

Abstract
A new class of poly-N-vinylpyrrolidinones containing an asymmetric center at C5 of the pyrrolidinone ring were synthesized from L-amino acids. The polymers, particularly 17, were used to stabilize nanoclusters such as Pd/Au for the catalytic asymmetric oxidations of 1,3- and 1,2-cycloalkanediols and alkenes, and Cu/Au for C-H oxidation of cycloalkanes. It was found that the bulkier the C5 substituent in the pyrrolidinone ring, the greater the optical yields produced. Both oxidative kinetic resolution of (±)-1,3- and 1,2-trans-cycloalkanediols and desymmetrization of meso cis-diols took place with 0.15 mol% of Pd/Au (3:1)-17 under oxygen atmosphere in water to give excellent chemical and optical yields of (S)-hydroxy ketones. Various alkenes were oxidized with 0.5 mol% of Pd/Au (3:1)-17 under 30 psi. of oxygen in water to give the dihydroxylated products in >93% ee. Oxidation of (R)-limonene at 25oC occurred at the C-1,2-cyclic alkene function yielding (1S,2R,4R)-dihydroxylimonene 49 in 92% yield. Importantly, cycloalkanes were oxidized with 1 mol% of Cu/Au (3:1)-17 and 30% H2O2 in acetonitrile to afford chiral ketones in very good to excellent chemical and optical yields. Alkene function was not oxidized under the reaction conditions. Mechanisms were proposed for the oxidation reactions and observed stereo- and regio-chemistry were summarized.
Keywords: catalytic asymmetric C-H oxidation; catalytic asymmetric oxidations of diols and alkenes; chiral substituted poly-N-vinylpyrrolidinones; Cu/Au, nanoclusters; Pd/Au
INTRODUCTION
Bimetallic nanoclusters which comprised of two metals have distinct properties from those comprised of single metals and bulk molecules. For this reason, they have attracted great interest in recent years.1,2 However, asymmetric oxidations of diols, alkenes, and cycloalkanes using bimetallic nanoclusters have not yet been reported previously.
In Au-bimetallic nanoclusters, such as Pd/Au, it has been suggested that Au pulls electron density from Pd leading to a greater interaction of Pd(δ+) with the substrates.3,4 Nanoclusters are generally stabilized by inorganic oxides, glutathione,5 or organic polymers including poly-N-vinylpyrrolidinone (PVP),3 poly(amidoamine) (PAMAM),6 chitosan,7 and others,8 which prevent the nanoparticles from aggregation and oxidation.9 The use of chiral polymers as chiral inducers with bimetallic nanoclusters in the catalytic asymmetric oxidation reactions is unprecedented.
Organic transformation reactions of bimetallic nanoclusters1 in the oxidation of alcohols,3,10,11 formic acid oxidation,12 aldehyde oxidation,13 oxidation of tetralin,14 Ullmann coupling,15 Suzuki coupling reaction,16 tandem oxidation-Michael addition reaction,17 have been reported. However, these reports do not include studies of enantio- or stereo-selectivity. Although desymmetrization of meso diols and diol derivatives using various organic reagents and enzymes have appeared,18–21 reports on the enantioselective oxidation of a mixture of racemic diols via kinetic resolution are limited, with the exception of oxidation of racemic benzylic vicinal diols.22 Hence, asymmetric oxidation of racemic or meso diols using bimetallic nanoclusters and chiral stabilizer may shed light onto the chemistry of the stabilizer and enhance the synthetic applicability of the method.
Catalytic asymmetric syn-dihydroxylation reactions have been intensely pursued, and the Sharpless asymmetric dihydroxylation process is a major breakthrough in asymmetric catalysis.23 However, there are a few shortfalls in the methodology and they include: a lower enantioselectivity in some Z-1,2-disubstituted alkenes, alkenes containing stereogenic centers, and the toxicity and volatility of osmium tetraoxide catalyst.23 Various improvements on the enantioselectivity of syn-dihydroxylation using osmium as catalyst have been made.24,25 Other asymmetric dihydroxylation reactions with osmium-free catalysts26 including palladium-catalyzed difunctionalization of alkenes,27 RuO4,28 KMnO4,29 and iron complexes30 have also been investigated. Asymmetric syn- and anti-dihydroxylation of alkenes using bimetallic nanoparticles and chiral polymers have not been disclosed previously. Avoidance of the use of toxic osmium metal and improving the enantioselectivity in the oxidation of alkenes would advance the asymmetric synthesis of diols from alkenes.
Regio- and enantioselective C-H oxidation of cycloalkanes is one of the most challenging transformations in organic synthesis. The preferred C-H oxidative sites often took place because of the weaker C-H bond dissociation energy, through-bond electronic effect, steric effect, conjugation and hyperconjugation, and releasing strain.31 The uses of homogeneous catalysts such as iron complexes,32–34 gold reagents,35 and others31,36–38 in C-H oxidation have appeared. Due to various possible reactive sites in even medium-size organic molecules, the regio- and enantio-selective C-H oxidations inevitably require directing or activating groups such as heteroatoms and allylic moiety.39–41 Although simple C-H oxidation of cyclohexane with Au nanoclusters on hydroxyapatite support42 or with Pt/TiO243 has been studied, no reports on the use of bimetallic nanoclusters and chiral supports in the catalytic asymmetric C-H oxidation have surfaced. C-H oxidation is a highly atom-economic process, and regioselective catalytic asymmetric C-H oxidation would provide a powerful tool for organic synthesis.
We report herein the synthesis of a class of chiral substituted poly-N-vinylpyrrolidinones (CSPVP) and their chiral induction by chelating Pd/Au or Cu/Au nanoclusters in enantioselective oxidation reactions of diols, alkenes, and cycloalkanes.
RESULTS AND DISCUSSION.
In the reported study of Pd/PVP nanoparticles using extended X-ray absorption fine structure spectroscopy, it has been suggested that the amide function of PVP chelated with Pd and stabilized the nanoparticles.14,44 We hypothesize that in the complex of substituted PVP and nanoparticles, a substituent on the pyrrolidinone ring would affect the stereochemical outcome of the reactions, resulting in asymmetric oxidations.
Synthesis of Chiral Substituted Poly-N-vinylpyrrolidinones (CSPVPs or substituted PVPs containing an asymmetric center).
To investigate the effects of the polymers on the bimetallic nanoparticles in oxidation reactions, we synthesized four CSPVPs 15 - 18 from optically pure amino acids (Scheme 1) and utilized them as stabilizers in the asymmetric oxidation reactions. Our synthesis of CSPVPs started from the polymerization of chiral 5-substituted N-vinylpyrrolidinones 9 – 11, which were prepared from the vinylation of the corresponding 5-substituted-2-pyrrolidinones, 6 – 8, respectively. Chiral 6 - 8 were made from L-amino acids by following a reported procedure.45–47 Hence, N-Boc L-(S)-amino acids, 1 and 2, were separately coupled with Meldrum’s acid followed by reduction of the ketone function with sodium borohydride, cyclization under heat,45 and removal of the Boc group with trifluoroacetic acid (TFA) in CH2Cl2 to give 5 and 7, respectively. The N-Boc protecting group of 3 can be removed selectively in the presence of O-tert-butyl group by TFA/CH2Cl2 at 25oC providing 6. From our studies (vide infra), it appears that a bulky substituent at C5 of the polymer would afford a higher enantioselectivity. Consequently, (R)-5-benzhydryloxymethyl-2-pyrrolidinone (8) was synthesized from the alkylation of compound 5 with sodium hydride and benzhydryl bromide. N-Vinylation of 6 – 8 with Na2PdCl4 and vinyl acetate48 gave the corresponding N-vinylpyrrolidinones 9 - 11, respectively.
To prepare uniform micron-size monodisperse particles,49 we utilized a dispersion polymerization method50 to prepare polymers 15 – 17 (Scheme 1). The dispersants, poly(5-substituted N-vinylpyrrolidinone-co-vinyl acetate) 12 – 14 were prepared from the copolymerization reactions of respective monomers 9 – 11 and 1 equiv. of vinyl acetate in the presence of a catalytic amount of azobisisobutyronitrile (AIBN) in refluxing acetone. Polymerization of 9 – 11 separately with a catalytic amount of AIBN and in the presence of 1% of respective copolymer 12 – 14 in DMF at 120oC for 7 days gave polymers 15 – 17. Polymer 18 was prepared through the removal of the tert-butyl group of polymer 15 with TFA. Polymers 15 - 18 are soluble in water and their molecular weights were determined by gel permeation chromatography [see Figure S1 in Supporting Information (SI)]. Average molecular weights of 15 - 18 are listed (Scheme 1). Both R- and S-stereochemistry are likely presented in the stereogenic center (marked with *) in the polymer alkane backbone, and they are not identifiable. Presumably, this stereogenic center of the polymers can be isotactic, atactic, and syndiotactic,51 and likely has lesser effect on the asymmetric oxidation reactions. The average sizes, size distribution, and shapes of the polymers were also studied by atomic force microscopy (AFM) and dynamic light scattering (DLS). AFM images of the polymers such as 17 in aqueous solution showed spheroid and ellipsoid shapes with diameter ~20 – 30 nm (Figures S2A-S2B). DLS graphs of 17 showed a major ensemble of 12 – 20 nm sizes particles with an average size of 18.6 nm along with a few 152 nm-sized particles, suggesting that different shapes and few large aggregates were presented in the aqueous solution (Figures S3A - S3B). On the contrary, nanoclusters Pd/Au-17 showed more discrete sizes in solution by DLS (vide infra).
Synthesis of Bimetallic Nanoclusters-CSPVP.
Nanoclusters can be prepared by a number of methods including molecular beams, chemical reduction, thermal decomposition, ion implantation, electrochemical synthesis, radiolysis, sonochemical synthesis, and biosynthesis, and are characterized by various analytical techniques.52 We have synthesized various bimetallic nanoclusters including Pd/Au and Cu/Au, using the chemical reduction method3 in the presence of chiral polymer 15, 16, 17, or 18. We used gold due to its high catalytic activity and synergistic electronic effects.3–9 For example, a solution of Na2PdCl4 (3 equiv.), HAuCl4 (1 equiv.), and CSPVP 17 (0.11 equiv.; based on the amount of Au) in water was treated with NaBH4 at 25oC for 30 min (Scheme 1) to give a light brown to dark grey solution depending on the concentrations (Figure S4). The resulting solution was used in the subsequent catalytic reactions without further manipulation. Similarly, Cu/Au (3:1)-17 was also prepared using CuCl (3 equiv.), HAuCl4 (1 equiv.), and CSPVP 17 (0.11 equiv.). The aforementioned nanocluster solution was filtered through a Vivaspin 20 centrifugal filter device (3,000 MWCO), washed with deionized water twice, and lyophilized to give a powder, which was used for various characterization including inductively coupled plasma-mass spectrometry (ICP-MS; see SI), transmission electron microscopy (TEM; Figure S5), X-ray photoelectron spectroscopy (XPS; Figure S6), AFM (Figures S2C - S2D), DLS (Figures S3C - S3D), and IR. Results of the ICP-MS showed a ratio of 3.12 ± 0.58 of Pd and Au in the Pd/Au (3:1)-17 complex, and a ratio of 3.0 ± 0.4 of Cu and Au in the Cu/Au (3:1)-17 sample. This is in agreement of the 3:1 ratio of the two metals used in preparation of the nanoclusters. TEM images revealed average diameters of 3.44 ± 1.63 nm and 3.66 ± 1.95 nm for Pd/Au (3:1)-17 and Cu/Au (3:1)-17, respectively (Figure S5). The nano-sizes (2 – 4 nm) of the nanoclusters are similar to those reported for Pd/Au using PVP as a stabilizer.3,14 XPS spectra of the Pd/Au (3:1)-17 showed the binding energies of 84.1 eV and 335.1 eV derived from 4f7/2 Au and 3d5/2 Pd, respectively (Figure S6).14 The spectra of Cu/Au (3:1)-17 displayed binding energies of 84.1 eV and 932.7 eV for respective 4f7/2 Au and 2p3/2 Cu (Figure S6). The binding energy values are consistent with those reported in the XPS database. From the AFM images of Pd/Au (3:1)-17, 50 – 200 nm in diameters and ~5.8 nm in heights of spherical particles were found, and in DLS, 100 – 140 nm sized nanoparticles were revealed (Figure S2C, 2D), suggesting the size of polymer 17 wrapped particles was ~5 times larger than polymer 17 alone, and the measured sizes of the polymer-particles from AFM and DLS were similar. As reported previously, AFM does not resolve subtle shape differences from the diameters, but can provide relatively accurate height of the nanoparticles.53 The ~5.8-nm height revealed by AFM is similar to that found by TEM. A small amount of ~15.7 nm-sized particles found in DLS of Pd/Au-17 sample may derive from free polymer 17 without complexing Pd/Au in the aqueous solution. The approximate total numbers of metal atoms and molecules of polymer in a spherical nanocluster can be calculated utilizing the magic-cluster sizes Ntotal = 1/3 (10n3 - 15n2 + 11n – 3), where n is the number of layers of shell in the metallic nanoparticles with face-centered cubic close-packed structure.54 Since the average sizes of Pd/Au (3:1)-17 and Cu/Au (3:1)-17 nanoclusters are 3.44 and 3.66 nm, respectively (from TEM), there are respectively ~727 and 1,151 atoms of Pd/Au (3:1) and Cu/Au (3:1) in a nanocluster calculating from the aforementioned equation and particle sizes (see SI for calculation). Each Pd/Au and Cu/Au nanoclusters are stabilized by approximately 20 and 32 molecules of polymers, respectively (see SI).4 Because of the numbers of molecules and extended structure of the chiral polymer, the sizes of the bimetallic nanocluster/polymer found by AFM and DLS are much larger than those in the nanoclusters found by TEM (the polymer cannot be observed by TEM). The amide C=O absorption band of the pyrrolidinone ring at 1650 cm−1 of polymer 17 in IR spectrum shifted to 1641 cm−1 for Pd/Au (3:1)-17, implying a greater character of δ−O-C=Nδ+ of the amide group in the nanoclusters due to chelation with the metals.
Catalytic asymmetric oxidation of cycloalkanediols.
After screening different ratios of Pd versus Au such as 1:1, 2:1, 3:1 of the bimetallic nanoclusters, Pd/Au (3:1)-CSPVP appears to provide the highest catalytic activity and chemical yields3 in the aerobic catalytic asymmetric oxidation of cycloalkanediols. Initially, (±)-trans-1,3-cyclohexanediol (20) was used to screen the enantioselectivity of various CSPVPs such as 15 – 18, and results are summarized in Table 1. For instance, treatment of (±)-20 with 0.15 mol% of Pd/Au (3:1)-CSPVP 15, 16, 17, or 18 (the amount of catalyst is based on the total moles of Pd and Au) and 0.3 equiv of K2CO3 in water under 1 atmospheric oxygen at 60oC for 7 days gave 46 – 49% chemical yields [or 92 – 97.5% yields based on reacted (S,S)-20] and 70 – 99% ee optical yields of (S)-3-hydroxycyclohexanone (23) along with the recovered (R,R)-20 (Table 1, entries 1 – 4). Results suggest that a bulkier C5-substituent on the pyrrolidinone ring of the polymer provided a greater optical yield. Since polymer 17 provided the highest chemical and optical yields, it was used for all other oxidation reactions including oxidations of alkenes and cycloalkanes. Diol (R,R)-20 was recovered in 50% yield and 92% ee from the reaction with Pd/Au (3:1)-17, suggesting that it reacted much slower than (S,S)-20 under the reaction conditions, i.e., a kinetic resolution is resulted. Hydroxyl ketone (S)-23 did not undergo oxidation, revealing an electron-withdrawing ketone group impeded further oxidation. The rate of the oxidation reaction was enhanced in the presence of 0.3 equiv of K2CO3. Using 0.15 mol% of Pd/Au (3:1)-17, other five- and seven-membered cycloalkanes, such as 1,3-trans diols (±)-19 and (±)-21 and 1,2-trans diols (±)-28 - (±)-30, were similarly oxidized to give excellent chemical and optical yields. Results are presented in Table 1 (entries 5 and 6, and entries 10 – 12). When the amounts of catalysts were increased to 0.30 mol% (double) or 0.60 mol% (quadruple) of Pd/Au (3:1)-17, the oxidations of (±)-20 were respectively completed in 6 and 5 days, with chemical yields of 47% (99% ee) and 49% (99% ee) of (S)-23. Hence, an increase of the amounts of catalyst shortens the reaction time. Oxidation of (±)-20 with 0.15 mol% of Pd-17 without Au under similar reaction conditions (at 60oC for 7 days) gave only 29% yield of (S)-23 in 95% ee, indicating bimetallic Pd/Au provided higher chemical and optical yields in the catalytic oxidation reactions. Catalyst Au-17 alone gave no oxidized product. When the temperature of the reaction was increased from 60oC to 80oC, the rate of the reaction did not change (7 days and 47% yield was found), whereas the optical yield of (S)-23 decreased to 94% ee. Further increasing the temperature to 100oC resulted in completion of the reaction in 6 days with a 47% yield of (S)-23 in 89% ee, indicating that higher reaction temperature lowers the optical yield. The recyclability of the Pd/Au (3:1)-17 catalyst was also examined. Hence, the catalyst, recovered from the oxidation reaction of (±)-20, was reused under similar reaction conditions for a second time, and 39% yield (99% ee) of (S)-23 was isolated. The catalyst was further recycled for a third time, but only 18% yield (98% ee) of (S)-23 was obtained, showing the catalyst can be reused albeit with lower catalytic activities. The turnover number (TON) of the catalytic oxidation reaction after three cycles is 706 (SI). The absolute configurations of (S)-22, (S)-23 and (S)-31 – (S)-33 were determined by comparison of the sign of the reported specific rotations (see SI).55–58 Optical purities were measured through HPLC/chiral column of their benzoyl derivatives, which were synthesized by the treatment of the hydroxyl ketones with benzoyl chloride and pyridine (SI). In all cases, PVP was used in place of CSPVP under similar reaction conditions to provide the racemic products for HPLC/chiral column analyses. The selectivity factor values of the HPLC/chiral column separation are 1.10 – 1.28 (see Table S1 in SI), indicating an acceptable separation. The specific rotation of (S)-24 has not been reported previously and its absolute configuration was assumed based on the retention time of its benzoate derivative in HPLC/chiral column. The (R)-enantiomer has a lower retention time than that of (S)-enantiomer, which is in agreement with benzoate derivatives of (S)-22 and (S)-23 (see SI). The absolute configurations and optical purities of the unreacted diols (R,R)-19 – (R,R)-21 and (R,R)-28 – (R,R)-30 were similarly determined by comparison of the sign of the reported specific rotations59,60 (SI) and through HPLC/chiral column of their dibenzoate derivatives obtained from the reactions of the diols with benzoyl chloride and pyridine (SI).
Table 1.
Catalytic asymmetric oxidation of 1,3-diols and 1,2-diols.
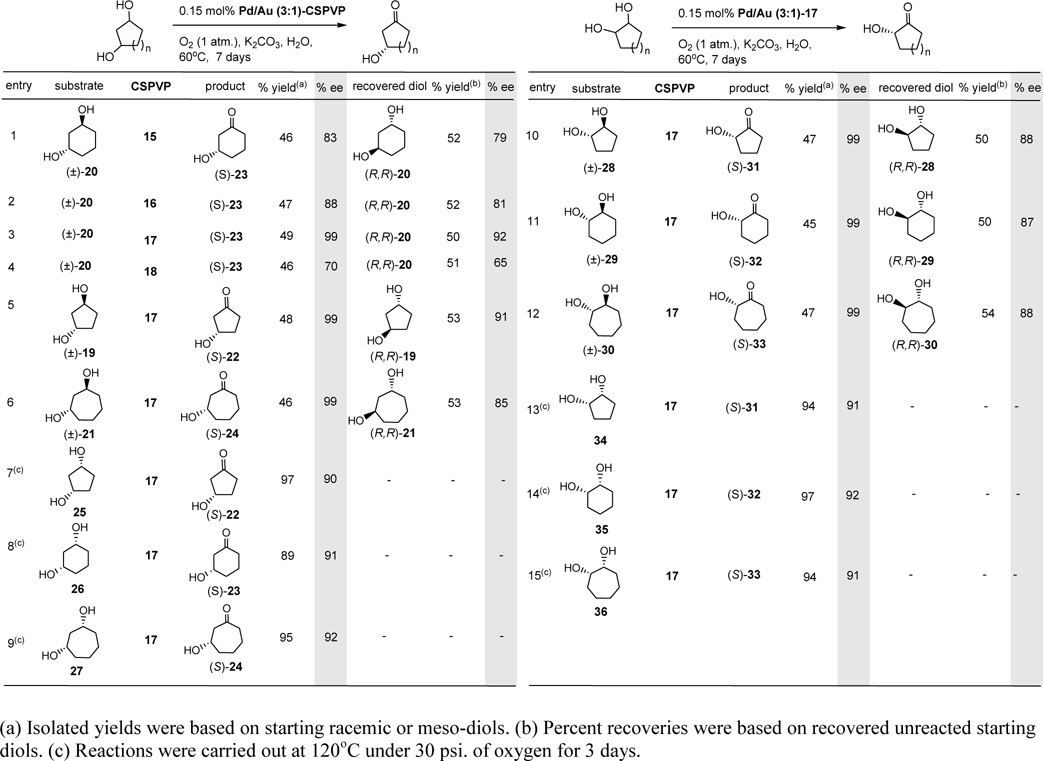 |
To our surprise, the meso 1,3-cis and 1,2-cis-cycloalkanediols did not undergo oxidation reactions under similar reaction conditions as those of trans-diols. It may due to the fact that cis-diols form stable complexes with the nanoparticles.61 However, under elevated pressure of oxygen (30 psi.) and temperature (120oC), the reactions took place in 3 days to give 89 – 97% yields and 90 – 92% ee of the oxidized (S)-hydroxyl ketones (Table 1, entries 7 – 9 and entries 13 – 15). The higher reaction temperature (120oC) lowers the optical yields in the catalytic oxidation reactions. No changes in reaction rate and chemical and optical yields in the oxidation reaction of meso-35 were found when doubling the amount of 17 but maintaining the same amount of Pd/Au (3:1; 0.15 mol%). This oxidative desymmetrization of meso-diols is synthetically useful, since it provides the chiral hydroxyketones without the recovery of starting diols. Importantly, both trans- and cis-diols gave only the (S)-hydroxy ketones.
Scheme 2 outlined a proposed mechanism and oxidative sites for the enantioselective oxidation reactions of diols. A η2-peroxido PdII (peroxopalladium) species II62 may form similar to that reported.63 A diol such as (S,S)-20 attacks Pd of II leading to complex III, which undergoes proton transfer to give IV. Subsequent removal of C1-H by K2CO3 leads to (S)-23 and I and hydrogen peroxide. Presumably, the C5(R)-bulky substituent of pyrrolidinone ring directs the stereo- and regio-chemistry of the oxidation reactions. The oxidative sites in trans- and cis-diols are depicted in VI and VII, respectively.
The pro-S-configurated carbon center of trans-diols (1,3- and 1,2-diols) of five-, six-, and seven-membered ring oxidizes much slower than the pro-R-configurated carbon center. Notably, the transition states are difficult to discern given the complexity of the polymer structures. Oxidation of I with H2O2 gives II.64 Nanocluster Pd/Au-17 provides excellent enantioselectivities (91 – 99% ee) in the oxidation of diols, and results of the enantioselectivity from the oxidation of trans-diols are comparable to those reported using enzymes.65,66
Catalytic asymmetric oxidation of alkenes.
The success in catalytic asymmetric oxidation of diols led us to examine the oxidation of alkenes. Although no oxidation of alkenes took place at 1 atmospheric of oxygen, various alkenes were readily oxidized at 30 psi. of oxygen in excellent to good chemical yields and excellent optical yields. Results of the oxidation are summarized in Table 2 (entries 1 – 7). For example, treatment of 1,2-dihydronaphthalene (37) with 0.5 mol% of Pd/Au (3:1)-17 in water at 25oC under 30 psi. of oxygen for 3 days gave (1S,2R)-38 in 86% yield and 99% ee (Table 2, entry 1). Only a single product was obtained in the reaction. The Sharpless dihydroxylation of 37 using DP-PHAL gave 56% ee of (1R,2S) enantiomer of 38.67 Other disubstituted alkenes including trans- and cis-β-methylstyrene (39 and 39A, respectively; Table 2, entries 2 and 3) also underwent oxidation reactions at 25oC. In the oxidation of trans-39, the syn-adduct (1S,2S)-40 was isolated in 87% yield and 99% ee along with a small amount of anti-adduct (1R,2S)- 41 in 6% yield and 97% ee. Similarly, in the oxidation of cis-39A, the syn-adduct (1S,2R)-41A was isolated in 90% yield and 98% ee as well as 8% yield of anti-adduct (1R,2R)-40A in 98% ee. Notably, the dihydroxylation takes place from the re face of the alkenes (in regarding to C1 of 37 or 39). Trisubstituted alkenes such as (R)-(+)-limonene (48), containing a stereogenic center, also underwent stereoselective oxidation at 25oC to give a single stereoisomer (1S,2R,4R)-49. No other stereoisomers were detected. The oxidation of alkenes containing an electron-withdrawing group such as trans-cinnamic acid ester 42 and indene (45) only proceeded at 50oC giving the syn-adducts (2R,3S)-43 (82% yield; 99% ee) and (1S,2R)-46 (67% yield; 93% ee), respectively, as the major products. The respective anti-adducts, (2R,3R)-44 (3% yield; 97% ee) and (1R,2R)-47 (11% yield; 94% ee) were also isolated as minor products. Since (1R,2R)-47 was obtained in 11% yield, we questioned whether this anti-adduct could be produced as the major product. As expected, in the presence of 0.3 equiv of K2CO3 at 70oC for 5 h, (1R,2R)-47 was isolated in 59% yield and 91% ee. However, only syn-addut (1S,2R)-38 was isolated when 0.3 equiv of K2CO3 was added to the oxidation reaction of 37 at 25oC. The absolute configurations were determined by comparing the sign and specific rotations of the respective reported molecules (see SI).65,66,68,69 Optical purities of all dihydroxylated molecules were measured using HPLC/chiral column (SI). To determine the stereochemistry of 49 (which has not been reported previously), we independently prepared (1S,2R,4R)-49 and its (1R,2S,4R)-diastereomer by the reaction of (R)-48 with OsO4 (catalytic amount)-NMO followed by silica gel column chromatography (SI). The cyclic trisubstituted alkene reacted preferentially compared to the acyclic terminal disubstituted olefin group, suggesting electron-donating groups such as alkyl and aryl enhance the reactivity towards the nanoclusters. The formation of (1S,2R,4R)-49 from the dihydroxylation reaction of limonene is remarkable, since the dihydroxyl functions were delivered from the same side of the bulky isopropenyl group. The highest reported % ee’s of the dihydroxylation products of 39, 39A, 42 (the ethyl ester analog), and 45 from Sharpless asymmetric dihydroxylation reactions are 98,70 81,71 99,69 and 89,72 respectively. Hence, the Pd/Au-17 provided higher or similar enantiomeric selectivities in the catalytic asymmetric dihydroxylation reactions.
Table 2.
Catalytic asymmetric oxidation of alkenes and cycloalkanes.
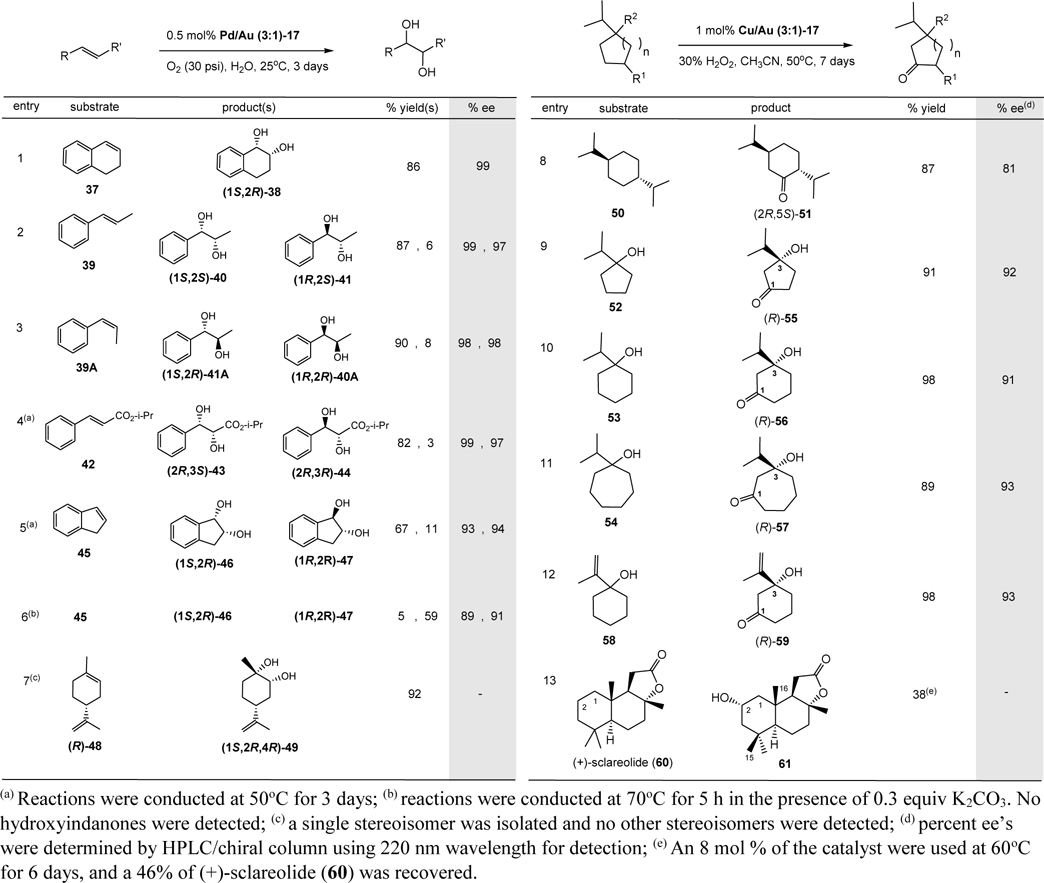 |
A proposed mechanism and stereo- and regio-chemistry for the enantioselective oxidation of alkenes62,73 are depicted in Scheme 3. Similar to that described in Scheme 2, η2-peroxido PdII (peroxopalladium) species II undergoes syn-addition reaction with alkenes via transition state VIII under high oxygen pressure, where Pd adds to C1 containing larger aryl or alkyl group (RL) and oxygen adds to C2 containing smaller alkyl group (RS), resulting in metallo-1,2-dioxolane IX.63 C1 containing either an aromatic ring (entries 1 – 6) or more substituted carbon (entry 7) is better able to accommodate the partial positive charge in the transition state (vide infra). Carbon-1 of IX undergoes a 1,2-alkyl shift from Pd to oxygen (path a) leading to intermediate X, which hydrolyzes to give the syn-adducts. In 37 and (R)-48 (Table 2, entries 1 and 7), the 1,2-alkyl shift takes place rapidly and only the syn-adducts were found. In other investigated alkenes (39 – 45), in addition to major path a, water attacks C1 of intermediate IX from the backside, path b, providing anti-addition diols as the minor products (Table 2, entries 2 – 5). In supporting this hypothesis, an addition of 0.3 equiv. of K2CO3, a stronger nucleophile than water, resulted in the formation of anti-adduct (1R,2R)-47 being the major product (Table 2, entry 6). Structures of the respective metallo-1,2-dioxolanes derived from the oxidation of benzocycloalkenes, trans- and cis-alkenes are depicted in XIIa, XIIb, XIIIa, and XIIIb, which resulted from the addition reactions of η2-peroxido PdII from the Re-face of the alkenes. For trisubstituted alkene such as (R)-limonene, the addition took place from the Si-face (structure XIIc).
The asymmetric aerobic oxidation of alkenes by Pd/Au-17 at ambient temperature or 50oC provided excellent chemical and optical yields of the syn-dihydroxylated products and in certain cases where higher temperature (such as 70oC) is required, an addition of a weak base such as K2CO3 afforded anti-dihydroxylated molecules as the major products.
Catalytic asymmetric oxidation of alkanes.
To explore the scope of the oxidation reactions, a more challenging catalytic asymmetric C-H oxidation was investigated. We found that Cu/Au (3:1)-17 provided greater chemical and optical yields in C-H oxidation than Pd/Au (3:1)-17. Results of the catalytic asymmetric C-H oxidation reactions of various substituted achiral cycloalkanes using 1 mol% Cu/Au (3:1)-17 and H2O2 at 50oC are summarized in Table 2. Without hydrogen peroxide, under oxygen atmosphere (30 psi.), no oxidation products were detected. For example, oxidation of meso-trans-1,4-diisopropylcyclohexane (50) gave an 87% yield of (2R,5S)-51 in 81% ee (Table 2, entry 8). The specific rotation and 1H and 13C NMR spectral data of (2R,5S)-51 were in agreement with those of the reported compound synthesized from (S)-perillaldehyde.74 The four methine C-H’s of 50, possessing weaker bond dissociation energies,31 and four sterically less hindered methyls were not oxidized under the reaction conditions, suggesting rigid or less freely rotated methylene groups are oxidized faster than methine and methyl groups. Higher ee’s were obtained when a directing group such as hydroxyl was presented. Hence, oxidation of alcohols 52 - 54, and 1-isopropenylcyclohexanol (58) gave excellent chemical and optical yields of (R)-55 - (R)-57, and (R)-59, respectively (Table 2, entries 9 – 12). Likely, under the reaction conditions, the alcohols were initially formed and they subsequently oxidized to the ketones. Importantly, in the absence of elevated pressure of oxygen, the alkene function of 58 was not oxidized at 50oC, despite the presence of an activating allylic hydroxyl group. This is significant since alkene functional group is more reactive than the alkane moiety. The regiochemistry of (R)-55 - (R)-57, and (R)-59 were analyzed by 2D COSY spectroscopy. In the COSY spectrum of (R)-55, the signal at δ 2.41 – 2.30 ppm assigned for C2-hydrogens has no correlation with other protons, while the signals at ~2.25 and 2.05 ppm assigned for C5 hydrogens show correlation with C4 hydrogens at 2.01 and 1.60 ppm. Similar correlations were found in the 2D COSY spectra of (R)-56, (R)-57, and (R)-59 (SI). Moreover, (R)-56 and (R)-57 were independently synthesized from (S)-23 and (S)-24, respectively, by silylation of the C3-hydroxyl group followed by addition reaction with isopropylmagnesium bromide, removal of the silicon protecting group, and oxidation with IBX-DMSO (see SI). Their NMR spectra and specific rotations were similar to those obtained from the C-H oxidations. A medium-sized natural product, (+)-sclareolide (60), also underwent regioselective oxidation to give 2S-2-hydroxysclareolide (61) in 38% yield and 3% yield of 1-oxosclareolide along with 46% recovery of 60 after silica gel column chromatographic separation. No other regioisomers were found. 1H and 13C NMR spectral data of 61 and 1-oxosclareolide are identical to those reported.75,34 Notably, unlike the reported iron complex-catalyzed oxidation,34 the C2-equatorial hydroxyl function of 61 does not undergo further oxidation, likely due to steric hindrance (C2-axial-H is shielded by C15-β- and C16-methyls75).
Based on the previously suggested mechanism of copper complexes bearing trispyrazolylborate ligands,76 a mechanism for the C-H oxidation reactions involving Cu/Au-17 was proposed and the stereo- and regio-chemistry of the major C-H oxidative sites were depicted in Scheme 4. Cuo/Au XIV reacts with H2O2 to give copper(II) oxo complex XV, which abstracts a hydrogen atom from cycloalkanes such as 50 to generate hydroxyl copper(I) and cycloalkyl radical (likely not a free radical76) in a cage-like complex XVI. A rapid combination of the cycloalkyl radical with hydroxyl radical in XVII takes place and subsequently releases the alcohol and regenerates XIV. The alcohols oxidized under the reaction conditions to give ketones.76 Oxidative sites for the C-H oxidation reactions are summarized in XIX - XXI. The two isopropyl substituents orient at the equatorial positions and copper(II) oxo XV likely approaches the C3 equatorial hydrogen, resulting in the observed 2R,5S configuration in 51. In the hydroxyl directed oxidation, C1 hydroxyl group coordinates with copper(II) oxo XV and abstraction of the C3 equatorial or axial hydrogen will lead to the observed C3-R configuration.
CONCLUSION.
Various substituted poly-N-vinylpyrrolidinones containing a stereogenic center were synthesized from L-amino acids and used in the catalytic asymmetric oxidations of cyclic diols, dihydroxylation of alkenes, and C-H oxidation of cycloalkanes. Polymer 17 afforded the highest enantiomeric selectivities. Excellent chemical yields and enantioselectivity were found. Both (±)-trans and meso-cis diols were oxidized under oxygen atmosphere with Pd/Au-CSPVP to give (S)-hydroxy ketones. Trans- and cis-alkenes oxidized under 30 psi. of oxygen to afford syn-diols as the major products. The resulting diols do not undergo further oxidation. Various substituted cycloalkanes including 1,4-disubstituted cyclohexane and 1-substituted 1-cycloalkanols were oxidized by Cu/Au-17 and H2O2 to furnish chiral cycloalkanones. Remarkably, disubstituted terminal alkene function was not oxidized under the C-H oxidation reaction conditions, which demonstrates a high potential for the synthetic methodology.
Supplementary Material
Scheme 1.

Syntheses of poly-N-vinylpyrrolidinones containing an asymmetric center at C5 and preparations of Pd/Au (3:1)-17 and Cu/Au (3:1)-17.
Scheme 2.

Proposed mechanism and oxidative sites for the catalytic asymmetric oxidation of racemic and meso 1,3- and 1,2-diols.
Scheme 3.

Proposed mechanism and stereo- and regio-chemistry of the enantioselective oxidation of alkenes.
Scheme 4.

Proposed mechanism and oxidative sites of the enantioselective catalytic oxidation of alkanes.
ACKNOWLEDGMENT
We thank Johnson Cancer Research Center, Kansas State University for partial support of this project, NIH National Institute of General Medical Sciences (P20 GM103418) for supporting A.M., NSF REU program (CHE1460898) for supporting V.N., and SUROP program, Kansas State University for supporting S.N.S. of a summer research program. D.H.H. thanks the Japan Society for the Promotion of Science (ID No. L14527) for a Visiting Professorship at the Institute for Molecular Science and Osaka University, Japan. We thank Professor Hidehiro Sakurai and Ms. Setsiri Haesuwannakij for helpful discussion and analysis of initial nanoclusters, and Dr. Hongwang Wang and Prof. Christopher Sorensen for assisting B.H. in obtaining and discussion of DLS data, respectively.
ABBREVIATIONS
- AFM
atomic force microscopy
- AIBN
azobisisobutyronitrile
- Au
gold
- COSY
correlation spectroscopy
- CSPVP
chiral substituted poly-N-vinylpyrrolidinone
- Cu
copper
- DLS
dynamic light scattering
- IBX
2-iodoxybenzoic acid
- ICP-MS
inductively coupled plasma-mass spectrometry
- NMO
N-methylmorpholine N-oxide
- PAMAM
poly(amidoamine)
- Pd
palladium
- PVP
poly-N-vinylpyrrolidinone
- TEM
transmission electron microscopy
- TFA
trifluoroacetic acid
- XPS
X-ray photoelectron spectroscopy
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. Molecular weight determination, ICP-MS, TEM, AFM and DLS images, XPS spectra, experimental procedures, HPLC/chiral column graphs, 1H and 13C NMR spectra of the described molecules, and 2D COSY spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Zhang H; Watanabe T; Okumura M; Haruta M; Toshima N Nature Mater 2012, 11, 49. [DOI] [PubMed] [Google Scholar]
- 2.Miyamura H; Kobayashi S Acc. Chem. Res 2014, 47, 1054. [DOI] [PubMed] [Google Scholar]
- 3.Hou W; Dehm NA; Scott RWJ J. Catal 2008, 253, 22. [Google Scholar]
- 4.Toshima N; Yonezawa T New J. Chem 1998, 22, 1179. [Google Scholar]
- 5.Yao H; Kobayashi R Colloid Interface Sci 2014, 419, 1. [DOI] [PubMed] [Google Scholar]
- 6.Kanaoka S; Yagi N; Fukuyama Y; Aoshima S; Tsunoyama H; Tsukuda T; Sakurai H J. Am. Chem. Soc 2007, 129, 12060. [DOI] [PubMed] [Google Scholar]
- 7.Sophiphun O; Wittayakun J; Dhital RN; Haesuwannakij S; Murugadoss A; Sakurai H Aust. J. Chem 2012, 65, 1238. [Google Scholar]
- 8.Lu J; Toy PH Chem. Rev 2009, 109, 815. [DOI] [PubMed] [Google Scholar]
- 9.Deng D; Jin Y; Cheng Y; Qi T; Xiao F ACS Appl. Mater. Interfaces 2013, 5, 3839. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H; Toshima N J. Nanosci. & Nanotechnol 2013, 13, 5405. [DOI] [PubMed] [Google Scholar]
- 11.Nishimura S; Yakita Y; Katayama M; Higashimine K; Ebitani K Catal. Sci. & Technol 2013, 3, 351. [Google Scholar]
- 12.Zhang S; Shao Y; Liao H; Liu J; Aksay IA; Yin G; Lin Y Chem Mater 2011, 23, 1079. [Google Scholar]
- 13.Wang F; Jiang Y; Wen X; Xia J; Sha G; Amal R ChemCatChem 2013, 5, 3557. [Google Scholar]
- 14.Murugadoss A; Okumura K; Sakurai HJ Phys. Chem. C 2012, 116, 26776. [Google Scholar]
- 15.Dhital RN; Kamonsatikul C; Somsook E; Bobuatong K; Ehara M; Kranjit S; Sakurai HJ Am. Chem. Soc 2012, 134, 20250. [DOI] [PubMed] [Google Scholar]
- 16.Dhital RN; Sakurai H Chem. Lett 2012, 41, 630. [Google Scholar]
- 17.Yoo W–J; Miyamura H; Kobayashi S J. Am. Chem. Soc 2011, 133, 3095. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki T Desymmetrization of meso diols. Yamamoto H; Carreira E, Eds. Comprehensive chirality, Elsevier: Amsterdam, 2012, volume 5 (21), page 502–533. [Google Scholar]
- 19.Meng S–S; Liang Y; Cao K–S; Zou L; Lin X–B; Yang H; Houk KN; Zheng W–H J. Am. Chem. Soc 2014, 136, 12249. [DOI] [PubMed] [Google Scholar]
- 20.Moritani J; Hasegawa Y; Kayaki Y; Ikariya T Tetrahedron Lett 2014, 55, 1188. [Google Scholar]
- 21.Mandal SK; Jensen DR; Pugsley JS; Sigman MS J. Org. Chem 2003, 68, 4600. [DOI] [PubMed] [Google Scholar]
- 22.Jakka K & Zhao C–G Org. Lett 2006, 8, 3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noe MC; Letavic MA; Snow SL Asymmetric dihydroxylation of alkenes. Organic Reactions; Overman LE Ed., John Wiley & Sons: Hoboken, New Jersey, 2005, Vol. 66, 109–625. [Google Scholar]
- 24.Konieczny S; Leurs M; Tiller JC ChemBioChem 2015, 16, 83. [DOI] [PubMed] [Google Scholar]
- 25.Shilpa N; Manna J; Rana RK Eur. J. Inorg. Chem 2015, 29, 4965. [Google Scholar]
- 26.Bataille CR; Donohoe TJ Chem. Soc. Rev 2011, 40, 114. [DOI] [PubMed] [Google Scholar]
- 27.Stoltz BM Chem. Lett 2004, 33, 362. [Google Scholar]
- 28.Neisius NM; Plietker BJ Org. Chem 2008, 73, 3218. [DOI] [PubMed] [Google Scholar]
- 29.de Boer JW; Browne WR; Harutyunyan SR; Bini L; Tiemersma-Wegman TD; Alsters PL; Hage R; Feringa BL Chem. Commun 2008, 3747. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C; Liu Y; Xu Z–J; Tse C–W; Guan X; Wei J; Huang J–S; Che C–M Angew. Chem. Int. Ed 2016, 55, 10253. [DOI] [PubMed] [Google Scholar]
- 31.Newhouse T; Baran PS Angew. Chem. Int. Ed 2011, 50, 3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gormisky PE; White MC J. Am. Chem. Soc 2013, 135, 14052. [DOI] [PubMed] [Google Scholar]
- 33.Osberger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Nature 2016, 537, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canta M; Font D; Gomez L; Ribas X; Costas M Adv. Synth. Catal 2014, 356, 818. [Google Scholar]
- 35.Boorman TC; Larrosa I Chem. Soc. Rev 2011, 40, 1910. [DOI] [PubMed] [Google Scholar]
- 36.Gutekunst WR; Baran PS Chem. Soc. Rev 2011, 40, 1976. [DOI] [PubMed] [Google Scholar]
- 37.Lu H; Zhang XP Chem. Soc. Rev 2011, 40, 1899. [DOI] [PubMed] [Google Scholar]
- 38.Collet F; Lescot C; Dauban P Chem. Soc. Rev 2011, 40, 1926. [DOI] [PubMed] [Google Scholar]
- 39.Covell DJ; White MC Angew. Chem. Int. Ed 2008, 47, 6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hoke T; Herdtweck E; Bach T Chem. Commun 2013, 49, 8009. [DOI] [PubMed] [Google Scholar]
- 41.Wei X–H; Wang G–W; Yang S–D Chem. Commun 2015, 51, 832. [DOI] [PubMed] [Google Scholar]
- 42.Liu Y; Tsunoyama H; Akita T; Xie S; Tsukuda T ACS Catal 2011, 1, 2. [Google Scholar]
- 43.Rekkab-Hammoumraoui I; Choukchou-Braham A; Pirault-Roy L; Kappenstein C Bull. Mater. Sci 2011, 34, 1127. [Google Scholar]
- 44.Weir MG; Knecht MR; Frenkel AI; Crooks RM Langmuir 2010, 26, 1137. [DOI] [PubMed] [Google Scholar]
- 45.Smrcina M; Majer P; Majerova E; Guerassina TA; Eissenstat MA Tetrahedron 1997, 53, 12867. [Google Scholar]
- 46.Kerr MS; de Alaniz JR; Rovis T J. Org. Chem 2005, 70, 5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward BD; Risler H; Weitershaus K; Bellemin-Laponnaz S; Wadepohl H; Gade LH Inorg. Chem 2006, 45, 7777. [DOI] [PubMed] [Google Scholar]
- 48.Digenis GA; McClannahan JS; Chen P–L J. Labelled Comp. Radiopharm 1993, 33, 11. [Google Scholar]
- 49.Kawaguchi S; Ito K Adv. Polym. Sci 2005, 175, 299. [Google Scholar]
- 50.Zhai L; Shi T; Wang H Front. Chem. China 2009, 4, 83. [Google Scholar]
- 51.Flebbe T; Hentschke R; Hadicke E; Schade C Macromol. Theory Simul 1998, 7, 567. [Google Scholar]
- 52.Ferrando R; Jellinek J; Johnston RL Chem. Rev 2008, 108, 850. [DOI] [PubMed] [Google Scholar]
- 53.Mulvaney P; Giersig M J. Chem. Soc., Faraday Trans 1996, 92, 3137. [Google Scholar]
- 54.Poole CP Jr.; Owens FJ Introduction to Nanotechnology; John Wiley & Sons: New Jersey, 2003. Pages 12–15. [Google Scholar]
- 55.Kobayashi S; Xu P; Endo T; Ueno M; Kitanosono T Angew. Chem. Int. Ed 2012, 51, 12763. [DOI] [PubMed] [Google Scholar]
- 56.Chen B–S; Hanefeld U J. Mol. Catal. B: Enz 2013, 85–86, 239. [Google Scholar]
- 57.Lifchits O; Mahlau M; Reisinger CM; Lee A; Fares C; Plyak I; Gopakumar G; Thiel W; List B J. Am. Chem. Soc 2013, 135, 6677–6693. [DOI] [PubMed] [Google Scholar]
- 58.Zhang J; Xu T; Li Z Adv. Synth. Catal 2013, 355, 3147. [Google Scholar]
- 59.Liu Y; Li R–R; Lu X–B Macromol 2015, 48, 6941. [Google Scholar]
- 60.Singh G; Aube J Org. Biomol. Chem 2016, 14, 4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lehtonen A; Kivekas R; Sillanpaa R Polyhedron 2002, 21, 1133. [Google Scholar]
- 62.Stahl SS, Thorman JL, Nelson RC; Kozee MA J. Am. Chem. Soc 2001, 123, 7188. [DOI] [PubMed] [Google Scholar]
- 63.Stahl SS Angew. Chem. Int. Ed 2004, 43, 3400. [DOI] [PubMed] [Google Scholar]
- 64.Talsi EP; Bryliakov KP Coord. Chem. Rev 2012, 256, 1418. [Google Scholar]
- 65.Boyd DR; Sharma ND; Berberian MV; Cleij M; Hardacre C; Ljubez V; McConville G; Stevenson PJ; Kulakov LA; Christopher CRA Adv. Synth. Catal 2015, 357, 1881. [Google Scholar]
- 66.Kihumbu D; Stillger T; Hummel W; Liese A Tetrahedron: Asym 2002, 13, 1069. [Google Scholar]
- 67.Becker H; King SB; Taniguchi M; Vanjesscje PM; Sharpless KB J. Org. Chem 1995, 60, 3940. [Google Scholar]
- 68.Jin Y; Yao Z; Zhang S; Jiang R; Sun X Chinese J. Org. Chem 2007, 27, 602. [Google Scholar]
- 69.Sengupta S; Mondal S Tetrahedron Lett 1999, 40, 3469. [Google Scholar]
- 70.Ahrgren L; Sutin L Org. Proc. Res. Dev 1997, 1, 425. [Google Scholar]
- 71.Fuji K; Tanaka K; Miyamoto H Tetrahedron Lett 1992, 33, 4021. [Google Scholar]
- 72.Reddy SM; Srinivasulu M; Reddy YV; Narasimhulu M; Venkateswarlu Y Tetrahedron Lett 2006, 47, 5285. [Google Scholar]
- 73.Wu W; Jiang H Acc. Chem. Res 2012, 45, 1736. [DOI] [PubMed] [Google Scholar]
- 74.Pisoni D. d. S.; da Costa JS; Gamba D; Petzhold CL; Borges A. C. de A.; Ceschi MA; Lunardi P; Goncalves CAS Eur. J. Med. Chem 2010, 45, 526. [DOI] [PubMed] [Google Scholar]
- 75.Choudhary MI; Musharraf SG; Sami A; Raman A. –u. Helv. Chim. Acta 2004, 87, 2685. [Google Scholar]
- 76.Conde A, Vilella L, Balcells D, Diaz-Requejo MM, Lledos A; Perez PJ J. Am. Chem. Soc 2013, 135, 3887. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.