

Abstract
Objective
Ketohexokinase (KHK), a primary enzyme in fructose metabolism, has two isoforms, namely, KHK-A and KHK-C. Previously, we reported that renal injury was reduced in streptozotocin-induced diabetic mice which lacked both isoforms. Although both isoforms express in kidney, it has not been elucidated whether each isoform plays distinct roles in the development of diabetic kidney disease (DKD). The aim of the study is to elucidate the role of KHK-A for DKD progression.
Materials and Methods
Diabetes was induced by five consecutive daily intraperitoneal injections of streptozotocin (50 mg/kg) in C57BL/6J wild-type mice, mice lacking KHK-A alone (KHK-A KO), and mice lacking both KHK-A and KHK-C (KHK-A/C KO). At 35 weeks, renal injury, inflammation, hypoxia, and oxidative stress were examined. Metabolomic analysis including polyol pathway, fructose metabolism, glycolysis, TCA (tricarboxylic acid) cycle, and NAD (nicotinamide adenine dinucleotide) metabolism in kidney and urine was done.
Results
Diabetic KHK-A KO mice developed severe renal injury compared to diabetic wild-type mice, and this was associated with further increases of intrarenal fructose, dihydroxyacetone phosphate (DHAP), TCA cycle intermediates levels, and severe inflammation. In contrast, renal injury was prevented in diabetic KHK-A/C KO mice compared to both wild-type and KHK-A KO diabetic mice. Further, diabetic KHK-A KO mice contained decreased renal NAD+ level with the increase of renal hypoxia-inducible factor 1-alpha expression despite having increased renal nicotinamide (NAM) level.
Conclusion
These results suggest that KHK-C might play a deleterious role in DKD progression through endogenous fructose metabolism, and that KHK-A plays a unique protective role against the development of DKD.
Keywords: Diabetic kidney disease, Fructose metabolism, Ketohexokinase, Tubular injury, Oxidative stress, NAD metabolism
1. Introduction
Incidence of diabetes is increasing worldwide, and diabetic kidney disease (DKD) is the leading cause of end-stage renal disease [1]. Multiple mechanisms for the pathogenesis of DKD including polyol pathway [2, 3], hexosamine pathway [4, 5], protein kinase C pathway [6, 7], and involvement of advanced glycation end-products [8, 9] have been identified. However, with current treatment strategies for DKD such as management of hyperglycemia, dyslipidemia, and blood pressure by blocking renin– angiotensin–aldosterone system, a portion of DKD patients still reach end-stage renal disease. Therefore, further understanding of the pathogenesis of DKD is warranted.
Intake of dietary fructose from sucrose and high fructose corn syrup has dramatically increased in the last hundred years, and epidemiologically associated with the increased incidence of obesity, metabolic syndrome, and diabetes [1]. Fructose is primarily metabolized by fructokinase (also known as ketohexokinase, KHK) that has two isoforms, namely, KHK-A and KHK-C. The gene encoding KHK in humans and mice comprise nine exons, of which exon 3a or 3c are subjected to splicing into mRNA that encodes KHK-A or KHK-C, respectively [10]. Previously, we reported that dietary fructose-induced obesity, hyperinsulinemia, and hepatic steatosis were prevented in mice lacking both KHK-A and KHK-C (KHK-A/C KO mice), but were exacerbated in mice lacking only KHK-A (KHK-A KO mice) [11].
KHK-C is expressed in the liver, intestine, and kidney and rapidly phosphorylates fructose to fructose-1-phosphate (F1P). In the liver, rapid fructose metabolism induces transient intracellular adenosine triphosphate (ATP) depletion, adenosine monophosphate (AMP) generation, and subsequent nucleotide degradation. In contrast, KHK-A is expressed in various tissues, including the kidney [12], and slowly metabolizes fructose without significant ATP consumption. Thus, KHK-A protects against KHK-C-induced metabolic syndrome by reducing hepatic fructose metabolism. A recent study reported that KHK-A also functions as a protein kinase [13]. KHK-A phosphorylates and activates phosphoribosyl pyrophosphate synthetase 1 (PRPS1) to promote pentose phosphate pathway-dependent de novo nucleic acid synthesis and drives hepatocellular carcinoma formation. However, the role of respective isoforms, KHK-A and KHK-C in the kidney has not been elucidated to date.
Dietary fructose induces tubular injury in the kidney [14] and accelerates the progression of chronic kidney disease in a remnant kidney rat model [15]. KHK-dependent fructose metabolism induces monocyte chemoattractant protein-1 (MCP-1), a proinflammatory marker, by activating xanthine oxidase in human proximal tubular HK-2 cells [16]. Fructose can also be generated endogenously from glucose through the polyol pathway, in which glucose is first converted to sorbitol and then to fructose [17]. High serum levels of glucose activate the polyol pathway in diabetes. Previously, we reported that blocking endogenous fructose metabolism by KHK prevented renal injury by the study using diabetic mice lacking both isoforms [18]. However, it is unclear whether KHK-A and KHK-C play different roles in DKD development. In the present study, we investigated whether KHK-A prevented or exacerbated DKD by using two genetically modified mouse models, i.e., KHK-A KO and KHK-A/C KO mouse models.
2. Materials and Methods
2.1. Animals
KHK-A/C KO mice, which lacked both KHK-A and KHK-C, and KHK-A KO mice, which lacked KHK-A alone, were generated using C57BL/6 mice, as described previously [10]. Male wild-type (WT), KHK-A/C KO homozygous mice, and KHK-A KO homozygous mice (age, 8–12 weeks; weight, 24–25 g) were used. The mice were housed every two mice per cage and maintained in temperature- and humidity-controlled specific pathogen-free conditions, with a 12-/12-hour dark/light cycle. DKD was induced by intraperitoneally injecting 50 mg/kg streptozotocin (STZ) (Sigma, St. Louis, MO) once each day for 5 consecutive days. Streptozotocin was dissolved in 10 mM fresh citrate buffer (pH 4.5). The same lot number of streptozotocin was used for all mice. Six-hour fasting blood glucose levels (tail vein) were determined measured using a portable glucose meter ANTSENSE II (Horiba, Kyoto, Japan). Mice with blood glucose levels of >300 mg/dL at 1 week after the last STZ injection were used. Mice were assigned into six groups: diabetic WT mice (n=11), diabetic KHK-A/C KO mice (n=9), diabetic KHK-A KO mice (n=10), control WT mice (n=9), control KHK-A/C KO mice (n=4), and control KHK-A KO mice (n=7). Normal mice (non-diabetic mice) were defined as the control mice in each genotype. All the mice were fed CE-2 diet (CLEA, Japan) and tap water ad libitum. Body weight was measured weekly, and urine samples were collected using metabolic cages at 2–3 weeks before euthanization. Systolic and diastolic blood pressure were measured as a mean value of 20 consecutive measurements by using a tail-cuff sphygmomanometer (BP-98A; Softron, Japan) as manufacturer instruction. All the mice were euthanized after fasting for 6 hours at 22–24 weeks after the last STZ injection. All animal experiments were approved by the Animal Care and Use Committee of the Nagoya University Graduate School of Medicine.
2.2. Biochemical analysis
Urine albumin was measured using Albuwell M (Exocell, Philadelphia, PA), and urine creatinine was measured using an enzymatic method with the Creatinine Assay Kit (Diazyme Laboratories, Poway, CA). Urinary NGAL level was measured using a mouse NGAL assay kit (R&D systems, Minneapolis, MN). Serum creatinine, urea nitrogen, LDL cholesterol, HDL cholesterol, triglyceride, and uric acid levels were measured using an automated chemistry analyzer (LSI Medience, Tokyo, Japan). Serum insulin level was determined using the Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem, Downers Grove, IL).
2.3. Immunohistochemical analysis
Formalin or methyl Carnoy solution-fixed, paraffin-embedded sections were used. Periodic aid-Schiff (PAS) staining, Masson’s trichrome, and Sirius Red (Polyscience Inc., Warrington, PA) were done in all diabetic mice and all control mice. Next, immunohistochemical analysis was performed, as described previously [18]. For antibody information, see the supplemental methods. To evaluate glomerular size, 40 glomeruli in each section were randomly selected and analyzed. To quantitate F4/80 positive area, 20 low-power fields in each section of each mouse were randomly selected and analyzed (Each diabetic group, n = 9-11. Each control group, n = 4-9). Digital images were analyzed using image scope software (Aperio Technologies, Vista, CA).
2.4. Quantitative RT-PCR
Total RNA from the whole kidney was extracted using RNeasy mini kit (Qiagen) and was reverse transcribed using a cDNA synthesis kit (Qiagen). Quantitative PCR of mouse KHK-A, KHK-C (Each control group, n = 4-9), and F4/80 (Each diabetic group, n = 9-11. Each control group, n = 4-9) was performed as described previously [18]. Quantitative PCR were performed using TaqMan® method (Applied Biosystems) as shown in Supplemental methods.
2.5. Oxidative stress marker measurement
Urine lipid peroxide level was measured using a thiobarbituric acid reactive substance (TBARS) assay kit (Cayman Chemicals, Ann Arbor, MI) and was normalized using urine creatinine level. Xanthine oxidase activity in kidney tissues was measured using a xanthine oxidase activity kit (Cayman Chemicals) to investigate upregulations of nucleotide degradation pathway and oxidative stress. Values were normalized using protein concentration measured by the BCA protein assay (Pierce).
2.6. Western blotting analysis
Proteins were extracted from renal cortex tissues using RIPA lysis buffer (Santa Cruz Biotechnology) supplemented with 2 mM PMSF, 2 mM sodium orthovanadate, and protease inhibitor. SDS-PAGE and western blotting were performed as described previously [18]. Immunoblotting was performed by overnight incubation with the following antibodies at 4°C: anti-mouse KHK antibody (1:500; Sigma) and anti-mouse β-actin antibody (1:10000; Sigma), followed by incubation with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody for 1 hour at room temperature (Each diabetic group, n = 9-11). The western blot was performed by three times independently.
2.7. Metabolome analysis
Metabolite extraction from kidney samples were performed as described previously [19]. Urine samples (10 μl) were mixed with 10 μl of Milli-Q water containing internal standards (2 mmol/l each of methionine sulfone and D-camphor 10-sulfonic acid, 3-aminopyrrolidine and trimesic acid) and 80 μl of Milli-Q water. The solution was transferred to a 5-kDa cutoff centrifugal filter tube and directly used for CE-TOFMS analysis. CE-MS-based metabolomic profiling and data analysis were performed essentially as described [20–24]. All diabetic and control mice were analyzed (Each diabetic group, n = 9-11. Each control group, n = 4-9).
2.8. Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis was performed using GraphPad Prism software (GraphPad Software, San Diego, CA). Data without indications were analyzed by student’s t-test. P < 0.05 was considered statistically significant.
3. Results
3.1. Kidney function was worse in diabetic KHK-A KO mice
Diabetes in WT, KHK-A/C KO, and KHK-A KO mice was induced by repeatedly injecting low-dose STZ. Deletion of both KHK-A and KHK-C in KHK-A/C KO mice, and deletion of KHK-A alone in KHK-A KO mice was confirmed by quantitative PCR (Figure 1a, b). Serum and urinary glucose levels were significantly higher and body weight was significantly lower in diabetic mice than in control mice, whereas these parameters were not significantly different among diabetic WT, KHK-A/C KO, and KHK-A KO mice (Table 1). Whereas serum insulin level was not significantly different in all control mice or all diabetic mice, it was significantly decreased in diabetic mice compared to control mice (Table 1), indicating similar degree of diabetes was induced. In addition, energy intake was not significantly different among all diabetic mice (Table 1), although those were higher than in normal WT, KHK-A/C KO, and KHK-A KO mice which were obtained in our previous report [11]. (WT; 10.6 ± 0.4 kcal/day, KHK-A/C KO; 10.0 ± 0.2 kcal/day, KHK-A KO; 10.6 ± 0.4 kcal/day, n=9, mean ± S.E). Kidney weight was significantly higher in diabetic mice than in control mice. However, kidney weight of diabetic WT and KHK-A KO mice was significantly higher than that of diabetic KHK-A/C KO mice (Figure 1c). Furthermore, increased serum creatinine level and decreased creatinine clearance indicated impaired renal function in KHK-A-KO mice compared to those in diabetic WT and KHK-A/C KO mice (Figure 1d, e). Blood pressure was not significantly different among diabetic and control WT, KHK-A/C KO, and KHK-A KO mice (Table 1), indicating blood pressure did not contribute to kidney injury in diabetic WT and KHK-A/C KO mice.
Figure 1.
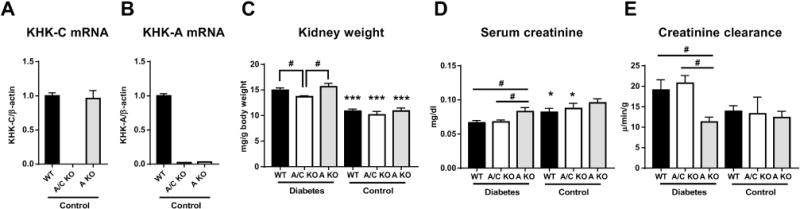
Kidney function and kidney weight of streptozotocin-induced diabetic and control mice. (A, B) Quantitative PCR for mouse KHK-C (A), mouse KHK-A (B) of all control mice. β-actin was used as an internal control. (Each control group, n = 4-9) (C) Kidney weight corrected by body weight. (D) Serum Creatinine levels. (E) Creatinine clearance. (C-E, Each diabetic group, n = 9-11. Each control group, n = 4-9). Data represent means ± SEM. *P < 0.05, *** P < 0.001 versus the respective diabetes group. # P < 0.05.
Table 1.
Characteristics of diabetic and control mice
| WT | A/C KO | A KO | WT | A/C KO | A KO | |
|---|---|---|---|---|---|---|
|
| ||||||
| n = 11 | n = 9 | n = 10 | n = 9 | n = 4 | n = 7 | |
|
|
||||||
| Diabetes | Control | |||||
| Body weight (33w, g) | 27 ± 0.7 | 28 ± 0.3 | 27 ± 0.9*** | 32 ± 0.6 | 31 ± 1.0 | 33 ± 0.8 |
| Δ Body weig ht (33w, g) | +2.25 ± 0.6 | +3.3 ± 0.4 | +1.5 ± 1.0 | |||
| SBP (mmHg) | 108 ± 1.2 | 107 ± 1.3 | 106 ± 1.7 | 108 ± 1.2 | 105 ± 2.3 | 110 ± 1.9 |
| DBP (mmHg) | 79 ± 1.6 | 75 ± 1.7 | 73 ± 1.9 | 85 ± 5.6 | 76 ± 2.9 | 81 ± 1.4 |
| Urinary glucose (mg/min) | 405 ± 69 | 379 ± 48 | 309 ± 44 | 0.24 ± 0.04 | 0.17 ± 0.04 | 0.21 ± 0.02 |
| Urinary fructose (mmol/gCr) | 7.7 ± 0.6*** | 123 ± 13a,*** | 8.0 ± 0.9*** | 0.86 ± 0.4 | 23.0 ± 3.2b | 0.93 ± 0.3 |
| Serum glucose (mg/dl) | 451 ± 25 | 442 ± 29*** | 437 ± 31*** | 142 ± 8.0 | 153 ± 27 | 129 ± 14 |
| Serum uric acid (mg/dl) | 2.7 ± 0.2 | 2.8 ± 0.2 | 3.7 ± 0.5 | 2.5 ± 0.2 | 2.4 ± 0.1 | 2.8 ± 0.3 |
| BUN (mg/dl) | 27.3 ± 1.2 | 25.3 ± 0.7 | 28.2 ± 1.6 | 24.0 ± 0.9 | 26.5 ± 1.2 | 24.0 ± 1.6 |
| Triglyceride (mg/dl) | 43 ± 6.5** | 32 ± 3.2 | 31 ± 6.4 | 18 ± 5.6 | 15 ± 1.7 | 16 ± 2.6 |
| LDL cholesterol (mg/dl) | 9.5 ± 2.4 | 5.4 ± 0.2 | 15.7 ± 5.1 | 5.2 ± 0.3 | 5.2 ± 0.3 | 6.0 ± 0.4 |
| HDL cholesterol (mg/dl) | 66 ± 2.5 | 61 ± 1.7 | 61 ± 8.6 | 61 ± 3.1 | 51 ± 5.8 | 50 ± 6.3 |
| Serum insulin (ng/ml) | 0.20 ± 0.04 | 0.26 ± 0.04 | 0.16 ± 0.03 | 0.61 ± 0.08*** | 0.69 ± 0.09*** | 0.76 ± 0.12 |
| Energy intake (kcal/day) | 19.9 ± 0.4 | 18.1 ± 1.0 | 19.6 ± 0.8 | N/A | N/A | N/A |
SBP, Systolic blood pressure. DBP, Diastolic blood pressure. BUN, blood urea nitrogen. LDL, low density lipoprotein HDL, high density lipoprotein.
p < 0.05,
p < 0.01,
p < 0.001, vs control mice.
, p < 0.001 vs diabetes WT and diabetes A KO.
, p < 0.001 vs control WT and control A KO. N/A, not applicable.
3.2. Evaluation of tubular and glomerular injuries
As mentioned above, kidney function was worse in diabetic KHK-A KO mice. It has been reported that KHK is expressed in the renal proximal tubules by immunohistochemistry. Further, both KHK-A and KHK-C mRNAs are expressed in the kidney [12, 18]. To confirm expressions of KHK-A and KHK-C in the renal proximal tubules, the isolation of renal proximal tubular cells was done. By qPCR, we confirmed expressions of both KHK-A and KHK-C in WT mice, and deletions of both KHK-A and KHK-C in KHK-A/C KO mice, and the deletion of KHK-A alone in KHK-A KO mice in proximal tubules (Supplementary Figure 1). In the present study, pathological changes and tubular damage were evaluated by performing PAS staining and by measuring urinary NGAL level, a known marker of tubular injury. The tubular lumen was significantly dilated in diabetic mice compared to that in respective control mice (Figure 2a, b). Tubular dilation, tubular epithelial cell degeneration, and vacuolization were mainly observed in the renal cortex, and were the most severe in KHK-A KO mice (Figure 2c, and Supplementary Figure 2), followed by that in diabetic WT mice and then in KHK-A/C KO mice. Consistently, urinary NGAL level was increased in all diabetic mice compared to respective control mice, and was increased in both diabetic WT and KHK-A KO mice compared to that in diabetic KHK-A/C KO mice; however, significant increase in urinary NGAL level was only observed in diabetic KHK-A KO mice (Figure 2d).
Figure 2.
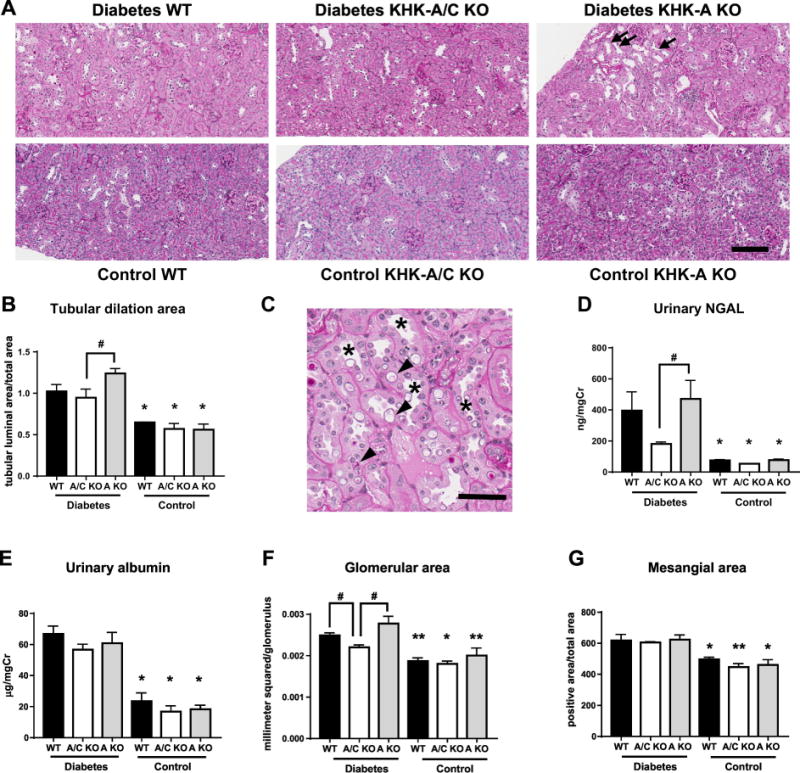
Analysis of Tubular and glomerular injuries of streptozotocin-induced diabetic and control mice. (A) Representative images of Periodic acid-Schiff (PAS) stained kidney. Scale bar = 100 μm. Arrows indicate dilated tubules. (B) Quantification of dilated tubular area. (C) Representative image of higher magnification of PAS stained kidney of diabetic KHK-A KO mice. Scale bar = 50 μm. Asterisks indicate dilated tubules. Arrowheads indicate tubular cell degeneration and vacuolization. (D) Urinary NGAL corrected for urine creatinine. (E) Urinary albumin corrected for urine creatinine. (F, G) Quantification of glomerular size (F) and mesangial area (G). (B, D-G, Each diabetic group, n = 9-11. Each control group, n = 4-9). Digital images were analyzed with quantitative methods using image scope software (Aperio Technologies, Vista, CA). Data represent means ± SEM. *P < 0.05 versus the respective diabetes group. # P < 0.05.
Urinary albumin excretion increased significantly in diabetic mice compared to control mice. However, no significant difference in urinary albumin level was observed among all diabetic mice (Figure 2e). Diabetes induced significant increase of glomerular size and mesangial expansion (evaluated by type IV collagen immunohistochemical staining) in diabetic mice compared to control mice (Figure 2f, g and supplemental figure 3). While glomerular size increased significantly in both diabetic WT and KHK-A KO mice compared to that in diabetic KHK-A/C KO mice, mesangial area was not significantly different among all diabetic mice (Figure 2f, g and supplemental figure 3). These results suggest that the deterioration of kidney function in diabetic KHK-A KO mice compared to WT mice is likely related to tubular injury rather than glomerular injury, and these changes were prevented in the KHK-A/C KO mice.
3.3. Inflammation and fibrosis
It has been recognized that inflammation plays an important role for deterioration of DKD [26], and that renal macrophage infiltration [27] and increased inflammatory cytokines such as MCP-1 [28], TNF-α [29], and iNOS [30] in DKD has been reported. In this study, interstitial macrophage infiltration (determined by the immunohistochemical analysis of F4/80) and renal F4-/80 expression significantly increased in diabetic KHK-A KO mice compared to that in diabetic WT and KHK-A/C KO mice (Figure 3a, b). Macrophage infiltration, renal F4/80, MCP-1 mRNA expression and renal MCP-1 protein expression significantly increased in all diabetic mice compared to respective control mice, with diabetic KHK-A KO mice showing significantly higher expressions of these markers, TNF-α, and iNOS compared to than diabetic WT mice (Figure 3c, d, e, f, g).
Figure 3.
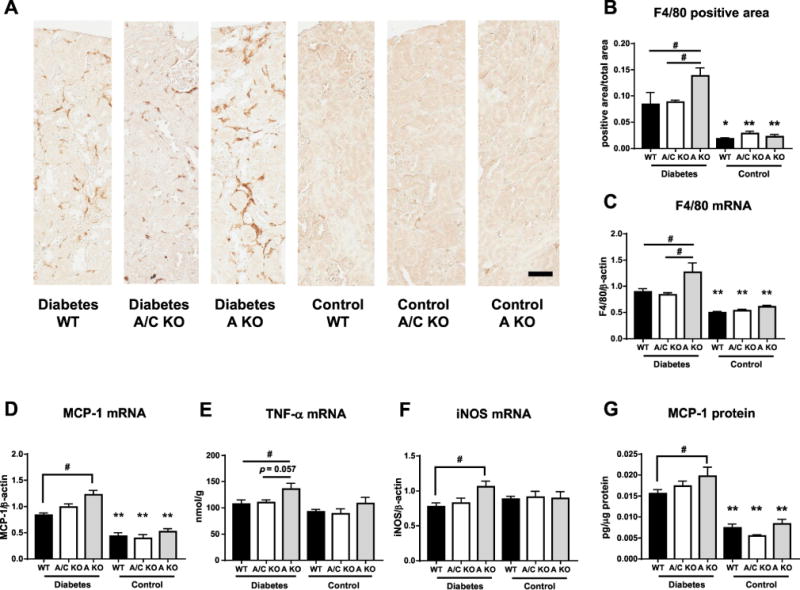
Inflammation marker in kidney of streptozotocin-induced diabetic and control WT mice. (A) Representative images of F4/80 stained kidney. Scale bar = 50μm (B) Quantification of F4/80 positive area in all diabetic groups. (C-F) Quantitative PCR for mouse F4/80 (C), mouse monocyte chemotactic protein-1(MCP-1, D), mouse tumor necrosis factor α (TNF-α, E), mouse inducible nitric oxide synthase (iNOS, F), in kidney. β-actin was used as an internal control. (G) MCP-1 protein in kidney corrected for protein concentration. (A-G, Each diabetic group, n = 9-11. Each control group, n = 4-9). Data represent means ± SEM. *P < 0.05, **P < 0.01 versus the respective diabetes group. # P < 0.05.
Interstitial fibrosis, as determined by Sirius Red staining, tended to, but not significantly, increase in all diabetic mice compared to respective control mice (Supplemental Figure 4a), whereas no difference was observed among all diabetic mice. Renal expression levels of COL4A3 [31], TGF-β [32, 33], and CTGF [34], which are associated with renal fibrosis, were similar among all diabetic mice or all control mice, however, these levels were significantly increased in all diabetic mice compared to respective control mice (Supplemental Figure 4b, c, d).
3.4. Polyol pathway and fructose metabolism
Activation of the polyol pathway induces the conversion of glucose to sorbitol by aldose reductase, conversion of sorbitol to fructose by sorbitol dehydrogenase, and metabolism of fructose to F1P by KHK. The mouse AKR1B3 gene is orthologous to human AKR1B1, both coding for aldose reductase, and have similar tissue distribution including kidney [35]. It has been reported that renal AKR1B3 expression was upregulated in STZ-induced diabetic mice [18]. In present study, all the diabetic mice showed significant and similar increase in renal glucose levels and AKR1B3 expression compared to those of respective control mice, suggesting the activation of the polyol pathway (Figure 4a, b). Although no difference was observed in renal sorbitol levels in all the groups (Figure 4c), fructose and F1P levels increased in the kidney of all diabetic mice compared to those of control mice (Figure 4d, e), suggesting that activation of the polyol pathway in diabetes promoted endogenous fructose production. KHK protein levels were not significantly different between diabetic WT and KHK-A KO mice (Figure 4f). Notably, in KHK-A/C KO mice, fructose (kidney, serum, and urine) and sorbitol (serum and urine) levels were markedly higher (Figure 4d, g, h and Table 1), and renal F1P levels were markedly lower than in WT and KHK-A KO mice regardless of their diabetic status, indicating that KHK-A/C KO mice had almost no ability to metabolize fructose (Figure 4e). Of note, renal fructose levels were significantly increased in diabetic KHK-A KO mice compared to diabetic WT mice (Figure 4d).
Figure 4.
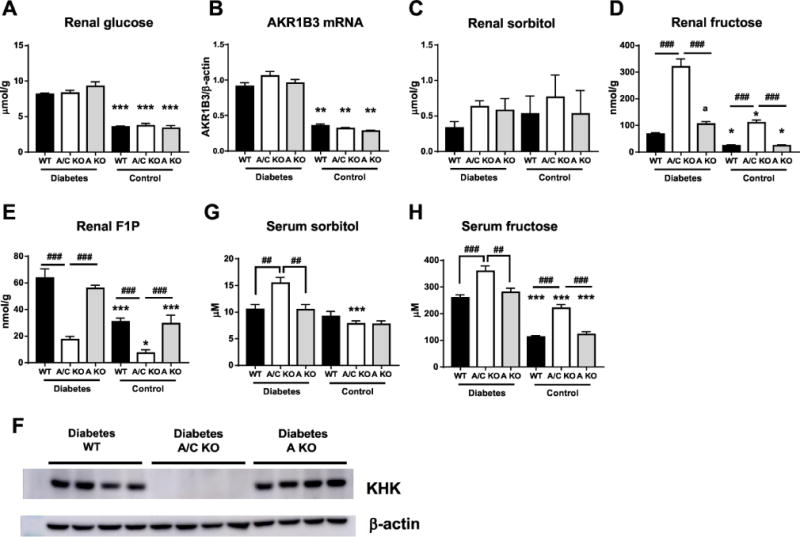
Polyol pathway and endogenous fructose metabolism of streptozotocin-induced diabetic and control mice. (A) Intrarenal glucose level. (B) Quantitative PCR for mouse AKR1b3 in kidney. β-actin was used as an internal control. (C-E) Intrarenal sorbitol (C), fructose (D), fructose-1-phosphate (F1P, E) level. (F) Representative image of KHK protein from renal cortex homogenates. (Each diabetic group, n = 9-11). (G) Serum sorbitol levels. (H) Serum fructose levels. (A-E, G, H. Each diabetic group, n = 9-11. Each control group, n = 4-9). Data represent means ± SEM. *P < 0.05, *** P < 0.001 versus the respective diabetes group. a, P < 0.01 vs diabetic WT mice. ## P < 0.01, ### P < 0.001.
3.5. Oxidative stress and nucleotide degradation pathway
KHK catalyzes the phosphorylation of fructose to F1P, which is an ATP-requiring process. Rapid fructose metabolism causes ATP depletion and AMP generation and activates the nucleotide degradation pathway, including uric acid generation by xanthine oxidase, which induces inflammation and oxidative stress (Supplemental Figure 5) [36–39]. In humans, uric acid is the final product of the nucleotide degradation pathway because of the absence of uricase, which converts uric acid to allantoin, whereas uric acid is converted to allantoin in mice [40]. Level of urinary TBARS, a marker of lipid peroxidation, and activity of renal xanthine oxidase were significantly elevated in all diabetic mice compared to control mice. Moreover, urinary TBARS level and renal xanthine oxidase activity were further increased in diabetic WT and KHK-A KO mice compared to diabetic KHK-A/C KO mice (Figure 5a, b) with a tendency to be higher in diabetic KHK-A KO mice than in diabetic WT mice. The expression of osteopontin (OPN), which was reported to upregulate by the increase of reactive oxygen species generation in diabetic kidney [41], showed similar changes as urinary TBARS and xanthine oxidase activity (Figure 5c). Although serum and renal uric acid levels were not different among all the mice (Table 1 and data not shown), urinary allantoin level was significantly increased in diabetic WT and KHK-A KO mice compared to diabetic KHK-A/C KO mice (Figure 5d). Further, urinary allantoin level was significantly correlated with renal AMP level (Figure 5e) and urinary NGAL level (Figure 5f). Moreover, although serum xanthine oxidase activity increased in all the diabetic mice, there was no significant difference among these mice (Figure 5g). The observed difference between renal and serum xanthine oxidase activity suggested that the nucleotide degradation pathway was further activated in the kidney rather than systemically, that contributed to renal tubular impairment in diabetic WT and KHK-A KO mice.
Figure 5.

Analysis of oxidative stress and nucleotide degradation pathway in streptozotocin-induced diabetic and control mice. (A) Urinary TBARS correlated for urine creatinine. (B) Renal xanthine oxidase (XO) activity. (C) Quantitative PCR for mouse osteopontin (OPN) in kidney. β-actin was used as an internal control. (D) Urinary allantoin levels corrected for urine creatinine. (E) Relationship between renal AMP and urinary allantoin in all groups. (F) Relationship between urinary NGAL and urinary allantoin in all groups. (G) Serum XO activity. (A-G, Each diabetic group, n = 9-11. Each control group, n = 4-9). Data represent means ± SEM. *P < 0.05, **P < 0.01, *** P < 0.001 versus the respective diabetes group. # P < 0.05, ## P < 0.01.
3.6. Downstream metabolites of fructose
Fructose is converted to F1P, and then to dihydroxyacetone phosphate (DHAP) and glyceraldehyde, which are intermediates of glycolysis. Elevations of DHAP levels are involved in PKC-β activation through increase of de novo synthesis of diacylglycerol (DAG) in diabetes [42] (Supplemental Figure 5). Results of the metabolomics analysis demonstrated that renal fructose, F1P, and DHAP levels was significantly increased in diabetic WT and KHK-A KO compared to those in respective control mice, and further increases of renal fructose and DHAP were observed in diabetic KHK-A KO mice compared to diabetic WT mice (Figure 4d, e and 6a), indicating that fructose metabolism was further enhanced in diabetic KHK-A KO mice than diabetic WT mice. Renal PKC-β expression was significantly upregulated in diabetic KHK-A KO compared to control KHK-A KO (Figure 6b). Among all control mice, there was no significantly difference in renal DHAP and renal PKC-β expression (Figure 6a, b). Glyceraldehyde measurement was not included in our metabolomics analysis.
Figure 6.
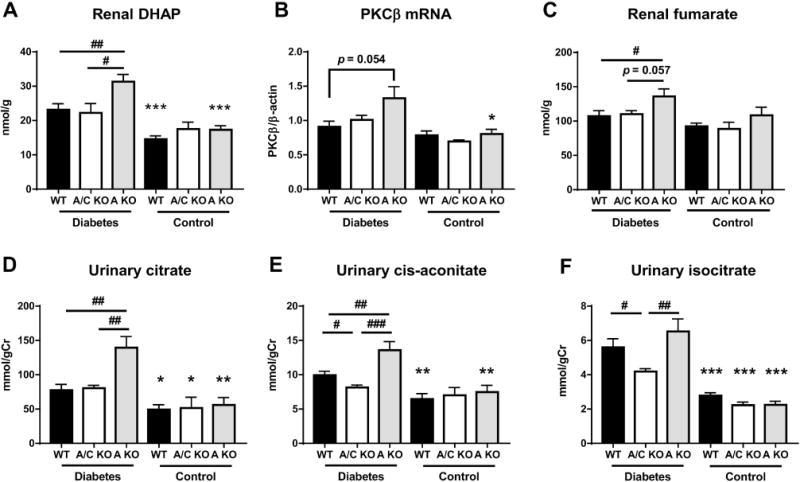
Downstream metabolites of fructose in streptozotocin-induced diabetic and control mice. (A) Renal dihydroxyacetone phosphate (DHAP) level. (B) Quantitative PCR for mouse protein kinase C-β (PKC-β) in kidney. β-actin was used as an internal control. (C) Renal fumarate level. (D) Urinary citrate concentration corrected for urine creatinine. (E) Urinary cis-aconitate concentration corrected for urine creatinine. (F) Urinary isocitrate concentration corrected for urine creatinine. (A-F, Each diabetic group, n = 9-11. Each control group, n = 4-9). Data represent means ± SEM. *P < 0.05, **P < 0.01, *** P < 0.001 versus the respective diabetes group. # P < 0.05, ## P < 0.01, ### P < 0.001.
Levels of TCA cycle intermediates including citrate, cis-aconitate, and fumarate have been reported to be elevated in diabetes and are correlated with albuminuria and kidney dysfunction in animal models of diabetes [43]. In addition, fumarate levels were increased in the kidney in STZ-induced diabetic rats [44]. In the present study, we found that no significant change of renal and urinary TCA cycle intermediates among all control mice (Figure 6c-f and Supplemental Figure 6, 7). However, fumarate levels were increased in the kidney of diabetic KHK-A KO mice compared to other diabetic mice (Figure 6c). Furthermore, in diabetic KHK-A KO mice, urinary levels of five TCA cycle intermediates, namely, citrate, cis-aconitate, isocitrate, 2-oxoglutarate and succinate were significantly increased compared to those of diabetic KHK-A/C KO mice (Figure 6d-f and Supplemental Figure 7), of which urinary citrate, cis-aconitate, 2-oxoglutarate were also significantly increased compared to those of diabetic WT mice (Figure 6d, e and Supplemental Figure 7). Diabetic WT mice also showed significant increases of urinary cis-aconitate and isocitrate compared to diabetic KHK-A/C KO mice (Figure 6e, f and Supplemental Figure 7). Given these results, it is likely that KHK-A depletion enhanced fructose and its downstream metabolism in diabetes, thus exacerbating inflammation and kidney injury.
3.7. Nicotinamide metabolism
Hyperglycemia reduces nicotinamide adenine dinucleotide (NAD+) level, coupled with increased urinary lactate level, decreased NAD+-dependent protein deacetylase SIRT1 activity, and the activation of HIF1α, thus inducing hyperglycemic pseudohypoxia [45–47]. We observed that renal NAD+ levels significantly decreased with significant increased renal HIF1α expression in diabetic KHK-A KO mice compared to diabetic WT and KHK-A/C KO mice (Figure 7a, d). Immunohistochemical staining detected HIF1α in the renal tubules showing apparent degeneration and vacuolization, which were more extensive in diabetic KHK-A KO mice than in diabetic WT mice (Supplemental Figure 8). A recent study reported that KHK-A also functions as a protein kinase [13]. KHK-A activates PRPS1, thus promoting the generation of phosphoribosyl pyrophosphate (PRPP) in the liver. In the salvage pathway of NAD+ biosynthesis, nicotinamide mononucleotide (NMN) is produced from nicotinamide (NAM) and PRPP and is converted to NAD+ (Supplemental Figure 9). In this study, inconsistent with the decrease of renal NAD+, renal NMN did not change, and renal NAM was significantly increased in diabetic KHK-A KO mice (Figure 7b and c). Expression of SIRT1 mRNA was significantly decreased in all diabetic mice compared to that of respective control mice, but did not differ among diabetic mice (Figure 7e). Furthermore, urinary lactate level was significantly elevated in diabetic KHK-A KO mice (Figure 7f). These findings suggest the possibility that insufficient PRPP generation because of KHK-A depletion impairs the conversion of NAM to NMN in diabetic KHK-A KO mice (Figure 7g).
Figure 7.
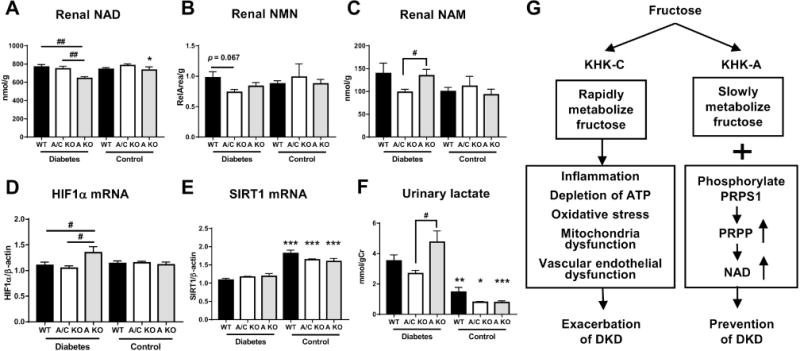
Nicotinamide adenine dinucleotide (NAD) related metabolites in streptozotocin-induced diabetic and control mice. (A) Renal NAD level. (B) Renal nicotinamide mononucleotide (NMN) level. (C) Renal nicotinamide (NAM) level. (D, E) Quantitative PCR for mouse HIF1α (D), mouse Sirt1 (E) in kidney. β-actin was used as an internal control. (F) Urinary lactate concentration corrected for urine creatinine. (A-F, Each diabetic group, n = 9-11. Each control group, n = 4-9). Data represent means ± SEM. *P < 0.05, **P < 0.01, *** P < 0.001 versus the respective diabetes group. # P < 0.05, ## P < 0.01.
4. Discussion
Tubulointerstitial injury is a characteristic of DKD [48]. Dietary fructose induces proinflammatory changes with uric acid generation in proximal tubular cells, and the inflammation can be prevented using NAPDH oxidase and xanthine oxidase inhibitors [16]. In diabetes, the polyol pathway is activated in the kidney, including the renal cortex, which induces endogenous fructose production [18], and kidney injury was attenuated in diabetic KHK-A/C KO mice with decreased uric acid and ROS production, and inflammation in kidney compared to diabetic WT mice [18]. In the present study, enhanced endogenous fructose production in the kidney of all diabetic mice was confirmed by the increase in renal aldose reductase expression and fructose level. Diabetes induced tubular injury in both diabetic WT and KHK-A KO mice with evidence for increased fructose metabolism (renal F1P), nucleotide degradation (renal AMP and urinary allantoin), and oxidative stress (urinary TBARS, renal xanthine oxidase activity), but those were attenuated in diabetic KHK-A/C-KO mice, despite similar energy intake, body weight, blood pressure, and serum and renal glucose levels among all diabetic mice. Glomerular size was larger in both diabetic WT and KHK-A KO mice than in diabetic KHK-A/C KO mice. However, mesangial expansion and albuminuria were not different among all diabetic mice. In the kidney, KHK is localized in the proximal tubules and not in the glomeruli [18], and mRNAs of both isoforms of KHKs, KHK-A and KHK-C, were expressed in the isolated renal proximal tubular cells (Supplementary figure 1). These results indicate that worse kidney damage in both diabetic WT and KHK-A KO mice than KHK-A/C KO was primarily caused by tubular injury rather than glomerular injury. Moreover, both WT and KHK-A KO mice express KHK-C which has higher affinity for fructose than KHK-A. Although kidney damage was yet observed in diabetic KHK-A/C KO mice indicating the inhibition of KHKs does not completely repress the progression of DKD, it was suggested that endogenous fructose metabolism exerted deleterious effects in DKD which was mediated probably by KHK-C.
The primary finding of the present study was that kidney injury was more advanced with severe inflammation (macrophage infiltration and inflammatory cytokines) in diabetic KHK-A KO mice than in WT mice. Although tubular damage was observed in both diabetic WT and KHK-A KO mice, pathological alterations in the renal tubules, kidney function, and inflammation were worse in diabetic KHK-A KO mice compared to diabetic WT mice. These findings indicate that KHK-A has a protective role in the development of DKD. Results of the metabolomic analysis showed increased intrarenal fructose and DHAP levels in diabetic KHK-A KO mice compared to diabetic WT mice. DHAP is a downstream metabolite of fructose and a precursor of DAG, which activates PKC leading to NF-κB activation and MCP-1 production in DKD [42]. The TCA cycle has been reported to be activated in DKD of the rodent models of type II diabetes, as evidence by increased urinary TCA cycle intermediates, including citrate, cis-aconitate and fumarate, with cis-aconitate correlating with albuminuria [21, 28]. The elevation of urinary TCA cycle intermediates was also confirmed in the fa/fa rat model of type II diabetes [43, 49, 50]. Hyperglycemia increases the proton gradient as a result of overproduction of electron donors by the TCA cycle, which in turn causes a marked increase in the production of superoxide in endothelial cells [17]. The enhancement of the TCA cycle may relate to systemic stress caused by hyperglycemia or local effects on kidney tubular transport and impairment of mitochondrial function [43]. In the present study, urinary cis-aconitate and isocitrate levels were increased in both diabetic WT and KHK-A KO mice. Moreover, diabetic KHK-A KO mice showed elevation of other TCA cycle intermediates such as citrate, 2-oxoglutarate, and fumarate. Compared to KHK-C, KHK-A has lower affinity for fructose, but distribute ubiquitously. Our results suggest that the deficiency of ubiquitously expressed and slow metabolizer KHK-A in diabetes enhances fructose metabolism by KHK-C in the kidney, thus increasing downstream metabolism (DHAP production and TCA cycle), which is responsible for inflammation, oxidative stress, and tubular dysfunction (Figure 7g). Fumarate accumulation increased HIF1α expression by inhibiting prolyl hydroxylase activity and upregulating transcription of HIF1α in podocytes [51], mouse embryonic fibroblasts [52], and renal cell carcinoma cells [53]. Longitudinal sustained HIF1α expression might influence the progression for tubular fibrosis. In our study, renal fumarate levels were elevated in diabetic KHK-A KO mice. Although the role of fumarate accumulation in tubular injury has not been elucidated to date, it might be correlated with the upregulation of HIF1α expression in diabetic KHK-A KO mice, resulting in kidney injury.
NAD+ is produced through the de novo and salvage pathways. Although tryptophan is the main source for NAD+ production from the de novo pathway, no significant difference was observed in renal tryptophan levels among all diabetic mice. In the salvage pathway, NMN is produced from NAM and PRPP by NAMPT (nicotinamide phosphoribosyltransferase) and is then converted to NAD+ [54, 55]. In our study, metabolomic analysis showed that renal NAD+ was reduced in diabetic KHK-A KO mice compared to other diabetic mice. In contrast, renal NAM was increased in kidney of diabetic WT and KHK-A KO mice. Furthermore, renal NMN was not increased in diabetic KHK-A KO mice, despite that in diabetic WT mice tended to have higher level. These results indicated that the generation of NMN from NAM and PRPP might be impaired in diabetic KHK-A KO mice. Because KHK-A also functions as a protein kinase to activate PRPS1 for promoting PRPP generation, it might be possible that KHK-A deficiency mediated lower activity of PRPS1, subsequent paucity of PRPP and NMN generation, resulting in reduced NAD+ with increased HIF1α expression and urinary lactate; that is pseudohypoxia in the kidney of diabetic KHK-A-KO mice. Because these findings were obtained by analyzing in vivo kidney samples, additional investigations, including in vitro studies, are needed to determine the role of KHK-A as an activator of PRPS1 in the kidney, especially tubular cells.
This study has some limitations. First, this study did not include mice lacking KHK-C alone. Therefore, the role of KHK-A in fructose metabolism in vivo in the absence of KHK-C remained to be determined. A second limitation is that the degree of renal injury in WT mice in this study was mild compared to our previous study [18]. That might be induced by high altitude where the previous study was done [18]. It has been known renal hypoxia has a central role in the progression in patients with chronic kidney disease and reduction of inspired oxygen at high altitude is closely related to reduction of intrarenal oxygen levels [56]. Previous report showed that dwellers at high altitude have worse kidney function, a higher prevalence of proteinuria compared to people living at sea level [57]. Furthermore, deteriorated renal function at high altitude was also reported in DKD, which demonstrated lower ambient oxygen levels at high-altitude affected the NAD/NADH ratio and energy metabolism including increased lactate production in diabetic rat kidney, which implicated in the accelerated development of diabetic nephropathy at high altitude [58]. Indeed, while there were features suggested that DKD was prevented in KHK-A/C KO mice compared to WT mice in the present study at low-altitude, the relatively mild nature of kidney injury in WT mice is likely why this was hard to document.
In conclusion, this study demonstrated the different role of KHK-A and KHK-C in DKD development. In streptozotocin-induced diabetes, kidney injuries, mainly involved in tubules, was exacerbated in mice lacking KHK-A with increased fructose, DHAP, TCA cycle metabolism, and decreased renal NAD+ with inflammation compared to WT mice, while those were prevented in mice lacking both isoforms. The results of the present study suggest that KHK-C might play an important deleterious role in DKD progression and that KHK-A plays a unique protective role against the development of DKD. Further studies are needed to investigate the role of the KHK isoforms, especially the distinct role of KHK-A, in the etiology of DKD.
Supplementary Material
KHK-A KO mice developed severe renal injury with inflammation compared to WT mice.
Fructose and its downstream metabolism were enhanced in diabetic KHK-A KO than WT.
Renal NAD decreased and renal HIF1α expression increased in diabetic KHK-A KO mice.
Opposite to KHK-C, KHK-A may have a protective role in the development of DKD.
Acknowledgments
We thank Noriyuki Suzuki, Naoko Asano, and Yuriko Sawa for their excellent technical assistance.
Disclosure
This work was partly funded by Aichi kidney foundation, Japan. AH was funded by JSPS KAKENHI Grant Number JP15K21364. RJJ and MAL report funding by NIH grants 1RO1DK109408 and DK108859. RJJ discloses he is an inventor on patents related to lowering uric acid as it relates to blood pressure, insulin resistance and diabetic renal disease. He also has equity in XORT therapeutics, Inc. and Colorado Research Partners LLC, both which are startup companies interested in developing novel xanthine oxidase inhibitors and fructokinase inhibitors, respectively. RJJ has also received honoraria from Danone, Astra Zeneca and is on the Scientific Board of Amway and Kibow, Inc.
Abbreviations
- AKR1B3
aldo-keto reductase family 1 member 3
- COL4
collagen 4
- CTGF
connective tissue growth factor
- DAG
diacylglycerol
- DHAP
dihydroxyacetone phosphate
- DKD
diabetic kidney disease
- F1P
fructose-1-phosphate
- HIF1α
hypoxia inducible factor 1, alpha subunit
- iNOS
inducible nitric oxide synthase
- KHK
ketohexokinase
- MCP-1
monocyte chemoattractant protein-1
- NAD
nicotinamide adenine dinucleotide
- NAM
nicotinamide
- NGAL
neutrophil gelatinase-associated lipocalin
- NMN
nicotinamide mononucleotide
- OPN
osteopontin
- PAS
periodic aid-Schiff
- PKCβ
protein kinase C-beta
- PRPP
phosphoribosyl pyrophosphate
- PRPS1
phosphoribosyl pyrophosphate synthetase 1
- SIRT1
sirtuin 1
- STZ
streptozotocin
- TBARS
thiobarbituric acid reactive substances
- TCA
tricarboxylic acid cycle
- XO
xanthine oxidase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
T.D. conducted experiments, analyzed data, wrote and edited the manuscript. T.I. designed research, analyzed data, wrote and edited the manuscript. T.H. conducted experiments. N.K., T.K., N.T., M.A.L., supervised and reviewed the study. S.I., M.H., A.H., T.S. performed metabolomic analysis. R.J.J., K.K. and S.M. supervised and edited the manuscript. T.I. and S.M. are the guarantors of this work and, as such, had full access to all the data in the study.
References
- 1.Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M, et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307–15. doi: 10.2337/db12-1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moczulski DK, Scott L, Antonellis A, Rogus JJ, Rich SS, Warram JH, et al. Aldose reductase gene polymorphisms and susceptibility to diabetic nephropathy in Type 1 diabetes mellitus. Diabetic medicine : a journal of the British Diabetic Association. 2000;17:111–8. doi: 10.1046/j.1464-5491.2000.00225.x. [DOI] [PubMed] [Google Scholar]
- 3.Hodgkinson AD, Sondergaard KL, Yang B, Cross DF, Millward BA, Demaine AG. Aldose reductase expression is induced by hyperglycemia in diabetic nephropathy. Kidney international. 2001;60:211–8. doi: 10.1046/j.1523-1755.2001.00788.x. [DOI] [PubMed] [Google Scholar]
- 4.James LR, Tang D, Ingram A, Ly H, Thai K, Cai L, et al. Flux through the hexosamine pathway is a determinant of nuclear factor kappaB- dependent promoter activation. Diabetes. 2002;51:1146–56. doi: 10.2337/diabetes.51.4.1146. [DOI] [PubMed] [Google Scholar]
- 5.Schleicher ED, Weigert C. Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney international Supplement. 2000;77:S13–8. doi: 10.1046/j.1523-1755.2000.07703.x. [DOI] [PubMed] [Google Scholar]
- 6.Langham RG, Kelly DJ, Gow RM, Zhang Y, Cox AJ, Qi W, et al. Increased renal gene transcription of protein kinase C-beta in human diabetic nephropathy: relationship to long-term glycaemic control. Diabetologia. 2008;51:668–74. doi: 10.1007/s00125-008-0927-x. [DOI] [PubMed] [Google Scholar]
- 7.Kelly DJ, Chanty A, Gow RM, Zhang Y, Gilbert RE. Protein kinase Cbeta inhibition attenuates osteopontin expression, macrophage recruitment, and tubulointerstitial injury in advanced experimental diabetic nephropathy. Journal of the American Society of Nephrology : JASN. 2005;16:1654–60. doi: 10.1681/ASN.2004070578. [DOI] [PubMed] [Google Scholar]
- 8.Morcos M, Sayed AA, Bierhaus A, Yard B, Waldherr R, Merz W, et al. Activation of tubular epithelial cells in diabetic nephropathy. Diabetes. 2002;51:3532–44. doi: 10.2337/diabetes.51.12.3532. [DOI] [PubMed] [Google Scholar]
- 9.Forbes JM, Cooper ME, Oldfield MD, Thomas MC. Role of advanced glycation end products in diabetic nephropathy. Journal of the American Society of Nephrology : JASN. 2003;14:S254–8. doi: 10.1097/01.asn.0000077413.41276.17. [DOI] [PubMed] [Google Scholar]
- 10.Diggle CP, Shires M, McRae C, Crellin D, Fisher J, Carr IM, et al. Both isoforms of ketohexokinase are dispensable for normal growth and development. Physiological genomics. 2010;42A:235–43. doi: 10.1152/physiolgenomics.00128.2010. [DOI] [PubMed] [Google Scholar]
- 11.Ishimoto T, Lanaspa MA, Le MT, Garcia GE, Diggle CP, Maclean PS, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:4320–5. doi: 10.1073/pnas.1119908109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diggle CP, Shires M, Leitch D, Brooke D, Carr IM, Markham AF, et al. Ketohexokinase: expression and localization of the principal fructose-metabolizing enzyme. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 2009;57:763–74. doi: 10.1369/jhc.2009.953190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Qian X, Peng LX, Jiang Y, Hawke DH, Zheng Y, et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nature cell biology. 2016;18:561–71. doi: 10.1038/ncb3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakayama T, Kosugi T, Gersch M, Connor T, Sanchez-Lozada LG, Lanaspa MA, et al. Dietary fructose causes tubulointerstitial injury in the normal rat kidney. American journal of physiology Renal physiology. 2010;298:F712–20. doi: 10.1152/ajprenal.00433.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gersch MS, Mu W, Cirillo P, Reungjui S, Zhang L, Roncal C, et al. Fructose, but not dextrose, accelerates the progression of chronic kidney disease. American journal of physiology Renal physiology. 2007;293:F1256–61. doi: 10.1152/ajprenal.00181.2007. [DOI] [PubMed] [Google Scholar]
- 16.Cirillo P, Gersch MS, Mu W, Scherer PM, Kim KM, Gesualdo L, et al. Ketohexokinase-dependent metabolism of fructose induces proinflammatory mediators in proximal tubular cells. Journal of the American Society of Nephrology : JASN. 2009;20:545–53. doi: 10.1681/ASN.2008060576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 18.Lanaspa MA, Ishimoto T, Cicerchi C, Tamura Y, Roncal-Jimenez CA, Chen W, et al. Endogenous fructose production and fructokinase activation mediate renal injury in diabetic nephropathy. Journal of the American Society of Nephrology : JASN. 2014;25:2526–38. doi: 10.1681/ASN.2013080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tando T, Hirayama A, Furukawa M, Sato Y, Kobayashi T, Funayama A, et al. Smad2/3 Proteins Are Required for Immobilization-induced Skeletal Muscle Atrophy. The Journal of biological chemistry. 2016;291:12184–94. doi: 10.1074/jbc.M115.680579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics : Official journal of the Metabolomic Society. 2010;6:78–95. doi: 10.1007/s11306-009-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soga T, Igarashi K, Ito C, Mizobuchi K, Zimmermann HP, Tomita M. Metabolomic profiling of anionic metabolites by capillary electrophoresis mass spectrometry. Analytical chemistry. 2009;81:6165–74. doi: 10.1021/ac900675k. [DOI] [PubMed] [Google Scholar]
- 22.Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer research. 2009;69:4918–25. doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
- 23.Klampfl CW, Buchberger W. Determination of carbohydrates by capillary electrophoresis with electrospray-mass spectrometric detection. Electrophoresis. 2001;22:2737–42. doi: 10.1002/1522-2683(200108)22:13<2737::AID-ELPS2737>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 24.Soga T, Heiger DN. Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Analytical chemistry. 2000;72:1236–41. doi: 10.1021/ac990976y. [DOI] [PubMed] [Google Scholar]
- 25.Marumo T, Yagi S, Kawarazaki W, Nishimoto M, Ayuzawa N, Watanabe A, et al. Diabetes Induces Aberrant DNA Methylation in the Proximal Tubules of the Kidney. Journal of the American Society of Nephrology : JASN. 2015;26:2388–97. doi: 10.1681/ASN.2014070665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nature reviews Nephrology. 2011;7:327–40. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 27.Chow F, Ozols E, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney international. 2004;65:116–28. doi: 10.1111/j.1523-1755.2004.00367.x. [DOI] [PubMed] [Google Scholar]
- 28.Wada T, Furuichi K, Sakai N, Iwata Y, Yoshimoto K, Shimizu M, et al. Up-regulation of monocyte chemoattractant protein-1 in tubulointerstitial lesions of human diabetic nephropathy. Kidney international. 2000;58:1492–9. doi: 10.1046/j.1523-1755.2000.00311.x. [DOI] [PubMed] [Google Scholar]
- 29.Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2002;16:438–40. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 30.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. Journal of the American Society of Nephrology : JASN. 2008;19:433–42. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 31.Udi S, Hinden L, Earley B, Drori A, Reuveni N, Hadar R, et al. Proximal Tubular Cannabinoid-1 Receptor Regulates Obesity-Induced CKD. Journal of the American Society of Nephrology : JASN. 2017;28:3518–32. doi: 10.1681/ASN.2016101085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, et al. Suppression of microRNA-29 expression by TGF-beta1 promotes collagen expression and renal fibrosis. Journal of the American Society of Nephrology : JASN. 2012;23:252–65. doi: 10.1681/ASN.2011010055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim DJ, Kang JM, Park SH, Kwon HK, Song SJ, Moon H, et al. Diabetes Aggravates Post-ischaemic Renal Fibrosis through Persistent Activation of TGF-beta1 and Shh Signalling. Scientific reports. 2017;7:16782. doi: 10.1038/s41598-017-16977-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rayego-Mateos S, Morgado-Pascual JL, Rodrigues-Diez RR, Rodrigues-Diez R, Falke LL, Mezzano S, et al. Connective tissue growth factor induces renal fibrosis via epidermal growth factor receptor activation. The Journal of pathology. 2017 doi: 10.1002/path.5007. [DOI] [PubMed] [Google Scholar]
- 35.Gimenez-Dejoz J, Weber S, Barski OA, Moller G, Adamski J, Pares X, et al. Characterization of AKR1B16, a novel mouse aldo-keto reductase. Chemico-biological interactions. 2017;276:182–93. doi: 10.1016/j.cbi.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 36.Desco MC, Asensi M, Marquez R, Martinez-Valls J, Vento M, Pallardo FV, et al. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51:1118–24. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- 37.Johnson RJ, Perez-Pozo SE, Sautin YY, Manitius J, Sanchez-Lozada LG, Feig DI, et al. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocrine reviews. 2009;30:96–116. doi: 10.1210/er.2008-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsumoto S, Koshiishi I, Inoguchi T, Nawata H, Utsumi H. Confirmation of superoxide generation via xanthine oxidase in streptozotocin-induced diabetic mice. Free radical research. 2003;37:767–72. doi: 10.1080/1071576031000107344. [DOI] [PubMed] [Google Scholar]
- 39.Perheentupa J, Raivio K. Fructose-induced hyperuricaemia. Lancet (London, England) 1967;2:528–31. doi: 10.1016/s0140-6736(67)90494-1. [DOI] [PubMed] [Google Scholar]
- 40.Johnson RJ, Titte S, Cade JR, Rideout BA, Oliver WJ. Uric acid, evolution and primitive cultures. Seminars in nephrology. 2005;25:3–8. doi: 10.1016/j.semnephrol.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 41.Hsieh TJ, Chen R, Zhang SL, Liu F, Brezniceanu ML, Whiteside CI, et al. Upregulation of osteopontin gene expression in diabetic rat proximal tubular cells revealed by microarray profiling. Kidney international. 2006;69:1005–15. doi: 10.1038/sj.ki.5000206. [DOI] [PubMed] [Google Scholar]
- 42.Sheetz MJ, King GL. Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. Jama. 2002;288:2579–88. doi: 10.1001/jama.288.20.2579. [DOI] [PubMed] [Google Scholar]
- 43.Wei T, Zhao L, Jia J, Xia H, Du Y, Lin Q, et al. Metabonomic analysis of potential biomarkers and drug targets involved in diabetic nephropathy mice. Scientific reports. 2015;5:11998. doi: 10.1038/srep11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Oliveira AA, de Oliveira TF, Bobadilla LL, Garcia CC, Berra CM, de Souza-Pinto NC, et al. Sustained kidney biochemical derangement in treated experimental diabetes: a clue to metabolic memory. Scientific reports. 2017;7:40544. doi: 10.1038/srep40544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou CK, Lee YT, Chen SM, Hsieh CW, Huang TC, Li YC, et al. Elevated urinary D-lactate levels in patients with diabetes and microalbuminuria. Journal of pharmaceutical and biomedical analysis. 2015;116:65–70. doi: 10.1016/j.jpba.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 46.Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–38. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–13. doi: 10.2337/diab.42.6.801. [DOI] [PubMed] [Google Scholar]
- 48.Gilbert RE. Proximal Tubulopathy: Prime Mover and Key Therapeutic Target in Diabetic Kidney Disease. Diabetes. 2017;66:791–800. doi: 10.2337/db16-0796. [DOI] [PubMed] [Google Scholar]
- 49.Li M, Wang X, Aa J, Qin W, Zha W, Ge Y, et al. GC/TOFMS analysis of metabolites in serum and urine reveals metabolic perturbation of TCA cycle in db/db mice involved in diabetic nephropathy. American journal of physiology Renal physiology. 2013;304:F1317–24. doi: 10.1152/ajprenal.00536.2012. [DOI] [PubMed] [Google Scholar]
- 50.Salek RM, Maguire ML, Bentley E, Rubtsov DV, Hough T, Cheeseman M, et al. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiological genomics. 2007;29:99–108. doi: 10.1152/physiolgenomics.00194.2006. [DOI] [PubMed] [Google Scholar]
- 51.You YH, Quach T, Saito R, Pham J, Sharma K. Metabolomics Reveals a Key Role for Fumarate in Mediating the Effects of NADPH Oxidase 4 in Diabetic Kidney Disease. Journal of the American Society of Nephrology : JASN. 2016;27:466–81. doi: 10.1681/ASN.2015030302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Flaherty L, Adam J, Heather LC, Zhdanov AV, Chung YL, Miranda MX, et al. Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Human molecular genetics. 2010;19:3844–51. doi: 10.1093/hmg/ddq305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer cell. 2005;8:143–53. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 54.Hershberger KA, Martin AS, Hirschey MD. Role of NAD+ and mitochondrial sirtuins in cardiac and renal diseases. Nature reviews Nephrology. 2017;13:213–25. doi: 10.1038/nrneph.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Canto C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell metabolism. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luks AM, Johnson RJ, Swenson ER. Chronic kidney disease at high altitude. Journal of the American Society of Nephrology : JASN. 2008;19:2262–71. doi: 10.1681/ASN.2007111199. [DOI] [PubMed] [Google Scholar]
- 57.Hurtado-Arestegui A, Plata-Cornejo R, Cornejo A, Mas G, Carbajal L, Sharma S, et al. Higher prevalence of unrecognized kidney disease at high altitude. Journal of nephrology. 2017 doi: 10.1007/s40620-017-0456-0. [DOI] [PubMed] [Google Scholar]
- 58.Laustsen C, Lycke S, Palm F, Ostergaard JA, Bibby BM, Norregaard R, et al. High altitude may alter oxygen availability and renal metabolism in diabetics as measured by hyperpolarized [1-(13)C]pyruvate magnetic resonance imaging. Kidney international. 2014;86:67–74. doi: 10.1038/ki.2013.504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.