

Abstract
The 1,2,4-triazolo[1,5-a]pyrimidine (TP) heterocycle, in spite of its relatively simple structure, has proved to be remarkably versatile as evidenced by the many different applications reported over the years in different areas of drug design. For example, as the ring system of TPs is isoelectronic with that of purines, this heterocycle has been proposed as a possible surrogate of the purine ring. However, depending on the choice of substituents, the TP ring has also been described as a potentially viable bio-isostere of the carboxylic acid functional group and of the N-acetyl fragment of ε-N-acetylated lysine. In addition, the metal-chelating properties of the TP ring have also been exploited to generate candidate treatments for cancer and parasitic diseases. In the present review article, we discuss recent applications of the TP scaffold in medicinal chemistry, and provide an overview of its properties and methods of synthesis.
Keywords: 1,2,4-triazolo[1,5-a]pyrimidine; heterocyclic chemistry; medicinal chemistry
Graphical Abstract

1. Introduction
The 1,2,4-triazolo[1,5-a]pyrimidine heterocycle (TP, Figure 1) was first reported in 1909 by Bulow and Haas. [1] Although this scaffold can be found in natural products, as exemplified by essramycin (1, Figure 1), a TP derivative that was isolated from the broth of the marine Streptomyces sp, [2] the vast majority of biologically active TP compounds described to date are not naturally occurring compounds. Over the years, the TP scaffold has found numerous applications in medicinal chemistry.
Figure 1.

The structure of the 1,2,4-triazolo[1,5-a]pyrimidine scaffold, the natural product, essramycin (1), and the drug Trapidil (2).
Due to the structural similarities of the TP heterocycle with the purine ring, different studies investigated TP derivatives as possible isosteric replacements for purines. However, as exemplified in the sections below, the TP heterocycle proved to be a versatile scaffold with applications beyond isosteric replacement strategies, often resulting in the identification of biologically active compounds with favorable ADME-PK properties. Important historical examples of biologically active TP derivatives include Trapidil (2, Figure 1), a platelet-derived growth factor antagonist originally developed as a vasodilator and anti-platelet agent [3, 4] that has been marketed in Japan and in other countries to treat patients with ischemic coronary heart, liver, and kidney disease. Furthermore, in recent years, several TP derivatives have been identified that demonstrated considerable potential in different therapeutic areas, such as cancer chemotherapy, neurodegenerative diseases and infectious diseases. In addition, several TP derivatives have also shown promise as agrochemicals. [5–8] This review article focuses on providing an overview of the properties and methods of synthesis, as well as an update of recent applications of the TP scaffold in the discovery/design of candidate therapeutics for the treatment of human diseases.
2. Overview of Properties and Methods of Synthesis
The 1,2,4-triazolo[1,5-a]pyrimidines are examples of aza-indolizines that have been described as aza-analogs of a delocalized 10-π electron system consisting of an electron-rich five-membered ring (6 π electrons) fused to an electron deficient six-membered ring (4 π electrons). [9] Although indolizines are generally known to possess a marked aromatic character, 1H-NMR methyl substituent effect studies suggested that the TP heterocycle exhibits rather limited degree of aromaticity. [10] In addition, although the TP heterocycle does not exhibit annular tautomerism, as exemplified in greater details in the sections below, appropriately substituted hydroxyl- or mercapto- derivatives exist in different tautomeric forms (e.g., oxo- or thioxo-). These characteristics are clearly important factors that determine the properties and chemical reactivity of this heteroaromatic scaffold. [9] For example, an important intrinsic characteristic of the TP scaffold is the ability to bind metal ions, which stems from the fact that three of the heterocyclic nitrogen atoms (i.e., N1, N3 and N4) present accessible electron pairs. TPs are known to be versatile ligands that can form different types of metal complexes, which typically involve the N3 position in monodentate complexes, or the N3 and N1/N4 positions in bidentate complexes. [11] Figure 2 shows representative examples of TP ligands that have been employed in complexes with metals.
Figure 2.

Selected representative examples of TP ligands (A) and their complexes with metals (B).
These metal chelating properties of the TP heterocycle have been exploited for different applications, including drug design as exemplified by the different Pt [12] and Ru [13] complexes that have been investigated as potential anti-cancer agents (e.g., see Figure 2B). In this context, the hydrogen-bonding properties of the TP heterocycle have been found to play an important role in determining the stability of the metal complex towards hydrolysis/aquation. Other interesting examples include a series of first-row transition metal-containing TP derivatives that have been identified as relatively potent and selective anti-parasitic agents against Leishmania and Trypanosoma cruzi, [14, 15] the causative pathogens of leishmaniasis and Chagas disease, respectively.
The synthetic methods most commonly employed to access variously substituted TPs have been reviewed previously [16–18] and generally involve annulation reactions starting from 1,2,4-aminotriazole or pyrimidine derivatives. In the former case, an appropriate 1,2,4-aminotriazole (10, Scheme 1A) is reacted with a 1,3-dicarbonyl or an α,β-unsaturated system to form TP derivatives of general structure 11 bearing a range of substitutions at positions 2, 5, 6, and 7. Alternatively, the TP heterocycle can be obtained in two steps starting with the cyclocondensation of the appropriate hydrazinylpyrimidine (12, Scheme 1B) with a carbonyl compound, followed by Dimroth rearrangement [19] of the resulting 1,2,4-triazolo[4,3- a]pyrimidine (13, Scheme 1B) under acidic conditions to give the corresponding TP. [20] Finally, substituted TP heterocycles can also be accessed through an oxidative cyclization of corresponding pyrimidin-2-yl-amidines (14, Scheme 1C). [21, 22]
Scheme 1.

Most common strategies for the synthesis of the TP heterocycle include: (A) cyclocondensation reactions of aminotriazoles and 1,3-dicarbonyl or α,β-unsaturated carbonyl compounds; (B) conversion of 1,2,4-triazolo[4,3-a]pyrimidine via Dimroth rearrangement; and (C) oxidative cyclization reactions from pyrimidin-2-yl-amidines.
3. 1,2,4-Triazolo[1,5-a]pyrimidines in (bio)-isosteric replacement strategies
3.1. As replacement of purines:
Like other aza-indolizines, the TP ring is isoelectronic with the purine heterocycle, which suggests that TPs may be considered as possible replacements, or bio-isosteres, [23] for purines in medicinal chemistry. However, unlike the purine ring, the TP heterocycle does not exhibit annular tautomerism, although as noted above, the presence of substituents (e.g., hydroxyl) can allow for different tautomeric forms (e.g., see 15, Scheme 2C).
Scheme 2.

(A) Tautomerism of the purine ring compared to the unsubstituted (B) and a substituted (C) 1,2,4-triazolo[1,5-a]pyrimidine ring (15).
The use of the TP scaffold as a purine surrogate has been documented over the years through different drug discovery programs. TP heterocycles have been investigated in the context of nucleoside analogs; [24, 25] however, these efforts rarely resulted in derivatives exhibiting significant biological activities. On the contrary, examples of successful deployment of the TP heterocycle as a purine surrogate have been reported for different non-nucleoside molecules. Such examples include a series of inhibitors of the cyclin-dependent protein kinase, CDK-2. [26] CDK-2 are important for cell cycle control as these enzymes are responsible for the activation of the cell cycle of quiescent cells, as well as for the G1/M and G2/S transitions of the cell cycle. Given the role of CDKs in the cell cycle and proliferation, these kinases have been recognized as valuable targets for the treatment of cancer. [27] Existing CDK inhibitors include the purine derivative, roscovitine (16, Scheme 3), which inhibits multiple CDKs (i.e., CDK-1, CDK-2, CDK-5 and CDK-7) through direct competition at the ATP-binding site. A focused screening campaign of >3,000 commercially available compounds led to the identification of pyrazolo[1,5- a]pyrimidine 17 (Scheme 3) with low μM IC50 value against CDK-2. [26] Although the corresponding TP derivative (18, Scheme 3) was found to be ~10x less potent against CDK-2 than 17, further SAR studies were conducted with the objective of improving both potency for CDK-2 as well as selectivity over glycogen synthase kinase-3β (GSK-3β). This effort ultimately led to more potent derivatives (e.g., 19, Scheme 3) with IC50 values in the sub-μM range, as well as improved selectivity against GSK3-β. [26] Interestingly, these studies confirmed that the TP derivatives interact with the ATP binding pocket of CDK-2. Indeed, X-ray and biological studies of closely related purine and TP congeners (e.g., 20 and 21, Figure 3) clearly confirmed that the TP heterocycle can be an effective replacement for purine resulting in biologically active derivatives that can maintain similar interactions within the ATP binding pocket of CDK-2 (Figure 3).
Scheme 3.

Schematic of chemical evolution of TP inhibitors of CDK-2.
Figure 3.

Overlay of purine (20) and TP (21) derivatives in the ATP binding pocket of CDK-2 reveals a similar binding mode (PDB codes: 2C6O and 2C6M).
Interestingly, although these studies clearly show that the TP heterocycle can be used to target the ATP binding site of kinases, as of today there are very few reports of TP derivatives identified as inhibitors of kinases. Such examples include TP-containing inhibitors of the phosphatidylinositol 3-kinases (PI3K), which exhibit potent activity and isoform selectivity (e.g., 22–26 Table 1). [28] In this case, however, there are no X-ray studies or additional direct evidence showing that the TP derivatives are targeting the ATP site of PI3K.
Table 1.
Structure and in vitro biological activity of a series of TP inhibitors of the PI3K. [28]
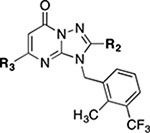 |
PI3K enzyme IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| Cpd. | R2 | R3 | α | β | δ | γ |
| 22 | CH3 | 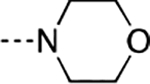 |
0.63 | 0.0006 | 0.020 | 0.79 |
| 23 | SCH3 |  |
0.32 | 0.0004 | 0.025 | 1.30 |
| 24 | cPr |  |
2.50 | 0.0013 | 0.040 | 1.60 |
| 25 | CH3 |  |
0.40 | 0.0013 | 0.010 | 0.16 |
| 26 | SCH3 |  |
1.3- | 0.0010 | 0.025 | 0.20 |
A further interesting example of TP as bio-isostere of purines includes a recent study in the area of methionine aminopeptidase-2 (MetAP-2) inhibitors. MetAP-2 is an enzyme believed to play a key role in angiogenesis. [29] Pharmacological inhibition of this enzyme with different classes of molecules was found to prevent the proliferation of vascular endothelial cells. [30–32] A recent screening campaign for novel reversible inhibitors of MetAP-2 led to the identification of a series of purine derivatives with promising low- to sub-μM activity in the in vitro assay. [33] Further hitto-lead optimization studies revealed that replacement of the purine ring with the TP could significantly boost in vitro biological activity while maintaining physicochemical properties in promising ranges. This is illustrated by the comparison of purine analog 27 (Scheme 4) and the corresponding TP derivative, 28, which exhibits improved in vitro biological activity, as well as acceptable properties, including thermodynamic water solubility, and permeability in the Caco-2 assay. [33]
Scheme 4.

The structure and in vitro activity of purine 27 and TP derivative 28 in the MetAP-2 assay.
Interestingly, superimposition of the X-ray structures of the enzyme-bound TP derivative 28 and a related purine analog 29 suggests that the mode of binding of the TP compound is different than the purine counterpart, as the TP heterocycle is flipped by approximately 180˚ relative to the purine inhibitor (Figure 4).
Figure 4.

Overlay of TP (28) and purine (29) derivatives bound to MetAP-2 reveals that the bicyclic scaffold of the former is flipped by ~180˚ relative to the purine compound (PDB codes: 5LYX and 5LYW).
While the examples above clearly illustrate the potential utility of the TP heterocycle as a purine isostere, as shown below, this scaffold has also been identified as a potentially viable replacement for other fragments and functional groups.
3.2. As carboxylic acid bio-isostere:
A program directed towards the discovery of novel noncarboxylated inhibitors of the fatty acid binding protein (FABP) identified selected TP derivatives in which the TP heterocycle act as a carboxylic acid bio-isostere. [34] The FABPs are intracellular lipid chaperones that are believed to play an important role in lipid-mediated biological processes and systemic metabolic homeostasis and, as such, have been recognized as potentially valuable therapeutic targets for a range of disorders, including obesity, diabetes and atherosclerosis. [35] Among the different FABP isoforms, FABP4 is one of the most well-characterized. Over the years, hundreds of inhibitors of FABP4 have been synthesized and evaluated as potential treatments for atherosclerosis and diabetes. [36] The vast majority of such inhibitors are characterized by the presence of a carboxylic acid moiety. In an effort to differentiate from the existing FABP4 inhibitors, and to achieve superior pharmacokinetic (PK) and cell permeability properties, a recent program by scientists at Merck focused on identifying non-carboxylic acid FABP4 inhibitors. [34] This effort, which involved a high-throughput screening (HTS) of a large library of compounds, led to the identification of a promising TP derivative (30, Figure 5), which displayed a Kd value of 0.86 μM for FABP4. Co-crystal structure of this hit with recombinant FABP4 revealed that all heteroatoms of the TP derivative form hydrogen bonds with the protein and with one structural water. Subsequent hit-to-lead optimization studies resulted in two improved analogs, 31 and 32 (Figure 5), which exhibited Kd values for FABP4 of 0.01 μM and 0.02 μM, respectively. Interestingly, the experimental pKa value of 31 (4.6) is similar to that of a carboxylic acid, suggesting that this heterocycle could serve as a carboxylic acid bio-isostere. [34, 37] When administered chronically to diet-induced obese mice, TP derivative 32 was found to be efficacious in improving dyslipidemia at all doses used in the studies (3, 10, and 30 mg/kg). [34]
Figure 5.

Structures of TP inhibitors of FABP4/5 and an example of a radiolabeled TP derivative (33) that was investigated as an imaging agent for FABP4.
Also of interest, these FABP4-interacting TP derivatives have also showed promise as potential starting points towards the development of selective imaging agents that could be utilized to monitor the expression of FABP4, which is believed to participate in disease malignancy of glioblastomas. [38] One such example is [125I]-TP derivative 33 (Figure 5), which showed high affinity for FABP4 (Kd value of ~69 nM) and promising in vivo properties.
3.3. As replacement of the N-acetyl fragment of ε-N-acetylated lysine:
An additional interesting example of TPs identified as possible bio-isosteres comes from a program directed to the identification of novel inhibitors of the bromodomain (BRD), in which the TP heterocycle was found to effectively mimic the N-acetyl fragment of ε-N-acetylated lysine (KAc). The BRDs are a diverse family of evolutionary conserved protein-interaction modules that bind KAc residues. Proteins that contain BRDs are thought to be potentially involved in the development of a variety of diseases and as a result, these proteins have been identified as promising drug targets for the treatment of cancer and other diseases. [39] Several small-molecule inhibitors have been identified to successfully act as KAc mimetics and bind within the BRD binding site. [40] Among these compounds, selected TP derivatives such as 34 (Figure 6A) have been identified through different screening efforts. [41, 42] Interestingly, crystal structures of these TP derivatives bound to BRD4 revealed good complementarity with the KAc binding site, suggesting that indeed the TP scaffold is an effective bio-isostere of the KAc motif (e.g., see Figure 6B).
Figure 6.

The structure of selected TP inhibitors (34 and 35) of BRD4 (A) and the X-ray structure (PDB code: 4MEN) of 34 bound within the KAc binding site (B).
4. 1,2,4-Triazolo[1,5-a]pyrimidines in anti-infectious agents
4.1. Anti-viral agents:
In recent years, different TP inhibitors of viral polymerases have been identified as part of drug discovery efforts against hepatitis C virus (HCV), human immunodeficiency virus type-1 (HIV-1), and influenza. The RNA-dependent RNA polymerase of HCV, NS5B, which is known to be considerably different than the human DNA and RNA polymerases, has long been recognized as a potentially promising therapeutic target for the development of novel and selective anti-HCV therapies. Optimization of a novel class of allosteric HCV NS5B dihydropyrone inhibitors (e.g., see 36, Figure 7) derived from a HTS campaign [43] led to the identification of promising TP containing compounds with low nM potency in a biochemical assay against the isolated HCV NS5B polymerase, and promising antiviral activities in a cell-based replicon assay (e.g., 37 and 38, Figure 7). [44] Moreover, TP candidates from this series of inhibitors that exhibit favorable PK profiles in multiple species have been reported. [45]
Figure 7.

Selected examples of TP containing inhibitors (37, 38) of HCV NS5B polymerase derived from HTS hit 36.
In the case of HIV-1, TP derivatives have been identified as novel examples of HIV-1 nonnucleoside reverse transcriptase inhibitors (NNRTIs) that are believed to interact at the diarylpyrimidine allosteric site, located approximately 10 Å away from the reverse transcriptase catalytic site. These compounds [46] (e.g., 39 and 40, Scheme 5), identified via structure-based core refining approaches [47] from closely related pyrrolopyrimidines [48] (e.g., 41, Scheme 5), were found to be highly potent NNRTIs against wild-type HIV-1 with EC50 values in the low nM range.
Scheme 5.

Representative examples of TP containing inhibitors of the reverse transcriptase of HIV-1 that have been identified through SAR and optimization studies of closely related pyrrolopyrimidines.
TP derivatives with promising anti-viral activity against influenza virus have also been identified. The viral RNA-dependent RNA polymerase of influenza virus, which is a heterotrimeric complex of independently folded subdomains with different functionalities, is highly conserved among different viral strains and as a result, has attracted considerable attention as a possible target for the development of anti-influenza agents of broad anti-viral activity. [49–54] The correct assembly of the three subunits (i.e., the PB1 subunit, which executes the polymerase activity; PB2, which is responsible for cap-binding of host cell pre-mRNA; and the PA subunit, which harbors the endonuclease function) is believed to be essential for both transcription and replication of the viral genome. Interestingly, the interaction of PA with PB1 has been characterized through X-ray crystallography studies. [55, 56] The availability of these structural data enabled a virtual screening of >3 million compounds, which resulted in the identification of a promising pyrazolo[1,5-a]pyrimidine (42, Scheme 6) that was confirmed to inhibit the formation of the PA-PB1 complex in vitro. [57] Further evaluation of the SAR revealed that the replacement of the pyrazolo[1,5-a]pyrimidine scaffold with the TP can result in analogues with improved in vitro anti-viral activity (e.g., 43 and 44, Scheme 6). [57, 58]
Scheme 6.

Representative examples of inhibitors of the RNA-dependent RNA polymerase of influenza virus.
4.2. Anti-bacterial agents:
Although the naturally occurring TP, essramycin [2] (1, Figure 1), which was reported to have relatively potent, broad spectrum activity against different Gram-positive and Gram-negative bacteria, was later found by different groups to be essentially devoid of anti-bacterial activity, [59, 60] several TP compounds have recently demonstrated promising results as anti-bacterial agents. For example, a series of TP compounds have been recently identified to exhibit potent activity against enterococcus faecium. [61] The E. faecium is responsible for approximately 20% of enterococcal infections in the United States, [62] and is known to be resistant to many commonly used antibiotics. Thus, the development of new and selective antibacterial agents against this organism is desirable. [61] Towards this end, in silico screening of 1.2 million compounds against the X-ray structure of a penicillin-binding protein (PBP), PBP2a, which is an essential enzyme in the biosynthesis of peptidoglycans, [63] resulted in the identification of selected TP derivatives, such as 45 (Scheme 7). [61] Interestingly, although 45 was confirmed experimentally to possess anti-bacterial activity against E. faecium (MIC of 8 μg/mL), this compound was found not to inhibit PBP2a. Nonetheless, an extensive SAR effort (Scheme 7B) identified a subset of TP congeners that exhibit both the desired narrow-spectrum antibacterial activity against E. faecium, as well as generally favorable ADME properties (Table 2). Mode of action studies suggested that the anti-bacterial TPs interfere with the cell-wall biosynthesis. [61]
Scheme 7.

(A) Structure of an anti-bacterial TP derivative (45) identified via in silico screening studies; (B) Schematic representation of SAR effort.
Table 2.
Structure, properties and anti-bacterial activity of selected TP antibiotics. [61]
 |
MIC of E. faecium C68 (μg/mL) | Half-life in human liver S9 (min) | Apparent intrinsic clearance (mLmin−1kg−1) | Plasma Protein Binding (%) | Hemolysis at 64 μg/mL (%) | ||
|---|---|---|---|---|---|---|---|
| Cpd | R1 | R3 | |||||
| 45 |  |
 |
16 | 56 ± 1 | 11.1 | 98.7 ± 0.7 | 22% |
| 46 |  |
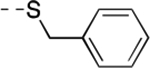 |
16 | 57 ± 8 | 10.9 | 99.9 ± 0.2 | <1% |
| 47 |  |
16 | >60 | <10.4 | 99.9 ± 0.4 | 46% | |
| 48 |  |
 |
4 | >60 | <10.4 | 97.6 ± 0.1 | 2% |
| 49 |  |
32 | >60 | <10.4 | 99.9 ± 0.5 | 23% | |
| 50 | 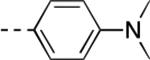 |
 |
4 | >60 | <10.4 | 96.8 ± 0.4 | 1% |
Other notable examples of TP derivatives endowed with anti-bacterial activity include examples with promising activity against Mycobacterium tuberculosis, the causative pathogen of tuberculosis (TB). [64, 65] Although several anti-TB agents have been developed over the years, TB remains one of the most devastating infectious diseases with >10 million cases and >1.5 million deaths reported worldwide in 2016, and the emergence of multi-drug resistant strains creates a further significant challenge. Thus, the discovery and evaluation of new therapeutic targets for the treatment of TB continues to be the focus of intense studies. In this context, the acetohydroxyacid synthase (AHAS), an anabolic enzyme involved in the branched-chain amino acid biosynthesis,[66] has been recognized as a promising drug target for discovering novel antitubercular compounds. [67–70] The branched-chain amino acid biosynthesis pathway is absent in humans but is essential for the survival of microbes, plants and fungi and thus provides an opportunity for effective and safe therapeutic intervention. [71] Interestingly, different TP derivatives, exemplified by Flumetsulam (51, Figure 8) and related compounds (e.g., Y6610, 52, Figure 8) that had been developed and marketed as herbicides, are known to exhibit AHAS inhibitory activity and compounds of this type have been proposed as possible leads for novel anti-TB agents. [72–74]
Figure 8.

Representative examples of TP sulfonamide inhibitors of AHAS with herbicidal activity.
Given the potential importance of AHAS in M. tuberculosis, efforts have been made, including structure-based drug design approaches [70] as well as HTS campaigns, [75] to identify potent inhibitors as candidate treatments for TB. In this context, a screening of 6,800 compounds resulted in the identification of several promising TP derivatives that were potent inhibitors of the AHAS of M. tuberculosis. Among these compounds, some were found to possess significant biological activity against different TB strains, including multidrug resistant (MDR-TB) and extensively drug-resistant (XDR-TB) strains (see 53 and 54, Table 3) while showing no cytotoxicity against mammalian cell lines. [75]
Table 3.
Structure and in vitro biological activity of representative TP derivatives with anti-tubercular activity. [75]
| Cpd | Structure | MIC (μM) against MDR-TB | MIC (μM) against XDR-TB | |||
|---|---|---|---|---|---|---|
| M22 | M23 | P887 | X24 | X59 | ||
| 53 |  |
2 | 2 | 2 | 2 | 2 |
| 54 |  |
0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
Interestingly, in addition to the TP sulfonamides that are known to inhibit AHAS, other examples of TP derivatives with relatively potent antitubercular activity have been reported recently. A phenotypic screening of compounds in a whole cell assay identified TP 55 (Figure 9) to have promising activity against M. tuberculosis. SAR studies identified improved congeners (e.g., 56 and 57, Figure 9) with sub-μM IC50 values against M. tuberculosis and no cytotoxicity against eukaryotic cells. [64] Although it is not clear whether the derivatives identified through phenotypic screens may be acting through inhibition of AHAS, the SAR data seem to suggest a distinct biological target.
Figure 9.

Additional examples of anti-tubercular TP derivatives.
4.3. Anti-parasitic agents:
Multiple research programs directed to the discovery of potential candidates for different parasitic diseases have identified several promising TP derivatives. For example, in recent years, TP derivatives that hold considerable promise as candidate treatments for malaria have been discovered. Malaria, a mosquito-borne parasitic disease that is especially widespread in tropical and subtropical regions, continues to be a significant threat. It is estimated that >200 million cases occurred worldwide in 2016 and that approximately 1,700 cases are diagnosed in the United States each year. Thus, the development of therapeutic strategies for the treatment and/or prevention of this disease remains an important priority. Dihydroorotate dehydrogenase (DHODH) is an enzyme involved in the de novo pyrimidine biosynthetic pathway and this enzyme has recently been identified as a promising and “druggable” target for the treatment of malaria. [76] HTS efforts aimed at the discovery of inhibitors of this enzyme resulted in the identification of potent TP inhibitors that generally feature an aromatic group linked at the C-7 position through a bridging nitrogen atom (e.g., DSM-1, 58, Scheme 8). X-ray studies with 58 (Figure 10) provided significant insight and revealed that the TP binding site comprises a hydrophobic pocked, which is occupied by the naphtyl substituent, and a H-bond pocket, which accommodates the TP ring. [76] Interestingly, the hydrophobic pocket is not highly conserved between different species and this provides a possible explanation for the >5,000-fold species selectivity that has been observed over the human enzyme. [77] Equally interesting, these studies highlighted an unexpected binding mode of the TP heterocycle within the H-bond pocket. As highlighted in Figure 10, the TP heterocycle engages in H-bonding interactions with the side chains of His185 and Arg265 in which the intrinsic dipole of the substituted TP ring is likely essential to the formation of relatively strong interactions with the imidazole (His185) and the guanidinium moiety (Arg265). Based on these structural data, an extensive SAR campaign resulted in the identification of improved analogs. [78–80] For example, optimization of the aromatic amine linked at C-7 resulted in derivatives, such as TP DSM-190 (59, Scheme 8), that combined good plasma exposure and better in vivo efficacy compared to previous TP congeners. [81] Further optimization of the substitution pattern around the TP heterocycle identified highly promising TP derivatives, such as DSM-265 (60) and DSM-421 (61, Scheme 8), with the latter exhibiting more favorable drug-like and in vivo properties, including improved solubility, lower intrinsic clearance and increased plasma exposure after oral dose (see Table 4). [80] Importantly, this compound produced excellent in vivo anti-parasitic activity against both P. falciparum and P. vivax in a mouse model of malaria.
Scheme 8.

Representative structures showing the chemical evolution of anti-malarial TP derivatives.
Figure 10.

The 2D (A) and 3D (B) image of the X-ray structure of 58 bound to DHODH (PBD code: 3I65). [76]
Table 4.
Selected activity and property data of anti-malarial TP derivatives. [80]
| Cpd | IC50 (μM) PfDHODH | IC50 (μM) hDHODH | Log D7.4 | Aqueous Solubility (μM) |
Human Plasma Protein Binding |
CLint human/mouse (μl/min/mg protein) |
|---|---|---|---|---|---|---|
| DSM-1 (58) |
0.047 | >200 | 3.2 | 45–90 | 93.8 % | 96/229 |
| DSM-190 (59) |
0.19 | >30 | 2.9 | 9–18 | 96.6 % | <5/<6 |
| DSM-265 (60) |
0.033 | >100 | 3.6 | 70–140 | 90.4 % | <5/10.1 |
| DSM-421 (61) |
0.014 | >100 | 2.5 | >100a | 50% | 0.045/5.1 |
In addition to the promising anti-malarial TP derivatives discussed above, other notable examples of anti-parasitic TPs include a series of broad-spectrum compounds against T. brucei, T. cuzi and Leishmania donovani. [82] Infections by these kinetoplastid parasites are responsible for Chagas disease (T. cruzi), African sleeping sickness (T. brucei) and leishmaniasis (L. donovani), which are believed to affect approximately 20 million people worldwide. A large screening effort of 3 million compounds against these parasites led to the identification of a promising azabenzoxazole hit (GNF5343, 62, Scheme 9). Further hit-to-lead optimization studies, which involved the design, synthesis and evaluation of approximately 3,000 analogs, revealed that scaffold replacement of the azabenzoxazole with C-6-substituted imidazo- and triazolopyrimidine cores (e.g., GNF2636, 63, and GNF3849, 64, Scheme 9) resulted in improved compounds. Optimization of the substituents flanking these heterocyclic compounds ultimately led to the identification of GNF6702 (65, Scheme 9), which combines broad-spectrum anti-parasitic activity with favorable ADME-PK properties. Interestingly, the anti-parasitic activity of these TP derivatives is linked to a non-competitive inhibition of the kinetoplastid proteasome. Moreover, as these molecules do not interfere with the mammalian proteasome, in vivo studies of 65 in mouse models of kinetoplastid infections revealed that the compound is both well-tolerated as well as effective against different parasitic infections. Notably, these studies included an evaluation of 65 in a mouse model of stage II sleeping sickness, which is caused by T. brucei infection in the central nervous system (CNS). Although 65 exhibits relatively low brain exposure (i.e., 0.1 brain-to-plasma ratio), administration of 100 mg/kg of the compound once daily for seven days caused sustained clearance of parasites, such that there were no detectable parasites in the brain of compound treated mice at termination of the experiment. [82]
Scheme 9.

Schematic of the evolution of kinetoplastid proteasome inhibitors from the initial phenotypic azabenzoxazole hit.
5. 1,2,4-Triazolo[1,5-a]pyrimidines in anti-cancer agents
In addition to the above mentioned instances of TP-containing inhibitors of cancer associated kinases (see CDK-2 and PI3-K inhibitors in Figure 3 and Table 1, respectively), the potential utility of TPs in the context of cancer chemotherapy is also clearly illustrated by the series of microtubule (MT)-targeting compounds from this class that have been reported over the past several years. MTs are polymeric structures that result from the assembly of α- and β-tubulin heterodimers and are involved in a multitude of structural and regulatory functions in all cell types. [83, 84] Tubulin/MTs proved to be valuable drug target especially in the context of cancer chemotherapy due to the key role of MTs in chromosome segregation during cell division. [85] The vast majority of MT-targeting agents are either natural products or derivatives of natural products that are often characterized by relatively complex chemical structures and sub-optimal physicochemical properties. In addition, the clinical utility of several of these compounds is often hindered by the occurrence of mechanisms of drug resistance, which can involve point mutations at α-tubulin and β-tubulin genes that ultimately reduce the binding affinity of the compound to tubulin/MTs. [86] Moreover, the expression of active transporters, such as the P-glycoprotein (Pgp), that can effectively efflux the MT-stabilizing drug out of cells is a frequent mechanism responsible for the occurrence of drug resistance. [87] For these reasons, over the past several years, efforts have been made in the identification of MT-active compounds with comparatively simpler structures and potentially more favorable drug-like properties that could also circumvent one or more mechanisms of drug resistance. In this context, several promising examples of MT-active TP derivatives have been identified.
The discovery of MT-targeting TP molecules originated from a screening campaign aimed at the identification of novel anti-fungal agents for agrochemical purposes. This effort led to the discovery of selected TP derivatives, such as BAS600F (66, Figure 11), which exhibited considerable activity against a relatively broad range of different plant diseases. [88] Subsequently, the pharmaceutical company Wyeth evaluated compounds from this class as potential chemotherapeutic agents. After comprehensive SAR studies, a preferred TP derivative, TTI-237 (also known as cevipabulin, 67, Figure 11), was identified to have potent anti-cancer activity both in vitro and in vivo. [89–92] In preclinical studies, the anticancer activity of cevipabulin was found to be comparable to that of other well characterized MT-stabilizing natural products, such as paclitaxel. However, unlike paclitaxel, 67 was found to retain anti-mitotic effects in taxol-resistant cells, including Pgp-overexpressing cancer cell lines. Moreover, 67 was found to be orally bioavailable (61%), metabolically stable (t1/2 = 13 h in female nude mice) and water-soluble (0.89 mg/mL). [89] Efforts to elucidate the mode of action indicated that, compared to MT-targeting natural products, 67 may be somewhat unusual. Competition-binding experiments showed that cevipabulin interferes with vincristine, a MT-depolymerizing agent, but not with taxol indicating that this TP may interact within the binding-site of the vinca alkaloids. [89] This possibility is bolstered by recent X-ray crystallography studies with a related TP congener (68, Figure 11 and Figure 12). [93] Cell-free tubulin polymerization and depolymerization experiments revealed that, at low molar ratios with tubulin heterodimers (approximately 1:30), 67 enhances the depolymerization kinetics of MTs in response to cold temperatures. However, at higher molar ratios (approximately 1:4) the compound stabilizes polymerized tubulin structures. [94] In more recent cell-based studies in which the effect of test compounds on MT stabilization is determined by monitoring known markers of stable MTs, such as acetylated α-tubulin and de-tyrosinated tubulin, [95] 67 was found to exhibit an unusual bell-shaped concentration-response relationship. [96] Moreover, this compound caused a proteasome-dependent degradation of α- and β-tubulin. These results indicated that TP 67 is a MT-active compound with properties that are clearly different from those of the classical vincaand taxane-site drugs. However, interestingly, further SAR and mode of action studies with a series of related TP congeners indicate that TP derivatives lacking the alkoxy side-chain at the para-position of the fluorinated phenyl ring (e.g., 68) act as MT-stabilizing compounds both in cell-free and cell-based experiments without causing a loss in total tubulin. [96, 97] These observed differences in the mode of action of MT-active TP compounds may have important implications in terms of therapeutic applications of compounds of this class. For example, whereas a disruption of MT integrity may be potentially desirable in those instances where compound treatment is intended to cause anti-mitotic effects, for other clinical indications (e.g., see example below concerning neurodegenerative tauopathies [98]), compounds able to stabilize MTs and preserve MT-integrity may be preferred. [96]
Figure 11.

Representative examples of MT-interacting TP derivatives that showed promise as agrochemicals (66), or as anti-cancer agents (67, and 68).
Figure 12.

2D schematics (A) and 3D image of X-ray structure of TP derivative 68 bound to a complex comprising two αβ-tubulin dimers, the stathmin-like protein RB3 and tubulin tyrosine ligase (PDB code: 5NJH). [93]
6. 1,2,4-Triazolo[1,5-a]pyrimidines as candidate treatments for diseases of the central nervous system
In light of the relatively small/simple structure, the generally favorable physicochemical properties, as well as the tunable mode of action, MT-active TP derivatives have been proposed as potential treatments for diseases of the CNS. [99–101] Interestingly, although 67 was found to have rather limited brain penetration as revealed by a brain-to-plasma ratio (B/P) of approximately 0.03 at 1h post i.p. administration in mice, several closely related congeners exhibit excellent brain exposure. [99] These findings provided an opportunity to investigate the potential utility of MT-active TP compounds for different CNS indications, including neurodegenerative diseases [96] and neuroparasitic infections. [101] For example, axonal transport deficits and axonal dystrophy are common features of multiple neurodegenerative diseases, including Alzheimer’s disease (AD), [102] and these axonal abnormalities are often thought to be associated with a loss of MT density in the axons of affected neurons. Thus, brain-penetrant MT-stabilizing agents have been proposed as candidate treatments for AD and related disorders. [103] Interestingly, recent in vivo efficacy and tolerability studies [104] with a MT-stabilizing TP derivative, CNDR-51657 (69, Figure 13), in aged PS19 tau transgenic mice that harbor tau pathology in the CNS, revealed that low doses (3 or 10 mg/Kg) administered twice-weekly for 3 months produce improvements in neuronal outcomes that resemble those previously observed upon treatment with MT-stabilizing natural products. [105–108] Moreover, the compound-treated mice showed no signs of compound intolerance during treatment, suggesting that MT-stabilizing TP compounds may be promising candidates for the treatment of AD as well as other neurodegenerative diseases.[109]
Figure 13.

Representative examples of TP MT-stabilizers (69) and inhibitors of AChE (70) under investigation as candidate therapeutics for AD.
In addition to the MT-stabilizing TP derivatives, other TP compounds [110, 111] have also been proposed as candidate treatments for AD. These molecules include multi-targeted ligands (e.g., 70, Figure 13), in which the TP unit was employed as an acetylcholine esterase (AChE)-inhibiting motif due to the anticipated π-π interaction with important residues at the peripheral anionic site (Tyr70 and Trp279) that are believed to regulate entry of substrate to the binding site of AChE. [112]
Finally, interesting examples of TP derivatives under development for CNS indications come from recent studies directed towards the discovery of phosphodiesterase 2a (PDE2a) inhibitors as candidate treatment of memory disorders. [100] PDEs are catabolizing enzymes responsible for the inactivation of important second messengers, such as cAMP and cGMP. As these second messengers are known to regulate signaling pathways implicated in long-term potentiation (LTP), PDEs have emerged as potential targets to treat memory disorders, [113–115] leading to a series of efforts in the search and development of selective inhibitors. [116] In this context, selected TP derivatives (e.g., 71 and 72, Table 5) with sub- μM IC50 values against PDE2a have been identified through a HTS campaign. Interestingly, the X-ray crystal structure of 72 bound to PDE2a (Figure 14) confirmed that these TP inhibitors bind within the active site of the enzyme and revealed that the TP heterocycle is involved in π-π stacking with Phe862. [100]
Table 5.
Structure and in vitro biological activity of representative TP inhibitors of PDE2a. [100]
| Cpd | Structure | IC50 (μM) against PDE2a |
|---|---|---|
| 71 |  |
0.17 |
| 72 |  |
0.14 |
| 73 |  |
0.001 |
| 74 |  |
0.008 |
Figure 14.

The X-ray crystal structure of 72 bound to PDE2a (PDB code: 5TZ3)
Efforts to improve the binding affinity and the potency of these TP derivatives focused on modulating the electronic properties of the TP heterocycle through alternative substitutions around the aromatic system. These studies led to the identification of closely related congeners, such as 73 (Table 5), that were found to be 2 orders of magnitude more potent than the initial HTS hits. Further refinement of ADME properties finally resulted in compound 74 (Table 5), which was selected as a lead compound with potentially favorable combination of PDE2a inhibition activity as well as in vitro ADME properties. [100]
7. Miscellaneous:
Additional interesting studies, schematically summarized in Table 6, that further illustrate the potential utility of the TP heterocycle in drug design have been reported in recent years. Scientists at Novartis reported a hit-to-lead optimization effort in which a pyrazolopyrimidine compound identified through a screening for antagonists of the chemokine receptor, CXCR2, led to the identification of an improved TP derivative 75 (Table 6). In this case, the replacement of the pyrazolopyrimidine core with the TP heterocycle led to selective antagonists with favorable combination of biological and PK properties. [117] In another study, Aghazadeh Tabrizi et al., reported the identification of a TP derivative (76, Table 6) found to act as an inverse agonist of the cannabinoid receptor, CB2. [118] In this case, the nature of the substituent at position 2 of the TP scaffold was found to play a crucial role in determining both the inverse agonist activity against the CB2 receptor, as well as the selectivity of CB2/CB1. [118] Finally, a TP derivative (WS-10, 77, Table 6) has recently been reported as a promising modulator of the ABCB1 active transporter, which is known to be involved in the development of multidrug resistance of chemotherapeutic agents. TP derivative WS-10, which was identified through a HTS campaign, was found to enhance the intracellular accumulation of paclitaxel in ABCB1-expressing cells, but did not affect the expression or localization of the active transporter. [119]
Table 6.
Other recent examples of biologically active TP derivatives.



8. Conclusions:
Depending on the nature and the arrangement of substituents, and whether or not these impart tautomerism, the TP heterocycle exhibit a relatively broad range of properties that ultimately led to several different applications in drug design. As illustrated in the sections above, a growing number of biologically active TP derivatives have been identified and developed in recent years, with many of these compounds characterized by generally favorable drug-like properties. Amongst the most recent examples, particularly promising are a series of anti-parasitic agents as candidate treatments for malaria and kinetoplastid infections, as well as a series of MT-targeting agents with potential therapeutic utility against cancer and/or neurodegenerative diseases.
Highlights.
TPs exhibit broad range of properties that led to several applications in drug design
TPs showed promise as bio-isosteric replacements of purines and other structural motifs
Several biologically active TPs are characterized by favorable drug-like properties
Different TPs have been identified as promising candidates for multiple CNS indications
Acknowledgments:
Financial support for this work was provided by NIH Grants AG044332 and AI13394.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bülow C, Haas K, Synthetische Versuche zur Darstellung von Derivaten des heterokondensierten, heterocyclischen 1.3-Triazo-7.0′-pyrimidins, Ber. Dtsch. Chem. Ges, 42 (1909) 4638–4644. [Google Scholar]
- [2].El-Gendy MM, Shaaban M, Shaaban KA, El-Bondkly AM, Laatsch H, Essramycin: a first triazolopyrimidine antibiotic isolated from nature, J. Antibiot. (Tokyo), 61 (2008) 149–157. [DOI] [PubMed] [Google Scholar]
- [3].Tenor E, Ludwig R, Drug chemical studies of the s-triazolo-1.5-a-pyrimidine series, Pharmazie, 26 (1971) 534–539. [PubMed] [Google Scholar]
- [4].Füller H, Hauschild F, Modersohn D, Thomas E, Pharmacology of 6-methyl-7-diethylamino-s-triazolo-(1,5-a) pyrimidine (Trapymin, Rocornal), a new compound with vasodilative effects, Pharmazie, 26 (1971) 554–562. [PubMed] [Google Scholar]
- [5].Lamberth C, Agrochemical lead optimization by scaffold hopping, Pest Manag. Sci, 74 (2018) 282–292. [DOI] [PubMed] [Google Scholar]
- [6].Liu Y-C, Qu R-Y, Chen Q, Yang J-F, Cong-Wei N, Zhen X, Yang G-F, Triazolopyrimidines as a New Herbicidal Lead for Combating Weed Resistance Associated with Acetohydroxyacid Synthase Mutation, J. Agric. Food Chem, 64 (2016) 4845–4857. [DOI] [PubMed] [Google Scholar]
- [7].Montel F, Lamberth C, Jung PMJ, First synthesis of 7-amido-[1,2,4]triazolo[1,5-a]pyrimidines using halogen–metal exchange, Tetrahedron, 64 (2008) 6372–6376. [Google Scholar]
- [8].Johnson TC, Mann RK, Schmitzer PR, Cast RE, de Boer GJ, Triazolopyrimidines, in: Krämer W, Schirmer U, Jeschke P, Witschel M (Ed.) Modern Crop Protection Compounds (2nd Edition), Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 99–117. [Google Scholar]
- [9].Tisler M, Structure and reactivity correlation of bicyclic 10-π electron systems with bridgehead nitrogen, Pure Appl. Chem, 52 (1980) 1611–1621. [Google Scholar]
- [10].Makisumi Y, Watanabe H, Tori K, Studies on Azaindolizine Compounds. XVIII. Proton Magnetic Resonance Spectra of s-Triazolo-[1, 5-a] pyrimidine and its Derivatives, Chem. Pharm. Bull, 12 (1964) 204–212. [DOI] [PubMed] [Google Scholar]
- [11].Salas JM, Angustias Romero M, Purificación Sánchez M, Quirós M, Metal complexes of [1,2,4]triazolo-[1,5-a]pyrimidine derivatives, Coord. Chem. Rev, 193–195 (1999) 1119–1142. [Google Scholar]
- [12].Łakomska I, Hoffmann K, Muzioł T, Sitkowski J, Multinuclear magnetic resonance and X-ray characterization of platinum(II) complexes with substituted-1,2,4-triazolo[1,5-a]pyrimidines, J. Mol. Struct, 1056–1057 (2014) 146–151. [Google Scholar]
- [13].Velders AH, Bergamo A, Alessio E, Zangrando E, Haasnoot JG, Casarsa C, Cocchietto M, Zorzet S, Sava G, Synthesis and chemical-pharmacological characterization of the antimetastatic NAMI-A-type Ru(III) complexes (Hdmtp)[trans-RuCl4(dmso-S)(dmtp)], (Na)[trans-RuCl4(dmso-S)(dmtp)], and [mer-RuCl3(H2O)(dmso-S)(dmtp)] (dmtp = 5,7-dimethyl[1,2,4]triazolo[1,5-a]pyrimidine), J. Med. Chem, 47 (2004) 1110–1121. [DOI] [PubMed] [Google Scholar]
- [14].Méndez-Arriaga JM, Esteban-Parra GM, Juárez MJ, Rodríguez-Diéguez A, Sánchez-Moreno M, Isac-García J, Salas JM, Antiparasitic activity against trypanosomatid diseases and novel metal complexes derived from the first time characterized 5-phenyl-1,2,4-triazolo[1,5-a]pyrimidi-7(4H)-one, J. Inorg. Biochem, 175 (2017) 217–224. [DOI] [PubMed] [Google Scholar]
- [15].Caballero AB, Rodríguez-Diéguez A, Quirós M, Salas JM, Huertas Ó, Ramírez-Macías I, Olmo F, Marín C, Chaves-Lemaur G, Gutierrez-Sánchez R, Sánchez-Moreno M, Triazolopyrimidine compounds containing first-row transition metals and their activity against the neglected infectious Chagas disease and leishmaniasis, Eur. J. Med. Chem, 85 (2014) 526–534. [DOI] [PubMed] [Google Scholar]
- [16].Fischer G, 1,2,4-Triazolo[1,5-a]pyrimidines, in: Katritzky AR (Ed.) Adv. Heterocycl. Chem, Academic Press, 1993, pp. 81–138. [Google Scholar]
- [17].Fizer M, Slivka M, Synthesis of [1, 2, 4] triazolo [1, 5-a] pyrimidine (microreview), Chem. Heterocycl. Com, 52 (2016) 155–157. [Google Scholar]
- [18].Fischer G, Recent Progress in 1,2,4-Triazolo[1,5-a]pyrimidine Chemistry, in: Katritzky AR (Ed.) Adv. Heterocycl. Chem, Academic Press, 2007, pp. 143–219. [Google Scholar]
- [19].Wahren M, Die Dimroth-Umlagerung — Platzwechsel zwischen Ring- und Seitenkettenatomen von Heteroaromaten oder ihren Dihydrostufen unter intermediärer Ringöffnung, Zeitschrift für Chemie, 9 (1969) 241–252. [Google Scholar]
- [20].Elkhadem HS, Kawai J, Swartz DL, Synthesis and rearrangements of imidazolo-diazines and triazolo-diazines, Heterocycles, 28 (1989) 239–248. [Google Scholar]
- [21].Song L, Tian X, Lv Z, Li E, Wu J, Liu Y, Yu W, Chang J, I2/KI-Mediated Oxidative N-N Bond Formation for the Synthesis of 1,5-Fused 1,2,4-Triazoles from N-Aryl Amidines, J. Org. Chem, 80 (2015) 7219–7225. [DOI] [PubMed] [Google Scholar]
- [22].Bartels B, Bolas CG, Cueni P, Fantasia S, Gaeng N, Trita AS, Cu-catalyzed aerobic oxidative cyclization of guanidylpyridines and derivatives, J Org Chem, 80 (2015) 1249–1257. [DOI] [PubMed] [Google Scholar]
- [23].Patani GA, LaVoie EJ, Bioisosterism: A Rational Approach in Drug Design, Chem. Rev, 96 (1996) 3147–3176. [DOI] [PubMed] [Google Scholar]
- [24].Revankar GR, Robins RK, Tolman RL, s-Triazolo[1,5-a]pyrimidine nucleosides. Site of N-glycosylation studies and the synthesis of an N-bridgehead guanosine analog, J. Org. Chem, 39 (1974) 1256–1262. [DOI] [PubMed] [Google Scholar]
- [25].Khalymbadzha IA, Deev SL, Shestakova TS, Rusinov VL, Chupakhin ON, Synthesis of acyclic nucleoside analogues based on 1,2,4-triazolo[1,5-a]pyrimidine-7-ones by one-step Vorbrüggen glycosylation, Chimica Techno. Acta, 2 (2015) 158–160. [Google Scholar]
- [26].Richardson CM, Williamson DS, Parratt MJ, Borgognoni J, Cansfield AD, Dokurno P, Francis GL, Howes R, Moore JD, Murray JB, Robertson A, Surgenor AE, Torrance CJ, Triazolo[1,5-a]pyrimidines as novel CDK2 inhibitors: Protein structure-guided design and SAR, Bioorg. Med. Chem. Lett, 16 (2006) 1353–1357. [DOI] [PubMed] [Google Scholar]
- [27].Asghar U, Witkiewicz AK, Turner NC, Knudsen ES, The history and future of targeting cyclin-dependent kinases in cancer therapy, Nat. Rev. Drug Discov, 14 (2015) 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sanchez RM, Erhard K, Hardwicke MA, Lin H, McSurdy-Freed J, Plant R, Raha K, Rominger CM, Schaber MD, Spengler MD, Moore ML, Yu H, Luengo JI, Tedesco R, Rivero RA, Synthesis and structure–activity relationships of 1,2,4-triazolo[1,5-a]pyrimidin-7(3H)-ones as novel series of potent β isoform selective phosphatidylinositol 3-kinase inhibitors, Bioorg. Med. Chem. Lett, 22 (2012) 3198–3202. [DOI] [PubMed] [Google Scholar]
- [29].Griffith EC, Su Z, Turk BE, Chen S, Chang Y-H, Wu Z, Biemann K, Liu JO, Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin, Chem. Biol, 4 (1997) 461–471. [DOI] [PubMed] [Google Scholar]
- [30].Tedman E, Scott F, Thomas Philip R, Haizhen AZ, David G, Jack A, Bowen JP, Methionine AminoPeptidase Type-2 Inhibitors Targeting Angiogenesis, Curr. Top. Med. Chem, 16 (2016) 1478–1488. [DOI] [PubMed] [Google Scholar]
- [31].Bernier SG, Taghizadeh N, Thompson CD, Westlin WF, H. G., Methionine aminopeptidases type I and type II are essential to control cell proliferation, J. Cell. Biochem, 95 (2005) 1191–1203. [DOI] [PubMed] [Google Scholar]
- [32].Wang J, Tucker LA, Stavropoulos J, Zhang Q, Wang Y-C, Bukofzer G, Niquette A, Meulbroek JA, Barnes DM, Shen J, Bouska J, Donawho C, Sheppard GS, Bell RL, Correlation of tumor growth suppression and methionine aminopetidase-2 activity blockade using an orally active inhibitor, Proc. Natl. Acad. Sci, 105 (2008) 1838–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Heinrich T, Buchstaller H-P, Cezanne B, Rohdich F, Bomke J, Friese-Hamim M, Krier M, Knöchel T, Musil D, Leuthner B, Zenke F, Novel reversible methionine aminopeptidase-2 (MetAP-2) inhibitors based on purine and related bicyclic templates, Bioorg. Med. Chem. Lett, 27 (2017) 551–556. [DOI] [PubMed] [Google Scholar]
- [34].Lan H, Cheng CC, Kowalski TJ, Pang L, Shan L, Chuang C-C, Jackson J, Rojas-Triana A, Bober L, Liu L, Voigt J, Orth P, Yang X, Shipps GW, Hedrick JA, Small-molecule inhibitors of FABP4/5 ameliorate dyslipidemia but not insulin resistance in mice with diet-induced obesity, J. Lipid Res, 52 (2011) 646–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Furuhashi M, Hotamisligil GS, Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets, Nat. Rev. Drug Discov, 7 (2008) 489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Floresta G, Pistarà V, Amata E, Dichiara M, Marrazzo A, Prezzavento O, Rescifina A, Adipocyte fatty acid binding protein 4 (FABP4) inhibitors. A comprehensive systematic review, Eur. J. Med. Chem, 138 (2017) 854–873. [DOI] [PubMed] [Google Scholar]
- [37].Ballatore C, Huryn DM, Smith AB III, Carboxylic acid (bio)isosteres in drug design, ChemMedChem, 8 (2013) 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nishigori K, Temma T, Onoe S, Sampei S, Kimura I, Ono M, Saji H, Development of a radioiodinated triazolopyrimidine probe for nuclear medical imaging of fatty acid binding protein 4, PloS one, 9 (2014) e94668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Muller S, Filippakopoulos P, Knapp S, Bromodomains as therapeutic targets, Expert Rev. Mol. Med, 13 (2011) e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fu LL, Tian M, Li X, Li JJ, Huang J, Ouyang L, Zhang Y, Liu B, Inhibition of BET bromodomains as a therapeutic strategy for cancer drug discovery, Oncotarget, 6 (2015) 5501–5516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vidler LR, Filippakopoulos P, Fedorov O, Picaud S, Martin S, Tomsett M, Woodward H, Brown N, Knapp S, Hoelder S, Discovery of novel small-molecule inhibitors of BRD4 using structure-based virtual screening, J. Med. Chem, 56 (2013) 8073–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Raux B, Voitovich Y, Derviaux C, Lugari A, Rebuffet E, Milhas S, Priet S, Roux T, Trinquet E, Guillemot JC, Knapp S, Brunel JM, Fedorov AY, Collette Y, Roche P, Betzi S, Combes S, Morelli X, Exploring Selective Inhibition of the First Bromodomain of the Human Bromodomain and Extra-terminal Domain (BET) Proteins, J. Med. Chem, 59 (2016) 1634–1641. [DOI] [PubMed] [Google Scholar]
- [43].Li H, Tatlock J, Linton A, Gonzalez J, Borchardt A, Dragovich P, Jewell T, Prins T, Zhou R, Blazel J, Parge H, Love R, Hickey M, Doan C, Shi S, Duggal R, Lewis C, Fuhrman S, Identification and structure-based optimization of novel dihydropyrones as potent HCV RNA polymerase inhibitors, Bioorg. Med. Chem. Lett, 16 (2006) 4834–4838. [DOI] [PubMed] [Google Scholar]
- [44].Li H, Linton A, Tatlock J, Gonzalez J, Borchardt A, Abreo M, Jewell T, Patel L, Drowns M, Ludlum S, Goble M, Yang M, Blazel J, Rahavendran R, Skor H, Shi S, Lewis C, Fuhrman S, Allosteric inhibitors of hepatitis C polymerase: discovery of potent and orally bioavailable carbon-linked dihydropyrones, J. Med. Chem, 50 (2007) 3969–3972. [DOI] [PubMed] [Google Scholar]
- [45].Li H, Tatlock J, Linton A, Gonzalez J, Jewell T, Patel L, Ludlum S, Drowns M, Rahavendran SV, Skor H, Hunter R, Shi ST, Herlihy KJ, Parge H, Hickey M, Yu X, Chau F, Nonomiya J, Lewis C, Discovery of (R)-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (PF-00868554) as a potent and orally available hepatitis C virus polymerase inhibitor, J. Med. Chem, 52 (2009) 1255–1258. [DOI] [PubMed] [Google Scholar]
- [46].Huang B, Li C, Chen W, Liu T, Yu M, Fu L, Sun Y, Liu H, De Clercq E, Pannecouque C, Balzarini J, Zhan P, Liu X, Fused heterocycles bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 3: optimization of [1,2,4]triazolo[1,5-a]pyrimidine core via structure-based and physicochemical property-driven approaches, Eur. J. Med. Chem, 92 (2015) 754–765. [DOI] [PubMed] [Google Scholar]
- [47].Wang L, Tian Y, Chen W, Liu H, Zhan P, Li D, Liu H, De Clercq E, Pannecouque C, Liu X, Fused heterocycles bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 2: discovery of novel [1,2,4]Triazolo[1,5-a]pyrimidines using a structure-guided core-refining approach, Eur. J. Med. Chem, 85 (2014) 293–303. [DOI] [PubMed] [Google Scholar]
- [48].Tian Y, Du D, Rai D, Wang L, Liu H, Zhan P, De Clercq E, Pannecouque C, Liu X, Fused heterocyclic compounds bearing bridgehead nitrogen as potent HIV-1 NNRTIs. Part 1: Design, synthesis and biological evaluation of novel 5,7-disubstituted pyrazolo[1,5-a]pyrimidine derivatives, Bioorg. Med. Chem, 22 (2014) 2052–2059. [DOI] [PubMed] [Google Scholar]
- [49].Suzuki T, Ainai A, Nagata N, Sata T, Sawa H, Hasegawa H, A novel function of the N-terminal domain of PA in assembly of influenza A virus RNA polymerase, Biochem. Biophys. Res. Commun, 414 (2011) 719–726. [DOI] [PubMed] [Google Scholar]
- [50].Yamada K, Koyama H, Hagiwara K, Ueda A, Sasaki Y, Kanesashi SN, Ueno R, Nakamura HK, Kuwata K, Shimizu K, Suzuki M, Aida Y, Identification of a novel compound with antiviral activity against influenza A virus depending on PA subunit of viral RNA polymerase, Microbes Infect, 14 (2012) 740–747. [DOI] [PubMed] [Google Scholar]
- [51].Muratore G, Goracci L, Mercorelli B, Foeglein A, Digard P, Cruciani G, Palu G, Loregian A, Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase, Proc. Natl. Acad. Sci. U. S. A, 109 (2012) 6247–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Massari S, Nannetti G, Goracci L, Sancineto L, Muratore G, Sabatini S, Manfroni G, Mercorelli B, Cecchetti V, Facchini M, Palu G, Cruciani G, Loregian A, Tabarrini O, Structural investigation of cycloheptathiophene-3-carboxamide derivatives targeting influenza virus polymerase assembly, J. Med. Chem, 56 (2013) 10118–10131. [DOI] [PubMed] [Google Scholar]
- [53].Pagano M, Castagnolo D, Bernardini M, Fallacara AL, Laurenzana I, Deodato D, Kessler U, Pilger B, Stergiou L, Strunze S, Tintori C, Botta M, The fight against the influenza A virus H1N1: synthesis, molecular modeling, and biological evaluation of benzofurazan derivatives as viral RNA polymerase inhibitors, ChemMedChem, 9 (2014) 129–150. [DOI] [PubMed] [Google Scholar]
- [54].Stubbs TM, Te Velthuis AJ, The RNA-dependent RNA polymerase of the influenza A virus, Future Virol, 9 (2014) 863–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Obayashi E, Yoshida H, Kawai F, Shibayama N, Kawaguchi A, Nagata K, Tame JR, Park SY, The structural basis for an essential subunit interaction in influenza virus RNA polymerase, Nature, 454 (2008) 1127–1131. [DOI] [PubMed] [Google Scholar]
- [56].He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, Ge R, Rao Z, Liu Y, Crystal structure of the polymerase PA(C)-PB1(N) complex from an avian influenza H5N1 virus, Nature, 454 (2008) 1123–1126. [DOI] [PubMed] [Google Scholar]
- [57].Lepri S, Nannetti G, Muratore G, Cruciani G, Ruzziconi R, Mercorelli B, Palu G, Loregian A, Goracci L, Optimization of small-molecule inhibitors of influenza virus polymerase: from thiophene-3-carboxamide to polyamido scaffolds, J. Med. Chem, 57 (2014) 4337–4350. [DOI] [PubMed] [Google Scholar]
- [58].Massari S, Nannetti G, Desantis J, Muratore G, Sabatini S, Manfroni G, Mercorelli B, Cecchetti V, Palu G, Cruciani G, Loregian A, Goracci L, Tabarrini O, A Broad Anti-influenza Hybrid Small Molecule That Potently Disrupts the Interaction of Polymerase Acidic Protein-Basic Protein 1 (PA-PB1) Subunits, J. Med. Chem, 58 (2015) 3830–3842. [DOI] [PubMed] [Google Scholar]
- [59].Tee EHL, Karoli T, Ramu S, Huang JX, Butler MS, Cooper MA, Synthesis of Essramycin and Comparison of Its Antibacterial Activity, J. Nat. Prod, 73 (2010) 1940–1942. [DOI] [PubMed] [Google Scholar]
- [60].Wang H, Hesek D, Lee M, Lastochkin E, Oliver AG, Chang M, Mobashery S, The Natural Product Essramycin and Three of Its Isomers Are Devoid of Antibacterial Activity, J. Nat. Prod, 79 (2016) 1219–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Wang H, Lee M, Peng Z, Blazquez B, Lastochkin E, Kumarasiri M, Bouley R, Chang M, Mobashery S, Synthesis and evaluation of 1,2,4-triazolo[1,5-a]pyrimidines as antibacterial agents against Enterococcus faecium, J. Med. Chem, 58 (2015) 4194–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Treitman AN, Yarnold PR, Warren J, Noskin GA, Emerging incidence of Enterococcus faecium among hospital isolates (1993 to 2002), J. Clin. Microbiol, 43 (2005) 462–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Lebar MD, May JM, Meeske AJ, Leiman SA, Lupoli TJ, Tsukamoto H, Losick R, Rudner DZ, Walker S, Kahne D, Reconstitution of peptidoglycan cross-linking leads to improved fluorescent probes of cell wall synthesis, J. Am. Chem. Soc, 136 (2014) 10874–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Zuniga ES, Korkegian A, Mullen S, Hembre EJ, Ornstein PL, Cortez G, Biswas K, Kumar N, Cramer J, Masquelin T, Hipskind PA, Odingo J, Parish T, The synthesis and evaluation of triazolopyrimidines as anti-tubercular agents, Bioorg. Med. Chem, 25 (2017) 3922–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bhatt JD, Chudasama CJ, Patel KD, Pyrazole clubbed triazolo[1,5-a]pyrimidine hybrids as an anti-tubercular agents: Synthesis, in vitro screening and molecular docking study, Bioorg. Med. Chem, 23 (2015) 7711–7716. [DOI] [PubMed] [Google Scholar]
- [66].McCourt JA, Duggleby RG, Acetohydroxyacid synthase and its role in the biosynthetic pathway for branched-chain amino acids, Amino Acids, 31 (2006) 173–210. [DOI] [PubMed] [Google Scholar]
- [67].Grandoni JA, Marta PT, Schloss JV, Inhibitors of branched-chain amino acid biosynthesis as potential antituberculosis agents, J. Antimicrob. Chemother, 42 (1998) 475–482. [DOI] [PubMed] [Google Scholar]
- [68].Sohn H, Lee K-S, Ko Y-K, Ryu J-W, Woo J-C, Koo D-W, Shin S-J, Ahn S-J, Shin AR, Song C-H, Jo E-K, Park J-K, Kim H-J, In vitro and ex vivo activity of new derivatives of acetohydroxyacid synthase inhibitors against Mycobacterium tuberculosis and non-tuberculous mycobacteria, Int. J. Antimicrob. Agents, 31 (2008) 567–571. [DOI] [PubMed] [Google Scholar]
- [69].Zohar Y, Einav M, Chipman DM, Barak Z.e., Acetohydroxyacid synthase from Mycobacterium avium and its inhibition by sulfonylureas and imidazolinones, Biochim. Biophys. Acta. Proteins Proteom, 1649 (2003) 97–105. [DOI] [PubMed] [Google Scholar]
- [70].Wang D, Zhu X, Cui C, Dong M, Jiang H, Li Z, Liu Z, Zhu W, Wang JG, Discovery of novel acetohydroxyacid synthase inhibitors as active agents against Mycobacterium tuberculosis by virtual screening and bioassay, J. Chem. Inf. Model, 53 (2013) 343–353. [DOI] [PubMed] [Google Scholar]
- [71].Singh V, Chandra D, Srivastava BS, Srivastava R, Biochemical and transcription analysis of acetohydroxyacid synthase isoforms in Mycobacterium tuberculosis identifies these enzymes as potential targets for drug development, Microbiology, 157 (2011) 29–37. [DOI] [PubMed] [Google Scholar]
- [72].Kleschick WA, Costales MJ, Dunbar JE, Meikle RW, Monte WT, Pearson NR, Snider SW, Vinogradoff AP, New herbicidal derivatives of 1,2,4-triazolo [1,5-a] pyrimidine, Pest. Sci, 29 (1990) 341–355. [Google Scholar]
- [73].Chen C-N, Chen Q, Liu Y-C, Zhu X-L, Niu C-W, Xi Z, Yang G-F, Syntheses and herbicidal activity of new triazolopyrimidine-2-sulfonamides as acetohydroxyacid synthase inhibitor, Bioorg. Med. Chem, 18 (2010) 4897–4904. [DOI] [PubMed] [Google Scholar]
- [74].Chen CN, Lv LL, Ji FQ, Chen Q, Xu H, Niu CW, Xi Z, Yang GF, Design and synthesis of N-2,6-difluorophenyl-5-methoxyl-1,2,4-triazolo[1,5-a]-pyrimidine-2-sulfonamide as acetohydroxyacid synthase inhibitor, Bioorg. Med. Chem, 17 (2009) 3011–3017. [DOI] [PubMed] [Google Scholar]
- [75].Jung IP, Ha NR, Lee SC, Ryoo SW, Yoon MY, Development of potent chemical antituberculosis agents targeting Mycobacterium tuberculosis acetohydroxyacid synthase, Int. J. Antimicrob. Agents, 48 (2016) 247–258. [DOI] [PubMed] [Google Scholar]
- [76].Deng X, Gujjar R, El Mazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA, Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds, J. Biol. Chem, 284 (2009) 26999–27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK, Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite Plasmodium falciparum, J. Med. Chem, 51 (2008) 3649–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jiménez-Díaz MB, Gamo F-J, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA, Structure-Guided Lead Optimization of Triazolopyrimidine-Ring Substituents Identifies Potent Plasmodium falciparum Dihydroorotate Dehydrogenase Inhibitors with Clinical Candidate Potential, J. Med. Chem, 54 (2011) 5540–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK, Lead Optimization of Aryl and Aralkyl Amine-Based Triazolopyrimidine Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase with Antimalarial Activity in Mice, J. Med. Chem, 54 (2011) 3935–3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Phillips MA, White KL, Kokkonda S, Deng X, White J, El Mazouni F, Marsh K, Tomchick DR, Manjalanagara K, Rudra KR, Wirjanata G, Noviyanti R, Price RN, Marfurt J, Shackleford DM, Chiu FC, Campbell M, Jimenez-Diaz MB, Bazaga SF, Angulo-Barturen I, Martinez MS, Lafuente-Monasterio M, Kaminsky W, Silue K, Zeeman AM, Kocken C, Leroy D, Blasco B, Rossignol E, Rueckle T, Matthews D, Burrows JN, Waterson D, Palmer MJ, Rathod PK, Charman SA, A Triazolopyrimidine-Based Dihydroorotate Dehydrogenase Inhibitor with Improved Drug-like Properties for Treatment and Prevention of Malaria, ACS Infect. Dis, 2 (2016) 945–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gujjar R, Marwaha A, El Mazouni F, White J, White KL, Creason S, Shackleford DM, Baldwin J, Charman WN, Buckner FS, Charman S, Rathod PK, Phillips MA, Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice, J. Med. Chem, 52 (2009) 1864–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Khare S, Nagle AS, Biggart A, Lai YH, Liang F, Davis LC, Barnes SW, Mathison CJN, Myburgh E, Gao M-Y, Gillespie JR, Liu X, Tan JL, Stinson M, Rivera IC, Ballard J, Yeh V, Groessl T, Federe G, Koh HXY, Venable JD, Bursulaya B, Shapiro M, Mishra PK, Spraggon G, Brock A, Mottram JC, Buckner FS, Rao SPS, Wen BG, Walker JR, Tuntland T, Molteni V, Glynne RJ, Supek F, Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness, Nature, 537 (2016) 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Gundersen GG, Cook TA, Microtubules and signal transduction, Curr. Op. Cell Biol, 11 (1999) 81–94. [DOI] [PubMed] [Google Scholar]
- [84].Desai A, Mitchison TJ, Microtubule polymerization dynamics, Annu. Rev. Cell. Dev. Biol, 13 (1997) 83–117. [DOI] [PubMed] [Google Scholar]
- [85].Jordan MA, Wilson L, Microtubules as a target for anticancer drugs, Nat. Rev. Cancer, 4 (2004) 253–265. [DOI] [PubMed] [Google Scholar]
- [86].Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS, Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization, J. Biol. Chem, 272 (1997) 17118–17125. [DOI] [PubMed] [Google Scholar]
- [87].Horwitz SB, Lothstein L, Manfredi JJ, Mellado W, Parness J, Roy SN, Schiff PB, Sorbara L, Zeheb R, Taxol: mechanisms of action and resistance, Ann. N. Y. Acad. Sci, 466 (1986) 733–744. [DOI] [PubMed] [Google Scholar]
- [88].Pees K-J, Albert G, Triazolopyrimidine derivatives with fungicidal activity, European Patent EP0550113, 1993. [Google Scholar]
- [89].Zhang N, Ayral-Kaloustian S, Nguyen T, Afragola J, Hernandez R, Lucas J, Gibbons J, Beyer C, Synthesis and SAR of [1,2,4]triazolo[1,5-a]pyrimidines, a class of anticancer agents with a unique mechanism of tubulin inhibition, J. Med. Chem, 50 (2007) 319–327. [DOI] [PubMed] [Google Scholar]
- [90].Beyer CF, Zhang N, Hernandez R, Vitale D, Lucas J, Nguyen T, Discafani C, Ayral-Kaloustian S, Gibbons JJ, TTI-237: a novel microtubule-active compound with in vivo antitumor activity, Cancer Res, 68 (2008) 2292–2300. [DOI] [PubMed] [Google Scholar]
- [91].Wang-Gillam A, Arnold SM, Bukowski RM, Rothenberg ML, Cooper W, Wang KK, Gauthier E, Lockhart AC, A phase I dose escalation study of TTI-237 in patients with advanced malignant solid tumors, Invest. New Drugs, 30 (2012) 266–272. [DOI] [PubMed] [Google Scholar]
- [92].Ayral-Kaloustian S, Zhang N, Beyer C, Cevipabulin (TTI-237): preclinical and clinical results for a novel antimicrotubule agent, Methods Find. Exp. Clin. Pharmacol, 31 (2009) 443–447. [DOI] [PubMed] [Google Scholar]
- [93].Saez-Calvo G, Sharma A, Balaguer FA, Barasoain I, Rodriguez-Salarichs J, Olieric N, Munoz-Hernandez H, Berbis MA, Wendeborn S, Penalva MA, Matesanz R, Canales A, Prota AE, Jimenez-Barbero J, Andreu JM, Lamberth C, Steinmetz MO, Diaz JF, Triazolopyrimidines Are Microtubule-Stabilizing Agents that Bind the Vinca Inhibitor Site of Tubulin, Cell Chem. Biol, 24 (2017) 737–750 e736. [DOI] [PubMed] [Google Scholar]
- [94].Beyer CF, Zhang N, Hernandez R, Vitale D, Nguyen T, Ayral-Kaloustian S, Gibbons JJ, The microtubule-active antitumor compound TTI-237 has both paclitaxel-like and vincristine-like properties, Cancer Chemother. Pharmacol, 64 (2009) 681–689. [DOI] [PubMed] [Google Scholar]
- [95].Fukushima N, Furuta D, Hidaka Y, Moriyama R, Tsujiuchi T, Post-translational modifications of tubulin in the nervous system, J. Neurochem, 109 (2009) 683–693. [DOI] [PubMed] [Google Scholar]
- [96].Kovalevich J, Cornec AS, Yao Y, James M, Crowe A, Lee VM, Trojanowski JQ, Smith AB, Ballatore C, Brunden KR, Characterization of brain-penetrant pyrimidine-containing molecules with differential microtubule-stabilizing activities developed as potential therapeutic agents for Alzheimer’s disease and related tauopathies, J. Pharmacol. Exp. Ther, 357 (2016) 432–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Oukoloff K, Kovalevich J, Cornec AS, Yao Y, Owyang ZA, James M, Trojanowski JQ, Lee VM, Smith AB III, Brunden KR, Ballatore C, Design, synthesis and evaluation of photoactivatable derivatives of microtubule (MT)-active [1,2,4]triazolo[1,5-a]pyrimidines, Bioorg. Med. Chem. Lett, 28 (2018) 2180–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lee VMY, Goedert M, Trojanowski JQ, Neurodegenerative Tauopathies, Annu. Rev. Neurosci, 24 (2001) 1121–1159. [DOI] [PubMed] [Google Scholar]
- [99].Lou K, Yao Y, Hoye AT, James MJ, Cornec AS, Hyde E, Gay B, Lee VM, Trojanowski JQ, Smith AB III, Brunden KR, Ballatore C, Brain-penetrant, orally bioavailable microtubule-stabilizing small molecules are potential candidate therapeutics for Alzheimer’s disease and related tauopathies, J. Med. Chem, 57 (2014) 6116–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gomez L, Massari ME, Vickers T, Freestone G, Vernier W, Ly K, Xu R, McCarrick M, Marrone T, Metz M, Yan YG, Yoder ZW, Lemus R, Broadbent NJ, Barido R, Warren N, Schmelzer K, Neul D, Lee D, Andersen CB, Sebring K, Aertgeerts K, Zhou X, Tabatabaei A, Peters M, Breitenbucher JG, Design and Synthesis of Novel and Selective Phosphodiesterase 2 (PDE2a) Inhibitors for the Treatment of Memory Disorders, J. Med. Chem, 60 (2017) 2037–2051. [DOI] [PubMed] [Google Scholar]
- [101].Monti L, Wang SC, Oukoloff K, Smith AB III, Brunden KR, Caffrey CR, Ballatore C, Brain-Penetrant Triazolopyrimidine and Phenylpyrimidine Microtubule Stabilizers as Potential Leads to Treat Human African Trypanosomiasis, ChemMedChem, 13 (2018) 1751–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Ballatore C, Smith AB III, Lee VM-Y, Trojanowski JQ, Brunden KR, Microtubule-Stabilizing Agents for Alzheimer’s and Other Tauopathies, in: Wlfe MS (Ed.), Alzheimer’s Disease II, Springer Berlin Heidelberg, Berlin, Heidelberg, 2016, pp. 1–21. [Google Scholar]
- [103].Ballatore C, Brunden KR, Huryn DM, Trojanowski JQ, Lee VMY, Smith AB III, Microtubule Stabilizing Agents as Potential Treatment for Alzheimer’s Disease and Related Neurodegenerative Tauopathies, J. Med. Chem, 55 (2012) 8979–8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Zhang B, Yao Y, Cornec A-S, Oukoloff K, James MJ, Koivula P, Trojanowski JQ, Smith AB III, Lee VM-Y, Ballatore C, Brunden KR, A brain-penetrant triazolopyrimidine enhances microtubule-stability, reduces axonal dysfunction and decreases tau pathology in a mouse tauopathy model, Mol. Neurodegener, 13 (2018) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Brunden KR, Zhang B, Carroll J, Yao Y, Potuzak JS, Hogan AM, Iba M, James MJ, Xie SX, Ballatore C, Smith AB III, Lee VM, Trojanowski JQ, Epothilone D improves microtubule density, axonal integrity, and cognition in a transgenic mouse model of tauopathy, J. Neurosci, 30 (2010) 13861–13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zhang B, Carroll J, Trojanowski JQ, Yao Y, Iba M, Potuzak JS, Hogan AM, Xie SX, Ballatore C, Smith AB III, Lee VM, Brunden KR, The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice, J. Neurosci, 32 (2012) 3601–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Makani V, Zhang B, Han H, Yao Y, Lassalas P, Lou K, Paterson I, Lee VMY, Trojanowski JQ, Ballatore C, Smith AB III, Brunden KR, Evaluation of the brain-penetrant microtubule-stabilizing agent, dictyostatin, in the PS19 tau transgenic mouse model of tauopathy, Acta Neuropathol. Commun, 4 (2016) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Barten DM, Fanara P, Andorfer C, Hoque N, Wong PYA, Husted KH, Cadelina GW, DeCarr LB, Yang L, Liu L, Fessler C, Protassio J, Riff T, Turner H, Janus CG, Sankaranarayanan S, Polson C, Meredith JE, Gray G, Hanna A, Olson RE, Kim SH, Vite GD, Lee FY, Albright CF, Hyperdynamic microtubules, cognitive deficits, and pathology are improved in tau transgenic mice with low doses of the microtubule-stabilizing agent BMS-241027 J. Neurosci, 32 (2012) 7137–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Brunden KR, Lee VM, Smith AB III, Trojanowski JQ, Ballatore C, Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs, Neurobiol. Dis, 105 (2016) 328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kumar J, Meena P, Singh A, Jameel E, Maqbool M, Mobashir M, Shandilya A, Tiwari M, Hoda N, Jayaram B, Synthesis and screening of triazolopyrimidine scaffold as multi-functional agents for Alzheimer’s disease therapies, Eur. J. Med. Chem, 119 (2016) 260–277. [DOI] [PubMed] [Google Scholar]
- [111].Jameel E, Meena P, Maqbool M, Kumar J, Ahmed W, Mumtazuddin S, Tiwari M, Hoda N, Jayaram B, Rational design, synthesis and biological screening of triazine-triazolopyrimidine hybrids as multitarget anti-Alzheimer agents, Eur. J. Med. Chem, 136 (2017) 36–51. [DOI] [PubMed] [Google Scholar]
- [112].Lewis WG, Green LG, Grynszpan F, Radić Z, Carlier PR, Taylor P, Finn MG, Sharpless KB, Click Chemistry In Situ: Acetylcholinesterase as a Reaction Vessel for the Selective Assembly of a Femtomolar Inhibitor from an Array of Building Blocks, Angew. Chem. Int. Ed, 41 (2002) 1053–1057. [DOI] [PubMed] [Google Scholar]
- [113].Frey U, Huang YY, Kandel ER, Effects of cAMP simulate a late stage of LTP in hippocampal CA1 neurons, Science, 260 (1993) 1661–1664. [DOI] [PubMed] [Google Scholar]
- [114].Xu Y, Zhang HT, O’Donnell JM, Phosphodiesterases in the central nervous system: implications in mood and cognitive disorders, Handb. Exp. Pharmacol, (2011) 447–485. [DOI] [PubMed] [Google Scholar]
- [115].Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J, Selective phosphodiesterase inhibitors: a promising target for cognition enhancement, Psychopharmacology, 202 (2009) 419–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Trabanco AA, Buijnsters P, Rombouts FJ, Towards selective phosphodiesterase 2A (PDE2A) inhibitors: a patent review (2010 - present), Expert Opin. Ther. Pat, 26 (2016) 933–946. [DOI] [PubMed] [Google Scholar]
- [117].Porter DW, Bradley M, Brown Z, Canova R, Charlton S, Cox B, Hunt P, Kolarik D, Lewis S, O’Connor D, Reilly J, Spanka C, Tedaldi L, Watson SJ, Wermuth R, Press NJ, The discovery of potent, orally bioavailable pyrazolo and triazolopyrimidine CXCR2 receptor antagonists, Bioorg. Med. Chem. Lett, 24 (2014) 72–76. [DOI] [PubMed] [Google Scholar]
- [118].Aghazadeh Tabrizi M, Baraldi PG, Ruggiero E, Saponaro G, Baraldi S, Poli G, Tuccinardi T, Ravani A, Vincenzi F, Borea PA, Varani K, Synthesis and structure activity relationship investigation of triazolo[1,5-a]pyrimidines as CB2 cannabinoid receptor inverse agonists, Eur. J. Med. Chem, 113 (2016) 11–27. [DOI] [PubMed] [Google Scholar]
- [119].Chang L, Xiao M, Yang L, Wang S, Wang S-Q, Bender A, Hu A, Chen Z-S, Yu B, Liu H-M, Discovery of a non-toxic [1,2,4]triazolo[1,5-a]pyrimidin-7-one (WS-10) that modulates ABCB1-mediated multidrug resistance (MDR), Bioorg. Med. Chem, 26 (2018) 5006–5017. [DOI] [PubMed] [Google Scholar]