform. For the wortmannin titration assays, wortmannin or an equivalent volume of DMSO was added to the protein and liposomes prior to the addition of ATP. Subsequent to reaction termination, a fraction of the non-aqueous portion of each sample was spotted on silica gel 60 TLC plates pretreated with oxylate. Lipid species were resolved in n-propanol:2M acetic acid (65:35) and the TLC plates were exposed to a phosphoscreen. Signal derived from [γ32P]-labeled products was imaged with a Typhoon 9400 scanner. Densitometry analysis of the obtained images (ImageJ) was used to determine the percentage inhibition by wortmannin. Each kinase assay was repeated independently three times, with the exception of the wortmannin titration assay which was repeated twice.
PIP ELISA Assay
Assays were purchased from Echelon (K-3000, K-4000K, K1000s) and performed according to protocols supplied by the manufacturer. For the detection of PI(3)P, KBZ buffer was used during the kinase reaction (Echelon). Proteins were added at the following concentrations: p110α, 20 nM; PI4KB, 2nM; OpiA in the K-3000 assay, 100 μM; OpiA in the K-4000K and K1000s assays, 1 mM; OpiAH261A, 1 mM. Absorbance at 405 nM was read using a Synergy H1 Hybrid Reader (Biotek). Two biological replicates, each with three technical replicates were performed for each assay. Measurements were initially normalized to protein concentration followed by normalization to the activity of the positive control (set to 100% relative activity). Data presented are the average of the two biological replicates.
Yeast Transformations
Plasmids were transformed into S. cerevisiae using the Lazy Bone transformation method (Burke et al., 2000) and plated on C-ura + 1 μg mL−1 doxycycline to repress expression of genes carried on pCM190 (Gari et al., 1997). Plates were incubated at 25°C for a minimum of 3 days before use in growth assays.
Yeast Growth Assay
Fresh transformants of S. cerevisiae were collect from C-ura + doxycycline plates and re-suspended in sterile water. Density of the suspensions was normalized to OD600=1 and 10-fold dilutions generated in sterile water were plated on C-ura with and without 1 μg mL−1 doxycycline for repressing and inducing conditions, respectively. Plates were incubated at 25°C and imaged after 5 days using a FluoroChemQ. Growth assays were repeated independently 3 times, each performed in duplicate.
Intramacrophage growth assays
5 × 105 RAW264.7 cells were seeded without antibiotic in 24-well plates and incubated overnight at 37°C. After ~16 hrs, the cells were washed and exposed to mid-log phase F. novicida at a multiplicity of infection (MOI) of 0.1. The infection was synchronized by centrifugation at 800 x g at 25°C for 15 min followed by a 45 min incubation at 37°C. Following this incubation, cells were washed to remove extracellular bacteria, treated with gentamycin for 30 min, rinsed again and returned to 37°C. At selected time points, cells were lysed by the addition of 0.1% (v/v) Triton X100 in PBS, then serially diluted in PBS and plated on TSAC for CFU enumeration. CFU counts from later time points were normalized to the number of CFUs obtained immediately post-infection. Additionally, averaged data from 2–3 biological replicates for each mutant strain were normalized to the average level of wild-type growth (set to 100%).
Secretion Assays
Overnight cultures of F. novicida grown in TSBC were washed and diluted to an OD600 of 0.1 in TSBC + 5% (w/v) potassium chloride to stimulate T6SSii activity (Clemens et al., 2015). Cultures were grown at 37°C until OD600=1 and then pelleted by centrifugation at 8,000 x g for 10 min. Supernatants were collected and centrifuged for a further 10min at 10,000 x g to remove any remaining bacterial cells. Proteins in resulting supernatant samples were concentrated using a 30kDa cutoff centrifugal filter unit (EMD Millipore) and then mixed 1:2 with SDS-PAGE sample loading buffer. Cell fractions were generated by re-suspending cells in buffer containing 500 mM NaCl2, 50 mM Tris pH 7.5 and 10% (v/v) glycerol followed by mixing 1:2 with SDS-PAGE sample loading buffer. Secretion assays were repeated independently 3 times.
Western Blotting
Anti-OpiA serum was generated by injecting rabbits with purified OpiA-6xHis, and polyclonal anti-OpiA antibodies were purified by antigen affinity (GenScript). To detect α-OpiA, an anti-rabbit antibody conjugated to horse radish peroxidase (HRP, Sigma Aldrich, 1:5000) was employed. ±-His-HRP conjugated primary (Qiagen, 1:2000) was used to detect purified LegA5–6xHis. Bacterial cell lysates were generated as described above for secretion assays. Samples in SDS-loading buffer were boiled at 95°C for 15 min, loaded at equal volumes to resolve using SDS-PAGE, then transferred to nitrocellulose membranes. Membranes were blocked in TBST (10 mM Tris-base pH 7.6, 150mM NaCl2, and 0.1% w/v Tween-20) with 3% (w/v) bovine serum albumin (BSA) for 30 min at 24°C, followed by incubation with primary antibodies diluted in blocking buffer for 2hrs at 24°C. The blots were developed using Radiance HRP substrate (Azure Biosystems) and visualized using the Azure Biosystems c600. Representative images were assembled using Adobe Illustrator CC 2015.
Mouse infections
Mice were exposed to aerosolized bacteria in a whole-body aerosol exposure chamber, as described (Walters et al., 2015). Briefly, stocks of each bacterial strain were grown to stationary phase at 37 °C in TSBC, diluted in 20% (v/v) glycerol, aliquoted, and stored at −80 °C. The post-freeze titer of each stock was determined to be >6 × 108 CFU/ml when cultured on TSAC. For each experiment, aliquots of bacteria were thawed and diluted in PBS to 108 CFU/ml prior to nebulization. Cohorts of mice were randomly assigned to experimental groups and exposed to aerosolized bacteria in a whole animal exposure chamber with a computer interface to control pressures and flows (Biaera Technologies, Hagerstown, MD). Bacterial aerosols were generated by mini-Heart nebulizers with a flow rate of 8 L/min at 40 psi. Dilution air was regulated at 11.5 L/min to maintain total chamber flow at 19.5 L/min during a 10-minute exposure. Actual bacterial deposition in each experiment was determined by quantitative culture of the homogenized left lungs of three sentinel mice euthanized immediately after completion of the aerosol exposure. The remaining mice were returned to their cages. Mice were observed and weighed daily. At serial time points after infection, mice were euthanized with pentobarbital, exsanguinated by cardiac puncture, and left lungs were harvested and homogenized in 1mL PBS. Serial dilutions were generated in PBS then plated on TSAC for quantification. N=10 mice per bacterial strain per time point were used over two independent experiments.
Lipid-binding assays
PIP Strips (Echelon) were utilized in lipid overlay assays according to the protocol supplied by the manufacturer. Briefly, membranes containing spotted lipid species were blocked for 1 hr with 3% (w/v) BSA in TBST.10 μg of purified protein (either OpiA-6xHis or LegA5–6xHis) was added to the membrane in blocking buffer and incubated at 25 °C for 1 hr. Membranes were then washed 3x for 10 min in TBST and then incubated with α-His-HRP (Qiagen, 1:2000) for 1 hr. Membranes were again washed 3x prior to addition of Radiance HRP substrate (Azure Biosystems) and imaged using the Azure Biosystems c600. Each binding assays was independently performed twice for each protein.
Bio-layer interferometry
Binding of proteins to phosphoinositol species was measured and analyzed using Bio-layer interferometry with an Octet Red 96. Streptavidin biosensors (ForteBio) were equilibrated in running buffer (500 mM NaCl, 50 mM Tris pH 7.5, 1 mM Tris(2-carboxyethyl) phosphine hydrochloride (TCEP), 0.05% Tween-20 (v/v) and loaded with 200 nM of biotin-labeled PI(3)P (Echelon). A duplicate sensor was run in buffer without lipid as a background binding control subtracted from experimental measurements. Binding assays were conducted at 30°C with shaking at 1000 rpm. The Kd of OpiA for PI(3)P was calculated by determining the kinetic fit of on and off rates for four protein concentrations (333, 111, 37, 12 nM) simultaneously using a 1:1 fit model. Data shown are a representative graph from one of two independent experiments.
Transfection and Immunofluorescence Staining of HeLa cells
5×104 HeLa cells were seeded on 12 mm coverslips, incubated 16hrs at 37°C, then transfected with pBABE-2xFYVE-mCherry (Florey et al., 2015), the appropriate pMSCVpuro vector, and pEGFP-mt target (in cells to be used for mitochondrial staining) using Lipofectamine 3000 according to the instructions from the manufacturer. 24hrs post-transfection, cells were gently washed x3 in PBS and fixed for 10min in 3% paraformaldehyde (v/v) in PBS at 37°C. Following fixation, coverslips were washed 3 additional times in PBS then incubated with 50 mM NH4CI in PBS for 10min to quench free aldehyde groups. Fixed cells were blocked and permeabilized in 10% (v/v) donkey or goat serum and 0.1% (v/v) saponin in PBS for 30min. Labeling was accomplished by inverting coverslips onto drops of the appropriate primary antibody diluted in blocking buffer and incubating at 25°C for 1 hour. Primary antibodies used were mouse α-Lamp1 (Abcam, 1:100), mouse α-EEA1 (Abcam, 1:50), rabbit α-calnexin (Cell Signaling Technologies, 1:50), rabbit α-pmp70 (Thermo Fisher, 1:100), and rabbit α-tgn46 (Abcam, 1:100). Samples were then incubated with a 1:500 dilution of the appropriate Alexa 488-conjugated secondary antibody (Invitrogen) for 45min in the dark. Coverslips were then washed twice with PBS + 0.1% (v/v) saponin, twice with PBS, and once in H2O followed by mounting onto slides using ProLong Diamond Antifade Moutant with DAPI. For wortmannin treatment, 24hr post-transfection cells were incubated 2x for 20min with fresh complete DMEM + 800 nM wortmannin. Samples were then washed and processed for immunofluorescence staining as described above. Confocal fluorescence microscopy was performed using a Leica TCS SP8X Microscope. Representative fluorescent confocal micrographs were acquired as 1024 × 1024 pixels images using a 63x/1.4 NA HC PL APO objective and assembled using ImageJ. To determine Pearson’s correlation coefficients, confocal micrographs were acquired as 3064 × 3064 pixels images using a 40x/1.24 HC PL APO objective and Leica analysis software was utilized to analyze a minimum of 75 individual cells over three independent replicates. Authors were blinded during quantitative analysis.
Intracellular growth assays in transfected HeLa cells
2 × 105 HeLa cells were seeded without antibiotic in 24-well plates, incubated at 37°C for ~16hrs, then transfected with either pCDNA3-eGFP-rab7_T22N or pCDNA3 (generated by cutting the rab7T22N plasmid with SalI and religating the empty vector) using Lipofectamine 3000 according to the instructions from the manufacturer (Sun et al., 2010). 24hrs-post transfection, cells were washed and exposed to mid-log phase F. novicida at an MOI of ~10. Infections were otherwise conducted as described above in the intramacrophage growth assay section.
Lentivirus Transduction
Lentiviral transductions of BMDMs were performed using pLenti_CMV_neo-mCherry-2xFYVE (Palibrk et al., 2014) and the packaging plasmids pCMVdeltaR8.2 and pMD2.G (plasmids 12263 and 12259; Addgene) which were gifts from D. Trono (EPFL, Lausanne, France). Lentiviral supernatants were generated as follows: 2×106 HEK 293T/17 cells were seeded in 10 cm tissue culture dishes in 20ml medium and transfected after 24 h with a mix of 800 μ1 DMEM, 16 μg DNA (1:1:1 molar ratio of plasmids), and 48 μ1 FuGene® 6 following the manufacturer’s protocol and incubated for 48 h prior to collection. Lentiviral supernatants filtered through a 0.45 μm filter were added to BMMs (2:5 ratio v/v), and lentiviral transduction proceeded for 72 h before В MM infections were performed.
Infection and Fluorescence microscopy of Primary Murine BMDMs
For BMDM infections, F. novicida strains were grown in Chamberlain’s modified medium at 37 °C under shaking to an OD600 of 0.3–0.5, washed once in DMEM by centrifugation for 5 min at 6,000 x g at 15 °C, diluted to an MOI of 100 in complete medium and added to ice-chilled BMDMs in 24-well plates. Bacteria were centrifuged onto BMDMs at 400 x g for 10 min at 4°C and plates were immediately placed on a 37 °C water tray in a CO2 incubator to rapidly initiate synchronized uptake. Samples were collected at indicated times post-infection for immunofluorescence analysis. For time points later than 30 min post infection, extracellular bacteria were removed at 30 min post-infection by 3 washings in pre-warmed DMEM followed by replenishing with complete medium, in order to avoid continued, asynchronous bacterial uptake.
mCherry-2xFYVE-expressing BMDMs seeded onto 12 mm glass coverslips and infected with GFP-expressing F. novicida strains were washed 3 times in 1 x PBS at specific times post infection and fixed in 3% (v/v) paraformaldehyde (EMD) in 1 x PBS for 10 min at 37°C. For LAMP1 staining, coverslips were processed as described above. Coverslips were then washed 3 times in 1 x PBS, rinsed in distilled H2O, and mounted on glass slides in Mowiol (Calbiochem). Samples were viewed with a Leica DM4000 epifluorescence upright microscope for quantitative analysis or a Leica SP8 confocal laser-scanning microscope for image acquisition. Authors were blinded during quantitative analysis. Representative confocal micrographs of 1024 × 1024 pixels were acquired and assembled using Adobe Photoshop CS6. Data shown represent the average of three biological repeats.
Information Related to Experimental Design
No data were excluded from the study. Authors were not blinded during experiments except where stated.
Quantitative and Statistical Analysis
Statistical analysis was performed using GraphPad Prism 7 software. Results are presented as means ± standard deviation, except for Pearson correlation coefficients which are presented as means ± standard measure of error. Statistical significance between treatment groups was determined using either an unpaired, two-tailed Student’s t-test, or for group analysis, using either two-way analysis of variance (ANOVA, on log-transformed data) with Tukey’s multiple-comparison test or one-way ANOVA with Dunnett’s multiple-comparison test. The values for N and the definition of N for each experiment is provided in the relevant methods section; statistical analyses performed for each experiment is indicated in appropriate figure legends. No particular method was used to determine whether the data met assumptions of the statistical approach.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-FTN_0131 | this paper | N/A |
| Mouse polyclonal anti-EEA1 | Abcam | Cat#Ab7051; RRID:AB_1603734; Lot#GR278239–1 |
| Mouse polyclonal anti-LAMP1 | Abcam | Cat#Ab25630; RRID:AB_470708; Lot#GR905059–2 |
| Mouse anti-penta-His HRP conjugated | Qiagen | Cat#34460; Lot#151045877 |
| Rabbit polyclonal anti-Calnexin | Cell Signaling Technology | Cat#2433S; RRID:AB_2243887; Lot#4 |
| Rabbit polyclonal anti-PMP70 | ThermoFisher Scientific | Cat#PA1–650; RRID:AB_221991 |
| Rabbit polyclonal anti-TGN46 | Abcam | Cat#Ab50595; RRID:AB_2203289 |
| Goat anti-Rabbit HRP conjugated | Sigma Aldrich | Cat#A6154; RRID:AB_258284; Lot#SLBP3451V |
| Alexa 488-conjugated goat anti-rabbit IgG | Invitrogen | Cat#A21202; RRID:AB_141607; Lot# 1796361 |
| Alexa 488-conjugated donkey anti-mouse IgG | Invitrogen | Cat#A11008; RRID:AB_13165; Lot# 1937184 |
| Alexa 594-conjugated donkey anti-mouse IgG | Invitrogen | Cat#R37115; RRID:AB_2556543 |
| Bacterial and Virus Strains | ||
| Francisella novicida U112 | Manoil lab | N/A |
| F. novicida ∆dotU | Eshraghi et al., 2016 | N/A |
| F. novicida ∆pdpC | Eshraghi et al., 2016 | N/A |
| F. novicida ∆pdpD | Eshraghi et al., 2016 | N/A |
| F. novicida ∆opiA | Eshraghi et al., 2016 | N/A |
| F. novicida ∆opiB | Eshraghi et al., 2016 | N/A |
| F. novicida ∆pdpC ∆opiA | This paper | N/A |
| F. novicida ∆pdpD ∆opiA | This paper | N/A |
| F. novicida ∆opiB ∆opiA | This paper | N/A |
| F. novicida ∆opiB ∆pdpC | This paper | N/A |
| F. novicida ∆pdpC ∆opiA + Tn7:opiA | This paper | N/A |
| F. novicida ∆pdpC ∆opiA + Tn7:opiAH261A | This paper | N/A |
| F. novicida + Tn7:Pbfr-sfGFP | This paper | N/A |
| F. novicida ∆pdpC + Tn7:Pbfr-sfGFP | This paper | N/A |
| F. novicida ∆opiA + Tn7:Pbfr-sfGFP | This paper | N/A |
| F. novicida ∆pdpC ∆opiA + Tn7:Pbfr-sfGFP | This paper | N/A |
| F. novicida MFN245 | Manoil lab | N/A |
| Escherichia coli strain DH5α | Thermo Fisher Scientific | Cat#18258012 |
| Escherichia coli strain BL21 | EMD Millipore | Cat#69450 |
| Saccharomyces cerevisiae BY4742 MATalpha his3∆1 leu2∆0 lys2∆0 ura3∆0 | Yeast Knockout Collection | N/A |
| S. cerevisiae BY4742 MATalpha his3∆1 leu2∆0 lys2∆0 ura3∆0 vps34∆::KanMX4 | Yeast Knockout Collection | N/A |
| S. cerevisiae BY4742 MATalpha his3∆1 leu2∆0 lys2∆0 ura3∆0 vps30∆::KanMX4 | Yeast Knockout Collection | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle’s Medium | Corning | Cat#15013 |
| Dulbecco’s Modified Eagle’s Medium with L-gluatamine and sodium pyruvate | Corning | Cat#10014 |
| Fetal Bovine Serum | Atlanta biologicals | Cat#S11550; Lot#K16116 |
| Fetal Bovine Serum | Gibco, Life Technologies | |
| Fetal Bovine Serum | Invitrogen | |
| Wortmannin | Santa Cruz Biotechnology | Cat#sc3505B |
| Doxycycline hyclate | Sigma-Aldrich | Cat#D9891 |
| Hygromycin B | Research Products International | Cat# H75020 |
| ProLong Diamond Antifade Moutant with DAPI | Thermo Fisher Scientific | Cat#P36962 |
| Mowiol | Calbiochem | |
| Biotin Phosphatidylinositol 3-phosphate | Echelon | Cat#C-03B6 |
| p110α/p85α | Promega | Cat#V1721 |
| PIK4B | Echelon | Cat#P32–10G |
| PIP5K1C | SignalChem | Cat#P16–102CG-05 |
| PI(3)P:PS liposomes | Promega | Cat#V1711 |
| PI(4,5)P2:PS liposomes | Promega | Cat#V1701 |
| PI(4)P:PS lipid substrate | SignalChem | Cat#P427–59 |
| Radian HRP substrate | Azure biosystems | Cat#AC2101 |
| Critical Commercial Assays | ||
| Class III PI3K Elisa Kit | Echelon | Cat#K-3000 |
| PI 4-Kinase Activity Assay | Echelon | Cat#K-4000K |
| PI3-Kinase Activity Assay | Echelon | Cat#K-1000s |
| PIP strip | Echelon | Cat#P-6001 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L30000015 |
| Experimental Models: Cell Lines | ||
| Mouse: RAW 264.7 (TIB-71) | ATCC | TIB-71; RRID:CVCL_0493 |
| Human: HeLa cells (CCL-2) | ATCC | CCL-2; RRID:CVCL_0030 |
| HEK293T/17 | ATCC | CRL-11268; RRID:CVCL_1926 |
| Experimental Models: Organisms/Strains | ||
| Mice: 8–10 week old Ffmale C57BL/6 | Jackson Laboratories (Bar Harbor, ME) | Cat# 000664 |
| Mouse: Bone Marrow Derived Macrophages (BMMs) from C57BL/6NHsd mice | Envigo | Cat# 4403F |
| Oligonucleotides | ||
| Primers used in this study are listed in Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pET28b-opiA | This paper | N/A |
| pET28b-opiAH261A | This paper | N/A |
| pET28b-legA5 | This paper | N/A |
| pET28b-legA5H178A | This paper | N/A |
| pEX18-pheS-km | Eshraghi et al., 2016 | N/A |
| pEX18-pheS-km-∆opiA | Eshraghi et al., 2016 | N/A |
| pEX18-pheS-km-∆opiB | Eshraghi et al., 2016 | N/A |
| pEX18-pheS-km-∆pdpC | Eshraghi et al., 2016 | N/A |
| pEX18-pheS-km-∆pdpD | Eshraghi et al., 2016 | N/A |
| pEX18-pheS-km-∆dotU | Eshraghi et al., 2016 | N/A |
| pMP720 | LoVullo et al., 2009 | N/A |
| pMP749 | LoVullo et al., 2009 | N/A |
| pMP749-opiA_comp (5’ end+337bp, 3’ end +169bp) | This paper | N/A |
| pMP749-opiAH261A_ comp (5’ end+337bp, 3’ end +169bp) | This paper | N/A |
| pMP749-Pbfr-sfGFP | This paper | N/A |
| pCM190 | Gari et al., 1997 | N/A |
| pCM190-opiA | This paper | N/A |
| pCM190-opiAH261A | This paper | N/A |
| pCM190-mcm1 | This paper | N/A |
| pEGFP-2xFYVE | Starr et al., 2012 | N/A |
| pBABE- 2xFYVE-mCherry | Gift from Dr. Oliver Florey; Florey et al., 2015 | N/A |
| pEGFP-mt target sequence from subunit VIII of human cytochrome oxidase | Gift from Dr. Suzanne Hoppins | N/A |
| pMSCVpuro | Clonetech | N/A |
| pMSCVpuro-opiA | This paper | N/A |
| pMSCVpuro-opiAH261A | This paper | N/A |
| plenti_CMV_neo_mCherry-2xFYVE | Gift from Dr. Schink; Palibrk et al., 2014 | N/A |
| pCDNA3-eGFP-rab7_T22N | Sun et al., 2010 | Addgene Cat# 28048 |
| pCDNA3 | This study | N/A |
| pCMVdeltaR8.2 | Gift from D. Trono | Addgene Cat# 12263 |
| pMD2.G | Gift from D. Trono | Addgene Cat# 12259 |
| Software and Algorithms | ||
| GraphPad Prism version 7.0 for Mac | GraphPad, Software, La Jolla, California, USA | www.graphpad.com; RRID:SCR_022798 |
| ImageJ | NIH | Imagej.nih.gov; RRID:SCR_003070 |
| Adobe Illustrator CC 2015 | Adobe Systems Incorporated, San Jose, California, USA |
www.adobe.com/products/illustrator; RRID:SCR_010279 |
| Adobe Photoshop CS6 | Adobe Systems Incorporated, San Jose, California, USA | www.adobe.com/products/photoshop; RRID:SCR_014199 |
| Leica LAS AF Image Acquisition Software | University of Washington | RRID:SCR_013673 |
| Geneious 10.0.9 | Geneious, Software, Newark, New Jersey, USA | www.geneious.com RRID:SCR_010519 |
| Other | ||
| 1mL HisTrap FF NI-TA cartridge | GE Healthcare | Cat#17–5319-01 |
Highlights.
The Francisella secreted effector OpiA is a bacterial phosphatidylinositol 3-kinase (PI3K)
OpiA generates PI(3)P on late endosome-like Francisella-containing phagosomes
Genetically blocking phagosome maturation restores growth of OpiA deficient Francisella
In conjunction with the effector PdpC, OpiA promotes bacterial escape into the cytoplasm
Acknowledgements
We thank Oliver Florey for providing the pBABE-2xFYVE-mCherry plasmid, Kay Oliver Schink for providing the plenti_CMV_neo_mCherry-2xFYVE plasmid, Leigh Knodler for providing the pEGFP-3xFYVE plasmid, and Suzanne Hoppins for providing the pEGFP-mt_target_sequence plasmid. Additionally, we would like to thank Jennifer Hyde for assistance with lentiviral production, Jennifer Cundiff for assistance with BMDM culturing, Thomas Hind and Ning Zheng for their assistance performing bio-layer interferometry, Michael Constanzo and Yuko Arita for investigating the function of OpiA, Nathaniel Peters for microscopy assistance in the Keck Center, Tamara O’Connor for insights relating to Legionella, and Simon Dove and Gene Nester for critical review of the manuscript. We acknowledge support from the NIH to JC (AI129992), AJM (GM077349), SJS (AI093646 and AI130798), and to the W. M. Keck Center for Advanced Studies in Neural Signaling (S10 0D016240). HDL and AE were supported by an NIH funded training grant (AI055396), and MA was supported by a Wenner-Grenn Foundation Postdoctoral Fellowship. JDM holds an Investigator in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund and is an HHMI Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Authors declare no competing interests.
References
- Backert S, and Meyer TF (2006). Type IV secretion systems and their effectors in bacterial pathogenesis. Current opinion in microbiology 9, 207–217. [DOI] [PubMed] [Google Scholar]
- Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, Liu J, Celli J, Arulanandam BP, and Klose KE (2009). The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 74,1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodmann M, Dreier RF, Broz P, and Basler M (2017). Francisella requires dynamic type VI secretion system and ClpB to deliver effectors for phagosomal escape. Nat Commun 8,15853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, and van Deurs B (2000). Rab7: a key to lysosome biogenesis. Mol Biol Cell 11,467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Dawson D, and Stearn T (2000). Methods in yeast genetics : a Cold Spring Harbor Laboratory course manual (Plainview, N.Y.: Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Byndloss MX, Rivera-Chavez F, Tsolis RM, and Baumler AJ (2017). How bacterial pathogens use type III and type IV secretion systems to facilitate their transmission. Current opinion in microbiology 35,1–7. [DOI] [PubMed] [Google Scholar]
- Chong A, and Celli J (2010). The francisella intracellular life cycle: toward molecular mechanisms of intracellular survival and proliferation. Front Microbiol 1,138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, and Celli J (2008). The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun 76, 5488–5499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou AY, Kennett NJ, Nix EB, Schmerk CL, Nano FE, and Elkins KL (2013). Generation of protection against Francisella novicida in mice depends on the pathogenicity protein PdpA, but not PdpC or PdpD. Microbes Infect 15, 816–827. [DOI] [PubMed] [Google Scholar]
- Choy CH, Han BK, and Botelho RJ (2017). Phosphoinositide Diversity, Distribution, and Effector Function: Stepping Out of the Box Bioessays 39, Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Clemens DL, Ge P, Lee BY, Horwitz MA, and Zhou ZH (2015). Atomic structure of T6SS reveals interlaced array essential to function. Cell 160, 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia FF, D’Onofrio A, Rejtar T, Li L, Karger BL, Makarova K, Koonin EV, and Lewis K (2006). Kinase activity of overexpressed HipA is required for growth arrest and multidrug tolerance in Escherichia coli. J Bacteriol 188, 8360–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene JO, Bertazzi DL, Bar S, and Friant S (2017). Phosphoinositides, Major Actors in Membrane Trafficking and Lipid Signaling Pathways Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Felipe KS, Glover RT, Charpentier X, Anderson OR, Reyes M, Pericone CD, and Shuman HA (2008). Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS pathogens 4, e1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong N, Niu M, Hu L, Yao Q, Zhou R, and Shao F (2016). Modulation of membrane phosphoinositide dynamics by the phosphatidylinositide 4-kinase activity of the Legionella LepB effector. Nat Microbiol 2,16236. [DOI] [PubMed] [Google Scholar]
- Escoll P, Mondino S, Rolando M, and Buchrieser C (2016). Targeting of host organelles by pathogenic bacteria: a sophisticated subversion strategy. Nat Rev Microbiol 14, 5–19. [DOI] [PubMed] [Google Scholar]
- Eshraghi A, Kim J, Walls AC, Ledvina HE, Miller CN, Ramsey KM, Whitney JC, Radey MC, Peterson SB, Ruhland BR, et al. (2016). Secreted Effectors Encoded within and outside of the Francisella Pathogenicity Island Promote Intramacrophage Growth. Cell host & microbe 20, 573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinet C, de la Torre MA, Aldea M, and Herrero E (1995). An efficient method to isolate yeast genes causing overexpression-mediated growth arrest. Yeast 11,25–32. [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Arndt W, Miller BL, Wheeler TJ, Schreiber F, Bateman A, and Eddy SR (2015). HMMER web server: 2015 update. Nucleic acids research 43, W30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsel I, Ragaz C, Hoffmann C, Harrison CF, Weber S, van Rahden VA, Johannes L, and Hilbi H (2013). The Legionella effector RidL inhibits retrograde trafficking to promote intracellular replication. Cell host & microbe 14, 38–50. [DOI] [PubMed] [Google Scholar]
- Florey O, Gammoh N, Kim SE, Jiang X, and Overholtzer M (2015). V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11,88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan JE, and Wolf-Watz H (2006). Protein delivery into eukaryotic cells by type III secretion machines. Nature 444, 567–573. [DOI] [PubMed] [Google Scholar]
- Gari E, Piedrafita L, Aldea M, and Herrero E (1997). A set of vectors with a tetracycline-regulatable promoter system for modulated gene expression in Saccharomyces cerevisiae. Yeast 13, 837–848. [DOI] [PubMed] [Google Scholar]
- Gaspar AH, and Machner MP (2014). VipD is a Rab5-activated phospholipase A1 that protects Legionella pneumophila from endosomal fusion. Proc Natl Acad Sci U S A 111, 4560–4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaullier JM, Simonsen A, D’Arrigo A, Bremnes B, Stenmark H, and Aasland R (1998). FYVE fingers bind PtdIns(3)P. Nature 394, 432–433. [DOI] [PubMed] [Google Scholar]
- Gehrmann T, and Heilmeyer LM Jr. (1998). Phosphatidylinositol 4-kinases. Eur J Biochem 253, 357–370. [DOI] [PubMed] [Google Scholar]
- Ghosh S, and O’Connor TJ (2017). Beyond Paralogs: The Multiple Layers of Redundancy in Bacterial Pathogenesis. Front Cell Infect Microbiol 7, 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray CG, Cowley SC, Cheung KK, and Nano FE (2002). The identification of five genetic loci of Francisella novicida associated with intracellular growth. FEMS Microbiol Lett 215, 53–56. [DOI] [PubMed] [Google Scholar]
- Guerra F, and Bucci C (2016). Multiple Roles of the Small GTPase Rab7 Cells 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond GR, and Balla T (2015). Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim Biophys Acta 1851, 746–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding CR, Mattheis C, Mousnier A, Oates CV, Hartland EL, Frankel G, and Schroeder GN (2013). LtpD is a novel Legionella pneumophila effector that binds phosphatidylinositol 3-phosphate and inositol monophosphatase IMPA1. Infect Immun 81,4261–4270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidtman M, Chen EJ, Moy MY, and Isberg RR (2009). Large-scale identification of Legionella pneumophila Dot/Icm substrates that modulate host cell vesicle trafficking pathways. Cellular microbiology 11,230–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood RD, Peterson SB, and Mougous JD (2017). From Striking Out to Striking Gold: Discovering that Type VI Secretion Targets Bacteria. Cell host & microbe 21, 286–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Zhu W, Brennan L, Tao L, Luo ZQ, and Mao Y (2012). Structural basis for substrate recognition by a unique Legionella phosphoinositide phosphatase. Proc Natl Acad Sci U S A 109, 13567–13572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivetac I, Munday AD, Kisseleva MV, Zhang XM, Luff S, Tiganis T, Whisstock JC, Rowe T, Majerus PW, and Mitchell CA (2005). The type Ialpha inositol polyphosphate 4-phosphatase generates and terminates phosphoinositide 3-kinase signals on endosomes and the plasma membrane. Mol Biol Cell 16, 2218–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jank T, Bohmer KE, Tzivelekidis T, Schwan C, Belyi Y, and Aktories K (2012). Domain organization of Legionella effector SetA. Cellular microbiology 14, 852–868. [DOI] [PubMed] [Google Scholar]
- Kadzhaev K, Zingmark C, Golovliov I, Bolanowski M, Shen H, Conlan W, and Sjostedt A (2009). Identification of genes contributing to the virulence of Francisella tularensis SCHU S4 in a mouse intradermal infection model. PLoS One 4, e5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingry LC, and Petersen JM (2014). Comparative review of Francisella tularensis and Francisella novicida. Front Cell Infect Microbiol 4, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. (2006). A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell 125, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Jian Y, Sun X, Yang C, Gao Z, Zhang Z, Liu X, Li Y, Xu J, Jing Y, et al. (2016). Negative regulation of phosphatidylinositol 3-phosphate levels in early-to-late endosome conversion. J Cell Biol 212, 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoVullo ED, Molins-Schneekloth CR, Schweizer HP, and Pavelka MS Jr. (2009). Single-copy chromosomal integration systems for Francisella tularensis. Microbiology 155, 1152–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu N, Shen Q, Mahoney TR, Neukomm LJ, Wang Y, and Zhou Z (2012). Two PI 3-kinases and one PI 3-phosphatase together establish the cyclic waves of phagosomal PtdIns(3)P critical for the degradation of apoptotic cells. PLoS Biol 10, e1001245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marat AL, and Haucke V (2016). Phosphatidylinositol 3-phosphates-at the interface between cell signalling and membrane traffic. EMBO J 35, 561–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nano FE, and Schmerk C (2007). The Francisella pathogenicity island. Ann N Y Acad Sci 1105, 122–137. [DOI] [PubMed] [Google Scholar]
- Nano FE, Zhang N, Cowley SC, Klose KE, Cheung KK, Roberts MJ, Ludu JS, Letendre GW, Meierovics AI, Stephens G, et al. (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol 186, 6430–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigorikawa K, Hazeki K, Sasaki J, Omori Y, Miyake M, Morioka S, Guo Y, Sasaki T, and Hazeki O (2015). Inositol Polyphosphate-4-Phosphatase Type I Negatively Regulates Phagocytosis via Dephosphorylation of Phagosomal PtdIns(3,4)P2. PLoS One 10, e0142091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor TJ, Boyd D, Dorer MS, and Isberg RR (2012). Aggravating genetic interactions allow a solution to redundancy in a bacterial pathogen. Science 338, 1440–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozanic M, Marecic V, Lindgren M, Sjostedt A, and Santic M (2016). Phenotypic characterization of the Francisella tularensis ApdpC and AiglG mutants. Microbes Infect 18, 768–776. [DOI] [PubMed] [Google Scholar]
- Palibrk V, Lang E, Lang A, Schink KO, Rowe AD, and Boe SO (2014). Promyelocytic leukemia bodies tether to early endosomes during mitosis. Cell Cycle 13, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patki V, Lawe DC, Corvera S, Virbasius JV, and Chawla A (1998). A functional PtdIns(3)P-binding motif. Nature 394, 433–434. [DOI] [PubMed] [Google Scholar]
- Pattai K, Jepson M, Stenmark H, and Banting G (2001). A PtdIns(3)P-specific probe cycles on and off host cell membranes during Salmonella invasion of mammalian cells. Curr Biol 11,1636–1642. [DOI] [PubMed] [Google Scholar]
- Pizarro-Cerda J, Kuhbacher A, and Cossart P (2015). Phosphoinositides and host-pathogen interactions. Biochim Biophys Acta 1851, 911–918. [DOI] [PubMed] [Google Scholar]
- Qiu J, and Luo ZQ (2017). Legionella and Coxiella effectors: strength in diversity and activity. Nat Rev Microbiol 15, 591–605. [DOI] [PubMed] [Google Scholar]
- Rapiteanu R, Davis LJ, Williamson JC, Timms RT, Paul Luzio J, and Lehner PJ (2016). A Genetic Screen Identifies a Critical Role for the WDR81-WDR91 Complex in the Trafficking and Degradation of Tetherin. Traffic (Copenhagen, Denmark) 17, 940–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidick C, Boutouja F, and Piatta HW (2017). The class III phosphatidylinositol 3-kinase Vps34 in Saccharomyces cerevisiae. Biol Chem 398, 677–685. [DOI] [PubMed] [Google Scholar]
- Renvoise A, Merhej V, Georgiades K, and Raoult D (2011). Intracellular Rickettsiales: Insights into manipulators of eukaryotic cells. Trends Mol Med 17, 573–583. [DOI] [PubMed] [Google Scholar]
- Russell AB, Wexler AG, Harding BN, Whitney JC, Bohn AJ, Goo YA, Tran BQ, Barry NA, Zheng H, Peterson SB, et al. (2014). A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell host & microbe 16, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M, and Suzuki A (2009). Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res 48, 307–343. [DOI] [PubMed] [Google Scholar]
- Sbrissa D, Ikonomov OC, and Shisheva A (2002). Phosphatidylinositol 3-phosphate-interacting domains in PIKfyve. Binding specificity and role in PIKfyve. Endomenbrane localization. J Biol Chem 277, 6073–6079. [DOI] [PubMed] [Google Scholar]
- Sun Q, Westphal W, Wong KN, Tan I, and Zhong Q (2010). Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A 107, 19338–19343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thi EP, Lambertz U, and Reiner NE (2012). Sleeping with the enemy: how intracellular pathogens cope with a macrophage lifestyle. PLoS pathogens 8, e1002551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulabi L, Wu X, Cheng Y, and Mao Y (2013). Identification and structural characterization of a Legionella phosphoinositide phosphatase. J Biol Chem 288, 24518–24527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters KA, Olsufka R, Kuestner RE, Wu X, Wang K, Skerrett SJ, and Ozinsky A (2015). Prior infection with Type A Francisella tularensis antagonizes the pulmonary transcriptional response to an aerosolized Toll-like receptor 4 agonist. BMC genomics 16, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber SS, Ragaz C, and Hilbi H (2009). Pathogen trafficking pathways and host phosphoinositide metabolism. Mol Microbiol 71, 1341–1352. [DOI] [PubMed] [Google Scholar]
- Weber SS, Ragaz C, Reus K, Nyfeler Y, and Hilbi H (2006). Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS pathogens 2, e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigele BA, Orchard RC, Jimenez A, Cox GW, and Alto NM (2017). A systematic exploration of the interactions between bacterial effector proteins and host cell membranes. Nat Commun 8, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West TE, Pelletier MR, Majure MC, Lembo A, Hajjar AM, and Skerrett SJ (2008). Inhalation of Francisella novicida AmglA causes replicative infection that elicits innate and adaptive responses but is not protective against invasive pneumonic tularemia. Microbes Infect 10, 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, and Panayotou G(1996). Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Molecular and cellular biology 16,1722–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung T, Heit B, Dubuisson JF, Faim GD, Chiu B, Inman R, Kapus A, Swanson M, and Grinstein S (2009). Contribution of phosphatidylserine to membrane surface charge and protein targeting during phagosome maturation. J Cell Biol 185, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaide G, Grosfeld H, Ehrlich S, Zvi A, Cohen O, and Shafferman A (2011). Identification and characterization of novel and potent transcription promoters of Francisella tularensis. Applied and environmental microbiology 77,1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.