

Abstract
Motivation
The Li and Stephens model, which approximates the coalescent describing the pattern of variation in a population, underpins a range of key tools and results in genetics. Although highly efficient compared to the coalescent, standard implementations of this model still cannot deal with the very large reference cohorts that are starting to become available, and practical implementations use heuristics to achieve reasonable runtimes.
Results
Here I describe a new, exact algorithm (‘fastLS’) that implements the Li and Stephens model and achieves runtimes independent of the size of the reference cohort. Key to achieving this runtime is the use of the Burrows-Wheeler transform, allowing the algorithm to efficiently identify partial haplotype matches across a cohort. I show that the proposed data structure is very similar to, and generalizes, Durbin’s positional Burrows-Wheeler transform.
1 Introduction
The genetic variation in a population of interbreeding individuals is highly structured. Kingman (1982) introduced the canonical model that describes this structure mathematically, known as Kingman’s coalescent, later extended by Hudson (1983) and Griffiths and Marjoram (1997) to include recombination. Although mathematically elegant, it is challenging to use these models directly for statistical inference. Li and Stephens (2003) introduced a model (LS) that is both a good approximation to the coalescent with recombination, and computationally tractable. As a result, LS now underpins a large range of key tools and scientific findings (Beaumont, 2010; Howie et al., 2009; The International HapMap Consortium, 2005; The Wellcome Trust Case Control Consortium, 2007). Depending on whether the input sequence is haploid or diploid, LS in its straightforward implementation as a hidden Markov model (HMM) runs in linear or quadratic time in the number of reference haplotypes. While this is orders of magnitude more efficient than algorithms based directly on Kingman’s coalescent or the ARG, the recent availability of affordable DNA sequencing technology has provided access to very large reference sets, on which even the LS model is intractable in its standard implementation, so that current implementations of LS use heuristics to cope with datasets encountered in practice (Howie et al., 2009).
A very different algorithm that is making an impact in genomics was introduced by Burrows and Wheeler (1994). Known as the Burrows-Wheeler transform (BWT), it permutes an arbitrary text in such a way that the original text can be recovered, while at the same time improving the compressibility of the transformed text by increasing simple repetitions. In addition, the transformed text, even in compressed form, serves as an index that allows rapid searching in the original text. In genomics this idea has so far been used mainly for fast alignment of short reads against a large and relatively repetitive reference genome (Langmead et al., 2009; Li and Durbin, 2009). More recently, Durbin (2014) introduced a variant of the BWT, termed the Positional Burrows-Wheeler Transform (PBWT), that exploits the additional structure that exists in a set of haplotypes in a population sample. These data, which are usually encoded as a series of 0 and 1 s representing the absence or presence in a sample of particular genetic variants along a reference sequence, have a natural representation as a matrix, where rows represent samples and columns represent the particular positions in a reference. Local matches between samples are only relevant at matching positions, and exploiting this restriction leads to improvements over a standard application of the BWT. The resulting data structure again allows for fast haplotype searches against a database, and achieves very high compression ratios.
2 Approach
There are two main results in this paper. First, I establish a formal connection between the standard and positional BWT, showing how the PBWT as introduced in Durbin (2014) is a special case of the BWT. This connection also shows how the PBWT can be slightly generalized to cope with the multiallelic case. Besides providing an additional perspective on the positional BWT algorithms, which helps to better understand them, it also provides a mechanical way to ‘lift’ existing algorithms operating on the BWT data structure to their positional equivalent, allowing the large literature on BWT algorithms to be applied to the current data structure. I show how this works by deriving the haplotype search algorithm from the equivalent BWT algorithm.
The second contribution consist of algorithms that implement the LS model on top of the BWT. More precisely, I present algorithms that compute maximum-likelihood (‘Viterbi’) paths through the LS hidden Markov model, providing a parsimonious description of a given sequence as an imperfect mosaic of reference haplotypes. The ability to efficiently identify matches in the database of reference haplotypes result in considerable improvements in runtime over the standard implementation, reducing the linear and quadratic asymptotic runtime to empirical constant time, independent of the number of reference haplotypes. More precisely, for H samples of n loci each, the standard implementation runs in time for a haploid input sequence, and for a diploid input sequence, while the proposed algorithms run in empirical O(n) time in both cases. This allows the Li and Stephens model to be used on very large reference panels, without recourse to approximations.
3 Materials and methods
3.1 Haplotype matching using the BWT
Let be H haplotype sequences, each consisting of n symbols from the alphabet A representing the possible allelic states at a locus; for simplicity I will often use in this paper. A straightforward way of identifying haplotype matches would be to use the BWT on the concatenation of haplotype sequences. It turns out that a more efficient algorithm is obtained, in terms of time and memory use, by embedding this sequence of characters into a sequence of characters taken from a much larger alphabet. The increase in sequence length and alphabet size is offset by the additional structure in the BWT that results from the chosen embedding. This in turn translates into better compression and a streamlined search algorithm.
I will write for the jth symbol in the sequence x, and for the subsequence starting at position j and ending at k – 1. I will also use to denote the half-open interval , and if Mij is a matrix, is the subsequence of the kth row of the matrix. Throughout this paper, all indices start at 0.
Let be n additional symbols in the alphabet, ordered such that . Introduce a new sequence X of length by inserting a symbol after each symbol and concatenating the resulting sequences into a single sequence of the form
| (1) |
Algorithm 1.
Calculating BWT(X)
Input: sequences , each of length n; alphabet A
Output: Block permutations .
1: ;
2: While i > 0:
3: ;
4: For j in :
5:
6: ;
Input: sequences , each of length n; alphabet
Output: Block permutations .
1: ;
2: while i > 0:
3:
4: For j in :
5: If :
6: ;
7: Else:
8: ;
(To impose a particular initial ordering I will later on replace the last symbol by H symbols , but to avoid cluttering the notation I ignore this detail for now.) Consider all cyclic shifts of X. Let M be the matrix obtained by writing Xk on the kth row of a square matrix, and sorting the resulting rows lexicographically. Let π be the permutation that sorts the rows, so that , and . The Burrows-Wheeler transform of X is the last column of this matrix: . Note that this is almost the traditional BWT of the sequence X, except that there is no special ‘end’ character. This character is used to identify the start of the sequence; here, the special structure of X is sufficient to navigate BWT(X).
Now consider how the matrix M may be constructed. The position symbols determine the coarse structure of M, which is independent of the data xi apart from the haplotype frequencies and (see Fig. 1). The fine-scale structure of M within each ‘block’ of H rows is determined by the data. More precisely, rows in the block starting at index iH are those cyclic shifts of X that start with symbol and end with for some , such that these rows are ordered lexicographically within the block. Let denote the permutation of that describes this order within block i, so that row iH + j ends with symbol . Determining M therefore boils down to determining the n permutations for , since these determine the top half of M, and those in turn determine the remaining rows (see Fig. 1 and the explanation).
Fig. 1.

Structure of the matrix Mij. The rows Mi are sorted lexicographically; in particular . The Burrows-Wheeler transform of X (see text) is the rightmost column of M, while the positional BWT of the sequences is the upper half of the same column (see text). The column indices are determined by , the allele frequency of symbol a at locus i, and , the cumulative frequency of symbol a across loci . Note that ordering of rows to is determined by the special position symbols , but to avoid cluttering the notation these are all written as
The permutations are determined recursively, working from backwards. Because we imposed the special ordering on the final position symbols, the permutation for block n – 1 is given by the identity permutation . Now suppose the permutation for block i has been determined. The sequences in block i – 1 are formed from those in block i by moving two characters from the end to the front. The first character in any sequence of this new block is , which does not influence the ordering within the block. The second character is an allele marker . To sort the sequences in block i – 1 in lexicographic order, it is therefore sufficient to list those sequences that start a 0 symbol first, followed by those starting with a 1 symbol (followed by other symbols if the locus is multiallelic), and otherwise leave the original order undisturbed. Doing this results in Algorithm 1.
To show that the proposed construction is equivalent to the positional Burrows-Wheeler transform, Algorithm 1 is given both for general alphabets A and specialized for the case , since that in that case the inner loop is precisely Algorithm 1 in Durbin (2014) (except that the proposed algorithm runs back-to-front, as is usual for BWT algorithms). As in the PBWT algorithm, the permutations play the role of the suffix array in the ordinary BWT algorithm. Note that the output includes a permutation , which encodes how the very first characters influence the permutation of the cyclic shifts Xk; this permutation is used in Algorithm 5. Following Durbin (2014) I now define the PBWT of as the first half of BWT(X), which is availably implicitly as . Figure 1 shows that the second half of BWT(X) is determined by the allele frequencies , which can be computed easily from the relevant block in the first half of BWT(X), so that the PBWT of is in fact equivalent to BWT(X).
3.2 Substring searching
Algorithm 1 calculates BWT(X) in linear time by exploiting the special structure of X, and is not a specialization of an existing, general algorithm to calculate the BWT. By contrast, Algorithm 2, which performs a substring search, can be derived directly from its analogous algorithm for a general BWT.
To describe the algorithm, let M be the sorted matrix of cyclic shifts of an arbitrary sequence X of length n, so that , and let (the ‘a-rank’ for row i) be the number of times that a appears in . This function can be calculated efficiently from BWT(X), particular if the data is stored in compressed form. Finally, let C(a) (the cumulative symbol frequency) be the number of symbols in X that are less than a. This notation makes it possible to write down Algorithm 2, for substring searching. (The symbol is used throughout to mark comments and invariants in the algorithms.)
To understand the algorithm, consider all rows of M that end with a symbol a. If these rows are cyclically shifted rightward, so that the last symbol becomes the first and all others are moved one position to the right, all rows will now start with a, and the relative order in which they appear in M (which they must as M contains all cyclic shifts of X) is the same as before the shift since they were ordered lexicographically to start with. Suppose that Mk is a row that ends with a, and that after right-shifting it ends up as row ; then the above observation means that the rank of the symbol a in Mk in the last column of M, is the same as the rank in the first column of M of the symbol a in . Because M is sorted lexicographically, the rows that start with a form a contiguous block in M, so that the first-column rank of the symbol a in row is , so that or
| (2) |
The function , mapping row k to the row corresponding to its right-shifted counterpart , is called the last-to-first mapping because it maps the last (rightmost) symbol of Mk to the corresponding symbol in the first (leftmost) position of . It is repeatedly used to identify the interval of rows corresponding to sequences that match one additional character of w.
Algorithm 2.
General subsequence search
Input: Sequence of sequence
Output: Indices s, e such that for
1:
2: While s < e and i > 0: ▹ matches for
3:
4:
5:
Note that the mapping is well-defined whether or not . This makes it possible to think of k as representing a possible location between two entries (k and k – 1) in M where a sequence (or sequence prefix) x not necessarily represented in M would be inserted; this is the view taken in the search algorithm. Alternatively, when k is thought of as a particular row in M, that row’s initial character a can be obtained from the function, and since the mapping (2) is invertible when restricted to the set of rows k ending in a, this makes the mapping invertible for all k. The existence of this inverse mapping also follows directly from the observation that it corresponds to rotating the sequence one position leftward; it could be called the first-to-last mapping, , and is used in Algorithm 5.
To derive the corresponding algorithm for matching a sequence in the PBWT data structure, it is enough to track the bounding variables for two steps through the standard BWT algorithm acting on the ‘lifted’ sequence X, matching a haplotype character and a position character. The first step identifies the new range depending on the haplotype character to be matched, and points these variables to the second half of the matrix. The next step moves the bounding variable back into the first half by moving a position character in front. Because of the regular form of BWT(X) (see Fig. 1), these two steps can be followed algebraically and combined into a single update step. The derivation, which is straightforward but requires additional notation, is presented in the Appendix. The resulting combined update step is given by a modified last-to-first mapping function, which now additionally depends on the current position i:
| (3) |
or for an arbitrary alphabet, . Here is the positional analogue of , and counts how often a appears in the first k rows of the ith block of , or equivalently, in , and is the (haplotype) frequency of a at position i. This leads to Algorithm 3.
Algorithm 3.
PBWT subsequence search
Input: Sequence , PBWT of
Output: Indices s, e such that for
1:
2: While s < e and i > 0: ▹ matches for
3:
4: ▹ see equation (3)
5:
3.3 Haploid Li and Stephens
The Li and Stephens (2003) model approximates the coalescent model describing the relationship between DNA sequences in a population, by generating a new sequence as a mosaic of imperfect copies of existing sequences The popularity of the model stems from the fact that it is both a good approximation to the full coalescent model with recombination, as well as fast to compute in its natural implementation as a hidden Markov model, running in time for H sequences of length n. However, for very large population samples this is still too slow in practice.
Here I describe an algorithm to compute the maximum likelihood path through the LS hidden Markov model (HMM) in empirical O(n) time. The approach is not to consider single sequences to copy from, but groups of sequences that share a common subsequence. Like the Viterbi algorithm for HMMs, the proposed algorithm traverses the sequence to be explained, but rather than using a dynamic programming approach, it uses a branch-and-bound approach considering (groups of) potential path prefixes to a maximum likelihood path. Where at each iteration the Viterbi algorithm must consider all possible sequences that a potential path prefix could end with, the proposed algorithm in principle considers all extensions of the current potential path prefixes (the ‘branch’ part), but ignores prefixes that cannot be part of an optimal path (the ‘bound’ part). For instance, if a prefix can be extended with a matching nucleotide, a recombination does not have to be considered, since the recombination can be postponed at no cost. Below I will show this more formally. This formal approach is perhaps not necessary (or even helpful) for the haploid case, but becomes useful when I introduce the diploid Li and Stephens algorithm.
Algorithm 4.
Haploid Burrows-Wheeler Li and Stephens
Input: Sequence , PBWT of , scores .
Output: Minimum path score under the Li and Stephens model
1: ;
2: While i > 0: ▹ st represent states of paths in full suffix set for
3: ; ;
4: For (s, e, score) in st:
5: If :
6: ;
7: If :
8:
9:
10: If score = gm:
11: If :
12: ;
13: If :
14:
15: If and extended = False: ▹ Never true on 1st iteration
16:
17:
18: ;
19: any of and
20: Return gm, , traceback
First some definitions. A placed character is a character c at a sequence position i; it is equivalent to a pair where is the position symbol introduced before. Two placed characters are contiguous if they occupy neighbouring positions; subsequences of placed characters are contiguous if every pair of neighbouring characters is; and two or more subsequences are contiguous if their concatenation is. A path π of m parts through a set of sequences is a contiguous sequence of m subsequences such that each si is a subsequence of some xj. I will write a path as
where ci is a character placed at position i, and are the recombination breakpoints identified by the symbol R (which is not part of the alphabet), and l is the length of the path. The (sequence) group associated with π is the set of all sequences for which the subsequences agree with the suffix that follows the last recombination in π. The extension (of length l + 1) is the path , if it exists; since by definition all subsequences that make up a path are subsequences of some xj, existence of an extension implies that its group is nonempty. The extension πR (of length l) is defined as , and always exists; its group is Ω. Finally, the path prefix is the path including any R symbols for recombinations between positions 0 and t – 1; a path prefix never ends with an R symbol.
Algorithm 5.
Haploid traceback
Input: Sequence , PBWT of , scores , minimum score gm, corresponding index , traceback list traceback.
Output: Representation path of a minimum-scoring path
1: Function FL(k, i): ▹ “First-to-last” mapping
2: ; ; if else 1
3: While lo < hi: ▹ and
4:
5: If :
6: Else:
7: Return
8: ;
9: For in reverse(traceback):
10: While :
11:
12: If :
13:
14: If :
15: ; ;
16: Return path
For a given sequence x and a path π, the Li and Stephens model assigns a joint likelihood to the event that π occurred and gave rise to sequence x. If π has m parts and has k mismatches to x, this likelihood is
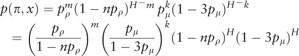 |
where is the probability of recombining into a particular other sequence, and is the probability of a mutation to one of the three other nucleotides. The negative log likelihood takes a particularly simple form,
where C is a constant, and . This motivates defining the path score as , where m and k are defined as above. I drop the subscript x from when this is possible without creating confusion.
Suppose we want to calculate a path π that minimizes . This can be done by iteratively constructing path prefixes , so that at each step one of them is a prefix of a full path π that minimizes . Note that the minimum score achievable by a path π that has as its prefix depends on the prefix score and the prefix group , but not on the rest of the prefix. This is because is the set of sequences the Li and Stephens model could be copying from at the end of , and the Markov property of the model implies that the minimum score only depends on the sequence being copied from (and the prefix score). This justifies the definition of state of a path (prefix) to be the pair .
The key observation for the algorithm is that some states (G, s) can be ignored, because any of their extensions give rise to paths and scores that are also achievable via other states. To make this precise I need one more definition. A set S of path prefixes, all of length l, is a full prefix set for if for any sequence whose prefix agrees with , there exists a path π that achieves the minimum score (i.e. ) and whose prefix is in S. If we can somehow find a way to iteratively construct full prefix sets of increasing length, the problem of finding a minimum-score path is solved, because the required path will be an element of the full prefix set for the full-length sequence x. The following theorem shows how to do this:
Theorem 1. Suppose S is a full prefix set fora set of prefixes of length l + 1, and letand. Thenis a full prefix set forif the following conditions hold:
a For alland allso that πa is an extension andwe have; and
b If there is noso thatandis an extension, thencontains a path of the formwith.
In other words, certain extensions are not required to be in : extensions πa whose score exceed the minimum plus ρ can be left out (since a recombination from the minimum-scoring prefix would give a path that is at least as good), and recombinations can be ignored altogether as long as any current lowest-scoring path has a matching extension (since otherwise postponing the recombination would again be at least as good) – and if not, only a single recombination from a lowest-scoring path needs to be considered.
Algorithm 4 implements these ideas. It does not actually construct prefix sets of paths, but sets of states of paths in prefix sets. This is sufficient since the state determines how paths can be extended. By using the PBWT, these states can be represented efficiently, using just the score and a pair of indices into the PBWT that correspond to a set of subsequence matches to sequences in Ω, similar to how the variables s and e in Algorithm 3 represent the interval corresponding to a set of subsequence matches. Another difference with the description above is that the algorithm scans the sequence back-to-front, extending partial matches leftward, so that the invariant refers to the full suffix set, rather than the full prefix set.
The algorithm computes , and keeps a running minimum score that bounds , ignoring states whose new score are not less than . At the end of an iteration, states whose score are not lower than the now updated plus ρ are not immediately removed, but are instead ignored in the next iteration. The algorithm implicitly considers both score bounds implied by gm and , but in each situation uses only the tighter bound of the two to decide which states to ignore.
It is possible for different paths to result in overlapping or identical states, resulting in duplicate or otherwise redundant entries in the st array. Although redundant entries do not impact the correctness of the algorithm, they can dramatically reduce efficiency. A practical implementation therefore includes a step that occasionally removes redundant states.
The algorithm can be generalized a little by allowing the mutation score to depend on the position. The path score is then defined as . Theorem 1 continues to hold, and so does Algorithm 4, with the obvious changes. The current approach does not lend itself easily to generalize to a position-dependent recombination probability, as the proof of Theorem 1 relies on delaying the recombination without changing the score, which is only possible if ρ is constant along the sequence.
Note that the algorithm can be simplified when , because a mismatch can always be circumvented by two recombinations (before and after the offending locus), so that only exact matches need to be considered. In human genetics polymorphisms are sparse, and recombinations can only be localized to within hundreds or thousands of positions. Even when a maximum likelihood path is sought it is natural to marginalize over these positions, and this makes the probability of a recombination between two polymorphic sites at least an order of magnitude higher than the probability of a mutation, so that . However, in the presence of phasing errors the probability of a mismatch can be much higher than that of a mutation, so that the regime is of practical importance.
Algorithm 4 only computes the optimal score, and to obtain an optimal-scoring path π itself a backtracking step is needed (Algorithm 5). Here it is useful that Algorithm 4 works in the backward direction, so that the result of the backtracking is oriented in the natural direction. To track an optimal path along a sequence, the PBWT index corresponding to that sequence can be tracked using the ‘first-to-last’ mapping, inverting the steps in lines 6 and 12 in Algorithm 4, and the minimum score of the remaining suffix is updated whenever a difference between this sequence and x is found. Recombinations are followed greedily, as it is always correct to follow a feasible recombination, and it is never clear whether a particular recombination is the last feasible one for a particular sequence. Algorithm 4 collects information about recombinations in the traceback list, and when a recombination and score is identified that forms a feasible suffix to the path so far, it is followed.
The naive implementation of Algorithm 5 is somewhat slower than the haploid Li and Stephens algorithm itself, due to the FL function which takes time in the implementation shown. In practice the PBWT will be stored in compressed form using run-length encoding, which allows a faster implementation of FL.
3.4 Diploid Li and Stephens
Where the haploid Li and Stephens algorithm computes a single haplotype path maximizing the probability of a given haploid sequence, the diploid Li and Stephens algorithm aims to find a pair of haplotype paths that maximizes the probability of a sequence of diploid genotypes under the same model. The approach used to derive the haploid algorithm also works in this case, but the details are more involved.
Let x be a sequence of genotypes, encoded as values 0, 1 or 2 at each position representing homozygous ancestral, heterozygous and homozygous derived genotypes. The aim is to compute a pair of paths α, β that minimizes a score. As before this score contains terms for recombinations and mismatches, but the mismatch term now considers genotypes rather than haplotypes. More precisely, the score associated to the pair is defined as , where represents the number of parts of path α, as before, and counts the number of mismatches of the paths α and β to the genotype sequence x.
The approach of the algorithm is similar to the haploid case, again sequentially building full prefix sets for ever longer sequence prefixes until a minimum path pair is found. To describe the approach, the definitions of sequence group, state and full prefix set need to be modified.
The sequence group associated to an unordered pair of paths is defined as . Similarly, using the same justification as before, the state of an (unordered) path pair is defined to be the pair . A full prefix set S for is defined as a set of (unordered) pairs of path prefixes such that for any sequence that extends , there exists a path pair that achieves the minimum score and whose prefix pair is in S. Finally, to formulate the theorem it is handy to introduce the notation to denote the set of ‘haplotype’ paths in S, or formally .
Theorem 2. Suppose S is a full prefix set forandis a set of prefixes of length l + 1. Let, and. Thenis a full prefix set forif:
For alland, so that αa and βb are both extensions and, we have; and
(If:) For allandwith, so that there is nosatisfyingandand both αa andare extensions, contains a path pair of the formwithand; and
(If:) For alland, so that there is nosatisfyingandand both αa andare extensions, contains a path pair of the formwithand; and
(If:) If there is no pairfor whichand eitheroris an extension, thencontains a path pair of the formwith.
Algorithm 6.
Diploid Burrows-Wheeler Li and Stephens
Input: , PBWT of , scores , .
Output: Minimum pair path score under diploid Li and Stephens model
1: Function ():
2: If c = 1: Return
3: Else: Return
4: ;
5:
6: While i > 0: ▹ st repr. states of path pairs in full suffix set for
7: ; ; ;
8:
9: For in :
10:
11: ; (j = 1, 2)
12: If or or and e1 = e2 and or or :
13: continue
14:
15: (j = 1, 2)
16:
17: If and :
18: (j = 1, 2)
19: For in :
20: ; ; ;
21:
22: ;
23: ;
24: If not or or or or :
25: continue
26: If :
27:
28: ()
29:
30:
31: ▹ Not !
32: If and and not and score = gm and
33: If :
34:
35:
36:
37: ; ;
38: any of and
39: Return gm, , traceback
Algorithm 6 implements these ideas. The core of the algorithm is formed by lines 11 and 14 that consider regular extensions with a pair of characters (a1, a2); lines 22–23 and 27 that consider single recombinations; and line 34 that considers simultaneous recombinations in both haplotypes. The remainder of the algorithm is concerned with implementing the conditions of Theorem 2 to ensure that redundant extensions are ignored. The variables gm and keep track of the current and next global minimum score and , while the associative arrays and keep track of and respectively. The associative array keeps track which paths α have a partner that achieves the minimum score , and for which both α and have extensions required in conditions b and c; whether the extension is appropriate is computed by the function . Finally, the variable is used to ensure that at most one double recombination is considered at every iteration.
The traceback algorithm for diploid Li and Stephens is similar to the haploid algorithm. Again, the traceback list contains records describing the recombinations that have been considered. These records now additionally contain a pair sx, ex that represent the range of PBWT indices corresponding to the sequence that does not undergo a recombination. As with the haploid algorithm, the traceback algorithm follows a recombination only if the path scores agree, but now also ensures that the index of the non-recombining path is contained in the range . Double recombinations are encoded by setting , and for such recombinations only the scores need to agree. A pseudocode implementation is given as Algorithm 7.
Algorithm 7.
Diploid traceback
Input: Sequence , PBWT of , scores , minimum score gm, corresponding indices , traceback list traceback.
Output: Representation of a minimum-scoring diploid path
1: Function FL(k, i): ▹ “First-to-last” mapping
2: ; ; if else 1
3: While lo < hi: ▹ and
4:
5: If :
6: Else
7: Return
8: ; ;
9: For in :
10: While :
11:
12:
13: ;
14: If :
15: If : ▹ Double recombination
16: ;
17: ;
18:
19: Else If :▹ Single rec. in path 2
20: ;
21:
22: Else if : ▹ Single rec. in path 1
23: ;
24:
25: Return
4 Performance
For testing the fastLS algorithms were implemented in C++, with all tables stored in uncompressed form in memory. To validate the implementations and to compare runtimes, standard Viterbi algorithms for the haploid and diploid LS model were also implemented. Traceback was included in the fastLS algorithms, but was excluded from the Viterbi implementations because of memory constraints. Two sets of simulations were performed. For the first, 30 Mb of sequence in populations of size 100 to 10 000 were simulated by scrm (Staab et al., 2015) using the ‘standard simulation’ model of Li and Durbin (2011) which roughly resembles the demography of the European population. For each population I simulated an additional 50 samples to serve as input sequences. This resulted in a number of segregating sites ranging from 129 945 for the 150-sample case, to 436 361 for 10 050 samples. For the second set, I simulated a single population of 100 000 samples under the same model (resulting in 621 156 segregating sites) and sub-sampled reference populations of 100 to 10 000 samples from these (Fig. 2).
Fig. 2.
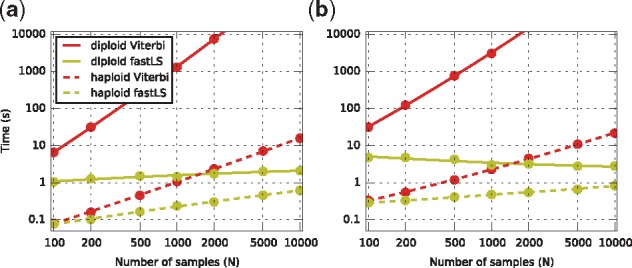
Running time for inferring inheritance patterns under the haploid (dashes) and diploid Li and Stephens model over a simulated reference set of n (horizontal axis) haploid sequences, using the Viterbi (red) and fastLS (green) algorithms, using . Dots represent measurements, curves show quadratic fits. (a) Results for a simulated reference population of n samples. (b) Results for a fixed simulated reference population of 100 000, subsampled to n samples
The run-times of the Viterbi algorithms show the expected linear and quadratic dependence on H. The fastLS algorithms show a weak dependence on H. In the case of the sub-sampled population, which have a fixed number of loci (not all of which segregate in the sample), the dependence on H is weakest, and in fact the diploid algorithm becomes faster for larger populations, probably because longer haplotype matches can be found in larger populations, resulting in more efficient pruning of the prefix sets.
A1. Appendix
A1.1 Derivation of Algorithm 3
To derive the PBWT algorithm for sequence matching we first need to describe the structure of M. From Figure 1 we see that
where is the number of symbols in X, and is the (haplotype) frequency of a at position i in . Let be the cumulative haplotype frequency across positions up to i – 1, and set . Then satisfies
| (4) |
| (5) |
| (6) |
I define so that
| (7) |
or equivalently, counts how often a appears in . To derive the PBWT sequence matching algorithm, it suffices to track one of the bounding variables, say s, for two steps through Algorithm 2. Assume that the subsequence matched so far starts at position i, so that , and that the next character to be matched is . The first step replaces s with
where the second equality follows from (7). The function returns the number of occurrences of a before the kth row within the block starting at row iH in M. This block includes all sequences that start with , so that for , and the conditions for (5) and (6) apply, allowing the result of the second step to be computed. The sequence now ends with the symbol , so that if a = 0, is replaced by
whereas if a = 1,
Since for , it follows that for , so that the last-to-first function mapping k to the new value satisfying is as defined in (3).
A1.2 Proof of Theorem 1
The key observation is that if contains a path with state , then does not need to contain any path π (of the same length) with state (G, s) if and . In this case I say that undercuts π, or symbolically . In addition, if I also say that , again because all scores that are achievable with π as prefix are also achievable with prefix .
Since S is a full prefix set for , a trivial full prefix set for is formed by the union of simple extensions , and recombination extensions . To prove that is also a full prefix set, we need to show that any path is undercut by some path . In the proof below I will identify for any such π a that strictly undercuts π (written as )—that is, either the score is strictly lower or the group is strictly larger—but which is not necessarily an element of . If an element is found that is not in , the process can be repeated, finding a , and so forth. This process has to stop eventually, with an element in , because s cannot decrease indefinitely and G cannot increase indefinitely.
Proof:
First consider an arbitrary element . Because we have . Consider with such that , then and , so that , and therefore .
Next, consider an arbitrary element of , say . We may assume that , as otherwise with strictly undercuts it. We may also assume that , since otherwise let πc be some extension of π (which must exist), then and so that and therefore . Finally, if πa exists, then and so that . This completes the proof.
A1.3 Proof of Theorem 2
The structure of this proof is identical to the previous one. The equivalent observation is that a full prefix set does not need to contain a path pair if already contains a path pair with and ; in this case I say that the path pair undercuts , or symbolically . I also write if any one of , or is true.
A trivial full prefix set for is formed by the union , where , and . The task is to prove that any path pair in or but not in is undercut by some element of , and again I do this by identifying for any a that strictly undercuts π.
Proof:
Consider an arbitrary not in , so that . Suppose first that , and let and be such that , then and , so , and so . Alternatively, suppose that , and let be a path so that and , then and , so that , and so . The case is similar.
Next, consider an arbitrary element . We may assume that as otherwise it is possible to undercut this pair by choosing β appropriately. We may also assume that no exists in S so that and αa and are extensions, for if such a pair exists, the pair undercuts as it achieves the same score and has a strictly larger group. Now suppose . If , for any extension of β we have and so that , as required. To deal with the case , say , suppose b = 1 and let be any extension, then so that , as required. The case is similar.
Finally, consider an arbitrary element . As before we may assume that . Let’s first deal with the case . If a = b then let be an arbitrary extension, then and so . If instead , then let and be arbitrary extensions. If then by a now familiar argument. If then is the required strictly undercutting path pair. If both and then and achieves the same score and a larger group, and therefore strictly undercuts . It remains to deal with the case , say . If either a = 1 or b = 1 (or both), say b = 1, then let be an arbitrary extension, then so that . So we can assume that . The argument in the case is similar. Finally, suppose there is a pair with and either or is an extension, say is, then as required. This completes the proof.
Acknowledgements
Thanks to Sorina Maciuca and Zam Iqbal who introduced me to the idea of position symbols which led directly to this work; and to Gil McVean and Jerome Kelleher for helpful comments on the manuscript.
Funding
This work was supported by Wellcome Trust grant 090532/Z/09/Z.
Conflict of Interest: none declared.
References
- Beaumont M.A. (2010) Approximate Bayesian Computation in evolution and ecology. Ann. Rev. Ecol. Evol. Syst., 41, 379–406. [Google Scholar]
- Burrows M., Wheeler D.J. (1994) A Block Sorting Lossless Data Compression Algorithm. Technical Report 12, Digital Equipment Corporation. [Google Scholar]
- Durbin R. (2014) Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics, 30, 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths R., Marjoram P. (1997) An ancestral recombination graph In: Donnelly P., Tavaré S. (eds) Progress in Population Genetics and Human Evolution, Volume 87 of IMA Volumes in Mathematics and Its Applications. Springer Verlag, Berlin. [Google Scholar]
- Howie B.N. et al. (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet., 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson R.R. (1983) Properties of a neutral allele model with intragenic recombination. Theor. Pop. Biol., 23, 183–201. [DOI] [PubMed] [Google Scholar]
- Kingman J.F.C. (1982) On the genealogy of large populations. J. Appl. Probab., 19, 27–43. [Google Scholar]
- Langmead B. et al. (2009) Ultrafast and memory-efficient alignment of short dna sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2011) Inference of human population history from individual whole-genome sequences. Nature, 475, 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Stephens M. (2003) Modeling linkage disequilibrium and identifying recombination hotspots using single-nucleotide polymorphism data. Genetics, 165, 2213–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staab P.R. et al. (2015) scrm: efficiently simulating long sequences using the approximated coalescent with recombination. Bioinformatics, 31, 1680–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International HapMap Consortium (2005) A haplotype map of the human genome. Nature, 437, 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Wellcome Trust Case Control Consortium (2007) Genome-wide association study of 14 000 cases of seven common diseases and 3000 shared controls. Nature, 447, 661–678. [DOI] [PMC free article] [PubMed] [Google Scholar]