

Summary
Zika virus (ZIKV), which emerged in regions endemic to Dengue virus (DENV), is vertically transmitted and results in adverse pregnancy outcomes. Antibodies to DENV can cross-react with ZIKV, but whether these antibodies influence ZIKV vertical transmission remains unclear. Here, we find that DENV antibodies increase ZIKV infection of placental macrophages (Hofbauer cells [HCs]) from 10% to over 80% and enhance infection of human placental explants. ZIKV-anti-DENV antibody complexes increase viral binding and entry into HCs but also result in blunted type I IFN, proinflammatory cytokine and antiviral responses. Additionally, ZIKV infection of HCs and human placental explants are enhanced in an IgG subclass-dependent manner, and targeting FcRn reduces ZIKV replication in human placental explants. Collectively, these findings support a role for pre-existing DENV antibodies in enhancement of ZIKV infection of select placental cell types and indicate that pre-existing immunity to DENV should be considered when addressing in ZIKV vertical transmission.
Keywords: Zika virus, placental macrophages, antibody-dependent enhancement, Dengue virus, cross-reactive antibodies, type I interferon, vertical transmission
Graphical Abstract

eTOC Blurb
Zimmerman et al. find that DENV cross-reactive antibodies enhance ZIKV infection of human placental macrophages and mid-gestation placental explants. ZIKV-anti-DENV antibody complexes increase viral entry but also result in blunted antiviral responses. These studies suggest that pre-existing immunity to DENV should be considered when addressing ZIKV vertical transmission.
Introduction
Zika virus (ZIKV) is a mosquito-borne flavivirus responsible for continuing epidemics of fetal congenital malformations within the Americas since its introduction to Brazil in 2015 (de Oliveira et al., 2017; Melo et al., 2017; Rasmussen et al., 2016). Symptoms include a self-limiting febrile illness accompanied by rash, conjunctivitis, arthralgias, myalgias, and fatigue; however, 80% of ZIKV infections are asymptomatic in healthy individuals (Lazear and Diamond, 2016). ZIKV is primarily transmitted through bites from infected Aedes mosquitos but can also be transmitted through sexual contact and blood transfusion (Lazear and Diamond, 2016). Notably, vertical transmission of ZIKV from mother to child in utero has been implicated in the rise of congenital microcephaly among neonates in ZIKV-endemic regions (Coyne and Lazear, 2016). Additionally, recent studies have discovered some infants with normal head circumference developed post-natal onset microcephaly, eye abnormalities, joint disorders, and sensorineural hearing loss following congenital ZIKV infection (Delaney et al., 2018; Fitzgerald et al., 2018; van der Linden et al., 2016). These studies demonstrate that congenital ZIKV infection has wide-ranging effects on infected fetuses, emphasizing the importance of understanding the mechanisms of vertical transmission.
The placenta acts as the sole physical and immunologic barrier between the maternal and fetal blood supply and is responsible for efficient gas, nutrient, and waste exchange during pregnancy (Coyne and Lazear, 2016). In humans, the placenta is composed of anchoring chorionic villi, which penetrate the uterine wall, as well as floating chorionic villi that are bathed in maternal blood pooling in the intervillous space (Arora et al., 2017). We and others have shown that ZIKV productively infects Hofbauer cells (HCs) and, to a lesser extent, cytotrophoblasts (CTBs) (Jurado et al., 2016; Quicke et al., 2016; Tabata et al., 2016). Following viral seeding of the placenta, ZIKV primarily infects HCs and persists within the placenta and fetal brain throughout pregnancy (Bhatnagar et al., 2017). However, the mechanisms of transplacental transmission of ZIKV and seeding of the placenta are not well understood. Syncytiotrophoblasts (STBs) maintain resistance to ZIKV infection through the constitutive secretion of IFN-λ, a type III IFN known for providing immunologic protection at anatomic barriers (e.g. blood-brain barrier, placenta, epithelial surfaces) during viral infection (Bayer et al., 2016; Lazear et al., 2015a; Lazear et al., 2015b). In mice, the IFN-λ-dependent antiviral response correlates with gestational age, specifically the development of the mature trophoblast barrier at later stages of pregnancy (Jagger et al., 2017). The inability of ZIKV to directly infect STBs suggests alternative routes for ZIKV transplacental transmission.
The emergence of ZIKV in the Americas overlaps with the regional distribution of dengue virus (DENV) seroprevalence, a related flavivirus that infects 50–100 million people per year (Bhatt et al., 2013). Numerous studies have shown that DENV antibodies can cross-react with ZIKV, which differs from DENV by 41–46% in the envelope protein, resulting in enhanced ZIKV infection in FcγR-expressing cells (Dejnirattisai et al., 2016; Paul et al., 2016; Priyamvada et al., 2016). To provide passive immunity to the developing fetus, transport of maternal IgG across the placenta starts by the 12th week of gestation and sharply increases between the second and third trimesters (Simister, 1998; Simister and Story, 1997). Furthermore, in DENV-seropositive pregnant mothers, DENV-specific IgG can be found at higher titers within the cord sera compared to the maternal serum by term birth (Castanha et al., 2016). However, it is not clear what role DENV-induced cross-reactive antibodies play in facilitating transplacental transmission of ZIKV.
Here, we evaluate the impact of cross-reactive DENV antibodies on ZIKV infection of the placenta. We demonstrate that the presence of DENV monoclonal antibodies (mAbs) increased ZIKV infection from approximately 10% to over 80% of HCs in culture. Despite enhanced ZIKV infection, we observed blunted type I IFN induction, antiviral gene expression, HC activation and pro-inflammatory cytokine production. We found that exogenous type I, but not type III IFN, significantly restricted ZIKV replication within ZIKV- and immune complex-infected HCs. Using mAbs with the identical binding site cloned onto different IgG Fc scaffolds, we determined that ZIKV complexed with IgG1 and IgG3 resulted in higher infection of HCs compared to IgG2 and IgG4. Finally, we found that immune-complexed ZIKV was more efficient at infecting human placental explant tissue than non-complexed virus. Additionally, ZIKV infection of HCs and human placental explants are enhanced in an IgG subclass-dependent manner and targeting FcRn reduced ZIKV replication in human placental explants. Collectively, these findings support a mechanism by which cross-reactive DENV antibodies may facilitate viral vertical transmission across the placental barrier and enhancement of ZIKV infection in HCs.
Results
Cross-reactive DENV antibodies enhance ZIKV infection of HCs.
The envelope (E) proteins between DENV and ZIKV are structurally similar, resulting in the generation of cross-reactive antibodies (Priyamvada et al., 2016; Sirohi et al., 2016). Thus, we determined whether DENV cross-reactive antibodies can enhance ZIKV infection of HCs. Primary HCs were isolated from full-term placenta and infected with ZIKV (PRVABC59) alone or complexed with anti-DENV2 mAbs (IgG1 subclass). We evaluated four DENV2 mAbs, which vary in ZIKV binding and neutralization (Priyamvada et al., 2016), along with a non-specific control mAb originally isolated from a patient with acute influenza infection (Wrammert et al., 2011) (Figure 1A). The three ZIKV cross-reactive DENV mAbs robustly enhanced ZIKV infection with >70% of cells infected at the highest mAb concentration (10 µg/mL) as compared to cells infected with ZIKV alone at a MOI of 1 (4%) or 10 (21%; Figure 1B). We found the most dramatic enhancement of ZIKV infection in HCs treated with the 33.3A06 mAb at 10 µg/mL (83.5%) as compared to the 31.3F03 (76.3%) and 33.3F05 (75.7%) mAbs. As previously reported, the non-cross-reactive 33.3E04 mAb and the non-specific control mAb (influenza virus EM4CO4 mAb) failed to enhance ZIKV infection (Priyamvada et al., 2016). We also performed 33.3A06 antibody titrations in HCs from three additional donors and observed a dose-dependent decrease in ZIKV infection, beginning at 1.6 × 10−2 µg/mL and reaching similar levels as ZIKV infection alone (MOI 1) by 1.28×10−4 µg/mL mAb (Figure 1C). We also observed the highest levels of infectious virus in the supernatants under conditions where ZIKV immune complexes were generated with 4×10−1 to 1.6X10−2 µg/mL of 33.3A06 mAb (Figure 1D-E). Compared to ZIKV infection alone (MOI 1 or 10) or ZIKV infection in the presence of the non-specific control mAb, we observed very little change in CD40, CD80, and CD86 upon ADE-ZIKV infection of HCs (Figure S1A-D). We also did not observe a substantial increase in cell death with either ZIKV infection alone or following infection with immune-complexed ZIKV as compared to mock infected cells (Figure S1E). Moreover, we treated HCs with Bafilomycin A1, which is a potent inhibitor of endosomal acidification (Wang et al., 2006), and observed reduced ZIKV infection in the context of ADE and non-ADE conditions (Figure S2). This suggests that immune complexed-ZIKV infects HCs through an endosomal-mediated process to infect HCs. Altogether, these findings demonstrate that cross-reactive DENV antibodies augment ZIKV infection of HCs with minimal effects on cellular activation or cell death.
Figure 1:

Cross-reactive DENV monoclonal antibodies (mAbs) enhance ZIKV infection of human placental macrophages (HCs). A) Binding and neutralization properties of mAbs. B) HCs were infected with ZIKV (MOI 1) in the presence of DENV cross-reactive mAbs or EM4CO4 non-specific control (five-fold dilutions starting at 10 µg/mL). Intracellular ZIKV E protein was assessed by 4G2 staining at 24 hours post-infection (hpi). Antibody dilutions were performed in singlicate. Control conditions are shown as the average of biological triplicates ±SD. Representative experiment from n=2 donors. Ab−, no mAb. ZIKV−, no ZIKV. C) HCs were infected with ZIKV (MOI 1) in the presence of mAb 33.3A06 (five-fold dilutions starting at 0.4 ug/ml) or EM4CO4 (0.4 ug/ml). Intracellular ZIKV E protein was assessed by 4G2 staining at 24 hpi (biological triplicates ±SD). Representative experiment from n=3 donors. D) Infectious virus in the supernatant was assessed by focus-forming assay (FFA) at 24 hpi (biological triplicates ±SD). Representative experiment from n=3 donors. FFU, focus-forming units. E) Representative FFA.
DENV mAb immune complexes increase ZIKV binding and entry in HCs.
To date, the mechanisms of ADE during flavivirus infection remain incompletely understood; however, it is hypothesized that enhancement of viral infection can involve two different, but not mutually exclusive, mechanisms: extrinsic or intrinsic ADE (Taylor et al., 2015). Extrinsic ADE is defined as sub-neutralizing concentrations of antibody binding to a virus and subsequently increasing attachment and entry into cells expressing Fc receptors. Intrinsic ADE involves negative regulation of innate immune signaling following binding of immune complexes to surface Fc receptors, thus making the cells more permissive to viral infection (Taylor et al., 2015). To establish the mechanism of ADE of ZIKV infection in HCs, we performed viral binding and entry assays on HCs infected with ZIKV alone (MOI 1 or 10) or in the presence of cross-reactive and non-specific mAbs (Figure 2A). As expected, we observed a log-fold increase in viral binding and entry in HCs between ZIKV at a MOI of 1 and 10 (Figure 2B). In ADE-ZIKV-infected HCs, we observed significantly increased viral binding as compared to ZIKV infection of HCs in the presence of the non-specific control mAb. Similarly, we also observed a log-fold increase in viral entry in ADE-ZIKV-infected cells as compared to ZIKV alone at a MOI of 1 or in the presence of non-specific control mAb. Despite similar levels of viral entry between cells infected with ZIKV at a MOI of 10 and ADE-ZIKV-infected cells, we consistently observed higher levels of infected cells as measured by viral E protein staining in ADE-ZIKV-infected HCs (Figure 1). These findings suggest that extrinsic ADE plays a significant role in enhancing ZIKV infection of HCs.
Figure 2:
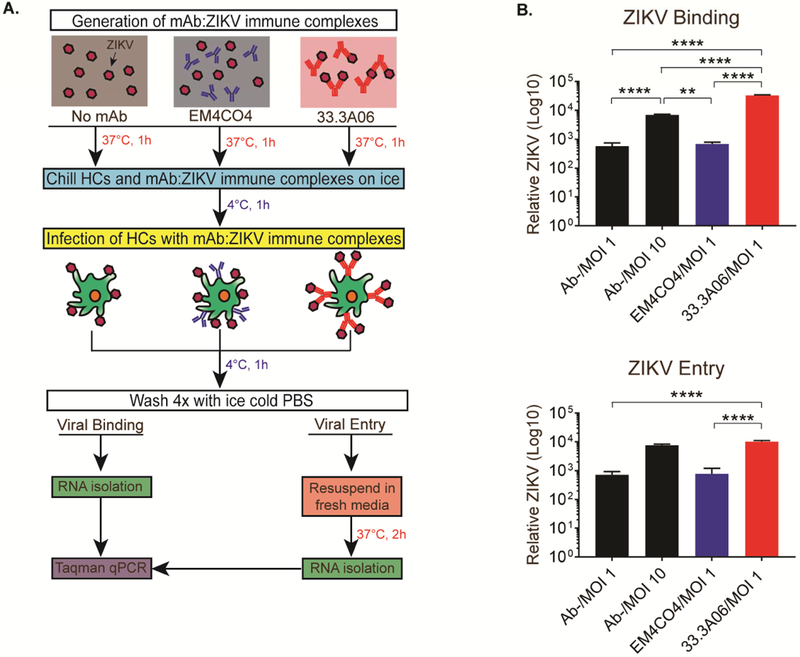
Infection with ZIKV in the presence of mAb 33.3A06 results in increased viral binding and entry of HCs. A) Binding/entry assay schematic. EM4CO4 and 33.3A06 mAbs were used at 0.4 µg/mL. B) ZIKV RNA from bound virus (top) or internalized virus (bottom) was measured by qRT-PCR. Representative experiment from n=2 donors (biological triplicates ±SD). Data were analyzed by 1-way ANOVA and Tukey’s multiple comparison test, **p<0.01, ***p<0.001, ****p<0.0001.
ADE of ZIKV infection induces IFN gene expression but dampens antiviral responses in HCs.
We next determined whether any differences exist in the induction of innate immune responses between non-ADE- and ADE-ZIKV-infected HCs. We evaluated the expression of type I and type III IFNs following ZIKV infection of HCs. HCs infected with ZIKV alone (MOI of 1 or 10) or in the presence of the non-specific control mAb displayed robust induction of type I and III IFN mRNAs, which corresponded with increased viral RNA as compared to time-matched mock-infected controls (Figure 3A). Similarly, robust increases in IFN-β (630-fold), IFN-α (267-fold) and IFN-λ (209-fold) transcript expression were observed in the highest ADE-ZIKV infected HCs as compared to time-matched mock-infected controls. Notably, we failed to detect IFN-β and IFN-λ protein in the supernatants of HCs infected under any condition (non-ADE and ADE-ZIKV infected HCs; Figure 3B). This finding is consistent with our previous observations in ZIKV-infected HCs and moDCs, in which we failed to observe IFN-β protein in cells or in the supernatants despite robust induction of IFN-β transcripts (Bowen et al., 2017; Quicke et al., 2016). We did observe basal IFN-α protein in the supernatants of mock-infected cells. Notably, we observed a significant reduction in IFN-α protein in ADE-ZIKV-infected HCs as compared to HCs infected with ZIKV alone (MOI 1) or in the presence of the non-specific control mAb (Figure 3B). These results indicate that ZIKV infection potently triggers transcription of type I and III IFNs, suggesting that ZIKV can block the translation/secretion of type I IFNs into the supernatant.
Figure 3:
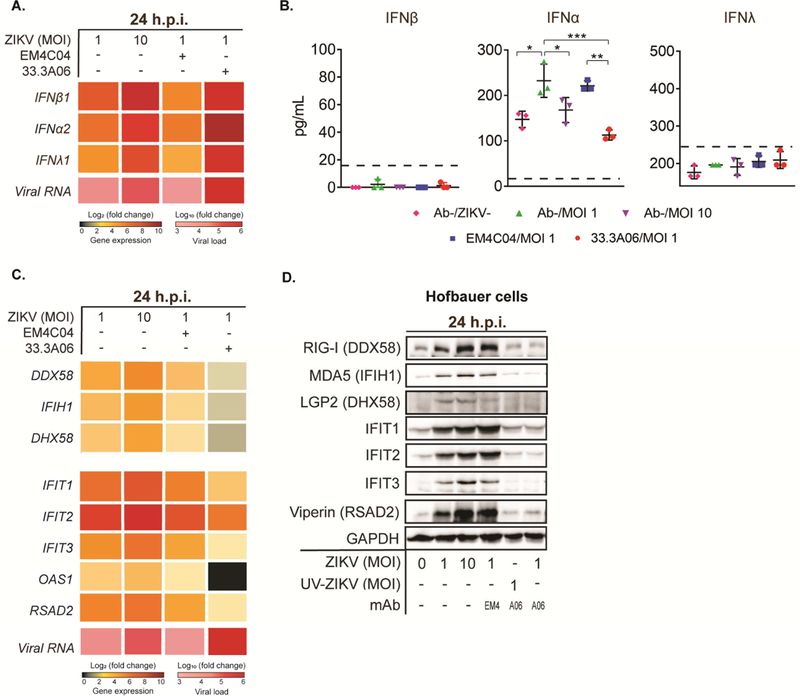
HCs infected with DENV mAb:ZIKV immune complexes result in blunted type I and type III IFN antiviral responses. A) Type I and type III IFN was measured by qRT-PCR at 24 hpi with ZIKV alone (MOI 1 and 10) or mAb:ZIKV (MOI 1) immune complexes. 33.3A06 and EM4CO4 were used at 0.4µg/mL. Representative experiment from n=2 donors. B) Type I and type III IFN was measured in the supernatant at 24 hpi (biological triplicates ±SD). Data were analyzed by 1-way ANOVA and Tukey’s multiple comparison test, *p<0.05, **p<0.01, ***p<0.001. Representative experiment from n=3 donors. Dashed line indicates lower limit of detection. C) Antiviral effector gene expression was measured by qRT-PCR at 24 hpi. Representative experiment from n=2 donors. D) Corresponding protein expression was measured at 24 hpi with the addition of mAb (0.4µg/mL): UV-inactivated ZIKV control (UV-ZIKV; MOI 1) immune complexes. Representative experiment from n=2 donors.
Next, we evaluated the effect of ADE-ZIKV infection on the induction of pro-inflammatory cytokines/chemokines. For this analysis, we performed a 25-plex cytokine/chemokine analysis on supernatants following ZIKV infection of HCs (non-ADE and ADE; Table S1). Infection of HCs with ZIKV at MOI 1 resulted in significant increases in MCP-1, MIP-1α and MIP-1β, and modest increases in IL-2R and IL-1Rα over mock-infected controls (Figure S3). Despite high levels of infection, we observed a lack of induction of MCP-1, MIP-1α, MIP-1β, IL-2R and IL-1Rα as well as other proinflammatory cytokines/chemokines (Table S1) in ADE-ZIKV-infected HCs over mock-infected cells. MCP-1 (CCL2), MIP-1α (CCL3), and MIP-1β (CCL4) are important for trafficking and infiltration of inflammatory myeloid cells and leukocytes to sites of flavivirus infection (Michlmayr and Lim, 2014). Notably, levels of IL-8, a cytokine canonically associated with neutrophil trafficking and degranulation but also non-inflammatory placental angiogenesis in HCs (Schliefsteiner et al., 2017), was reduced in ADE-ZIKV-infected HCs compared to mock infected controls (Figure S3). Furthermore, we failed to detect IL-10, which has been implicated in contributing to intrinsic ADE (Tsai et al., 2014), in either mock-infected cells or following ZIKV infection of HCs (Table S1).
We next measured the expression of the RIG-I-like receptors (RLRs), which play a critical role in triggering an innate immune response following ZIKV infection and antiviral effector genes that restrict ZIKV infection (Bowen et al., 2017). At the transcript level, we observed that DDX58 (RIG-I), IFIH1 (MDA5), and DHX58 (LGP2) expression were induced in HCs infected with ZIKV alone (MOI of 1 and 10) or in the presence of the non-specific control mAb as compared to time-matched mock-infected controls (Figure 3C). However, at the highest concentrations of 33.3A06 mAb immune-complexed ZIKV, which corresponded with robust ZIKV replication, we observed minimal induction of DDX58 (3.4-fold), IFIH1 (3.1-fold), and DHX58 (2.8-fold) as compared to time-matched mock-infected controls. In a similar manner, we observed minimal induction of antiviral effector genes IFIT1 (8.0-fold), IFIT2 (31.4-fold), IFIT3 (4.9-fold), RSAD2 (5.0-fold) and OAS1 (1.1-fold) in ADE-ZIKV-infected HCs as compared to cells infected with ZIKV alone (MOI of 1 and 10) or in the presence of the non-specific control mAb (Figure 3C). To determine whether ZIKV also blocks the translation of antiviral effector genes, we performed Western blot analysis on a parallel set of infected HC samples. ZIKV infection alone (MOI of 1 and 10) showed robust increases in expression of the RLRs, IFIT proteins, Viperin and OAS1 over mock-infected controls (Figure 3D). However, we observed a lack of RLRs and antiviral effector proteins in ADE-ZIKV-infected cells as compared to mock-infected cells. As a control, we did not observe any changes in GAPDH protein across the various conditions of infected HCs. Combined, these findings suggest that binding of ZIKV immune-complexes to Fc receptors induces intrinsic ADE by triggering a negative signal within HCs that blunts antiviral effector gene expression.
Type I IFN, but not Type III IFN, restricts ADE-ZIKV infection in HCs.
We next determined whether type I or III IFN can restrict ADE of ZIKV infection in HCs by performing an interferon inhibition assay (Bowen et al., 2017). In this assay, we infected HCs with ZIKV immune complexes for 1h followed by treatment with recombinant human IFN-λ1 or IFN-β (10 IU/mL or 100 IU/mL) for 24h. We measured both ZIKV infection by intracellular viral E protein staining and infectious virus production by FFA. Following IFN-λ1 treatment, we observed a modest, yet significant, increase in the percentage of ZIKV-infected cells as compared to untreated cells (Figure. 4A, top). Notably, we observed this increase in both non-ADE- and ADE-ZIKV-infected cells (Figure 4C). However, we did not observe an increase in infectious virus release following IFN-λ treatment (Figure. 4A, bottom). In contrast to IFN-λ, IFN-β treatment resulted in significant reduction in both the percentage of ZIKV infected cells and infectious virus in the supernatants (Figure. 4B). This reduction was consistent across non-ADE- and ADE-ZIKV-infected HCs (Figure 4C). These results indicate that HCs are highly responsive to IFN-β treatment and that IFN-β inhibits ZIKV replication in HCs despite the high levels of infection seen with ADE.
Figure 4:

Type I IFN, but not type III IFN, restricts ZIKV infection of HCs. A) HCs were infected with ZIKV (MOI 1) in the presence of 33.3A06 mAb (five-fold dilutions starting at 3.2×10−3 µg/mL) and subsequently treated with 10 or 100 IU/ml of IFN-λ (blue) or left untreated (black). B) ZIKV-infected HCs were treated with 10 or 100 IU/ml of IFN-β (red) or left untreated (black). C) HCs were infected with ZIKV alone at MOI 1 or 10, or in the presence of EM4CO4 (3.2×10−3 µg/mL). Cells were subsequently treated with 10 or 100 IU/ml of IFN-λ (blue) or IFN-β (red) or left untreated (black). Upper panels: Intracellular ZIKV E protein was evaluated by 4G2 staining at 24 hpi. Lower panels: Supernatants from infected HCs were collected 24 hpi and assessed by FFA (biological triplicates ±SD). Data were analyzed by 2-way ANOVA and Tukey’s multiple comparison test.*p<0.05, ***p<0.001. Representative experiment from n=3–4 donors.
IgG subclass influences infectivity of HCs during ADE.
Given that our work thus far employed an IgG1 subclass mAb, we next determined whether similar levels of enhanced ZIKV infection were observed across various IgG subclasses. We performed flow cytometry analysis on HCs isolated from full-term placenta and found that these cells express CD16, CD32 and CD64, albeit to varying levels (Figure. 5A). We observed that >98% of HCs expressed CD32 and CD64; however, <2% of cells expressed CD16. FcγRs bind to the Fc portion of different IgG subclasses with varying affinities (Smith and Clatworthy, 2010); therefore we assessed the ability of ZIKV complexed with IgG1, IgG2, IgG3 and IgG4 to infect HCs. We generated a panel of mAbs containing the Fab region from the cross-reactive 33.3A06 mAb but interchanged the Fc regions with the four human IgG subclasses. We also generated a mutant form of IgG1 containing a “LALA” modification in the Fc region, which is known to substantially inhibit FcγRI, FcγRII, and FcγRIII binding (Beltramello et al., 2010; Hessell et al., 2007). We generated ZIKV immune complexes using this panel of mAbs and observed similar levels of infection with IgG1, IgG3, and IgG4 subclasses with substantially reduced infection with IgG2 at the highest antibody concentration (Figure 5B). However, at lower Ab concentrations (1.6×10−2, 3.2×10−3 µg/mL), we observed distinct differences in viral infection using the panel of 33.3A06 IgG subclasses. IgG1 and IgG3 immune complexes displayed a higher percentage of infected cells (76.9% and 82% 4G2+ cells, respectively); IgG4 had an intermediate phenotype (64.5% 4G2+ cells), and IgG2 displayed low levels of infection (4.38% 4G2+ cells; Figure 5B). As previously shown, ADE of ZIKV infection is lost at the 6th-7th mAb dilutions for the entire panel of 33.3A06 mAb IgG subclasses. ZIKV immune complexes generated with the 33.3A06 IgG1-LALA mAb variant showed reduced infection across all antibody concentrations and reached similar levels as cells infected with ZIKV alone (MOI 1) or in the presence of non-specific control mAb. This demonstrates that ADE-ZIKV occurs through an Fc receptor-dependent mechanism. Collectively, the results demonstrate that HCs express specific FcγRs that promote binding of viral immune complexes and augment ZIKV infection in an IgG subclass-specific manner.
Figure 5:

Enhanced ZIKV infection of HCs is modulated in an IgG subclass-dependent manner. A) Flow cytometry plots showing expression of FcγRIII (CD16), FcγRII (CD32), and FcγRI (CD64) of uninfected HCs. Representative experiment from n=2 donors. B) HCs were infected with ZIKV (MOI 1) in the presence of different 33.3A06 IgG subclasses (five-fold dilutions starting at 8×10−2 µg/mL). Intracellular ZIKV E protein was assessed by 4G2 staining at 24 hpi. mAb:ZIKV conditions were performed in singlicate (left) and controls (right) as the average of biological triplicates ±SD. Representative experiment from n=3 donors.
Cross-reactive DENV mAbs enhance ZIKV infection of human mid-gestation placental explants.
We next determined the ability of cross-reactive DENV mAbs to enhance ZIKV infection of human mid-gestation placental explants. For these studies, we generated ZIKV immune complexes using the panel of 33.3A06 IgG subclasses and infected second-trimester human placental villous explants with the immune complexes, ZIKV alone, or in the presence of non-specific control mAb. In explants treated with the 33.3A06 mAb immune complexes, we detected increased levels of positive- and negative-sense ZIKV RNA as compared to ZIKV infection alone or in the presence of non-specific control mAb (Figure 6A). Specifically, we found that infection with IgG1 and IgG3 displayed increased infection as compared to IgG4 and IgG2. We observed a mAb dose-dependent increase in ZIKV replication as ZIKV immune complexes generated with low 33.3A06 mAb concentrations (0.001 µg/mL) displayed similar levels of positive and negative-strand viral RNA as that of ZIKV alone, or in the presence of the non-specific control mAb. Additionally, we observed a log-fold increase in ZIKV RNA in in the supernatants from 33.3A06 IgG1 immune complex-infected explants and a 1.5 log-fold increase with IgG3-ZIKV immune complexes as compared to the control samples (Figure 6B). Similarly, we found increased infectious virus in the supernatants from placental explants infected with IgG1-ZIKV immune complexes but not in the explants infected with ZIKV alone or in the presence of the non-specific control mAb (Figure S4). Altogether, these data show that cross-reactive DENV mAbs, particularly IgG1 and IgG3 subclasses, enhance ZIKV infection of human placental explants.
Figure 6:
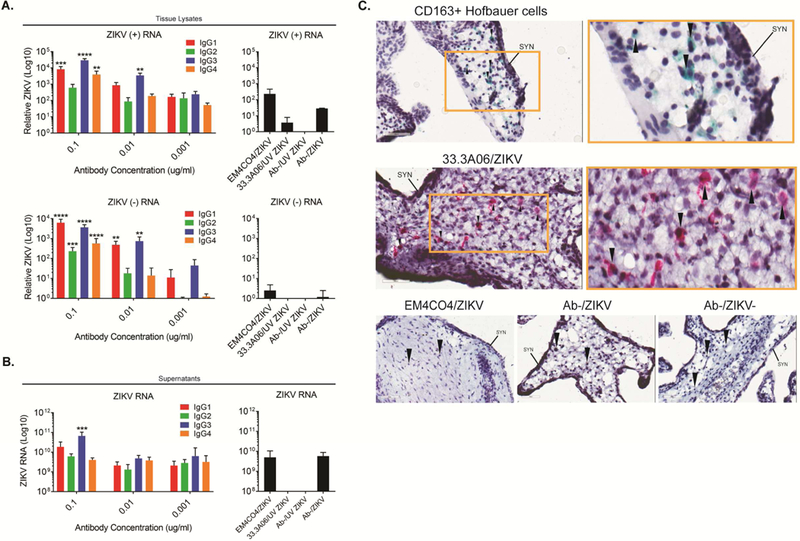
IgG1 and IgG3 subclasses preferentially enhance ZIKV infection of human placental explants. A) Human placental explants were infected with ZIKV (5×105 PFU/ml) in the presence of 33.3A06 mAb IgG subclasses (0.1, 0.01, 0.001 µg/mL). Viral replication within tissues was assessed by strand-specific qRT-PCR at 24 hpi (biological triplicates ±SD). Control conditions used ZIKV alone and UV-ZIKV at 5×105 PFU/ml and mAbs at 0.1 ug/ml. Representative experiment from n=3 donors. B) Supernatants from human placental explants infected as in (A) were collected at 24 hpi and ZIKV RNA measured by qRT-PCR (biologic triplicates). N=2 donors. Data were analyzed by 1-way ANOVA and Dunnett’s multiple comparison test comparing log-transformed ZIKV RNA with 33.3A06 IgG subclass ZIKV RNA levels to the log-transformed non-specific antibody control, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. C) Explants were infected with ZIKV (5×105 PFU/ml) in the presence of 33.3A06 (0.4 ug/ml) or EM4CO4 (0.4 ug/ml). HCs (top) and ZIKV E protein (middle and bottom) were visualized by chromogenic staining with anti-CD163 and anti-4G2 antibodies, respectively. Magnification is at 40x (left) and 80x (right, yellow box). Controls are shown across the bottom (40x magnification). SYN, syncytiotrophoblast layer. Arrows indicate HCs. N=2 donors.
We next performed immunohistochemical analyses on placental explants infected with the 33.3A06 ZIKV immune complex, the EM4CO4 non-specific antibody or ZIKV alone. In the explants treated with ZIKV immune complexes, we demonstrated that ZIKV E protein (red deposition) was predominantly found within cells with similar morphology and localization within the villous stroma as CD163+ HCs (green deposition; Figure 6C). Markedly, no ZIKV E protein was detected within the STB or CTB layers. In contrast, explants infected with ZIKV alone or in the presence of a non-specific antibody control showed no evidence of ZIKV E protein throughout the tissue.
Targeting IgG-FcRn interactions reduces ZIKV infection of human placental explants.
ZIKV infection of HCs within the villous stroma coupled with the lack of STB or CTB infection in mid-gestation explants indicates that these viral immune complexes can transcytose across the trophoblast layer. During pregnancy, maternal IgG is transported across the STB layer of the chorionic villi, providing passive immunity to the developing fetus. To determine what role FcRn plays in transplacental transport of ZIKV immune complexes, we pre-treated mid-gestation placental explants with Protein A from Staphylococcus aureus, a protein shown to bind the CH2-CH3 domains of IgG and inhibit binding to FcRn (Deisenhofer, 1981; Raghavan et al., 1994), and subsequently infected with ZIKV complexed with the 33.3A06 IgG1 enhancing antibody. Human IgG3 is not able to bind Protein A (Jendeberg et al., 1997; Lindmark et al., 1983); therefore, as a control, we also infected placental explants with ZIKV complexed to 33.3A06 IgG3 mAbs. Using strand-specific qPCR, we observed significant dose-dependent decreases in both positive- and negative-sense ZIKV RNA with increasing concentrations of Protein A (Figure 7A). In detail, we detected 14.3-fold and 33.3-fold decreases in positive-sense ZIKV RNA as well as 4.9-fold and 12.2-fold decreases in negative-sense ZIKV RNA in explants treated with 10 ug/ml and 100 ug/ml Protein A, respectively. Notably, explants infected with ZIKV IgG3 immune complexes showed minimal change in either positive- or negative sense ZIKV RNA in the presence of Protein A. We also found that treatment of mid-gestation explants with DVN24, a mAb that specifically targets and blocks the IgG-binding region of FcRn (Christianson et al., 2012), significantly inhibited ZIKV replication by 16.5-fold (Figure S5A).
Figure 7:
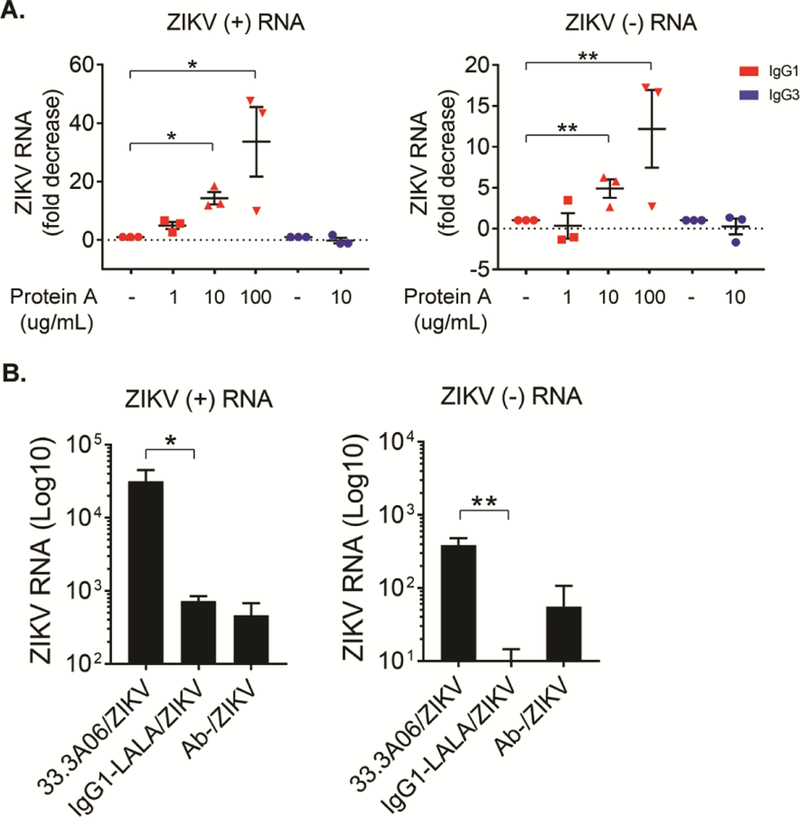
Targeting FcRn reduces ZIKV infection of human placental explants. A) Human placental explants were treated with 1, 10 or 100 ug/ml of Protein A and subsequently infected ZIKV (5×105 PFU/ml) in the presence of either 33.3A06 IgG1 or IgG3 mAb (0.4 ug/ml). Viral replication was assessed by strand-specific qRT-PCR at 24 hpi (biological triplicates ±SEM). Data were analyzed by 1-way ANOVA and Dunnett’s multiple comparison test, *p<0.05, **p<0.01. Representative experiment from n=3 donors. B) Human placental explants were infected with ZIKV (5×105 PFU/ml) in the presence of either 33.3A06 IgG1 or IgG1-LALA mutant mAbs. Viral replication was assessed by strand-specific qRT-PCR at 24 hpi (biologic triplicates ±SD). Data were analyzed by Student’s t-test, *p<0.05, **p<0.01, Representative experiment from n=4 donors.
Lastly, we determined whether ADE of ZIKV infection seen in mid-gestation explants was dependent on FcγR binding. In agreement with our earlier observations in HCs in vitro, we observed >1.5 log-fold decreases in both positive- and negative-sense ZIKV RNA in explants infected with ZIKV bound to 33.3A06 IgG1-LALA mAbs compared to ones infected with the 33.3A06 IgG1 viral immune complexes (Figure 7B). Additionally, explants infected with 33.3A06 IgG1-LALA immune complexes were analyzed by immunohistochemistry and probed for ZIKV E protein. Consistent with the lack of viral enhancement seen in HCs and explants, explants infected with 33.3A06 IgG1-LALA immune complexes did not exhibit appreciable ZIKV infection (Figure S5B). Overall, this data illustrates that ZIKV infection of placental explants are enhanced by cross-reactive DENV antibodies, and ZIKV infects and replicates in HCs within the villous stroma. Moreover, these studies also suggest that ZIKV immune complexes can utilize the FcRn IgG transport system to transcytose across the STB layer to infect HCs in an FcγR-dependent fashion.
Discussion
Our findings demonstrate that cross-reactive DENV mAbs can augment ZIKV infection of HCs isolated from full-term placenta in a dose-dependent manner. Mechanistically, we determined that cross-reactive DENV mAb immune complexes enhanced viral binding to HCs and increased viral entry. However, despite the high levels of infection seen in ADE-ZIKV-infected HCs, we observed minimal upregulation of costimulatory markers and production of pro-inflammatory cytokines. Although HCs induce type I and III IFN transcript during ADE-ZIKV infection, IFN protein secretion as well as expression of key antiviral effectors were severely diminished in ADE-ZIKV-infected cells. Notably, ADE-ZIKV infection was significantly reduced upon IFN-β treatment. We also found that enhancement of ZIKV infection in HCs is dependent on the IgG-subclass of the cross-reactive antibody with the strongest enhancement observed in the presence of IgG1 and IgG3 immune complexes. Consistent with these results, we observed enhanced ZIKV infection in human mid-gestation placental explants in an IgG-subclass specific manner and obtained data suggesting that ZIKV immune complexes can transcytose across the STB layer in an FcRn-dependent manner to target HCs within the villous stroma.
Although HCs have been found to be the primary cell type infected during ZIKV infection (Bhatnagar et al., 2017; Jurado et al., 2016; Quicke et al., 2016), a limitation of our study is the relevance of the observed immune complex-mediated increase of ZIKV infection in isolated human HCs in vitro. To address this, we examined the impact of cross-reactive DENV mAbs on enhancement of ZIKV infection in ex vivo human second-trimester placental explants. ZIKV infection of the developing fetus and subsequent ZIKV-associated congenital abnormalities can occur across all three trimesters during pregnancy (Shapiro-Mendoza et al., 2017). However, because transport of maternal IgG across the placenta is minimal during the first trimester and rises dramatically between 22–26 weeks of gestation (Palmeira et al., 2012; Simister and Story, 1997), we chose to focus our study on second-trimester human explants. Recent work has demonstrated that ZIKV replicates and persists within the placentas of ZIKV-infected women as well as the brains of the developing fetuses (Bhatnagar et al., 2017). In addition, replicative intermediate viral RNA appeared to correspond to HC staining in the placenta of ZIKV-infected women (75%; n=12) with adverse pregnancy outcomes during the first or second trimester. Given that ZIKV infection induces minimal HC death (Supplemental Figure 1E), ZIKV immune complexes target HCs (Figure 6C), and that ZIKV persists in HCs throughout pregnancy, continuous spillover of ZIKV into the fetal bloodstream could lead to continuous viral seeding of the fetus.
More recent work has shown that, in a cohort of pregnant women with possible ZIKV exposure, over half of the women with previous anti-flavivirus immunity who successfully gave birth were positive for ZIKV RNA in placental tissues (Reagan-Steiner et al., 2017). Others have shown that the presence of DENV IgG in ZIKV-infected pregnant women did not significantly increase the incidence of abnormal birth outcomes compared to DENV-IgG negative patients (Halai et al., 2017). However, this study focused on the role of previous flavivirus immunity on adverse pregnancy outcomes, not viral seeding of the placenta. Neither of these studies categorized the flavivirus-exposed, ZIKV-infected women based on DENV IgG titers nor prevalence of individual DENV IgG subclasses. Making these differentiations could potentially increase the observed incidence of viral seeding of the placenta and adverse pregnancy outcomes during ZIKV infection in flavivirus-exposed women. Our observations suggest that viral immune complexes can bypass the STB layer through an FcRn-dependent manner and establish infection within HCs localized within the villous core in an IgG-subclass dependent manner. Altogether, our data support that previous flavivirus immunity could influence viral seeding of the placenta.
DENV infection initiates a robust IgG response with peak levels occurring a few weeks after infection and persisting for a decade or longer (Wahala and Silva, 2011). Further characterization of this IgG response has shown skewed production of IgG1 and IgG3 in individuals who develop symptomatic dengue fever (Koraka et al., 2001). It has also been shown that >100-fold increased DENV-specific IgG1 can be found within the cord blood of infants born to DENV-experienced mothers compared to DENV-specific IgG4 (Castanha et al., 2016). This correlates with our findings that increased negative-sense ZIKV RNA and infectious virus release was detected in HCs and placental explants treated with cross-reactive IgG1 and IgG3 mAbs complexed to ZIKV compared to IgG4 and IgG2 mAb subclasses (Figure 6A-B). Translocation of IgG across the placenta is a normal physiologic process facilitated by FcRn, a specialized Fc receptor expressed by STBs, to provide passive immunity to the developing fetus (Coyne and Lazear, 2016; Simister and Story, 1997; Simister et al., 1996; Story et al., 1994). Our results could reflect that FcRn preferentially binds immune complexes comprised of different IgG subclasses. Studies examining the kinetics of monomeric human IgG (hIgG) binding to human FcRn (hFcRn) have determined that hIgG1 binds to hFcRn with greater affinity than hIgG3, hIgG4 and hIgG2, with hIgG2 exhibiting the lowest affinity for FcRn binding (Abdiche et al., 2015; Ober et al., 2001). This correlates with our observations that the highest levels of viral enhancement were observed in IgG1 and IgG3 immune complexes.
Mechanistically, we observed that DENV mAb:ZIKV immune complexes increased viral binding and entry in HCs, a phenomenon known as “extrinsic ADE” (Figure 2B). Moreover, we discovered that increased ZIKV infection of HCs correlated with reduced expression of RLRs (DDX58, IFIH1, DHX58) as well as key antiviral effectors (IFIT1, IFIT2, IFIT3, OAS1, RSAD2) (Figure 3). This may reflect the ability of ZIKV to block type I IFN signaling through mechanisms involving inhibition of STAT phosphorylation (Bowen et al., 2017) and/or STAT2 degradation (Grant et al., 2016). An alternative hypothesis is that binding of cross-reactive DENV mAb immune complexes to FcγRs on HCs dampens innate immune responses through a cell autonomous process termed “intrinsic ADE.” Fc γRII, which is highly expressed on HCs (Figure 5A), is differentiated into two subclasses, FcγRIIa and FcγRIIb, each of which express cytoplasmic Ig gene family tyrosine activation or inhibitory motifs (ITAMs and ITIMS), respectively (Smith and Clatworthy, 2010). Previous studies have demonstrated that binding DENV-Ab complexes to FcγRIIa, but not FcγRIIb, enhances DENV infection of a monocytic cell line. Modifying the ITAM to an ITIM domain on FcγRIIa ablated enhanced DENV infection (Boonnak et al., 2013). However, the downstream effects of switching the ITAM and ITIM domains on modulating antiviral immunity is unclear. Our data revealed that the IgG isotype directly affects viral infection of HCs: (highest to lowest) 33.3A06 IgG3>IgG1>IgG4>> IgG2 (Figure 5B). This order is similar to the optimal Ab binding affinities of FcγRIIb, which is known to exert downstream inhibitory signals through its ITIM cytoplasmic domain (Smith and Clatworthy, 2010). This suggests that intrinsic ADE, in addition to extrinsic ADE, may be an important determinant in Ab-mediated augmentation of ZIKV infection in HCs.
Trafficking of IgG immune complexes coupled with high levels of cellular FcγR expression in HCs within the chorionic villi provides an ideal environment for virus immune complexes to evade the antiviral nature of the placental barrier and infect resident cells. Several congenital viruses, including hCMV and HIV-1, are known to utilize maternal antibodies to transcytose through the trophoblast layer and enter the fetal compartment through FcRn (Maidji et al., 2006; Toth et al., 1994). Similarly, we have demonstrated that DENV cross-reactive mAbs bound to ZIKV undergo FcRn-mediated transcytosis across the placenta to productively infect HCs within the villous stroma (Figures 6–7). These findings could have large implications concerning the role of serologic cross-reactivity on viral infections of the placenta. Past work has shown that related flaviviruses, including West Nile virus (WNV), Japanese encephalitis virus (JEV), and DENV, can be detected within the placenta in vivo and have been associated with adverse outcomes such as spontaneous abortions, microcephaly, and post-natal growth defects (Centers for Disease and Prevention, 2002; Chaturvedi et al., 1980; O’Leary et al., 2006; Ribeiro et al., 2017). Our results presented here, along with previous studies concerning neurotropic flavivirus infection during pregnancy, underpin the importance of understanding of the effects of antibody-mediated viral transport in the placenta and the developing fetus.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author Mehul Suthar (msuthar@emory.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics statement.
Second trimester human placentae were obtained from consented donors who elected to terminate normal pregnancies between weeks 14–20 of gestation. Tissues were received from the University of Pittsburgh Health Sciences Tissue Bank via an honest broker system as approved by the University of Pittsburgh Institutional Review Board and in accordance with the University of Pittsburgh anatomical tissue procurement guidelines. Human term placentae (>37 weeks gestation) were collected from hepatitis B, HIV-1 seronegative women (>18 years of age) immediately after elective cesarean section without labor from Emory Midtown Hospital, Atlanta, GA. This study was approved by the Emory University Institutional Review Board (IRB 000217715). Written informed consent was acquired from all donors before cesarean section and sample collection. Samples were de-identified before being transferred to laboratory personnel for primary HC isolation.
Viruses and cells.
Zika virus (ZIKV) strain PRVABC59 was used for all experiments. PRVABC59 was initially isolated in 2015 from a patient infected while in Puerto Rico. We obtained this strain from the Centers for Disease Control and Prevention in Fort Collins, CO. The virus used in these experiments has undergone a total of 5 passages in Vero cells. Viral titers were determined by plaque assay on Vero cells (ATCC®CCL-81™). ZIKV was UV-inactivated (UV-ZIKV) by exposing virus to UV light in a Spectroline UV Crosslinker for 1 hour. Vero cells were cultured in complete DMEM medium consisting of 1x DMEM (Corning Cellgro), 10% FBS, 25mM HEPES Buffer (Corning Cellgro), 2mM L-glutamine, 1mM sodium pyruvate, 1x Non-essential Amino Acids, and 1x antibiotics, and were maintained at 37°C and 5% CO2.
Hofbauer cell model.
HCs were isolated from membrane-free villous placenta as previously described (Johnson and Chakraborty, 2012). On average, the purity was >95%. After isolation, HCs were cultured in complete RPMI medium consisting of 1x RPMI (Corning Cellgro), 10% FBS (Optima, Atlanta Biologics), 2mM L-glutamine (Corning Cellgro), 1mM sodium pyruvate (Corning Cellgro), 1x Non-essential Amino Acids (Corning Cellgro), 1x antibiotics (penicillin, streptomycin, amphotericin B; Corning Cellgro) at 37°C and 5% CO2. HCs were infected immediately following isolation. Monoclonal antibodies (mAb) were diluted in 1x PBS to the desired concentrations and mixed 1:1 with ZIKV at MOI of 1. No antibody (Ab-) conditions received 1x PBS; no virus (ZIKV-) conditions received RPMI. DENV mAb:ZIKV immune complexes were incubated at 37°C for 1 hour. HCs were then infecte d in 200ul mAb:ZIKV complexes, or with ZIKV alone at MOI of 1 or 10, as indicated, at 37°C for 1 hour. HCs were washed once with warm RPMI to remove residual immune complexes and re-suspended in complete RPMI medium. Infected cells were incubated at 37°C.
Human placental explant model.
Chorionic villi were dissected from placental tissue and maintained in DMEM/F12 medium (ThermoFisher Scientific, Gibco) with 10% FBS and penicillin/streptomycin at 37°C and 5% CO2. Villi were separated into individual wells of a 48-well plate each containing 800ul of DMEM/F12 medium for subsequent experiments. Following isolation of human placental explants, diluted mAbs were mixed 1:1 with 5×105 PFU/ml ZIKV. After the 1 hour incubation, 200ul of explant medium was removed from each well before addition of 200ul mAb:ZIKV complexes. Tissues were incubated at 37°C for 2 hours then washed 2x with w arm complete DMEM/F12 medium to remove residual immune complexes and re-supplied with 800ul complete DMEM/F12 medium. Infected explant tissues were incubated at 37°C and 5% CO2.
Antibodies.
The human monoclonal antibodies (mAbs) used in these experiments were generated as previously described (Priyamvada et al., 2016). Briefly, plasmablasts were isolated from DENV-infected patients and single cell sorted for use in expression cloning. Immunoglobulin (Ig) genes were amplified by RT-PCR and inserted into IgG1 expression vectors. IgG1 vectors were transiently expressed in expi293F cells and secreted IgG antibodies were purified from supernatants using protein A coupled sepharose beads (Pierce). Antibodies were stored in 1x PBS with 0.05% sodium azide. The pan-flavivirus anti-envelope protein 4G2 mAb (mouse IgG1) was isolated from the supernatant of mouse hybridoma D1-4G2-4-15 (ATCC; HB-112) using a protein G column (GE Life Sciences). IgG2, IgG3, and IgG4 variants of mAb 33.3A06 were generated by subcloning the heavy chain variable domain into the appropriate IgG subclass vector by restriction digest (AgeI and SalI) and ligation. IgG3 antibodies were purified using protein G coupled sepharose beads (Pierce). The IgG1-LALA variant of mAb 33.3A06 was generated by replacing the constant region of the wild-type IgG1 heavy chain expression vector with a gene synthesized construct (Integrated DNA Technologies), containing a leucine (L) to alanine (A) substitution at amino acid positions 234 and 235 of the IgG1 constant region by restriction digest (SalI and HindIII) and ligation.
Interferon treatment.
IFN-β (PBL Assay Science) and IFN-λ (PBL Assay Science) were diluted in complete RPMI medium and added to HCs at 10 IU/ml or 100 IU/ml following the 1-hour infection incubation.
Flow cytometry.
Most conditions were run with biological triplicate samples, and 2×105 HCs were used per sample. HCs were blocked for 10min on ice with 0.25ul/sample Human TruStain FcX (BioLegend) in FACS buffer (1x PBS, 0.1% BSA, 1mM EDTA) and stained for surface markers for 20min on ice using 0.25ul/sample of the following anti-human antibodies from BioLegend in FACS buffer: CD14 (M5E2), CD80 (2D10), CD86 (IT2.2), CD40 (5C3), and HLA-DR (G46–6; BD Biosciences); or CD16 (3G8), CD32 (FUN-2), CD64 (10.1), and Ms IgG Isotype Control (C1.18.4; TONBO Biosciences). Cells were also live/dead stained for 20min on ice with 0.1ul/sample either Ghost 780 or Ghost 510 viability dye (TONBO Biosciences) in 1x PBS. HCs were fixed with 1x Transcription Factor Fix/Perm (diluted in Transcription Factor Fix/Perm Diluent; TONBO Biosciences) for 20min on ice and permeabilized by washing twice with 1x Flow Cytometry Perm Buffer (diluted in ddiH2O; TONBO Biosciences). HCs were re-blocked for 5min on ice with 0.25ul/sample Human TruStain FcX and 0.25ul/sample normal mouse serum (ThermoFisher Scientific) in Perm Buffer and stained for ZIKV E protein for 20min on ice using 0.5ul/sample of a 4G2-APC antibody in Perm Buffer. Unconjugated monoclonal 4G2 antibody was conjugated to APC using a Novus Lighting-Link kit per the manufacturer’s instructions. Flow cytometry samples were re-suspended in 1x PBS and run on an LSR-II flow cytometry machine.
Focus-forming assay.
Focus-forming assay (FFA) was performed on Vero cells with supernatants from ADE-ZIKV-infected HCs (2×105 cells per condition) or human placental explants and accompanying controls. Supernatants were initially diluted 1:10 in DMEM with 1% FBS followed by 10-fold serial dilution. Vero cells were plated in a 96-well plate and infected with 50ul diluted supernatant for 1 hour at 37°C. Cells and inoculum were then overlaid with methylcellulose (DMEM [Corning Cellgro], 1% antibiotic, 2% FBS, 2% methylcellulose [Sigma Aldrich]) and incubated at 37°C for 72 hours. Methylcellulose was aspirated, and cells were washed 3x with 1x PBS and fixed/permeabilized with a 1:1 mixture of acetone and methanol. Cells were washed once with 1x PBS and blocked with 5% milk in 1x PBS for 20min at RT. Cells were incubated with primary mouse 4G2 antibody (1µg/mL) in 5% milk in 1x PBS for 2 hours at RT and washed 2x with 1x PBS. Goat anti-mouse HRP-conjugated secondary antibody was applied at 1:3000 dilution in 5% milk in 1x PBS for 1 hour at RT. Cells were washed 2x with 1x PBS and foci were developed with TrueBlue Peroxidase Substrate (KPL). Plates were read on a CTL50 ImmunoSpot S6 Micro Analyzer and spots were counted manually using ImageJ.
Binding and entry assay.
DENV mAb:ZIKV immune complexes were prepared as described above and incubated for 1 hour at 37°C. m Ab:ZIKV complexes and HCs were then chilled on ice for 1 hour prior to infection. HCs were infected with mAb:ZIKV complexes for 1 hour on ice and then washed 4x with ice cold 1x PBS. To assess virus binding to the cell surface, HCs were immediately lysed in RNA lysis buffer after washes as previously described (Boonnak et al., 2013). To assess viral entry into cells, HCs were re-suspended in pre-warmed complete RPMI medium and incubated at 37°C for 2 hours. HCs were then washed 4x with ice cold 1x PBS and lysed in RNA lysis buffer as previously described (Bowen et al., 2017). ZIKV genomic RNA levels were assessed by qRT-PCR as described above.
Quantitative real time-PCR.
HCs infected with DENV mAb:ZIKV immune complexes and control cells (1×105 cells per condition) were lysed in RNA Lysis Buffer. Total RNA was isolated from cells using the Quick-RNA MiniPrep Kit (Zymo Research) per the manufacturer’s instructions. For human placental explants, tissues infected with DENV mAb:ZIKV immune complexes and control conditions were suspended in TRI reagent and mechanically homogenized using ceramic bead tubes (Omni International) on a Beadruptor Homogenizer. Total RNA was isolated from homogenized tissues using the Direct-zol RNA MiniPrep Plus Kit (Zymo Research) per the manufacturer’s instructions. Purified RNA was reverse transcribed using random primers with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). HC gene expression and ZIKV viral RNA levels were quantified by qRT-PCR using PrimeTime Gene Expression Master Mix (Integrated DNA Technologies), ZIKV-specific primers and probe set (see Table S2) (Lanciotti et al., 2008) and TaqMan gene expression assays (ThermoFisher) for host genes (see Table S3): Gapdh (Hs02758991_g1), Ifna2 (Hs00265051_s1), Ifnb1 (Hs01077958_s1), Ifnl1 (Hs00601677_g1), Ifit1 (Hs03027069_s1), Ifit2 (Hs01922738_s1), Ifit3 (Hs01922752_s1), Ddx58 (Hs01061436_m1), Ifih1 (Hs00223420_m1), Dhx58 (Hs01597843_m1), Oas1 (Hs00973637_m1), and Rsad2 (Hs00369813_m1). CT values were normalized to the reference gene Gapdh and represented as fold change over values from time-matched mock samples using the formula 2−∆∆CT. All primers and probes were purchased from Integrated DNA Technologies (IDT). qRT-PCR was performed in 384-well plates and run on an Applied Biosystems 7500 HT Real-Time PCR System.
ZIKV strand-specific qRT-PCR.
Purified RNA was reverse transcribed using oligo(dT) and a ZIKV-specific cDNA primer. The two ZIKV-specific cDNA primers are complementary to either the positive-strand or negative-strand and include a unique 5’ tag (see Table S2). Two cDNA and qRT-PCR reactions were run for each sample, one for positive-strand and one for negative-strand. For ZIKV strand-specific detection, a ZIKV-specific primer and tag-specific primer were used for targeted amplification of the tagged cDNA in addition to the ZIKV-specific probe. CT values were normalized to the reference gene Gapdh and represented as fold change over values from time-matched mock samples using the formula 2−∆∆CT.
qRT-PCR of viral RNA from supernatants.
Total RNA was isolated from the supernatants of infected HCs and human placental explants using the QIAamp Viral RNA Mini Kit (QIAGEN) per the manufacturer’s instructions. ZIKV RNA standard was generated by annealing two oligonucleotides spanning the target ZIKV prM-E gene region and performing in vitro transcription using the MEGAscript SP6 Transcription Kit (Ambion). For ZIKV RNA quantification in supernatants, a standard curve was generated using tenfold serial dilutions of ZIKV RNA standard, and qRT-PCR was performed using ZIKV-specific primers and probe (see Table S2) (Lanciotti et al., 2008) Viral RNA copies were interpolated from the standard curve using the sample CT value and represented as copies per mL of supernatant.
Multiplex bead assay.
Type I IFN and cytokine concentrations in the supernatants of ADE-ZIKV-infected HCs (2×105 cells per condition) and accompanying controls were assessed using a human cytokine 25-plex panel (Novex) and a ProcartaPlex human IFN-beta simplex kit (Invitrogen) per the manufacturers’ instructions. Plates were read on a Luminex 100 Analyzer.
Western blot.
ADE-ZIKV-infected HCs and control cells (1.2×106 cells per condition) were washed 2x with 1x PBS with 1mM EDTA and lysed with modified RIPA buffer (10mM Tris, 150mM NaCl, 1% NA-deoxycholate, 1% Triton X-100, 1x protease inhibitor cocktail [ThermoFisher Scientific], 1x phosphatase inhibitor cocktail [ThermoFisher Scientific]). Protein concentrations were determined by Bradford assay – 2ul cell lysate in 200ul 1x Bradford Reagent (BioRad) and read on a SynergyH1 Hybrid Reader (BioTek). Proteins were denatured with 1x loading buffer (0.25M Tris, 40% glycerol, 20% β-ME, 9.2% SDS, 0.04% Bromophenol Blue) and boiling for 15min. Lysates were then run on SDS-PAGE gel and transferred to nitrocellulose membrane for Western blotting. Blots were blocked in 5% milk in PBST (1xPBS, 0.1% Tween-20) and rinsed with ddiH2O. Blots were incubated with the following primary antibodies in PBST with 10% FBS: Rb anti-IFIT1 (1:1000; Cell Signaling), Ms anti-IFIT2 (1:1000; Cell Signaling), Rb anti-IFIT3 (1:10,000; kindly provided by Dr. Ganes Sen), Rb anti-RIG-I (1:1000; Cell Signaling), Rb anti-MDA5 (1:1000; Cell Signaling), Rb anti-LGP2 (1:100; IBL), Rb anti-Viperin (1:1000; Cell Signaling), and Rb anti-GAPDH (1:2500; Cell Signaling). Blots were washed with PBST and incubated for 10min with anti-mouse or anti-rabbit HRP-conjugated secondary antibodies at 1:750 dilution in PBST with 1% FBS. Blots were washed again with PBST and developed with ThermoScientific SuperSignal West Femto Maximum Sensitivity Substrate. Blots were imaged on a BioRad ChemiDocXRS+.
Endosomal pathway inhibitor analysis.
Human HCs were treated with 100nM Bafilomycin A1 (Cayman Chemical Company) 1 hour prior to infection with immune complexes. Infection was performed as described above and without the removal of Bafilomycin A1. RNA was extracted from tissues and analyzed by ZIKV strand-specific qRT-PCR as described above.
FcRn blocking analysis.
Protein A treatment.
Human placental explants were treated with 1, 10 or 100 ug/ml of Protein A from Staphylococcus aureus (ThermoFisher Scientific) prior to infection with immune complexes. Infection was performed as described above and without the removal of Protein A. Upon removal of viral inoculum and re-suspension in complete DMEM/F12, 1, 10 or 100 ug/ml of Protein A was added back into cell supernatant. RNA was extracted from tissues and analyzed by ZIKV strand-specific qRT-PCR as described above.
FcRn blocking Ab.
Human placental explants were treated with 1 ug/ml of an anti-FcRn blocking antibody (DVN24; Aldevron) prior to infection with immune complexes. Infection was performed as described above and without the removal of the blocking antibody. Upon removal of viral inoculum and re-suspension in complete DMEM/F12, 1 ug/ml anti-FcRn blocking antibody was added back into cell supernatant. RNA was extracted from tissues and analyzed by ZIKV strand-specific qRT-PCR as described above.
Chromogenic staining and imaging.
Human placental explants were fixed in 4% PFA and incubated overnight in 30% sucrose solution. Tissues were then flash frozen in OCT media (Sakura) using 2-methylbutane (ACROS Organics) cooled on dry ice. 20 µm slices were made from tissue blocks and affixed to slides in chilled acetone. Tissues were washed once in 1x TBS Buffer (Abcam) for 5min and permeabilized with 0.3% Triton-X 100 (Fisher Scientific) in TBS for 20min at RT. After washing 3 times in 1x TBS, antigen retrieval was performed by submerging tissue slides in 1x DIVA Decloaker solution (Biocare Medical) and boiling in vegetable steamer for 30min. Tissues were washed 3 times in 1x TBS and incubated with primary antibodies (made up in TBS with 1% BSA and 0.05% Tween-20) overnight at 4°C – Ms an ti-4G2 (3 ug/ml), Rb anti-CD163 (1:300; Abcam). Next, tissues were washed and incubated with either Mach 2 mouse AP polymer (Biocare Medical) or Mach 2 rabbit HRP polymer (Biocare Medical) for 30min at RT. Slides were washed and stained with Warp Red (for AP polymer; Biocare Medical) or Vina Green (for HRP polymer; Biocare Medical) for approximately 5min. Counterstain was performed with Hematoxylin (Vector Laboratories) for 30sec and washing in ddH2O. Slides were dried for 3min at 60°C and mounted w ith Cytoseal (VWR). Slides were imaged using an Aperio Slide Scanner (Leica Biosystems) at 40x magnification.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses.
Viral binding and entry assays were analyzed using 1-way ANOVA and Tukey’s multiple comparison test, p<0.05. Cytokine protein data (multiplex bead assay) and viral binding/entry data were analyzed by 1-way ANOVA followed by Tukey’s test for multiple comparisons, p<0.05. Interferon treatment data were analyzed by 2-way ANOVA followed by Tukey’s test for multiple comparisons, p<0.05. Viral RNA data for both the IgG subclass infections and Protein A treatments of mid-gestation placental explants were analyzed by 1-way ANOVA and Dunnett’s multiple comparison test, p<0.05. Viral RNA data from HCs treated with Bafilomycin A and explants infected with the 33.3A06 IgG1-LALA mutant mAb were analyzed by Student’s t-test, p<0.05. All statistical analysis was performed using GraphPad Prism software. In each of the main and supplemental figure legends, “N” represents the number of patient placental donors from which HCs or explants were derived. Further experimental statistical details can be found in the Figure legends.
Supplementary Material
Highlights:
Cross-reactive DENV antibodies enhance ZIKV infection in human Hofbauer cells
Enhanced ZIKV infection in mid-gestation explants is IgG subclass-dependent
ZIKV immune complexes target Hofbauer cells within the villous stroma
Acknowledgements:
We thank the Department of Pediatrics flow cytometry core and the Emory Vaccine Center flow cytometry core, which is supported in part by the National Institutes of Health grant P30A050509). We also thank Siddhartha Bhaumik for providing us with anti-DENV monoclonal antibodies, and Dr. Hedda Wardemann for providing the IgG subclass vectors.
Funding: This work was funded in part by National Institutes of Health grants U19AI083019 (M.S.S), U01AI131566 (M.S.S. and R.C.), 2U19AI057266 (M.S.S.), ORIP/OD P51OD011132 (M.S.S), Emory Department of Pediatrics CCVI Pilot project (M.S.S and J.W.), 5U19AI057266–13REVIS Supplement (M.S.S and J.W.), Emory University Department of Pediatrics Junior Faculty Focused Award (M.S.S), Children’s Healthcare of Atlanta (M.S.S.), Emory Vaccine Center (M.S.S.), and The Georgia Research Alliance (M.S.S). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: The authors declare no competing interests.
References
- Abdiche YN, Yeung YA, Chaparro-Riggers J, Barman I, Strop P, Chin SM, Pham A, Bolton G, McDonough D, Lindquist K, et al. (2015). The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. MAbs 7, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora N, Sadovsky Y, Dermody TS, and Coyne CB (2017). Microbial Vertical Transmission during Human Pregnancy. Cell Host Microbe 21, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer A, Lennemann NJ, Ouyang Y, Bramley JC, Morosky S, Marques ET Jr., Cherry S, Sadovsky Y, and Coyne CB (2016). Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 19, 705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramello M, Williams KL, Simmons CP, Macagno A, Simonelli L, Quyen NT, Sukupolvi-Petty S, Navarro-Sanchez E, Young PR, de Silva AM, et al. (2010). The human immune response to Dengue virus is dominated by highly cross-reactive antibodies endowed with neutralizing and enhancing activity. Cell Host Microbe 8, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar J, Rabeneck DB, Martines RB, Reagan-Steiner S, Ermias Y, Estetter LB, Suzuki T, Ritter J, Keating MK, Hale G, et al. (2017). Zika Virus RNA Replication and Persistence in Brain and Placental Tissue. Emerg Infect Dis 23, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, et al. (2013). The global distribution and burden of dengue. Nature 496, 504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonnak K, Slike BM, Donofrio GC, and Marovich MA (2013). Human FcgammaRII cytoplasmic domains differentially influence antibody-mediated dengue virus infection. J Immunol 190, 5659–5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, and Suthar MS (2017). Zika Virus Antagonizes Type I Interferon Responses during Infection of Human Dendritic Cells. PLoS Pathog 13, e1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castanha PM, Braga C, Cordeiro MT, Souza AI, Silva CD Jr., Martelli CM, van Panhuis WG, Nascimento EJ, and Marques ET (2016). Placental Transfer of Dengue Virus (DENV)-Specific Antibodies and Kinetics of DENV Infection-Enhancing Activity in Brazilian Infants. J Infect Dis 214, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease, C., and Prevention (2002). Intrauterine West Nile virus infection--New York, 2002. MMWR Morb Mortal Wkly Rep 51, 1135–1136. [PubMed] [Google Scholar]
- Chaturvedi UC, Mathur A, Chandra A, Das SK, Tandon HO, and Singh UK (1980). Transplacental infection with Japanese encephalitis virus. J Infect Dis 141, 712–715. [DOI] [PubMed] [Google Scholar]
- Christianson GJ, Sun VZ, Akilesh S, Pesavento E, Proetzel G, and Roopenian DC (2012). Monoclonal antibodies directed against human FcRn and their applications. MAbs 4, 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne CB, and Lazear HM (2016). Zika virus - reigniting the TORCH. Nat Rev Microbiol 14, 707–715. [DOI] [PubMed] [Google Scholar]
- de Oliveira WK, de Franca GVA, Carmo EH, Duncan BB, de Souza Kuchenbecker R, and Schmidt MI (2017). Infection-related microcephaly after the 2015 and 2016 Zika virus outbreaks in Brazil: a surveillance-based analysis. Lancet 390, 861–870. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J (1981). Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 20, 2361–2370. [PubMed] [Google Scholar]
- Dejnirattisai W, Supasa P, Wongwiwat W, Rouvinski A, Barba-Spaeth G, Duangchinda T, Sakuntabhai A, Cao-Lormeau VM, Malasit P, Rey FA, et al. (2016). Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat Immunol 17, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney A, Mai C, Smoots A, Cragan J, Ellington S, Langlois P, Breidenbach R, Fornoff J, Dunn J, Yazdy M, et al. (2018). Population-Based Surveillance of Birth Defects Potentially Related to Zika Virus Infection - 15 States and U.S. Territories, 2016. MMWR Morb Mortal Wkly Rep 67, 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald B, Boyle C, and Honein MA (2018). Birth Defects Potentially Related to Zika Virus Infection During Pregnancy in the United States. JAMA [DOI] [PMC free article] [PubMed]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sanchez-Seco MP, Evans MJ, Best SM, et al. (2016). Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 19, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halai UA, Nielsen-Saines K, Moreira ML, de Sequeira PC, Pereira JP, Zin AA, Cherry J, Gabaglia CR, Gaw SL, Adachi K, et al. (2017). Maternal Zika Virus Disease Severity, Virus Load, Prior Dengue Antibodies, and Their Relationship to Birth Outcomes. Clin Infect Dis 65, 877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CM, Landucci G, Forthal DN, Parren PW, et al. (2007). Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449, 101–104. [DOI] [PubMed] [Google Scholar]
- Jagger BW, Miner JJ, Cao B, Arora N, Smith AM, Kovacs A, Mysorekar IU, Coyne CB, and Diamond MS (2017). Gestational Stage and IFN-lambda Signaling Regulate ZIKV Infection In Utero. Cell Host Microbe 22, 366–376 e363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendeberg L, Nilsson P, Larsson A, Denker P, Uhlen M, Nilsson B, and Nygren PA (1997). Engineering of Fc(1) and Fc(3) from human immunoglobulin G to analyse subclass specificity for staphylococcal protein A. J Immunol Methods 201, 25–34. [DOI] [PubMed] [Google Scholar]
- Johnson EL, and Chakraborty R (2012). Placental Hofbauer cells limit HIV-1 replication and potentially offset mother to child transmission (MTCT) by induction of immunoregulatory cytokines. Retrovirology 9, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado KA, Simoni MK, Tang Z, Uraki R, Hwang J, Householder S, Wu M, Lindenbach BD, Abrahams VM, Guller S, et al. (2016). Zika virus productively infects primary human placenta-specific macrophages. JCI Insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koraka P, Suharti C, Setiati TE, Mairuhu AT, Van Gorp E, Hack CE, Juffrie M, Sutaryo J, Van Der Meer GM, Groen J, et al. (2001). Kinetics of dengue virus-specific serum immunoglobulin classes and subclasses correlate with clinical outcome of infection. J Clin Microbiol 39, 4332–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanciotti RS, Kosoy OL, Laven JJ, Velez JO, Lambert AJ, Johnson AJ, Stanfield SM, and Duffy MR (2008). Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg Infect Dis 14, 1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Daniels BP, Pinto AK, Huang AC, Vick SC, Doyle SE, Gale M Jr., Klein RS, and Diamond MS (2015a). Interferon-lambda restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med 7, 284ra259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, and Diamond MS (2016). Zika Virus: New Clinical Syndromes and Its Emergence in the Western Hemisphere. J Virol 90, 4864–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazear HM, Nice TJ, and Diamond MS (2015b). Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 43, 15–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindmark R, Thoren-Tolling K, and Sjoquist J (1983). Binding of immunoglobulins to protein A and immunoglobulin levels in mammalian sera. J Immunol Methods 62, 1–13. [DOI] [PubMed] [Google Scholar]
- Maidji E, McDonagh S, Genbacev O, Tabata T, and Pereira L (2006). Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal Fc receptor-mediated transcytosis. Am J Pathol 168, 1210–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo ASO, Chimelli L, and Tanuri A (2017). Congenital Zika Virus Infection: Beyond Neonatal Microcephaly-Reply. JAMA Neurol 74, 610–611. [DOI] [PubMed] [Google Scholar]
- Michlmayr D, and Lim JK (2014). Chemokine receptors as important regulators of pathogenesis during arboviral encephalitis. Front Cell Neurosci 8, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DR, Kuhn S, Kniss KL, Hinckley AF, Rasmussen SA, Pape WJ, Kightlinger LK, Beecham BD, Miller TK, Neitzel DF, et al. (2006). Birth outcomes following West Nile Virus infection of pregnant women in the United States: 2003–2004. Pediatrics 117, e537–545. [DOI] [PubMed] [Google Scholar]
- Ober RJ, Radu CG, Ghetie V, and Ward ES (2001). Differences in promiscuity for antibody-FcRn interactions across species: implications for therapeutic antibodies. Int Immunol 13, 1551–1559. [DOI] [PubMed] [Google Scholar]
- Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, and Carneiro-Sampaio M (2012). IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immunol 2012, 985646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul LM, Carlin ER, Jenkins MM, Tan AL, Barcellona CM, Nicholson CO, Michael SF, and Isern S (2016). Dengue virus antibodies enhance Zika virus infection. Clin Transl Immunology 5, e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyamvada L, Quicke KM, Hudson WH, Onlamoon N, Sewatanon J, Edupuganti S, Pattanapanyasat K, Chokephaibulkit K, Mulligan MJ, Wilson PC, et al. (2016). Human antibody responses after dengue virus infection are highly cross-reactive to Zika virus. Proc Natl Acad Sci U S A 113, 7852–7857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quicke KM, Bowen JR, Johnson EL, McDonald CE, Ma H, O’Neal JT, Rajakumar A, Wrammert J, Rimawi BH, Pulendran B, et al. (2016). Zika Virus Infects Human Placental Macrophages. Cell Host Microbe 20, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan M, Chen MY, Gastinel LN, and Bjorkman PJ (1994). Investigation of the interaction between the class I MHC-related Fc receptor and its immunoglobulin G ligand. Immunity 1, 303–315. [DOI] [PubMed] [Google Scholar]
- Rasmussen SA, Jamieson DJ, Honein MA, and Petersen LR (2016). Zika Virus and Birth Defects--Reviewing the Evidence for Causality. N Engl J Med 374, 1981–1987. [DOI] [PubMed] [Google Scholar]
- Reagan-Steiner S, Simeone R, Simon E, Bhatnagar J, Oduyebo T, Free R, Denison AM, Rabeneck DB, Ellington S, Petersen E, et al. (2017). Evaluation of Placental and Fetal Tissue Specimens for Zika Virus Infection - 50 States and District of Columbia, January-December, 2016. MMWR Morb Mortal Wkly Rep 66, 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro CF, Lopes VGS, Brasil P, Pires ARC, Rohloff R, and Nogueira RMR (2017). Dengue infection in pregnancy and its impact on the placenta. Int J Infect Dis 55, 109–112. [DOI] [PubMed] [Google Scholar]
- Schliefsteiner C, Peinhaupt M, Kopp S, Logl J, Lang-Olip I, Hiden U, Heinemann A, Desoye G, and Wadsack C (2017). Human Placental Hofbauer Cells Maintain an Anti-inflammatory M2 Phenotype despite the Presence of Gestational Diabetes Mellitus. Front Immunol 8, 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro-Mendoza CK, Rice ME, Galang RR, Fulton AC, VanMaldeghem K, Prado MV, Ellis E, Anesi MS, Simeone RM, Petersen EE, et al. (2017). Pregnancy Outcomes After Maternal Zika Virus Infection During Pregnancy - U.S. Territories, January 1, 2016-April 25, 2017. MMWR Morb Mortal Wkly Rep 66, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simister NE (1998). Human placental Fc receptors and the trapping of immune complexes. Vaccine 16, 1451–1455. [DOI] [PubMed] [Google Scholar]
- Simister NE, and Story CM (1997). Human placental Fc receptors and the transmission of antibodies from mother to fetus. J Reprod Immunol 37, 1–23. [DOI] [PubMed] [Google Scholar]
- Simister NE, Story CM, Chen HL, and Hunt JS (1996). An IgG-transporting Fc receptor expressed in the syncytiotrophoblast of human placenta. Eur J Immunol 26, 1527–1531. [DOI] [PubMed] [Google Scholar]
- Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, and Kuhn RJ (2016). The 3.8 A resolution cryo-EM structure of Zika virus. Science 352, 467–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KG, and Clatworthy MR (2010). FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol 10, 328–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story CM, Mikulska JE, and Simister NE (1994). A major histocompatibility complex class I-like Fc receptor cloned from human placenta: possible role in transfer of immunoglobulin G from mother to fetus. J Exp Med 180, 2377–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, Petitt M, Puerta-Guardo H, Michlmayr D, Wang C, Fang-Hoover J, Harris E, and Pereira L (2016). Zika Virus Targets Different Primary Human Placental Cells, Suggesting Two Routes for Vertical Transmission. Cell Host Microbe 20, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A, Foo SS, Bruzzone R, Dinh LV, King NJ, and Mahalingam S (2015). Fc receptors in antibody-dependent enhancement of viral infections. Immunol Rev 268, 340–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth FD, Mosborg-Petersen P, Kiss J, Aboagye-Mathiesen G, Zdravkovic M, Hager H, Aranyosi J, Lampe L, and Ebbesen P (1994). Antibody-dependent enhancement of HIV-1 infection in human term syncytiotrophoblast cells cultured in vitro. Clin Exp Immunol 96, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai TT, Chuang YJ, Lin YS, Chang CP, Wan SW, Lin SH, Chen CL, and Lin CF (2014). Antibody-dependent enhancement infection facilitates dengue virus-regulated signaling of IL-10 production in monocytes. PLoS Negl Trop Dis 8, e3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden V, Pessoa A, Dobyns W, Barkovich AJ, Junior HV, Filho EL, Ribeiro EM, Leal MC, Coimbra PP, Aragao MF, et al. (2016). Description of 13 Infants Born During October 2015-January 2016 With Congenital Zika Virus Infection Without Microcephaly at Birth - Brazil. MMWR Morb Mortal Wkly Rep 65, 1343–1348. [DOI] [PubMed] [Google Scholar]
- Wahala WM, and Silva AM (2011). The human antibody response to dengue virus infection. Viruses 3, 2374–2395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JP, Liu P, Latz E, Golenbock DT, Finberg RW, and Libraty DH (2006). Flavivirus activation of plasmacytoid dendritic cells delineates key elements of TLR7 signaling beyond endosomal recognition. J Immunol 177, 7114–7121. [DOI] [PubMed] [Google Scholar]
- Wrammert J, Koutsonanos D, Li GM, Edupuganti S, Sui J, Morrissey M, McCausland M, Skountzou I, Hornig M, Lipkin WI, et al. (2011). Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J Exp Med 208, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.