

Abstract
Effects of Alzheimer’s disease (AD) risk factors on brain volume changes may partly explain what happens during the pre-clinical AD stage in people who develop subsequent cognitive impairment (SI). We investigated predictors of neurodegeneration, measured by MRI-based volume loss, in older adults prior to diagnosis of cognitive impairment. There were 623 cognitively normal and 65 SI Baltimore Longitudinal Study of Aging participants (age 55–92 years) enrolled in the neuroimaging substudy from 1994 to 2015. Mixed-effects regression was used to assess the associations of AD risk factors (age, APOE e4 carrier status, diabetes, hypertension, obesity, current smoking, and elevated cholesterol) with brain regional volume change among the overall sample and by diagnostic status. Older age, APOE e4 carrier status, hypertension, and HDL cholesterol were predictors of volumetric change. Among SI participants only, hypertension, obesity, and APOE e4 carrier status were associated with greater declines in selected brain regions. SI individuals in pre-clinical AD stage are vulnerable to risk factors that have either a protective or null effect in cognitively normal individuals.
Keywords: neurodegeneration, cognitive impairment, cardiovascular risk factors
1. Introduction
The pathophysiological process of Alzheimer’s disease (AD) likely begins decades before symptom onset. A well-established AD biomarker is atrophy on structural MRI, including global and hippocampal volume loss and ventricular volume expansion (DeCarli et al., 2007; Desikan et al., 2009; Dickerson et al., 2012; Fleisher et al., 2005; Vemuri et al., 2009). Longitudinal MRI-based measures provide estimates of trajectories of brain atrophy, a proxy for neurodegeneration. Brain atrophy is correlated with neuronal loss (Schuster et al., 2015). While longitudinal volume loss and ventricular expansion are observed in both cognitively normal and subsequently impaired (SI) individuals, the latter group shows greater rates of atrophy (Fjell et al., 2013; Pacheco et al., 2015), signaling impending cognitive impairment.
Longitudinal MRI-based trajectories of brain structural changes can be used to examine the associations of AD risk factors on annual rates of regional volumetric change. Cardiovascular risk factors (Kivipelto et al., 2001; Snyder et al., 2015) are associated with increased AD risk. Age (Jack Jr et al., 2017), sex (Fleisher et al., 2005; Jack Jr et al., 2017; Li and Singh, 2014), and the Apolipoprotein E e4 (APOE e4) risk allele (Fleisher et al., 2005) have also been implicated in the risk of dementia and AD, specifically. Yet, there is limited information regarding how these risk factors affect longitudinal brain volumetric changes.
To identify patterns of predictors of neurodegeneration during the preclinical stage, we investigated a sample of 623 cognitively normal and 65 SI Baltimore Longitudinal Study of Aging (BLSA) participants with structural MRIs collected from 1994 onward, making this data rich with repeated measures of older adults prior to symptom onset (Driscoll et al., 2009; Resnick et al., 2003). We first characterized and compared the trajectories of volumetric change of brain regions of interest (ROIs) in the overall sample and by diagnostic status (SI vs. cognitive normal). Second, we identified predictors of volumetric change in the overall sample. Lastly, we examined associations of predictors with volumetric change in selected ROIs, stratified by diagnostic status.
2. Materials and Methods
2.1. Characteristics of the Study Sample
There were 889 participants from the BLSA neuroimaging substudy who were followed from February 1994 to December 2015. The BLSA imaging and visit schedules have varied over time. Participants in the original imaging study had annual imaging assessments from 1994–2004, and they were enrolled based on enrollment procedures described elsewhere (Resnick et al., 2003). Thereafter, participants aged 60 to 79 years had biennial BLSA and imaging visits, while participants aged 80 years and older had annual visits. Participants were excluded, based on significant health conditions that could affect brain structure or function (i.e. stroke, closed head injury, cranial/brain surgery, malignant cancer, gliomas, intracranial cysts with brain tissue displacement, seizure and bipolar disorders; n=30). There were three participants with myocardial infarction and two participants with angioplasty prior to enrollment in the MRI study. These participants were included in main analyses, but we performed a sensitivity analysis excluding them. The results were unchanged. Supplementary Figure 1 shows the inclusion and exclusion criteria of the current study. The final sample was 688 participants with 2,137 scans.
The procedures for diagnostic status determination have been detailed previously (Resnick et al., 2003), and these are detailed in Appendix I. Briefly, BLSA participants’ serial clinical and neuropsychological data were reviewed at each consensus case conference if they had ≥4 errors on the Blessed Information-Memory-Concentration Test (Fuld, 1978), or if their Clinical Dementia Rating Scale total combined score was ≥0.5 (Morris, 1993). Diagnostic criteria for Mild Cognitive Impairment (MCI) and dementia are included in Supplemental Figure 1 and Appendix I. The local Institutional Review Board approved the research protocol for this study, and written informed consent was obtained at each visit from all participants.
2.2. Predictors of Neurodegeneration
Demographic characteristics.
Demographic characteristics included baseline mean-centered age, sex, diagnostic status, race (white vs. non-white), mean-centered years of education, and APOE e4 carrier status (≥1 vs. 0 e4 alleles).
Vascular burden.
Baseline vascular burden (Gottesman et al., 2017), the cumulative burden of cardiovascular risk factors, was defined by the following: current smoking status (current vs. former/never), hypertension diagnosis, diabetes diagnosis, obesity (body mass index ≥30 kg/m2 vs. <30 kg/m2), and elevated total cholesterol (≥200 mg/dl vs. <200 mg/dl) (Gottesman et al., 2017). Detailed definitions of hypertension and diabetes are listed in Table 1. Total cholesterol was calculated using Friedewald’s formula: the sum of high-density lipoprotein (HDL) and low-density lipoprotein (LDL) cholesterol and 20% of triglycerides (Friedewald et al., 1972). We also examined each component of total cholesterol. Because few had 3–4 cardiovascular risk factors at most, vascular burden was categorized as 0, 1, or 2+ cardiovascular risk factors.
Table 1.
Sample Characteristics from the Baltimore Longitudinal Study of Aging (N=688)
| Overall | Cognitively Normal |
Subsequently Impaired |
p-value for difference by diagnostic status |
|
|---|---|---|---|---|
| Baseline Characteristics | N=688 | N=623 | N=65 | |
| Age, in years, mean(SD) | 71.4 (8.6) | 71.20 (8.7) | 73.83 (7.7) | 0.019 |
| Male, n(%) | 332 (48.3) | 295 (47.4) | 37 (56.9) | 0.181 |
| White, n(%) | 513 (74.6) | 456 (73.2) | 57 (87.7) | 0.016 |
| Education, in years, mean(SD) | 16.8 (2.6) | 16.87 (2.5) | 16.17 (3.4) | 0.037 |
| APOE e4 allele, n(%) | 148 (27.3) | 129 (26.8) | 19 (30.6) | 0.627 |
| APOE e2 allele, n(%) | 84 (12.2) | 76 (12.2) | 8 (12.3) | 1.000 |
| Hypertension*, n(%) | 158 (23.0) | 133 (21.3) | 25 (38.5) | 0.003 |
| Diabetes**, n(%) | 28 (4.1) | 28 (4.5) | 0 (0.0) | 0.157 |
| Elevated Cholesterol, n(%) | 268 (39.2) | 232 (37.4) | 36 (57.1) | 0.004 |
| Obese, n(%) | 171 (25.0) | 155 (25.0) | 16 (25.0) | 1.000 |
| Current Smoker, n(%) | 24 (3.5) | 20 (3.2) | 4 (6.2) | 0.381 |
| Vascular Burden, n(%) | 0.005 | |||
| 0 conditions | 236 (34.3) | 221 (35.5) | 15 (23.1) | |
| 1 condition | 287 (41.7) | 263 (42.2) | 24 (36.9) | |
| 2+ conditions | 165 (24.0) | 139 (22.3) | 26 (40.0) | |
| Glucose, mean(SD) | 97.6 (14.2) | 97.4 (14.1) | 99.0 (15.6) | 0.403 |
| HDL Cholesterol, mean(SD) | 59.0 (17.6) | 59.8 (17.5) | 50.7 (15.6) | <0.001 |
| LDL Cholesterol, mean(SD) | 106.3 (32.2) | 105.3 (32.0) | 115.7 (32.1) | 0.013 |
| Triglycerides, mean(SD) | 100.0 (56.4) | 99.62 (55.95) | 103.8 (61.1) | 0.569 |
| Intracranial Volume, mean(SD) | 1393.1 (144.0) | 1392.6 (143.2) | 1398.2 (152.5) | 0.763 |
| Follow-up Time, mean (SD) | 3.7 (4.7) | 3.5 (4.7) | 4.6 (4.3) | <0.001 |
| Number of Follow-up Visits, n(%) | <0.001 | |||
| 1 | 688 (100.0) | 623 (100) | 65 (100) | |
| 2 | 422 (61.3) | 372 (59.7) | 50 (76.9) | |
| 3 | 218 (31.7) | 175 (28.1) | 43 (66.2) | |
| 4 | 143 (20.8) | 102 (16.4) | 41 (63.1) | |
| 5 | 124 (18.0) | 84 (13.5) | 40 (61.5) | |
| 6 | 112 (16.3) | 74 (11.9) | 38 (58.5) | |
| 7 | 104 (15.1) | 68 (10.9) | 36 (55.4) | |
| 8 | 83 (12.1) | 60 (9.6) | 23 (35.4) | |
| 9 | 71 (10.3) | 50 (8.0) | 21 (32.3) | |
| 10 | 55 (8.0) | 42 (6.7) | 13 20.0) | |
| 11 | 40 (5.8) | 34 (5.5) | 6 (9.2) | |
| 12+ | 77 (11.2) | 63 (10.1) | 14 (21.5) | |
SD – standard deviation, ICV – intracranial volume, HDL – high-density lipoprotein, LDL – low-density lipoprotein
Note: We used t-tests for continuous variables and chi-squared tests for categorical variables. There were 145 (21.1%) missing for APOE e4 genotype, 6 (0.9%) missing for baseline diabetes, 5 (0.7%) missing for baseline elevated cholesterol, and 4 (0.6%) missing for baseline obesity status.
Hypertension diagnosis was defined as a systolic blood pressure ≥140 mm Hg and/or diastolic blood pressure ≥90 mm Hg or treatment with antihypertensive medications.
Diabetes diagnosis was defined by fasting glucose >125 mg/dL, a pathologic oral glucose tolerance test, or a positive history of a diagnosis plus treatment with oral anti-diabetic drugs or insulin.
2.3. Image Acquisition
Scanning was performed on a General Electric (GE) Signa 1.5 T scanner (Milwaukee, WI) or a 3T Philips Achieva. GE 1.5-T scans used a high-resolution volumetric spoiled gradient recalled acquisition in a steady state (GRASS) series (axial acquisition, repetition time=35msec, echo time=5msec, flip angle=45°, field of view=24 cm, matrix=256×256, number of excitations=1, voxel dimensions=0.94×0.94×1.5 mm slice thickness). T1-weighted magnetization-prepared rapid gradient echo (MPRAGE) scans were acquired on a 3T Philips Achieva (repetition time [TR]=6.8msec, echo time [TE]=3.2msec, flip angle=8°, image matrix=256×256, 170 slices, pixel size=1×1mm, slice thickness=1.2mm). There were 152 participants with 1,035 1.5-T scans and 536 participants with 1,102 3-T scans.
2.4. Harmonization of MUSE Anatomical Labels across 1.5-T SPGR and 3-T MPRAGE
A new automated labeling method specifically designed to achieve a consistent parcellation of brain anatomy in longitudinal MRI studies with scanner and imaging protocol differences was used to harmonize BLSA MRI data. This method combines the MUSE anatomical labeling approach (Doshi et al., 2016) with harmonized acquisition-specific atlases (Erus et al., 2018). The approach is described in more detail in Erus et al. (2018). Briefly, using 35 labeled 3-T MPRAGE brain MRIs from the OASIS data set (available for download at https://masi.vuse.vanderbilt.edu/workshop2012) as atlases, we first performed the MUSE labeling method on 3-T MPRAGE images for 32 BLSA participants with 1.5-T spoiled gradient recalled (SPGR) images at an earlier time point. Then, for each participant, we deformably registered their 1.5-T SPGR image to their 3-T MPRAGE image using a robust registration strategy that combines an ensemble of registrations obtained using two different algorithms and multiple smoothness parameters. From these steps, we obtained 32 pairs of 1.5-T SPGR and 3-T MPRAGE images in the same space with common anatomical labels. These then served as atlases in the MUSE approach to obtain labels on the entire BLSA collection of 1.5-T SPGR and 3-T MPRAGE images. This workflow for anatomical labeling has been extensively validated on the BLSA MRI data set (Erus et al., 2018). Stability measures for longitudinal volumes were consistent over time, with intraclass correlations ranging from 0.89 to 0.99.
2.5. Statistical Analysis
We characterized the sample using means and percentages, and we evaluated differences of baseline sample characteristics by diagnostic status, using two-sample t-tests for continuous variables and χ2 tests for categorical variables. Type I error level was set to 0.05 for ROI analyses, and we applied a more stringent level of p<0.001 for multiple comparisons adjustment. Stata SE 15.0 (StataCorp, 2017) was used for all analyses.
2.5.1. Longitudinal Brain Volumetric Change as Function of Diagnostic Status
Linear mixed-effects models were used to compare longitudinal changes in global and lobar regions as well as in specific brain structures in the overall sample. Our base model consisted of fixed effects, i.e., baseline intracranial volume (ICV), image type (1.5-T SPGR vs. 3-T MPRAGE) (Erus et al., 2018), age, sex, diagnostic status, race, time, and two-way interactions of image type, age, sex, diagnostic status, and race with time, and random effects (intercept and time) with unstructured covariance. Random effects allowed individual-specific baseline brain volumes and rates of volumetric change to vary.
Annual rates of change were estimated from the base model. Also, we further evaluated differences in change of unstandardized and standardized ROI volumes by diagnostic status. Each ROI volume was converted into z-score standardized to volume at baseline visit (both mean and standard deviation [SD]) across all participants. Effect sizes (ES) for difference in rates of ROI volumetric changes by diagnostic status were calculated by dividing the estimated difference in annual rates of change by the estimated SD of the between-subject rates of change. Given that this analysis was exploratory, all results are reported in tables to help guide future research.
2.5.2. Predictors of Volumetric Change
To evaluate the associations of predictors with annual ROI volumetric change in the overall sample, we added each predictor and its interaction with time (predictor*time) to the base model for each brain region. If predictor*time was significant, we used likelihood ratio tests to determine whether model fit improved with the inclusion of the terms. Also, we modeled three-way interactions among each predictor, diagnostic status, and time to determine whether diagnostic status modified the association between the predictor and rate of volumetric change. We included all significant predictors, the two-way interactions of each predictor with time, and three-way interactions of predictors, diagnostic status, and time to the base model. This model became our final model that was used for examining associations of predictors of volumetric change in the overall sample. We tested the two-way and three-way interactions that allowed for the longitudinal change to accelerate or decelerate as a function of the covariates of interest.
2.5.3. Comparison of Predictors of Volumetric Change by Diagnostic Status
We then stratified the final model by diagnostic status. ROIs were selected by the significance of the interaction of diagnostic status with time. Additional ROIs were selected from a previous BLSA study on longitudinal patterns of ROI volumetric changes (Driscoll et al., 2009). These were whole brain volume, ventricles, temporal gray matter (GM), orbitofrontal cortex, and temporal association cortices, including the hippocampus. As a sensitivity analysis, we excluded those with myocardial infarction (n=3) and angioplasty (n=2) prior to baseline MRI in these analyses to determine whether these conditions confounded the main findings.
3. Results
3.1. Characteristics of Study Sample
Table 1 shows the sample characteristics for the overall sample and by diagnostic status. On average, SI (n=65) were older and had fewer years of education, lower HDL cholesterol, higher LDL cholesterol, and more follow-up time on study than cognitively normal (n=623) (Table 1). Also, SI were more likely to be white and have hypertension, greater vascular burden, and elevated cholesterol than cognitively normal. Distributions of sex, APOE e4 allele, current smoker status, diabetes, obesity, glucose, triglycerides, and ICV were similar between SI and cognitively normal groups (Table 1). The average follow-up time for the overall sample was 3.7 years (Standard Deviation, [SD]=4.7 years). The mean follow-up time for participants with more than one visit (N=266) was 5.5 (SD=5.3) years in the overall sample, 5.1 (SD=5.3) years among cognitively normal participants, and 8.3 (SD=4.7) years among SI participants. The average time between the last imaging assessment included in the study and date of MCI/dementia onset was 3.4 (SD=3.2) years.
3.2. Longitudinal Brain Volumetric Change as Function of Diagnostic Status
Table 2 presents the annual rates of change in global and lobar brain volumes, amygdala, hippocampus, entorhinal cortex, and parahippocampal gyrus in the overall sample and by diagnostic status. There were significant longitudinal declines in volumes of global and lobar regions and ventricular enlargement in the overall sample (Table 2).
Table 2.
Annual rates of change in regional brain volumes (cm3) in the Baltimore Longitudinal Study of Aging (N=688)
| Unstandardized Brain Volumes | Standardized Brain Volumes | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall Sample | SI | Cognitively Normal |
Difference between SI and Cognitively Normal |
SI | Cognitively Normal |
Difference between SI and Cognitively Normal |
||||||||||||||
| Brain Regions of Interest |
β | SE | p- value |
β | SE | β | SE | β | SE | p- value |
Effect Size |
β | SE | β | SE | β | SE | p- value |
Effect Size |
|
| Total Brain | −4.31 | 0.38 | <0.01 | −4.67 | 0.49 | −4.27 | 0.38 | −0.40 | 0.37 | 0.28 | −0.21 | −0.038 | 0.004 | −0.035 | 0.003 | −0.003 | 0.003 | 0.28 | −0.21 | |
| GM | −3.79 | 0.29 | <0.01 | −4.29 | 0.38 | −3.73 | 0.30 | −0.56 | 0.29 | 0.05 | −0.34 | −0.064 | 0.006 | −0.056 | 0.004 | −0.008 | 0.004 | 0.05 | −0.34 | |
| WM | −1.59 | 0.16 | <0.01 | −1.72 | 0.22 | −1.58 | 0.17 | −0.14 | 0.16 | 0.39 | −0.16 | −0.032 | 0.004 | −0.030 | 0.003 | −0.003 | 0.003 | 0.39 | −0.16 | |
| Ventricles | 1.25 | 0.11 | <0.01 | 1.63 | 0.17 | 1.21 | 0.11 | 0.42 | 0.14 | <0.01 | 0.44 | 0.078 | 0.008 | 0.058 | 0.005 | 0.020 | 0.007 | <0.01 | 0.44 | |
| Amygdala | −0.02 | 0.00 | <0.01 | −0.03 | 0.00 | −0.01 | 0.00 | −0.01 | 0.00 | <0.01 | −0.78 | −0.090 | 0.011 | −0.052 | 0.008 | −0.038 | 0.008 | <0.01 | −0.78 | |
| Hippocampus | −0.05 | 0.01 | <0.01 | −0.07 | 0.01 | −0.05 | 0.01 | −0.02 | 0.01 | <0.01 | −0.61 | −0.079 | 0.008 | −0.054 | 0.006 | −0.025 | 0.006 | <0.01 | −0.61 | |
| Entorhinal Cortex | −0.02 | 0.01 | <0.01 | −0.04 | 0.01 | −0.02 | 0.01 | −0.02 | 0.01 | <0.01 | −0.78 | −0.066 | 0.011 | −0.030 | 0.009 | −0.035 | 0.009 | <0.01 | −0.79 | |
| Parahippocampal Gyrus | −0.03 | 0.01 | <0.01 | −0.04 | 0.01 | −0.03 | 0.01 | −0.02 | 0.01 | <0.01 | −0.48 | −0.048 | 0.009 | −0.030 | 0.007 | −0.018 | 0.006 | <0.01 | −0.48 | |
| Frontal Lobe | −2.05 | 0.15 | <0.01 | −2.17 | 0.19 | −2.04 | 0.15 | −0.13 | 0.14 | 0.35 | −0.19 | −0.054 | 0.005 | −0.050 | 0.004 | −0.003 | 0.004 | 0.36 | −0.20 | |
| Frontal GM | −1.33 | 0.10 | <0.01 | −1.44 | 0.13 | −1.32 | 0.10 | −0.12 | 0.10 | 0.23 | −0.22 | −0.069 | 0.006 | −0.063 | 0.005 | −0.006 | 0.005 | 0.23 | −0.22 | |
| Frontal WM | −0.68 | 0.07 | <0.01 | −0.70 | 0.09 | −0.68 | 0.07 | −0.02 | 0.07 | 0.75 | −0.06 | −0.033 | 0.004 | −0.032 | 0.003 | −0.001 | 0.003 | 0.75 | −0.06 | |
| Temporal Lobe | −0.97 | 0.08 | <0.01 | −1.18 | 0.10 | −0.94 | 0.08 | −0.23 | 0.07 | <0.01 | −0.56 | −0.049 | 0.004 | −0.040 | 0.003 | −0.010 | 0.003 | <0.01 | −0.56 | |
| Temporal GM | −0.67 | 0.06 | <0.01 | −0.84 | 0.08 | −0.65 | 0.06 | −0.19 | 0.06 | <0.01 | −0.56 | −0.067 | 0.006 | −0.052 | 0.005 | −0.015 | 0.005 | <0.01 | −0.57 | |
| Temporal WM | −0.28 | 0.04 | <0.01 | −0.31 | 0.06 | −0.27 | 0.04 | −0.03 | 0.04 | 0.40 | −0.19 | −0.025 | 0.005 | −0.022 | 0.003 | −0.003 | 0.003 | 0.40 | −0.19 | |
| Parietal Lobe | −0.89 | 0.08 | <0.01 | −0.99 | 0.10 | −0.87 | 0.08 | −0.12 | 0.08 | 0.13 | −0.30 | −0.049 | 0.005 | −0.043 | 0.004 | −0.006 | 0.004 | 0.13 | −0.30 | |
| Parietal GM | −0.59 | 0.06 | <0.01 | −0.67 | 0.07 | −0.58 | 0.06 | −0.09 | 0.06 | 0.12 | −0.32 | −0.062 | 0.007 | −0.054 | 0.005 | −0.008 | 0.005 | 0.12 | −0.32 | |
| Parietal WM | −0.27 | 0.04 | <0.01 | −0.29 | 0.05 | −0.27 | 0.04 | −0.02 | 0.04 | 0.55 | −0.10 | −0.027 | 0.005 | −0.025 | 0.004 | −0.002 | 0.004 | 0.55 | −0.11 | |
| Occipital Lobe | −0.55 | 0.06 | <0.01 | −0.69 | 0.09 | −0.53 | 0.06 | −0.16 | 0.06 | 0.01 | −0.39 | −0.045 | 0.006 | −0.035 | 0.004 | −0.010 | 0.004 | 0.01 | −0.39 | |
| Occipital GM | −0.43 | 0.06 | <0.01 | −0.53 | 0.07 | −0.42 | 0.06 | −0.11 | 0.06 | 0.05 | −0.36 | −0.053 | 0.007 | −0.042 | 0.006 | −0.011 | 0.006 | 0.05 | −0.36 | |
| Occipital WM | −0.10 | 0.03 | <0.01 | −0.13 | 0.04 | −0.09 | 0.03 | −0.04 | 0.03 | 0.17 | −0.23 | −0.022 | 0.006 | −0.016 | 0.005 | −0.006 | 0.004 | 0.17 | −0.23 | |
SI – subsequently impaired, GM – gray matter, WM – white matter, SE = standard error, Note: All bolded values mean p≤0.01. Linear mixed-effects models that included baseline ICV, scan type, age, sex, diagnostic status, race, time, and two-way interactions of scan type, age, sex, diagnostic status, and race with time were used to determine annual rates of change. Continuous variables were mean-centered.
There were differences in rates of volumetric declines by diagnostic status. Compared to cognitively normal participants, SI participants had steeper rates of annual ventricular enlargement (β=0.42, standard error, [SE]=0.14, p<0.01) and steeper rates of annual decline in amygdala (β=−0.01, SE=0.00, p<0.01), hippocampus (β=−0.02, SE=0.01, p<0.01), entorhinal cortex (β=−0.02, SE=0.01, p<0.01), parahippocampal gyrus (β=−0.02, SE=0.01, p<0.01), temporal lobe (β=−0.23, SE=0.07, p<0.01), temporal GM (β=−0.19, SE=0.06, p<0.01), and occipital lobe (β=−0.16, SE=0.06, p=0.01) (Table 2). Standardized ES to evaluate the differences by diagnostic status ranged from −0.79 for the entorhinal cortex to 0.44 for the ventricles. The greatest ES were in the entorhinal cortex (ES=−0.79), amygdala (ES=−0.78), and inferior temporal gyrus (ES=−0.64). Supplementary Table 1 contains the annual rates of change in all ROI volumes in the overall sample and by diagnostic status.
3.3. Predictors of Neurodegeneration
We assessed demographic characteristics and vascular burden as predictors of neurodegeneration in the overall sample. Table 3 shows the adjusted associations of each predictor with annual rates of ROI volumetric change, and Supplementary Figure 2 shows associations of each predictor with volumetric change in certain ROIs. Baseline age, sex, diagnostic status, race, hypertension, elevated cholesterol, and APOE e4 carrier status had significant interactions with time. Interaction of elevated cholesterol with time was also significant, but HDL could have been affecting rate of volumetric change.
Table 3.
Predictors of neurodegeneration for the overall sample in Baltimore Longitudinal Study of Aging (N=688)
| Age*Time | Hypertension*Time | Obesity*Time | APOE e4 Carrier Status*Time |
HDL Cholesterol*Time | Race*Time | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain Regions of Interest |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
| Whole Brain | −0.0375 | 0.0204 | 0.066 | −0.8075 | 0.3236 | 0.013 | −0.2374 | 0.2302 | 0.303 | −0.2334 | 0.3305 | 0.480 | −0.0048 | 0.0103 | 0.642 | −0.7118 | 0.3981 | 0.074 |
| Gray Matter | −0.0745 | 0.0164 | <0.001 | −0.4890 | 0.2601 | 0.060 | −0.1631 | 0.1872 | 0.383 | −0.1753 | 0.2651 | 0.508 | 0.0012 | 0.0082 | 0.885 | −0.6076 | 0.3193 | 0.057 |
| White Matter | 0.0091 | 0.0090 | 0.312 | −0.3045 | 0.1419 | 0.032 | −0.0045 | 0.1056 | 0.966 | −0.0620 | 0.1443 | 0.667 | −0.0020 | 0.0045 | 0.664 | −0.2345 | 0.1751 | 0.180 |
| Ventricles | 0.0340 | 0.0067 | <0.001 | −0.0174 | 0.1141 | 0.879 | −0.0235 | 0.0417 | 0.573 | 0.1156 | 0.1192 | 0.332 | −0.0037 | 0.0034 | 0.269 | 0.2370 | 0.1295 | 0.067 |
| Amygdala | −0.0005 | 0.0001 | <0.001 | 0.0001 | 0.0021 | 0.959 | 0.0010 | 0.0013 | 0.404 | −0.0042 | 0.0021 | 0.050 | 0.0001 | 0.0001 | 0.300 | −0.0004 | 0.0025 | 0.881 |
| Hippocampus | −0.0018 | 0.0003 | <0.001 | 0.0042 | 0.0046 | 0.363 | 0.0023 | 0.0030 | 0.444 | −0.0130 | 0.0048 | 0.007 | 0.0001 | 0.0001 | 0.636 | −0.0143 | 0.0057 | 0.012 |
| Entorhinal Cortex | −0.0008 | 0.0003 | 0.016 | 0.0005 | 0.0051 | 0.925 | 0.0012 | 0.0035 | 0.730 | −0.0042 | 0.0052 | 0.428 | 0.0003 | 0.0002 | 0.045 | −0.0119 | 0.0062 | 0.057 |
| Parahippocampal Gyrus | −0.0012 | 0.0003 | <0.001 | 0.0038 | 0.0052 | 0.465 | 0.0013 | 0.0037 | 0.719 | −0.0021 | 0.0053 | 0.695 | 0.0004 | 0.0002 | 0.030 | −0.0142 | 0.0063 | 0.025 |
| Orbitofrontal Cortex | −0.0019 | 0.0007 | 0.004 | −0.0208 | 0.0105 | 0.047 | 0.0026 | 0.0084 | 0.753 | −0.0025 | 0.0105 | 0.813 | −0.0001 | 0.0003 | 0.871 | −0.0299 | 0.0129 | 0.021 |
| Superior Temporal Gyrus | −0.0010 | 0.0005 | 0.033 | −0.0062 | 0.0074 | 0.407 | −0.0047 | 0.0061 | 0.442 | −0.0016 | 0.0074 | 0.828 | 0.0000 | 0.0002 | 0.970 | −0.0166 | 0.0092 | 0.069 |
| Middle Temporal Gyrus | −0.0042 | 0.0011 | <0.001 | −0.0186 | 0.0180 | 0.303 | 0.0008 | 0.0133 | 0.953 | 0.0028 | 0.0183 | 0.881 | −0.0002 | 0.0006 | 0.728 | −0.0557 | 0.0222 | 0.012 |
| Inferior Temporal Gyrus | −0.0045 | 0.0010 | <0.001 | −0.0193 | 0.0154 | 0.211 | 0.0130 | 0.0120 | 0.279 | −0.0045 | 0.0155 | 0.773 | −0.0003 | 0.0005 | 0.600 | −0.0603 | 0.0189 | 0.001 |
| Fusiform | −0.0020 | 0.0007 | 0.004 | −0.0023 | 0.0110 | 0.834 | 0.0019 | 0.0077 | 0.808 | −0.0126 | 0.0112 | 0.261 | 0.0001 | 0.0003 | 0.771 | −0.0252 | 0.0135 | 0.061 |
SE – standard error, Note: All bolded values mean p<0.05. Linear mixed-effects models included each predictor and an interaction with that predictor and time. These models were adjusted by baseline ICV, scan type, age, sex, diagnostic status, race, time, and two-way interactions of scan type, age, sex, diagnostic status, and race with time. Continuous variables were mean-centered.
We tested whether there were three-way interactions between predictor, predictor*time, and predictor*time*diagnostic status when added to the base model. We found some three-way interactions at p<0.10 (Supplementary Table 2). For instance, obesity modified the association of diagnostic status with volume change in whole brain, orbitofrontal cortex, and fusiform, while APOE e4 carrier status modified the association of diagnostic status with volume change in ventricles, amygdala, hippocampus, and entorhinal cortex. Given these trends and limited power for detection of higher order interactions, we decided to implement the same model across analyses stratified by diagnostic status to determine whether the patterns of predictors of neurodegeneration differed by cognitive status.
Baseline age was associated with greater declines in GM, amygdala, hippocampus, entorhinal cortex, parahippocampal gyrus, orbitofrontal cortex, superior, middle, and inferior temporal gyri, and with greater ventricular enlargement (Table 3). Figure 1 shows the t-values <−3.0 for associations of baseline age and diagnostic status with volumetric declines on brain MRI.
Figure 1.
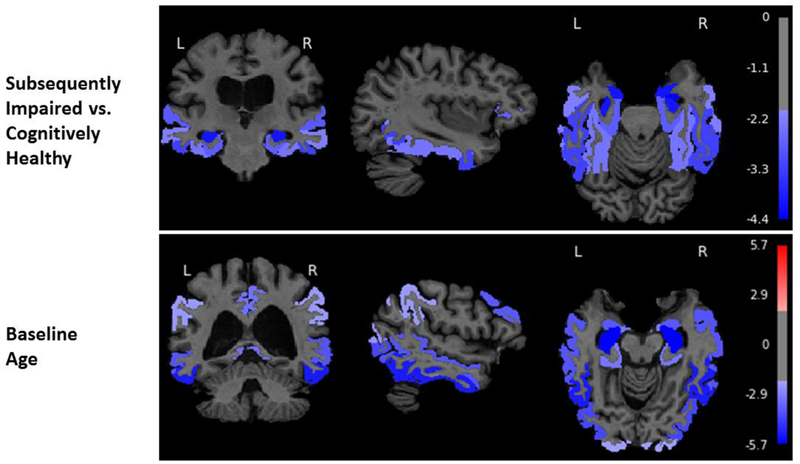
Associations of diagnostic status (subsequently impaired vs. cognitively healthy) and baseline age with gray matter volume change in the overall sample from the Baltimore Longitudinal Study of Aging (N=688). The color bar represents t-values from the results of the linear mixed effects models. These models consisted of fixed effects (baseline intracranial volume (ICV), image type [1.5-T SPGR vs. 3-T MPRAGE], age, sex, diagnostic status, race, time since first MRI, and two-way interactions of image type, age, sex, diagnostic status, and race with time) and random effects (intercept and time) with unstructured covariance. We used a threshold of ±1.96 to highlight areas of either volume expansion (positive t-values) or volume loss (negative t-values). Note that the colors are uniform within regional labels since the figures depict ROI rather than voxel-based analyses.
Hypertension, APOE e4 carrier status, HDL, and race were also associated with rates of volumetric change (Table 3). Hypertension was associated with steeper volumetric declines in total brain, WM, and orbitofrontal cortex (Table 3). APOE e4 carrier status was associated with steeper declines in the amygdala and hippocampus (Table 3). HDL was associated with less steep volume declines in the entorhinal cortex and parahippocampal gyrus. Race was associated with greater volumetric declines in hippocampus, parahippocampal gyrus, orbitofrontal cortex, and middle and inferior temporal gyri (Table 3). Obesity was not associated with volumetric change, but we used this predictor in the subsequent analysis (Table 3).
3.4. Predictors of Neurodegeneration Stratified by Diagnostic Status
The associations of predictors with annual rates of change in selected ROI volumes are presented by diagnostic status in Table 4. Among SI, older age was associated with steeper declines in total brain, GM, amygdala, and hippocampus. In the cognitively normal group, older age was associated with steeper declines in GM, amygdala, hippocampus, entorhinal cortex, parahippocampal gyrus, frontal GM, superior, middle, and inferior temporal gyri, and ventricular enlargement (Table 4). Among SI, hypertension was associated with greater declines in total brain, GM, WM and hippocampus, while hypertension was associated with less steep decline in hippocampus among the cognitively normal (Table 4). Among the SI only, obesity was associated with greater declines in total brain, GM, orbitofrontal cortex, and middle temporal gyrus and ventricular enlargement. APOE e4 carrier status was associated with greater declines in amygdala, hippocampus, and entorhinal cortex among SI only (Table 4). Among the cognitively normal only, higher HDL was associated with less steep decline in entorhinal cortex and parahippocampal gyrus, and race was associated with declines in orbitofrontal cortex and inferior temporal gyrus (Table 4). When we excluded those with myocardial infarction and angioplasty, the results were unchanged (results not shown).
Table 4.
Predictors of neurodegeneration stratified by diagnostic status in the Baltimore Longitudinal Study of Aging
| Age*Time | Hypertension*Time | Obesity*Time | APOE e4 Carrier Status*Time |
HDL Cholesterol*Time | Race*Time | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain Regions of Interest |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
β | SE | p- value |
| SUBSEQUENTLY IMPAIRED ONLY (n=65 with 390 observations) | ||||||||||||||||||
| Whole Brain | −0.1057 | 0.0424 | 0.013 | −1.3348 | 0.4918 | 0.007 | −1.2126 | 0.3925 | 0.002 | −0.2763 | 0.5958 | 0.643 | 0.0180 | 0.0157 | 0.251 | −1.3558 | 0.7702 | 0.078 |
| Gray Matter | −0.0955 | 0.0358 | 0.008 | −0.9912 | 0.4220 | 0.019 | −1.0919 | 0.3344 | 0.001 | −0.6616 | 0.5067 | 0.192 | 0.0052 | 0.0136 | 0.700 | −0.5064 | 0.6606 | 0.443 |
| White Matter | 0.0001 | 0.0195 | 0.995 | −0.5810 | 0.2274 | 0.011 | −0.1360 | 0.1729 | 0.432 | 0.0095 | 0.2750 | 0.973 | 0.0071 | 0.0073 | 0.331 | −0.6046 | 0.3564 | 0.090 |
| Ventricles | 0.0045 | 0.0200 | 0.821 | 0.3097 | 0.2524 | 0.220 | 0.2017 | 0.0878 | 0.022 | 0.4893 | 0.2979 | 0.100 | 0.0070 | 0.0084 | 0.408 | −0.1815 | 0.3850 | 0.637 |
| Amygdala | −0.0012 | 0.0004 | 0.001 | −0.0067 | 0.0043 | 0.123 | −0.0015 | 0.0026 | 0.571 | −0.0195 | 0.0052 | 0.000 | 0.0001 | 0.0001 | 0.699 | −0.0010 | 0.0068 | 0.877 |
| Hippocampus | −0.0028 | 0.0009 | 0.002 | −0.0212 | 0.0108 | 0.049 | 0.0000 | 0.0067 | 0.997 | −0.0401 | 0.0129 | 0.002 | 0.0000 | 0.0004 | 0.928 | −0.0216 | 0.0168 | 0.201 |
| Entorhinal Cortex | −0.0017 | 0.0009 | 0.061 | −0.0093 | 0.0110 | 0.394 | −0.0088 | 0.0073 | 0.225 | −0.0378 | 0.0132 | 0.004 | 0.0001 | 0.0004 | 0.819 | −0.0109 | 0.0172 | 0.527 |
| Parahippocampal Gyrus | −0.0015 | 0.0008 | 0.051 | −0.0014 | 0.0088 | 0.873 | 0.0006 | 0.0068 | 0.935 | −0.0158 | 0.0106 | 0.137 | −0.0003 | 0.0003 | 0.342 | −0.0137 | 0.0138 | 0.321 |
| Orbitofrontal Cortex | −0.0028 | 0.0016 | 0.078 | −0.0314 | 0.0190 | 0.099 | −0.0388 | 0.0157 | 0.014 | −0.0171 | 0.0227 | 0.452 | −0.0003 | 0.0006 | 0.587 | −0.0020 | 0.0295 | 0.945 |
| Superior Temporal Gyrus | −0.0001 | 0.0013 | 0.940 | 0.0041 | 0.0154 | 0.788 | −0.0145 | 0.0120 | 0.228 | 0.0001 | 0.0184 | 0.996 | −0.0001 | 0.0005 | 0.782 | 0.0096 | 0.0240 | 0.690 |
| Middle Temporal Gyrus | −0.0042 | 0.0028 | 0.127 | −0.0327 | 0.0321 | 0.308 | −0.0513 | 0.0256 | 0.045 | −0.0638 | 0.0389 | 0.101 | −0.0013 | 0.0010 | 0.203 | −0.0475 | 0.0503 | 0.345 |
| Inferior Temporal Gyrus | −0.0029 | 0.0030 | 0.333 | −0.0432 | 0.0356 | 0.225 | −0.0157 | 0.0255 | 0.538 | −0.0522 | 0.0428 | 0.223 | −0.0008 | 0.0012 | 0.507 | −0.0777 | 0.0559 | 0.164 |
| Fusiform | −0.0016 | 0.0017 | 0.350 | −0.0230 | 0.0196 | 0.239 | −0.0092 | 0.0142 | 0.518 | −0.0359 | 0.0236 | 0.128 | 0.0003 | 0.0006 | 0.693 | 0.0088 | 0.0307 | 0.775 |
| COGNITIVELY NORMAL ONLY (n=623 with 1,747 observations) | ||||||||||||||||||
| Whole Brain | −0.0227 | 0.0240 | 0.344 | −0.6766 | 0.4025 | 0.093 | 0.1692 | 0.2807 | 0.547 | −0.3498 | 0.4050 | 0.388 | −0.0087 | 0.0133 | 0.516 | −0.6363 | 0.4739 | 0.179 |
| Gray Matter | −0.0742 | 0.0192 | <0.001 | −0.3373 | 0.3238 | 0.298 | 0.1494 | 0.2263 | 0.509 | −0.0445 | 0.3250 | 0.891 | 0.0002 | 0.0107 | 0.986 | −0.5494 | 0.3801 | 0.148 |
| White Matter | 0.0157 | 0.0106 | 0.139 | −0.2080 | 0.1772 | 0.240 | 0.0869 | 0.1289 | 0.500 | −0.1798 | 0.1777 | 0.312 | −0.0024 | 0.0059 | 0.683 | −0.2477 | 0.2093 | 0.237 |
| Ventricles | 0.0389 | 0.0071 | <0.001 | −0.1445 | 0.1253 | 0.249 | −0.0716 | 0.0481 | 0.136 | 0.0238 | 0.1292 | 0.854 | −0.0067 | 0.0037 | 0.074 | 0.2351 | 0.1369 | 0.086 |
| Amygdala | −0.0005 | 0.0001 | <0.001 | 0.0012 | 0.0023 | 0.602 | 0.0016 | 0.0014 | 0.277 | −0.0013 | 0.0024 | 0.577 | 0.0001 | 0.0001 | 0.170 | 0.0004 | 0.0027 | 0.885 |
| Hippocampus | −0.0018 | 0.0003 | <0.001 | 0.0119 | 0.0049 | 0.015 | 0.0029 | 0.0033 | 0.392 | −0.0079 | 0.0049 | 0.112 | 0.0002 | 0.0002 | 0.183 | −0.0104 | 0.0058 | 0.070 |
| Entorhinal Cortex | −0.0009 | 0.0003 | 0.007 | 0.0005 | 0.0055 | 0.930 | 0.0023 | 0.0040 | 0.563 | 0.0028 | 0.0054 | 0.612 | 0.0004 | 0.0002 | 0.013 | −0.0101 | 0.0064 | 0.115 |
| Parahippocampal Gyrus | −0.0012 | 0.0004 | 0.001 | 0.0052 | 0.0063 | 0.416 | 0.0019 | 0.0045 | 0.671 | 0.0010 | 0.0064 | 0.879 | 0.0006 | 0.0002 | 0.006 | −0.0108 | 0.0075 | 0.147 |
| Orbitofrontal Cortex | −0.0015 | 0.0008 | 0.059 | −0.0169 | 0.0129 | 0.189 | 0.0189 | 0.0101 | 0.060 | −0.0077 | 0.0127 | 0.544 | 0.0000 | 0.0004 | 0.992 | −0.0371 | 0.0152 | 0.015 |
| Superior Temporal Gyrus | −0.0014 | 0.0005 | 0.007 | −0.0094 | 0.0081 | 0.245 | 0.0022 | 0.0070 | 0.747 | 0.0057 | 0.0079 | 0.468 | 0.0000 | 0.0003 | 0.988 | −0.0184 | 0.0096 | 0.054 |
| Middle Temporal Gyrus | −0.0047 | 0.0013 | <0.001 | −0.0148 | 0.0210 | 0.480 | 0.0207 | 0.0156 | 0.184 | 0.0261 | 0.0209 | 0.212 | −0.0001 | 0.0007 | 0.888 | −0.0406 | 0.0247 | 0.100 |
| Inferior Temporal Gyrus | −0.0053 | 0.0010 | <0.001 | −0.0139 | 0.0163 | 0.393 | 0.0241 | 0.0134 | 0.072 | 0.0128 | 0.0160 | 0.424 | −0.0001 | 0.0006 | 0.868 | −0.0467 | 0.0192 | 0.015 |
| Fusiform | −0.0023 | 0.0008 | 0.002 | 0.0009 | 0.0130 | 0.945 | 0.0033 | 0.0092 | 0.722 | −0.0044 | 0.0130 | 0.734 | 0.0000 | 0.0004 | 0.918 | −0.0293 | 0.0153 | 0.055 |
GM – gray matter, WM – white matter, SE – standard error, Note: All bolded values mean p<0.05. Linear mixed-effects models included each predictor and an interaction with that predictor and time. These models were adjusted by baseline ICV, scan type, age, sex, race, time, and two-way interactions of scan type, age, sex, and race with time. Continuous variables were mean-centered.
4. Discussion
Our current study extends prior work in the BLSA that examined differences of longitudinal patterns of ROI volumetric change by diagnostic status (Driscoll et al., 2009). The current study includes an expanded sample size and investigates potential risk factors associated with higher (or lower) rates of ROI volumetric change, providing potential mechanistic insights into drivers of neurodegeneration. First, we identified patterns of neurodegeneration by diagnostic status up to a 21-year follow-up period. Then, we determined predictors of these neurodegenerative patterns in the overall sample. We found distinctive patterns of volumetric change by diagnostic status and showed that observed changes were associated with AD risk factors, e.g., hypertension, in the overall sample. Finally, we found that patterns of these predictors of neurodegeneration varied by diagnostic group.
In identifying patterns of neurodegeneration, we found that trajectories of volume loss differed between SI and cognitively normal older adults. These associations are consistent with prior reports of greater age-related brain volumetric declines in SI than in cognitively normal (Driscoll et al., 2009; Karas et al., 2004; Solé-Padullés et al., 2009). SI had steeper rates of volumetric decline in amygdala, hippocampus, entorhinal cortex, parahippocampal gyrus, temporal and occipital lobes, along with higher rates of increased ventricular volumes, compared to cognitively normal. These findings are similar to those of a prior study that reported occurrence of GM volume loss in amygdala, hippocampus, and entorhinal cortex among amnestic MCI individuals (Whitwell et al., 2007). The effect sizes are reported as annual rates of change. Although some may be incrementally small, they can be substantial cumulatively over many years.
We next examined predictors of the patterns of neurodegeneration in the overall sample. Predictors of volume change included older age, male sex, APOE e4 carrier status, hypertension, lower HDL, and race. One study found that sex, APOE e4 status, and hypertension diagnosis affected brain shrinkage rates over a 30-month period (Raz et al., 2010). Another study examined the associations of cardiovascular risk factors with 10-year volumetric changes in total brain and temporal horn of the ventricles and found that hypertension and obesity were associated with increased rate of global and hippocampal atrophy (Debette et al., 2011). While we did not observe an association of hypertension with an increased rate of hippocampal atrophy, we found that hypertension was associated with greater declines in total brain, WM, and orbitofrontal cortex, which is consistent with previous studies that reported associations of hypertension with WM changes and pre-frontal areas (Basile et al., 2006; Raz et al., 2003). We also found that greater HDL cholesterol was associated with less steep declines in entorhinal cortex and parahippocampal gyrus, which suggests a potential protective effect of HDL in regions affected by early AD pathology.
Additionally, we found that whites had steeper volumetric declines in hippocampus, parahippocampal gyrus, orbitofrontal cortex, and middle and inferior temporal gyri than did African Americans. Both groups in our sample were of high education and had similar numbers of follow-up. Further, these findings were most likely driven by the group that remained cognitively normal throughout the follow-up period, since there was only a total of eight African Americans who were subsequently impaired and only six had more than a single MRI. Finally, we performed stratified analyses and compared patterns of associations of the predictors with ROI volumetric changes by diagnostic status. While hypertension, obesity, and APOE e4 carrier status were associated with ROI volumetric declines among SI, there were no associations of these predictors with volume declines among the cognitively normal. This suggests that SI are more vulnerable to potential effects of these predictors, since age, sex, hypertension, obesity, and APOE e4 status were associated with volumetric decline in a greater number of ROIs. Also, the associations of certain predictors, i.e., hypertension, obesity, and APOE e4 status, with volumetric decline in the overall sample could be driven predominantly by the SI. From these findings, the stratified analysis showed that a number of risk factors were more evident in the SI than cognitively normal group, despite more limited power.
Moreover, we found some surprising results in the stratified analysis. For instance, hypertension was associated with less steep hippocampal volume decline among only those who remained cognitively normal, which could be attributed to sample selection, since we had few individuals with severe cardiovascular disease at baseline. The estimated effects of baseline age were more widespread in the cognitively normal group than in the SI group. These results could be attributed to differences in sample sizes and observations, since the cognitively normal group had more serial assessments than the SI group.
The predictors of neurodegeneration identified in our analyses are known AD risk factors (Artero et al., 2008; Debette et al., 2011; Yaffe et al., 2009). These findings are consistent with a two-hit vascular hypothesis of AD. This hypothesis postulates that microvascular damage is the first insult through which blood-brain barrier dysfunction and/or diminished brain capillary flow generate secondary neuronal injury, which then leads to amyloid-beta accumulation (Zlokovic, 2011). Cardiovascular and genetic risk factors, i.e., hypertension, obesity, and APOE e4, can lead to microvascular damage, and these factors are associated with volumetric declines in structures of the medial temporal lobe, an area affected by early AD pathology, more among SI than among cognitively normal groups. Such findings are consistent with studies showing that clinical symptoms of dementia are more likely when vascular disease is present in additional to AD pathology (Schneider et al., 2007; Troncoso et al., 2008). Thereby, the cumulative effects of vascular disease and neuropathology possibly lead to increased liability to the clinical expression of disease. It is possible that vascular disease interacts with amyloid or tau deposition through other mechanisms, which may then lead to cognitive decline, or vascular disease may cause cognitive decline independently of amyloid, as suggested by the additive effects of these pathologies on cognitive impairment. Since some pathological effects are predominant in the medial temporal lobe, where tau tends to deposit, there also could be an association of vascular disease and tau, which could then lead to cognitive decline. The complex associations between vascular disease and AD pathology are an area of active investigation.
There are many strengths of this study. First, this study consists of an extensively characterized large sample of older adults with repeated measures up to 21 years. Second, we were able to examine the longitudinal brain changes during the preclinical period prior to cognitive impairment diagnosis. Third, our image processing pipeline uses state-of-the-art and validated multi-atlas approaches for regional definition, yielding high measurement stability over time.
There were also several limitations. First, our sample is highly educated, mostly Caucasian, and relatively healthy, thus limiting generalizability. We excluded participants with stroke and other significant health conditions that could affect brain structure and function. This may have contributed to the relatively low prevalence of hypertension and diabetes in the sample at baseline. Nevertheless, prior BLSA studies showed similar rates of AD onset (Kawas et al., 2000) and similar rates of brain changes over time, relative to other studies (Resnick et al., 2003). Second, since the BLSA is ongoing, 38.8% of the sample had a single assessment during the current analysis, but are included, as they contribute to stability of cross-sectional associations. Third, 34.4% of the SI group converted to MCI/dementia one year after the last visit, so group effects observed for the SI group could be underestimated. Fourth, we evaluated self-identified race in this study, as a way to adjust for differences in the prevalence of risk factors and socioeconomic exposures. These results neither infer causality nor racial biologic differences when none exist, and more research is needed to further assess these associations. Fifth, we did not evaluate the potentially beneficial effects of APOE e2 on volume change in our study, since we found no significant differences in e2 genotypes across cognitively normal and SI groups.
Sixth, the average follow-up times provided in the manuscript are the follow-up over time points included in the MRI study, which represent a snapshot in time. While we cannot rule out the fact that some who were cognitively normal at the last MRI visit will subsequently develop cognitive impairment, we would expect this to attenuate differences between cognitively normal and SI groups. Lastly, mean enrollment age was ≥65 years, so information on midlife risk factors is limited. The timing of the predictors in relation to age and dementia onset is crucial. Total cholesterol decreases with age (Postiglione et al., 1989), yet decreased cholesterol, influenced by APOE e4, could be a consequence of dementia (Duron and Hanon, 2008; Evans et al., 2000). Several years before dementia onset, blood pressure and BMI begin to decline (Panza et al., 2006). Through the lengthy prospective follow-up in our study, we were able to minimize, but not eliminate, the impact of preclinical disease.
In summary, age, sex, race, APOE e4 carrier status, and hypertension were associated with greater longitudinal declines in regional brain volumes in the overall sample. Hypertension, obesity, and APOE e4 status were associated with greater rates of neurodegeneration among the SI only, suggesting that there could be increased vulnerability to pathologic change with these risk factors among those in the pre-symptomatic stages of dementia. Greater understanding of the ways in which multiple risk factors interact together to increase dementia risk would help identify those most likely to benefit from lifestyle counseling and/or medications as preventative measures. Our findings highlight the importance of considering preclinical disease within cognitively normal samples.
Supplementary Material
Acknowledgments
We would like to thank the participants and staff of the BLSA, the neuroimaging staff of the Laboratory of Behavioral Neuroscience, and the staff of the Johns Hopkins and NIA MRI facilities.
Funding/Support:This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute on Aging and by NIH funding sources N01-AG-3–2124, R01-AG14971, and RF1-AG054409, and all funding for the study came from the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
Role of Sponsor: The authors of this manuscript include employees of the Intramural Research Program of the NIA. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Conflicts of Interest: The authors report no conflicts of interest.
References
- Artero S, Ancelin M-L, Portet F, Dupuy A, Berr C, Dartigues J-F, Tzourio C, Rouaud O, Poncet M, Pasquier F, Auriacombe S, Touchon J, Ritchie K, 2008. Risk profiles for mild cognitive impairment and progression to dementia are gender specific. Journal of Neurology, Neurosurgery & Psychiatry 79(9), 979–984. [DOI] [PubMed] [Google Scholar]
- Basile AM, Pantoni L, Pracucci G, Asplund K, Chabriat H, Erkinjuntti T, Fazekas F, Ferro JM, Hennerici MG, O’Brien J, Scheltens P, Visser MC, Wahlund LO, Waldemar G, Wallin A, Inzitari D, 2006. Age, hypertension, and lacunar stroke are the major determinants of the severity of age-related white matter changes. Cerebrovascular Diseases 21(5–6), 315–322. [DOI] [PubMed] [Google Scholar]
- Debette S, Seshadri S, Beiser A, Au R, Himali JJ, Palumbo C, Wolf PA, DeCarli C, 2011. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology 77(5), 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarli C, Frisoni G, Clark C, Harvey D, Grundman M, Petersen R, Thal L, Jin S, Jack C, Scheltens P, Alzheimer’s Disease Cooperative Study Group, 2007. Qualitative estimates of medial temporal atrophy as a predictor of progression from mild cognitive impairment to dementia. Archives of Neurology 64(1), 108–115. [DOI] [PubMed] [Google Scholar]
- Desikan R, Cabral H, Hess C, Dillon W, Glastonbury C, Weiner M, Schmansky N, Greve D, Salat D, Buckner R, Fischl B, Alzheimer’s Disease Neuroimaging Initiative, 2009. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer’s disease. Brain 132(8), 2048–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson B, Wolk D, On behalf of the Alzheimer’s Disease Neuroimaging Initiative, 2012. MRI cortical thickness biomarker predicts AD-like CSF and cognitive decline in normal adults. Neurology 78(2), 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi J, Erus G, Ou Y, Resnick SM, Gur RC, Gur RE, Satterthwaite TD, Furth S, Davatzikos C, The Alzheimer’s Neuroimaging Initiative, 2016. MUSE: MUlti-atlas region Segmentation utilizing Ensembles of registration algorithms and parameters, and locally optimal atlas selection. NeuroImage 127, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll I, Davatzikos C, An Y, Wu X, Shen D, Kraut M, Resnick SM, 2009. Longitudinal pattern of regional brain volume change differentiates normal aging from MCI. Neurology 72(22), 1906–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duron E, Hanon O, 2008. Vascular risk factors, cognitve decline, and dementia. Vascular Health and Risk Management 4(2), 363–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erus G, Doshi J, An Y, Verganelakis D, Resnick SM, Davatzikos C, 2018. Longitudinally and inter-site consistent multi-atlas based parcellation of brain anatomy using harmonized atlases. NeuroImage 166, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, Emsley C, Gao S, Sahota A, Hall K, Farlow M, Hendrie H, 2000. Serum cholesterol, APOE genotype, and the risk of Alzheimer’s disease: a population-based study of African Americans. Neurology 54(1), 240–240. [DOI] [PubMed] [Google Scholar]
- Fjell A, McEvoy L, Holland D, Dale A, Walhovd K, for the Alzheimer’s Disease Neuroimaging Initiative, 2013. Brain changes in older adults at very low risk for Alzheimer’s disease. The Journal of Neuroscience 33(19), 8237–8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher A, Grundman M, Jack C, Petersen R, Taylor C, Kim H, Schiller D, Bagwell V, Sencakova D, Weiner M, DeCarli C, DeKosky S, van Dyck C, Thal L, Alzheimer’s Disease Cooperative Study, 2005. Sex, apolipoprotein e ε4 status, and hippocampal volume in mild cognitive impairment. Archives of Neurology 62(6), 953–957. [DOI] [PubMed] [Google Scholar]
- Friedewald W, Levy R, Fredrickson D, 1972. Estimation of the concentration of Low-Density Lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clinical Chemistry 18(6), 499–502. [PubMed] [Google Scholar]
- Fuld PA, 1978. Psychological testing in the differential diagnosis of the dementias, in: Katzman R, Terry R, Bick K (Eds.), Alzheimer’s Disease: Senile Dementia and Related Disorders. Raven Press, New York, NY, pp. 185–193. [Google Scholar]
- Gottesman R, Schneider A, Zhou Y, Coresh J, Green E, Gupta N, Knopman D, Mintz A, Rahmim A, Sharrett A, Wagenknecht L, Wong D, Mosley T, 2017. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA 317(14), 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Wiste HJ, Weigand SD, Therneau TM, Knopman DS, Lowe V, Vemuri P, Mielke MM, Roberts RO, Machulda MM, Senjem ML, Gunter JL, Rocca WA, Petersen RC, 2017. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. The Lancet Neurology 16(6), 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karas GB, Scheltens P, Rombouts SARB, Visser PJ, van Schijndel RA, Fox NC, Barkhof F, 2004. Global and local gray matter loss in mild cognitive impairment and Alzheimer’s disease. NeuroImage 23(2), 708–716. [DOI] [PubMed] [Google Scholar]
- Kawas C, Gray S, Brookmeyer R, Fozard J, Zonderman A, 2000. Age-specific incidence rates of Alzheimer’s disease: The Baltimore Longitudinal Study of Aging. Neurology 54(11), 2072–2077. [DOI] [PubMed] [Google Scholar]
- Kivipelto M, Helkala E, Laakso M, Hänninen T, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A, 2001. Midlife vascular risk factors and Alzheimer’s disease in later life: longitudinal, population based study. BMJ 322(7300), 1447–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Singh M, 2014. Sex differences in cognitive impairment and Alzheimer’s disease. Frontiers in Neuroendocrinology 35(3), 385–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, 1993. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414. [DOI] [PubMed] [Google Scholar]
- Pacheco J, Goh JO, Kraut MA, Ferrucci L, Resnick SM, 2015. Greater cortical thinning in normal older adults predicts later cognitive impairment. Neurobiology of Aging 36(2), 903–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panza F, D’Introno A, Colacicco A, Capurso C, Pichichero G, Capurso S, Capurso A, Solfrizzi V, 2006. Lipid metabolism in cognitive decline and dementia. Brain Research Reviews 51(2), 275–292. [DOI] [PubMed] [Google Scholar]
- Postiglione A, Cortese C, Fischetti A, Cicerano U, Gnasso A, Gallotta G, Grossi D, Mancini M, 1989. Plasma lipids and geriatric assessment in a very aged population of south Italy. Atherosclerosis 80(1), 63–68. [DOI] [PubMed] [Google Scholar]
- Raz N, Ghisletta P, Rodrigue KM, Kennedy KM, Lindenberger U, 2010. Trajectories of brain aging in middle-aged and older adults: regional and individual differences. NeuroImage 51(2), 501–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Acker JD, 2003. Hypertension and the brain: vulnerability of the prefrontal regions and executive functions. Behavioral Neuroscience 117(6), 1169–1180. [DOI] [PubMed] [Google Scholar]
- Resnick S, Pham D, Kraut M, Zonderman A, Davatzikos C, 2003. Longitudinal magnetic resonance imaging studies of older adults: a shrinking brain. The Journal of Neuroscience 23(8), 3295–3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA, 2007. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69(24), 2197–2204. [DOI] [PubMed] [Google Scholar]
- Schuster C, Elamin M, Hardiman O, Bede P, 2015. Presymptomatic and longitudinal neuroimaging in neurodegeneration—from snapshots to motion picture: a systematic review. Journal of Neurology, Neurosurgery & Psychiatry 86(10), 1089–1096. [DOI] [PubMed] [Google Scholar]
- Snyder H, Corriveau R, Craft S, Faber J, Greenberg S, Knopman D, Lamb B, Montine T, Nedergaard M, Schaffer C, Schneider J, Wellington C, Wilcock D, Zipfel G, Zlokovic B, Bain L, Bosetti F, Galis Z, Koroshetz W, Carrillo M, 2015. Vascular contributions to cognitive impairment and dementia including Alzheimer’s disease. Alzheimer’s & Dementia 11(6), 710–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé-Padullés C, Bartrés-Faz D, Junqué C, Vendrell P, Rami L, Clemente IC, Bosch B, Villar A, Bargalló N, Jurado MA, Barrios M, Molinuevo JL, 2009. Brain structure and function related to cognitive reserve variables in normal aging, mild cognitive impairment and Alzheimer’s disease. Neurobiology of Aging 30(7), 1114–1124. [DOI] [PubMed] [Google Scholar]
- StataCorp, 2017. Stata Statistical Software: Release 15. StataCorp LLC, College Station, TX. [Google Scholar]
- Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O’Brien RJ, 2008. Effect of infarcts on dementia in the Baltimore Longitudinal Study of Aging. Annals of Neurology 64(2), 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste H, Weigand S, Shaw L, Trojanowski J, Weiner M, Knopman D, Petersen R, Jack C, On behalf of the Alzheimer’s Disease Neuroimaging Initiative, 2009. MRI and CSF biomarkers in normal, MCI, and AD subjects: Diagnostic discrimination and cognitive correlations. Neurology 73(4), 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitwell JL, Przybelski S, Weigand SD, Knopman DS, Boeve BF, Petersen RC, Jack CR, 2007. 3D Maps from multiple MRI illustrate changing atrophy patterns as subjects progress from MCI to AD. Brain 130(Pt 7), 1777–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, Fiocco A, Lindquist K, Vittinghoff E, Simonsick E, Newman A, Satterfield S, Rosano C, Rubin S, Ayonayon H, 2009. Predictors of maintaining cognitive function in older adults: The Health ABC Study. Neurology 72(23), 2029–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV, 2011. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature Reviews Neuroscience 12(12), 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.