

Abstract
Motivation
Peptides crosslinked with cleavable chemical crosslinkers are identified with mass spectrometry by independent database search of spectra associated with the two linked peptides. A major challenge is to combine together the evidence of the two peptides into an overall assessment of the two-peptide crosslink.
Results
Here, we describe software that models crosslink specific information to automatically validate XL-MS cleavable peptide crosslinks. Using a dataset of crosslinked protein mixtures, we demonstrate that it computes accurate and highly discriminating probabilities, enabling as many as 75% more identifications than was previously possible using only search scores and a predictable false discovery rate.
Availability and implementation
XLinkProphet software is freely available on the web at http://brucelab.gs.washington.edu.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Crosslinking in conjunction with mass spectrometry (XL-MS) has been used to establish distance constraints on protein residues, and thereby provide structural information for proteins and multi-protein complexes (Holding, 2015; Leitner et al., 2016). Cleavable crosslinkers such as BDP (Zhang et al., 2009), DSSO (Kao et al., 2011) and others (Kandur et al., 2015) are particularly useful for detecting protein interactions in situ in biological contexts such as tissue culture cells and tissues. Crosslinking studies of cells in phenotypic comparisons (Chavez et al., 2015), in response to drug treatment (Chavez et al., 2016), or other perturbations can provide unique insights into how protein interactions and conformations change over time, increasing understanding at the systems structural biology level. Identification of crosslinked peptides by XL-MS is simplified by the use of cleavable crosslinkers since each peptide of the crosslinked pair can be identified independently during database search. In contrast, the use of non-cleavable crosslinkers requires a database search of all peptide pairs which becomes impractical if not impossible, with large numbers of proteins.
Several workflows have been described for using cleavable crosslinkers in conjunction with XL-MS, and most require customized data analysis (Gotze et al., 2015; Holding, 2015; Liu et al., 2017). In contrast, MS3 spectra generated through ReACT (Weisbrod et al., 2013) and MS2 spectra subjected to Mango (Mohr et al., 2018), can be analyzed with any traditional search engine such as Comet (Eng et al., 2013) to identify the released crosslinked peptides. Nevertheless, it has been a challenge to combine together the independent evidence of the two peptides into an overall assessment of the two-peptide crosslink. Traditionally this has been done in a conservative manner by using the worse search engine score assigned to either of the two linked peptides, as was initially proposed in the context of non-cleavable crosslinkers (Trnka et al., 2014), despite not taking any additional crosslink specific information into consideration. Here, we present the XLinkProphet software that models several types of crosslink information, including the joint probabilities that both crosslinked peptides are correct search results, to compute accurate and discriminating probabilities that the crosslinked peptide pairs are correct identifications in the dataset.
2 Materials and methods
XLinkProphet can be applied to any cleavable crosslink XL-MS data with database search results validated by PeptideProphet (Keller et al., 2002), and a file indicating which pairs of spectra originated from the same crosslink and the crosslink parent mass and charge. With that information on hand, XLinkProphet models seven pieces of discriminating information that relate to each crosslinked peptide pair (Supplementary Material). Learning model distributions from the data by Expectation-Maximization (Dempster, 1977), it computes probabilities that each crosslinked peptide pair is a correct identification. Proteins corresponding to the two crosslinked peptides are prioritized based on ProteinProphet (Nesvizhskii et al., 2003) analysis of the input search results, giving preference when possible, for two identical proteins corresponding to an intra-protein crosslink. XLinkProphet also combines probabilities of multiple identifications of the same crosslink (unique peptide sequences and crosslink positions) in the data to a probability at the non-redundant crosslink level, corresponding to the likelihood that at least one observed instance is correct.
In order to evaluate XLinkProphet, we created a test dataset from samples of nine commercially available protein mixtures which were independently crosslinked with BDP-NHP (Weisbrod et al., 2013) and initially subjected to shotgun analysis to identify their protein contents. In total 65 proteins were identified with a ProteinProphet probability of 1 in the acquired data (Supplementary Table S1). The nine samples were independently subjected to ReACT and Mango XL-MS analysis. The spectra obtained, MS3 for ReACT and MS2 for Mango analysis, were searched with Comet using a Fasta database with sequences of the 65 standard proteins, 4185 B. subtilis proteins and equal numbers of reverse decoys. Crosslinks between two standard proteins identified by shotgun analysis in that sample were classified as true positives, whereas those between one or more B. subtilis proteins, or with a standard protein not in that sample, were classified as false positives (Supplementary Material).
3 Results
Database search results of ReACT and Mango spectra were validated with PeptideProphet followed by iProphet (Shteynberg et al., 2011). XLinkProphet was then run on the iProphet results, assigning probabilities that each identified crosslink is correct (Supplementary Table S2). Figure 1A illustrates that 589 crosslinks were identified with ReACT at 1% actual FDR using XLinkProphet probabilities to filter the data, 26% more than the 468 obtained using the traditional filter employing the greater (worse) of the two peptides’ Comet expect scores. Similarly, 903 crosslinks were identified with Mango at 1% FDR using the computed probabilities, 75% more than the 516 obtained using the maximum expect score. Supplementary Figure S1 shows a ROC plot demonstrating improved sensitivity over a wide range of FDR values using probabilities to filter the data rather than Comet expect scores. Good agreement was observed between the actual and modeled distributions learned from the data (Supplementary Table S3). It is evident that the models discriminate well between correct and incorrect results, particularly the ‘joint score’ (joint PeptideProphet or iProphet probability that both peptides are correct search results), ‘intra-link’ (whether non-identical peptides corresponding to the same protein were assigned to both ends of the crosslinker) and ‘nsx’ (the number of other confident crosslinks between the same two proteins) distributions.
Fig. 1.
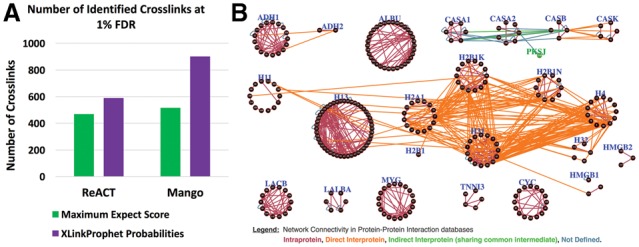
(A) Number of crosslinks identified at 1% FDR in the ReACT and Mango datasets when filtering by maximum Comet expect score versus XLinkProphet probability. (B) Protein-protein interaction network showing 709 non-redundant crosslinks at predicted 1% FDR in the combined ReACT and Mango datasets. Nodes indicate crosslink sites in proteins and are grouped together by Uniprot ID (blue label). Edges correspond to crosslinks and are colored based on known interactions, as indicated in the legend. False positive protein PKSJ_BACSU is indicated as a green node
XLinkProphet probabilities of both the ReACT and Mango results are close to their actual values, as determined by numbers of classified true and false positives, enabling accurate predictions of FDR based on the probabilities (Supplementary Figure S2). The ReACT and Mango results were combined together with iProphet, and their non-redundant crosslinks uploaded at a predicted 1% FDR to XLinkDB (Zheng et al., 2013), a publicly available web resource designed to integrate XL-MS data with databases of protein structure. There the protein–protein interaction network can be viewed and the crosslinks observed in the context of protein structure models. Figure 1B shows that 709 non-redundant crosslinks involving 23 of the 65 standard proteins in the Fasta database were identified, of which 65% were intra-protein. Identified intra-protein crosslinks of 99%, in the contexts of protein structures, had lysine–lysine Euclidean distances within the expected 35 Å (Navare et al., 2015), compared with 80% of random inter-lysine distances in those structures. Solvent accessible surface distance (SASD) has been shown to better predict the consistency of structures with identified crosslinks than Euclidean distance (Allen Bullock et al., 2016), and whereas 79% of intra-protein crosslinked residues had SASD within 35 Å, only 40% of random lysine pairs did (Supplementary Figure S3). Almost all of the identified inter-protein crosslinks are consistent with known yeast, bovine, or human direct interactions. For example, 181 identified inter-protein crosslinks involve histone protein components of the nucleosome core that are known to interact with one another in bovine or human cells (Miller and Costa, 2017), as well as the interaction between high mobility group protein B1 (HMGB1) and histone H3 (Watson et al., 2014). Two crosslinks spanned yeast ADH1 and ADH2, previously known to interact (Gao et al., 2010). Finally, several inter-protein crosslinks were identified among bovine caseins α-S1, α-S2, β, and κ. These proteins, major components of milk, are known to aggregate and form micelles under certain salt conditions (Phadungath, 2005), so likely interacted to an extent in the α-casein and β-casein samples.
XLinkProphet was applied to a large crosslinked human tissue culture dataset consisting of 99 raw files acquired with ReACT and searched with Comet (Chavez et al., 2016). This resulted in the identification at 1% decoy estimated FDR of 27 456 crosslinks (including redundancies), 12% more than the number identified sorting and filtering data based on the maximum expect score. It was also applied to published MS2 data acquired from HeLa lysate samples treated with a different crosslinker, DSSO (Liu et al., 2017) and analyzed with Mango. This led to 3859 identified crosslinks at a decoy estimated FDR of 1%, more than double the 1639 identified based on filtering with the maximum expect score (Supplementary Material).
4 Conclusion
XLinkProphet enables robust validation of crosslinked peptides identified in XL-MS employing cleavable chemical crosslinkers and independent database search to assign sequences to the two released peptides. Its computed probabilities are accurate and highly discriminating, enabling greater numbers of identifications at a predicted FDR when used to filter data relative to using search scores, particularly with MS2-based methods like Mango. Since cleavable crosslinkers and MS2-based methods are rapidly emerging as structural proteomics tools in many labs, XLinkProphet can have widespread utility within this community.
Funding
This work has been supported by NIH grants 5R01GM086688 and 5U19AI107775.
Conflict of Interest: none declared.
Supplementary Material
References
- Chavez J.D. et al. (2016) In vivo conformational dynamics of Hsp90 and its interactors. Cell Chem. Biol., 23, 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez J.D. et al. (2015) Quantitative interactome analysis reveals a chemoresistant edgotype. Nat. Commun., 6, 7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster A.P. et al. (1977) Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B (Methodological), 39, 1–38. [Google Scholar]
- Eng J.K. et al. (2013) Comet: an open-source MS/MS sequence database search tool. Proteomics, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- Gao Q. et al. (2010) Coupling protein complex analysis to peptide based proteomics. J. Chromatogr. A, 1217, 7661–7668. [DOI] [PubMed] [Google Scholar]
- Gotze M. et al. (2015) Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass Spectrom, 26, 83–97. [DOI] [PubMed] [Google Scholar]
- Holding A.N. (2015) XL-MS: protein cross-linking coupled with mass spectrometry. Methods, 89, 54–63. [DOI] [PubMed] [Google Scholar]
- Kandur W.V. et al. (2015) Design of CID-cleavable protein cross-linkers: identical mass modifications for simpler sequence analysis. Org. Biomol. Chem., 13, 9793–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao A. et al. (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell Proteomics, 10, M110 002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A. et al. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem., 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Leitner A. et al. (2016) Crosslinking and mass spectrometry: an integrated technology to understand the structure and function of molecular machines. Trends Biochem. Sci., 41, 20–32. [DOI] [PubMed] [Google Scholar]
- Liu F. et al. (2017) Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun., 8, 15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen Bullock J.M. et al. (2016) The importance of non-accessible crosslinks and solvent accessible surface distance in modeling proteins with restraints from crosslinking mass spectrometry. Mol. Cell Proteomics, 15, 2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T.C., Costa A. (2017) The architecture and function of the chromatin replication machinery. Curr. Opin. Struct. Biol., 47, 9–16. [DOI] [PubMed] [Google Scholar]
- Mohr J.P. et al. (2018) Mango: a general tool for collision induced dissociation-cleavable cross-linked peptide identification. Anal. Chem., 90, 6028–6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navare A.T. et al. (2015) Probing the protein interaction network of Pseudomonas aeruginosa cells by chemical cross-linking mass spectrometry. Structure, 23, 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A.I. et al. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem., 75, 4646–4658. [DOI] [PubMed] [Google Scholar]
- Phadungath C. (2005) Casein Micelle structure: a concise review. SongklanakarJ. Sci. Technol., 27, 201–212. [Google Scholar]
- Shteynberg D. et al. (2011) iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell Proteomics, 10, M111 007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trnka M.J. et al. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell Proteomics, 13, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson M. et al. (2014) Characterization of the interaction between HMGB1 and H3-a possible means of positioning HMGB1 in chromatin. Nucleic Acids Res., 42, 848–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod C.R. et al. (2013) In vivo protein interaction network identified with a novel real-time cross-linked peptide identification strategy. J. Proteome Res., 12, 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. (2009) Identification of protein-protein interactions and topologies in living cells with chemical cross-linking and mass spectrometry. Mol. Cell Proteomics, 8, 409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C. et al. (2013) XLink-DB: database and software tools for storing and visualizing protein interaction topology data. J. Proteome Res., 12, 1989–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.