

Abstract
Glia continuously survey neuronal health during development, providing trophic support to healthy neurons while rapidly engulfing dying ones. These diametrically-opposed functions necessitate a foolproof mechanism enabling glia to unambiguously identify those neurons to support versus those to engulf. To ensure specificity, glia are proposed to interact with dying neurons via a series of carefully choreographed steps. However, these crucial interactions are largely obscure. Here we show that dying neurons and glia communicate via Toll receptor-regulated innate immune signaling. Neuronal apoptosis drives processing and activation of the Toll-6 ligand, Spätzle5. This cue activates a dSARM-mediated Toll-6 transcriptional pathway in glia, which controls expression of the Draper engulfment receptor. Pathway loss drives early-onset neurodegeneration, underscoring its functional importance. Our results identify an upstream priming signal that prepares glia for phagocytosis. Thus, a core innate immune pathway plays an unprecedented role setting the valence of neuron-glia interactions during development.
Keywords: Toll, FoxO, SARM1, Spätzle, Furin, Draper, phagocytosis, glia, find-me signal, neurodegeneration
eTOC blurb
Dying neurons are proposed to interact with phagocytic glia via a series of carefully choreographed steps, but these signaling interactions are poorly understood. McLaughlin et al. define a Toll receptor-mediated innate immune signaling pathway that acts to ensure the speed and specificity of neuronal corpse engulfment during neurodevelopment.
Graphical Abstract
Introduction
Precise control of neuronal number and connectivity underlies the establishment and maintenance of neural circuits. However, developing nervous systems overproduce neurons, neurites, and synapses. Those that are not integrated into circuits are eliminated, either via complete removal of a neuron by programmed cell death or by selective elimination of projections. The amount of neuronal cell death during development is striking; roughly half of neurons undergo apoptosis in vertebrate nervous systems, while about one-third die during late Drosophila embryogenesis (Rogulja-Ortmann et al., 2007; Yeo and Gautier, 2004). Accordingly, refinement of neuronal number and connectivity generates a great deal of debris, which is cleared by phagocytes. Failure to promptly remove debris can trigger inflammation and is hypothesized to be detrimental to brain health and function.
Glia are the major phagocytic cell type in the central nervous system (CNS) and eliminate nearly all neuronal debris generated during development (Kurant, 2011; Sierra et al., 2013; Tasdemir-Yilmaz and Freeman, 2014). In mammals, microglia are dedicated phagocytes and prune synapses during circuit development (Neumann et al., 2009; Wu et al., 2015). Demonstrating the involvement of immune signaling components in circuit remodeling, microglia in the visual system utilize Complement Receptor 3 to recognize and phagocytose low-activity synapses tagged with Complement Component C3 ligand (Schafer et al., 2012). Astrocytes also have phagocytic ability (Chung et al., 2013; Tasdemir-Yilmaz and Freeman, 2014), and possess the remarkable capacity to switch between supportive and phagocytic roles. While molecular mechanisms promoting the generation of reactive glial in disease and inflammation have been identified (Fourgeaud et al., 2016; Liddelow et al., 2017; Perry and Holmes, 2014; Zhao et al., 2018), almost nothing is known about how glia toggle between supportive and degenerative functions during development.
Drosophila is an ideal system in which to elucidate molecular mechanisms directing glial phagocytosis of apoptotic neurons. Fly glia are experimentally tractable and have phagocytic capabilities akin to their mammalian counterparts. Yet unlike mammals, the Drosophila CNS does not have a dedicated phagocyte population. Instead, several glial subtypes contribute to debris clearance (Freeman, 2015; Logan, 2017). Astrocyte processes are found within the neuropil and prune exuberant neurites and synapses (Tasdemir-Yilmaz and Freeman, 2014), while cortex glia physically contact almost all neuronal cell bodies in an elaborate meshwork of process (Coutinho-Budd et al., 2017). Both astrocytes and cortex glia utilize the phagocytic receptor Draper (Drpr), a homolog of C. elegans CED-1 and mammalian MEGF10/Jedi-1, for engulfment (Etchegaray et al., 2016; Tasdemir-Yilmaz and Freeman, 2014).
Toll-like receptor (TLR) superfamily members are promising candidates to regulate phagocytic functions of glia during development. In the immune system, TLRs play essential roles recognizing invading pathogens and detecting endogenous cellular damage (Valanne et al., 2011; Barak et al., 2014; Lindsay and Wasserman, 2014; Valanne et al., 2011). In the vertebrate CNS, microglia and astrocytes utilize TLRs to trigger inflammatory pathways in response to disease or infection (Hanke and Kielian, 2011). Activation of TLR2 or 4, for example, enhances microglial phagocytosis of pathogens (Kochan et al., 2012; Ribes et al., 2010). While TLR activation promotes glial phagocytosis during infection, glia have not been proposed to use TLRs to clear apoptotic neurons in the healthy brain.
TLR signaling capabilities are dictated by conserved molecular domains. On the extracellular side, TLRs contain leucine rich repeats (Gay and Gangloff, 2007). Binding of Spätzle (Spz) ligands to TLR ectodomains activates downstream signaling (Valanne et al., 2011). On the intracellular side, cytoplasmic Toll/Interleukin-1 receptor (TIR) domains interact with TIR adaptor-containing proteins (O’Neill and Bowie, 2007). Vertebrates have five TIR adaptors, while Drosophila has two: dSARM and Myd88. Myd88 is central to innate immunity (Horng and Medzhitov, 2001; Tauszig-Delamasure et al., 2002), while dSARM/SARM1 is highly expressed in the CNS and is key for axon degeneration following injury (Gerdts et al., 2013; Osterloh et al., 2012). In support of a TLR-dSARM signaling link, we uncovered a Toll-6-dSARM pathway required for structural plasticity in motoneurons (McLaughlin et al., 2016).
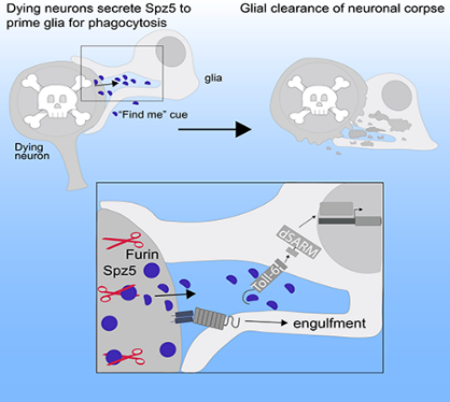
We were interested in testing the hypothesis that Toll-6 signals via dSARM in additional neurodevelopmental contexts. Here we demonstrate that Toll-6 and dSARM function in cortex glia to transduce a cue from dying neurons. Specifically, neuronal apoptosis activates the Toll-6 ligand, Spz5. Neuronal Spz5 drives Toll-6 signaling in glia, which promotes FoxO-dependent transcription of the Draper engulfment receptor and phagocytic clearance of apoptotic neurons. Finally, pathway loss leads to early-onset neurodegeneration. Our data define an innate immune-mediated interaction between glia and dying neurons required for engulfment of apoptotic debris. We propose that neuronal Spz5 serves as a type of “find-me” signal, upstream of “eat-me” signals, required to ensure the speed and specificity of corpse clearance.
Results
Cortex glial Toll-6 signaling limits apoptotic debris in the CNS
We recently defined a role for Toll-6 in developing motoneurons (McLaughlin et al., 2016), where we identified dSARM and FoxO as mediators of Toll-6 activity. Toll-6 is proposed to have neurotrophic function in the fly CNS as loss of Toll-6 leads to an accumulation of apoptotic neurons in the embryonic ventral nerve cord (VNC) (McIlroy et al., 2013). Our interest in molecular mechanisms of TLR signaling prompted us to test if dSARM and FoxO participate in Toll-6-mediated signal transduction in this additional context.
In line with published work, we find that loss of Toll-6 results in an 82% increase in apoptotic debris in the third-instar larval (L3) brain as visualized by cleaved Death caspase-1 (Dcp-1; Figure 1 A-B, E) (McIlroy et al., 2013). Similarly, loss of dSARM leads to a 68% increase in apoptotic debris in the brain (Figure 1 C, E), and foxO nulls display an 74% increase (Figure 1 D-E). All three mutants display similarly increased levels of apoptotic debris in the L3 ventral nerve cord (VNC) (Table S1). Consistent with a linear genetic pathway, Toll-6, foxO double mutants exhibit equivalent levels of apoptotic debris as individual single mutants (Figure 1 E). We attempted to assay apoptotic debris in dSARM, Toll-6 and dSARM, foxO double mutants, but they are embryonic lethal. Instead, we quantified debris in dSARM/+, Toll-6/+ and dSARM/+, foxO/+ double heterozygotes. While single heterozygotes do not exhibit Dcp-1 phenotypes, double heterozygotes exhibit Dcp-1 phenotypes analogous to individual single mutants (Figure 1 F). Thus, dSARM and FoxO cooperate with Toll-6 to limit apoptotic debris in the developing CNS.
Figure 1. Cortex glial Toll-6-FoxO signaling limits apoptotic debris in the CNS.

(A-D) Representative z-projections of one L3 brain lobe of indicated genotypes labeled with anti-Dcp-1. (E-H) Quantification of Dcp-1 particles indicates that Toll-6-FoxO signaling functions in glia. (I) Diagram of glial subtypes in the CNS and subtype-specific drivers. (J-K) Quantification of Dcp-1 particles indicates that cortex glia utilize Toll-6 signaling to limit apoptotic debris. (L-O) Representative z-projections of one L3 brain lobe of the indicated genotypes labeled with anti-Dcp-1. (P-Q) Quantification of Dcp-1 particles indicates that dSARM and FoxO function downstream of Toll-6 in cortex glia. Statistical measures above column are compared to control. (R) Diagram of TIR domain in wild-type Toll-6 and Toll-6TIRdead. Scale bar is 50 μm. n is noted at the bottom of each column. n.s., not significantly different. *, P<0.05; **, P<0.01; ***, P<0.001. Values for mean and SEM are located in Table S1.
Toll-6 is proposed to have neurotrophic activity based on analysis of Toll-6 nulls (Foldi et al., 2017; McIlroy et al., 2013). Yet, the specific cellular requirements for Toll-6 function were not investigated. We employed RNAi-mediated knock-down of Toll-6, dSARM, and foxO either in all neurons (ElavC155Gal4) or all glia (RepoGal4) and quantified Dcp-1 debris. Surprisingly, pan-neuronal RNAi-mediated knock-down of Toll6 pathway members does not alter the amount of apoptotic debris (Figure 1 G). In contrast, pan-glial RNAi-mediated knock-down of Toll-6, dSARM, and foxO mimics the LOF phenotypes (Figure 1 H), arguing that these proteins function in glia.
The Drosophila nervous system has a diversity of glial cell types (Figure 1 I). To map the relevant glial population, we took advantage of Gal4 drivers expressed in the major classes of CNS glia (Figure 1 I). RNAi-mediated Toll-6 knockdown in astrocyte-like glia, ensheathing glia, perineurial glia, or subperineurial glia does not recapitulate LOF phenotypes (Figure 1 I-J). However, Toll-6 knock-down in cortex glia phenocopies the increase in Dcp-1 debris seen in the nulls (Figure 1 I-J). Cortex glia are found throughout the brain and VNC at all stages of development and adulthood and are required for organismal survival (Awasaki et al., 2008; Coutinho-Budd et al., 2017). Unlike surface glia which form the blood brain barrier (BBB) and neuropil glia which associate with neuronal processes and synapses, cortex glia preferentially contact cell bodies (Figure 1 I).
Knock-down of dSARM or FoxO via the cortex glial NP577Gal4 driver similarly results in elevated apoptotic debris (Figure 1 K). Since NP577Gal4 is also expressed in a small subset of neurons, we utilized a split-Gal4 line expressed exclusively in cortex glia (Coutinho-Budd et al., 2017), here referred to as Ctxglia-splitGal4. When Toll-6, dSARM, and foxO are selectively removed in cortex glia via Ctxglia-splitGal4, we observe 95%, 95%, and 65% increases in Dcp-1 debris, respectively (Figure 1 L-P). Similar phenotypes are observed in the VNC (Table S1). Dcp-1 debris in these backgrounds appears randomly distributed throughout the brain and not in a stereotyped pattern. We conclude that Toll-6-mediated signaling acts in cortex glia to limit accumulation of apoptotic debris.
Our analyses indicate that Toll-6 engages a non-canonical pathway in cortex glia. However, they do not exclude the possibility of canonical TLR signaling. Thus, we investigated if loss of established TLR components results in excess apoptotic debris. We first asked if Toll-7, another TLR expressed in the CNS (McIlroy et al., 2013; Ward et al., 2015), similarly regulates apoptotic debris. However, we do not see a change in Dcp-1 puncta in two Toll-7 backgrounds (Table S1). We also assayed intracellular Toll signaling components. Traditionally, Toll receptors signal via adaptor proteins to regulate the transcription factors Dorsal and Relish (Lindsay and Wasserman, 2014; Valanne et al., 2011). Using LOF alleles, chromosomal deficiencies, and cortex glial RNAi-mediated knockdown we assayed Dcp-1 debris when MyD88, Relish, and Dorsal are lost. We do not observe significant increases in Dcp-1 debris in any of these backgrounds (Table S1), indicating that Toll-6 does not employ these TLR components to regulate Dcp-1 levels at the L3 stage. As a result, we focused on defining the functions of Toll-6, dSARM, and FoxO.
We next analyzed whether overexpression of Toll-6 signaling components is sufficient to reduce apoptotic debris. Indeed, Toll-6 overexpression reduces debris by 35% compared to controls (Figure 1 Q). This overexpression phenotype depends on dSARM and FoxO as Toll-6 overexpression in dSARM or foxO mutants results in excess apoptotic debris like that observed in the single mutants (Figure 1 Q; Table S1). We also overexpressed dSARM and FoxO in cortex glia. Overexpression of FoxO reduces Dcp-1 debris by 36% (Figure 1 Q). While dSARM overexpression results in a similar trend, it did not reach statistical significance (Figure 1 Q). These analyses indicate that Toll-6 is genetically upstream of dSARM and FoxO but do not speak to the directness of the interaction.
If Toll-6-dSARM signaling requires TIR-TIR interactions, we predicted that overexpressing a Toll-6 variant with a non-functional TIR domain (Toll-6TIRdead; Figure 1 R; Ward et al. 2015) would not yield an overexpression phenotype like that of wild-type Toll-6. Suggesting the mutant protein is stable, Toll-6TIRdead overexpression rescues Toll-6 null phenotypes in the olfactory system, where the TIR domain is dispensable for its function (Ward et al., 2015). Supporting a requirement for the TIR domain in cortex glial signaling, overexpression of Toll-6TIRdead does not result in a Dcp-1 phenotype (Figure 1 Q, Table S1). Thus, Toll-6’s TIR domain is required to control accumulation of apoptotic debris, supporting the involvement of dSARM. In further support of this pathway, Toll-6 and dSARM are detected in a physical complex (Foldi et al., 2017). Together, our data uncover a cortex glial Toll-6-dSARM-FoxO pathway necessary and sufficient to limit apoptotic debris during development.
Toll-6, dSARM, and FoxO are expressed in cortex glia
While Toll-6, dSARM, and FoxO are all expressed in neurons (McIlroy et al., 2013; Nechipurenko and Broihier, 2012; Osterloh et al., 2012), none were previously known to be either expressed in glia or have glial functions. To address the plausibility of this putative glial TLR pathway, we set out to better characterize the CNS expression profiles of Toll-6, dSARM, and FoxO.
We began with Toll-6. Since anti-Toll-6 antibodies were not readily available, we used a GFP exon trap inserted in Toll-6 coding sequence (Toll-6MIMICGFP ) (Venken et al., 2011), which recapitulates Toll-6 transcript expression (McIlroy et al., 2013). Toll-6MIMICGFP has a cytoplasmic distribution in the L3 brain and VNC (Figure 2 A”-A”‘, B”-B”‘). To determine if it is within cortex glia, membrane-bound Cherry was expressed via Ctxglia-splitGal4 (Ctxglia-split>mCD8::Cherry) in a Toll-6MIMICGFP background (Figure 2 A-B”“). We find that Toll-6MIMICGFP is present in cortex glial processes in dorsal brain (Figure 2 A’’-A”“, B’’-B”“; arrows). To validate this finding, we also marked cortex glia nuclei (Ctxglia-split>mCherry.NLS) in a Toll-6MIMICGFP background and find that Toll-6MIMICGFP is expressed in the cytoplasm of dorsally positioned cortex glia (Figure S1 A-B““; arrows). We conclude that Toll-6MIMICGFP is present in cortex glia.
Figure 2. Toll-6, dSARM, and FoxO are expressed in cortex glia.

(A-B) Representative 3 μm projections of L3 CNS of indicated genotype labeled with anti-GFP (green) and anti-mCherry (magenta). (A’, B’) Projections of one brain lobe. (A”-A”“, B”-B”“) Higher magnification projections of Toll-6MIMICGFP expression in cortex glia processes. (C) Representative 3 μm projections of L3 CNS of indicated genotype labeled with anti-mCherry (magenta) and anti-Repo (green). (C’) Projection of one brain lobe. (C”-C”“) Higher magnification projections of dSARM-positive nuclei and anti-Repo. (D) Representative 3 μm projection of L3 CNS of indicated genotype labeled with anti-ßgal (green) and anti-mCherry (magenta). (D’) Projection of one brain lobe. (D”-D”“) Higher magnification projections of cortex glial nuclei and dSARM-positive nuclei. (E) Representative 3 μm projection of L3 CNS of indicated genotype labeled with anti-GFP (green) and anti-mCherry (magenta). (E’) Projection of one brain lobe. (E”-E”“) Higher magnification projections of cortex glial nuclei and FoxOMIMICGFP. Scale bar for CNS images is 200 μm, for brain lobe images is 50 μm, and for higher magnification images is 13 μm.
Next, we turned to dSARM. While dSARM’s functions in axon degeneration are intensively studied, its expression pattern has remained enigmatic in large part because antibodies recognizing the endogenous protein do not exist. We identified a Gal4 insertion (dSARM[NP7460]) in an intron upstream of the exons coding for dSARM’s conserved domains (here called dSARMGal4). Arguing that this Gal4 line is expressed in the endogenous dSARM pattern, dSARM>dSARM RNAi leads to elevated levels of neuronal debris like that observed in dSARM LOF mutants (Figure S1 D-E). We find that dSARMGal4 is widely expressed in the L3 CNS. Notably, mCherry.NLS driven by dSARMGal4 labels many Repo-positive glia (Figure 2 C-C”“), as well as Repo-negative cells likely to be neurons. The widespread glial expression pattern of dSARM is noteworthy since dSARM was not previously thought to be expressed in glia.
To interrogate whether dSARM is specifically expressed in cortex glia, we used the Q system to express nuclear LacZ in cortex glia with the wrapper932iQF2 driver, since the use of dSARMGal4 precluded using a second Gal4 driver (Potter et al., 2010). Wrapper932iQF2 is largely specific to cortex glia but may also be expressed in some surface glia (Coutinho-Budd et al., 2017); therefore, we avoided imaging cells on the exterior of the CNS where surface glia reside. We detect striking co-localization of mCherry and LacZ in many regions of the brain and VNC, indicating cortex glial expression of dSARMGal4 (Figure 2 D-D”“, S1 C-C’“). We conclude that dSARM is broadly expressed in cortex glia.
We utilized a GFP exon trap inserted into a FoxO intron, FoxOMIMICGFP, as a FoxO reporter (Venken et al., 2011). FoxOMIMICGFP expression largely colocalizes with a FoxO antibody that our lab generated and validated (Figure S1 F-G’“; Nechipurenko & Broihier 2012). FoxOMIMICGFP marks cortex glial nuclei labeled with mCherry.NLS expressed via Ctxglia-splitGal4 (Figure 2 E-E”“). FoxO-positive cortex glia are primarily located in the dorsal region of the brain, but we also detect expression in VNC cortex glia (Figure 2 E’, data not shown). Collectively, these analyses show that Toll-6, dSARM, and FoxO are expressed in cortex glia in the L3 CNS.
Toll-6-FoxO signaling regulates Draper transcription in cortex glia
Cortex glia encapsulate neuronal cell bodies in an elaborate meshwork of thin processes extending from the BBB to the neuropil, providing trophic support to healthy neurons and regulating clearance of apoptotic ones (Coutinho-Budd et al., 2017; Etchegaray et al., 2016). Toll-6 is proposed to have neurotrophic activity (McIlroy et al., 2013; Foldi et al., 2017), so we tested if Toll-6 is required in glia to promote survival of neurons by quantifying the number of Hb9, Eve, and Dbx-positive neurons in Toll-6 pathway null and cortex glial-specific LOF backgrounds. However, we failed to detect appreciable changes in neuronal number in any of these genotypes (Table S2), arguing against a neurotrophic function. We also failed to detect a change in the overall number of cortex glia (Table S2), indicating that Toll-6 signaling does not regulate cortex glial survival. We further considered the possibility that cortex glia morphology might be disrupted, which could lead to disrupted trophic signaling (Coutinho-Budd et al., 2017). However, we did not observe any defects in gross morphology of cortex glial processes in cortex glial-specific LOF brains or VNCs (Figure S2 A-D). Thus, increased levels of apoptotic debris observed in pathway mutants at L3 do not appear to result from defects in Toll-6-mediated trophic signaling.
We proceeded to investigate the alternative possibility that Toll-6 signaling regulates phagocytosis. If so, it might regulate expression of the key phagocytic receptor Draper (Drpr), which is expressed in glia throughout lifespan and is the major receptor responsible for regulating clearance of cellular debris in the CNS (Freeman et al., 2003; MacDonald et al., 2006; Tasdemir-Yilmaz and Freeman, 2014). We first characterized Drpr expression and function in cortex glia. We find that Drpr is expressed in cortex glia (labeled with Ctxglia-split>mCD8::GFP; in region encompassed by dotted line) (Figure 3 A). Confirming antibody specificity, cortex glial RNAi-mediated knock-down of all Drpr isoforms results in a 49% decrease in Drpr levels (Figure 3 F), and cortex glial RNAi-mediated knockdown of Drpr-I, the isoform that promotes debris clearance, results in a 53% decrease (Figure 3 B, F; Logan et al. 2012). As expected, drpr nulls exhibit a vast excess of apoptotic debris (Figure 3 L, M) (Hilu-Dadia et al., 2018). And in line with a previous report (Etchegaray et al., 2016), we find that Drpr functions specifically in cortex glia since cortex glial-specific knockdown of all Drpr isoforms (Figure 3 K-N), or only Drpr-I (Figure 3 K,O) results in increased Dcp-1 debris. Cortex glia-specific reduction of Drpr does not alter gross morphology of cortex glia (Figure S2 E-F). Thus, cortex glia express and utilize the Drpr phagocytic receptor.
Figure 3. Toll-6-FoxO signaling regulates Draper transcription in cortex glia.

(A-E) Representative 3 μm z-projections of L3 brains of indicated genotypes labeled with anti-GFP (green) and anti-Drpr (magenta). Dashed lines signify area of cortex glial processes. (F-I) Quantification of mean Drpr intensity normalized to control indicates that cortex glial Toll-6, dSARM and FoxO promotes Drpr expression. (J) qRT-PCR analysis of relative Drpr mRNA levels in: wild type: 1.0 ± 0.28; FoxOΔ94: 0.29 ± 0.08. Error bars are SEM. (K) Quantification of Dcp-1 particles indicates that cortex glia utilize Drpr to clear apoptotic cells. (L-O) Representative z-projections of indicated genotypes labeled with anti-Dcp-1. (P) Quantification of Dcp-1 particles indicates that Drpr overexpression rescues cell clearance defects in Toll-6 pathway mutants. Error bars are min and max data points. Statistical measures directly above each column are compared to control. n is noted at the bottom of each column. n.s., not significantly different. *, P<0.05; **, P<0.01; ***, P<0.001. Scale bars are 50 μm. Values for mean and SEM are located in Table S3.
Having characterized Drpr expression in cortex glia, we quantified average Drpr intensity in cortex glia in pathway mutants. Cortex glial knock-down of Toll-6 using two RNAi lines results in 31% and 39% decreases in cortex glial Drpr (Figure 3 C, G). Similarly, knock-down of dSARM using two RNAi lines results in 16% and 35% reductions in Drpr (Figure 3 D, G), while knockdown of FoxO via two RNAi lines decreases Drpr by 29% and 25% (Figure 3 E, G). We observe similar reductions in Drpr levels using the NP77Gal4 driver to drive RNAi-mediated knockdown (Figure 3 H) as well as in null backgrounds (Figure 3 I). These data demonstrate that cortex glial Toll-6 signaling promotes Drpr expression. We next performed quantitative RT-PCR on RNA isolated from wild-type and foxO mutant L3 CNS to test if FoxO regulates Drpr transcription. We find that drpr mRNA is 71% downregulated in foxO nulls (Figure 3 J). We likewise tested if drpr mRNA is reduced in Toll-6 mutants and find that drpr mRNA levels are decreased 40% in the Toll-6 LOF CNS (data not shown). These data indicate that Drpr is either a direct or indirect transcriptional target of FoxO.
To analyze a function for Toll-6 signaling in phagocytosis via an independent assay, we characterized the expression of the late endosomal/lysosomal marker Rab7 using an endogenously tagged Rab7-YFP allele (Dunst et al., 2015). In controls, we observe both diffuse cytoplasmic Rab7 expression (Figure S2 G; insets) and pronounced Rab7 localization to distinct donut-shaped structures (Figure S2 H; insets & arrows). Strikingly, 89% of Rab7 donuts are located within cortex glial membranes (Figure S2 I). Hence, we asked if Rab7 donuts surround Dcp-1 debris, and could reflect sites of active engulfment. Indeed, 70% of Rab7 donuts encircle Dcp-1 debris in controls (Figure S2 J, M), while 10% wrap Elav-positive neurons (Figure S2 K, M), and 2% surround Repo-positive glia (Figure S2 L-M). Thus, Rab7 donuts in cortex glia usually form around apoptotic debris arguing they reflect enhanced expression/localization of Rab7 during corpse engulfment. Next, we assayed whether Toll-6 signaling regulates Rab7 donut formation. We find that cortex glia-specific knockdown of Toll-6, dSARM, and foxO results in 31%, 40%, and 34% decreases in the number of Rab7 donuts in cortex glia compared to controls (Figure S2 N-O). Likewise, Drpr-I knockdown leads to a 40% reduction in cortex glial Rab7 donuts (Figure S2 N-O). Impaired accumulation of Rab7 machinery in mutant backgrounds supports the idea that phagocytosis is defective.
Finally, we reasoned that if reduced phagocytosis is responsible for Dcp-1 accumulation in Toll-6 pathway mutants, then Drpr overexpression might rescue Dcp-1 phenotypes in the mutants. Importantly, overexpression of the pro-phagocytic Drpr-I isoform in an otherwise wild-type background does not yield a Dcp-1 phenotype (Figure 3 P), enabling us to interpret Drpr-I overexpression in Toll-6, dSARM, and foxO nulls. We find that cortex glial-specific Drpr-I overexpression completely corrects the Dcp-1 phenotype in Toll-6, dSARM, and foxO LOF mutants in both the brain and the VNC (Figure 3 P). Thus, Drpr is a crucial functional target of Toll-6 signaling in cortex glia. These studies uncover a critical role for Toll-6 signaling in promoting Draper-dependent glial phagocytosis of apoptotic debris.
Toll-6 signaling promotes cortex glial engulfment of dying neurons
Since Toll-6 signaling is upstream of Drpr, we hypothesized that it is required for the initial engulfment of apoptotic debris. In this event, we predicted decreased association of apoptotic debris with cortex glial processes. To test this model, we developed a live assay in a semi-intact preparation to analyze recognition of neuronal debris in L3 brain, similar to assays previously used in embryos (Shklover et al. 2015; Shklyar et al. 2014; Kurant et al. 2008). Cortex glia were visualized with membrane-bound GFP (Ctxglia-split>mCD8::GFP) and apoptotic debris labeled with an Annexin V-conjugated fluorophore. Annexin V has high affinity for phosphatidylserine which accumulates on the outer leaflet of apoptotic cell membranes (Vermes et al., 1995). Arguing that Annexin V labels apoptotic cells in this assay, we find an 80% increase in Annexin V positive particles following induction of apoptosis using 10 mM cycloheximide (CHX)(Figure S3 A; Bolkan et al., 2007). Verifying some BBB permeability in this preparation, 10 kDa 568 conjugated dextran crosses the BBB (labeled with Spg>mCD8::GFP; Figure S3 B-D; Stork et al., 2008).
Further supporting a function for Toll-6 signaling in phagocytosis, cortex glial knockdown of pathway members results in increased Annexin V labeling (Figure 4 F). To assay whether Annexin V particles are likely engulfed by cortex glia, we asked if they associate with the lysosomal marker, Rab7-YFP. Indeed, Annexin V particles in the cortical region of the brain extensively colocalize with Rab7-YFP in controls (Figure S3 E-G), suggesting cortex glial engulfment. We then quantified Annexin V particles within cortex glia membranes in control and mutant backgrounds. In controls, 75% of Annexin V particles appear inside cortex glial processes (dashed circles; 3D rendering) while 25% of particles are not cortex glia-associated (arrows), (Figure 4 A, G). Cortex glia-specific knockdown of Toll-6, dSARM, and foxO results in 28%, 25%, and 28% decreases in the percentage of Annexin V particles within cortex glial membranes (Figure 4 B-D, G). We also tested whether Drpr regulates engulfment of apoptotic cells by cortex glia, since Drpr can regulate multiple steps in phagocytosis including recognition, engulfment, and phagosome maturation (Etchegaray et al., 2016; Kurant et al., 2008; Tung et al., 2013). Cortex glial Drpr-I knockdown drives a 36% decrease in Annexin V particles within cortex glial membranes (Figure 4 E, G). We conclude that the Toll-6 pathway and Drpr are required for cortex glial engulfment of apoptotic neuronal cell bodies and not simply for degradation of debris following internalization.
Figure 4. Toll-6 signaling promotes cortex glial engulfment of dying neurons.

(A-E) Representative images of cortex glial membranes labeled with Ctxglia-split>mCD8::GFP (green) and Annexin V-594 (magenta) in indicated genotypes. Dashed circles denote Annexin V within cortex glial membranes and arrows indicate Annexin V particles outside of membranes. Orthogonal views are higher magnification of merged images. 3D volume renderings were made from merged data presented here. Scale bars are: 10 μm (merged images), 5 μm (orthogonal views) 3 μm (3D volume renderings). (F) Quantification indicates that Toll-6 pathway mutants display increased Annexin V particles. (G) Proportion of Annexin V engulfed by cortex glia is reduced in Toll-6 pathway mutants. n is noted at the bottom of each column; ***, P<0.001. Values for mean and SEM are located in Table S4.
Neuronal Spz5 is the ligand for glial Toll-6
We hypothesized that Toll-6 signaling enables initial interaction of dying neurons with glia. If so, the Dcp-1-positive debris is predicted to be of neuronal origin. However, cell corpses lose identifying markers, preventing us from double-labeling with Dcp-1 and neuron-specific antibodies. So we turned to a genetic strategy and reduced neuronal apoptosis in Toll-6 mutants by pan-neuronally overexpressing a baculoviral P35 transgene. P35 is a potent suppressor of apoptosis and inhibits caspase activation in flies (Hay et al., 1994). We find that pan-neuronal overexpression of P35 fully rescues the Dcp-1 phenotype in Toll-6 null animals (Figure 5 A), demonstrating that the cell corpses are largely of neuronal origin.
Figure 5. Neuronal Spz5 is the ligand for glial Toll-6.

(A) Quantification indicates that Dcp-1 particles in Toll-6 mutants is neuronal in origin. (B-C) Representative 3 μm z-projections of L3 brains of indicated genotypes labeled with anti-Drpr. Scale bar is 100 μm. (D-E) Quantification of mean Drpr intensity indicates that neuronal Spz5 regulates cortex glial Drpr expression. (F-G) Representative partial z-projections of indicated genotypes labeled with anti-Dcp-1. Scale bar is 50 μm. (H-L) Quantification of Dcp-1 particles indicates that neuronal Spz5 promotes cortex glial engulfment of apoptotic debris. Error bars are min and max data points. n is noted at the bottom of each column. n.s., not significantly different. *, P<0.05; **, P<0.01; ***, P<0.001. Values for mean and SEM are located in Table S5.
Encouraged by these results, we screened for a possible neuronal cue. Unlike mammalian TLRs which have a diverse array of activating ligands, Drosophila Toll ligands are members of the secreted Spätzle (Spz) family (Gay and Gangloff, 2007). We queried whether RNAi-mediated neuronal knockdown of any of the six Spz family members regulates Drpr levels in cortex glia and identified Spz5 (Drosophila neurotrophin 2, dNT2; Figure S4). Pan-neuronal spz5/dNT2 knockdown via two RNAi lines results in 29% and 43% decreases in Drpr levels in cortex glia (Figure S4, B-D). We verified this finding using a spz5 null allele (Figure 5 E). Neuronal knockdown of spz2/dNT1 using one RNAi line also yielded a decrease in Drpr levels, but follow-up functional studies indicated it is unlikely to be the relevant ligand (Figure S4; data not shown). Spz5/dNT2 has previously been shown to bind Toll-6 in vitro (Foldi et al., 2017), supporting its identification in our screen.
To probe a functional requirement, we asked if spz5/dNT2 knockdown in neurons mimics the Dcp-1 phenotype of Toll-6 pathway mutants. Pan-neuronal knockdown of spz5/dNT2 using three RNAi lines yields 84%, 94% and 93% increases in Dcp-1 debris in the brain (Figure 5 F-H). We do not find a change in Dcp-1 debris when spz5/dNT2 is knocked down in cortex glia (Figure 5 I), demonstrating cellular specificity. We observe a similar increase in Dcp-1 debris in spz5 LOF animals (Figure 5 J). Arguing against a widespread neurotrophic function for spz5/dNT2 at the L3 stage, neuronal knockdown does not alter neuronal number (Table S2). Thus, we now refer to this ligand by its original gene name, spz5.
The equivalent requirements for neuronal Spz5 and glial Toll-6 suggest that this ligand-receptor pair mediates clearance of apoptotic neurons. We utilized two approaches to test this hypothesis. First, we asked if Spz5 genetically interacts with Toll-6, dSARM and FoxO by quantifying Dcp-1 debris in Spz5/+, Toll-6/+; Spz5/+, dSARM/+; and Spz5/+, foxO/+ double heterozygotes. While single heterozygotes do not display Dcp-1 phenotypes (Figure 1 F and 5 J), double heterozygotes accumulate apoptotic debris in the brain (Figure 5 J). Second, we confirmed that excess Dcp-1 debris in spz5 LOF mutants stems from impaired Drpr-dependent engulfment. We find that cortex glia-specific Drpr-I overexpression corrects the Dcp-1 phenotype in spz5 LOF mutants (Figure 5 K). These results argue that neuronal Spz5 activates cortex glial Toll-6 to direct glial engulfment of apoptotic neurons.
These LOF analyses indicate that neuronal spz5 is necessary for clearance of apoptotic debris. We next assayed if increased Spz5 can reduce Dcp-1 debris. This experiment was of particular interest since Toll-6 overexpression results in a 35% reduction in Dcp-1 debris in the brain (Figure 1 Q), which we interpret to result from accelerated clearance of dying neurons. If Spz5 is released from dying neurons, neuronal overexpression of Spz5 might likewise speed debris clearance. We tested this hypothesis using a Spz5-HA construct (Bischof et al., 2013). The Dcp-1 phenotype observed in spz5 nulls is fully rescued when Spz5-HA is pan-neuronally expressed in a spz5 background (Figure 5 L), establishing its functionality. We next asked if overexpressing Spz5-HA in an otherwise wild-type background expedites Dcp-1 clearance. However, we do not detect a change in Dcp-1 accumulation with pan-neuronal overexpression of Spz5 (Figure 5 L), arguing that increased Spz5 expression alone is insufficient to accelerate debris clearance. This finding hinted at an additional layer of regulation of Spz5 in apoptotic neurons.
Neuronal apoptosis promotes Furin-mediated Spz5 processing
Like many secreted cues, Spz ligands are synthesized as inactive precursor proteins and subsequently cleaved into pro-domain and signaling fragments. Similar to a previous study (Foldi et al., 2017), we identified a predicted cleavage site for the proprotein convertase Furin between the pro-domain and signaling fragments of Spz5, suggesting Furin processing (Figure 6 J). We wondered whether Spz5 activity in dying neurons is regulated at the level of proteolysis. Accordingly, we tested if loss of neuronal Furin results in similar phenotypes as loss of neuronal Spz5. There are two Furin homologs in Drosophila, Furin1 (Fur1) and Furin2 (Fur2), and we assayed for a functional requirement of both. Reducing neuronal Fur1 expression via two independent RNAi lines results in modest decreases in cortex glial Drpr (Figure 6 A-B, D). In contrast, reducing neuronal Fur2 via three independent RNAi lines leads to more pronounced decreases of 28%, 28%, and 41% in cortex glial Drpr (Figure 6 A, C-D). These results indicate that glial Drpr is sensitive to neuronal Furin levels, in particular to levels of Fur2. Using a Gal4 insertion directly upstream of the Fur2 locus (Fur2[NP4074]), we investigated if Fur2 is likely expressed in neurons (Hayashi et al., 2002). Nuclear mCherry driven by Fur2Gal4 is predominantly neuronal (Figure S5 A-B““) consistent with our genetic analysis.
Figure 6. Neuronal apoptosis promotes Furin-mediated Spz5 processing.

(A-C) Representative 3 μm z-projections of L3 brains of indicated genotypes labeled with anti-Drpr. Scale bar is 100 μm. (D) Quantification of mean Drpr intensity indicates that neuronal Furins promote cortex glial Drpr expression. Error bars are SEM. (E-F) Representative 10 μm z-projections of indicated genotypes labeled with anti-Dcp-1. Scale bar is 50 μm. Quantification of Dcp-1 particles indicates that neuronal Fur2 is (G) necessary and (H) sufficient to limit apoptotic debris. Error bars are min and max data points. (I) Quantification of mean Drpr intensity indicates that neuronal Fur2 is sufficient to promote cortex glial Drpr expression. (J) Schematic representation of Spz5-HA protein domains. (K, M, Q, O) Representative Western blots depicting Spz5-HA cleavage in indicated genotypes. Black arrows denote full-length Spz5 and cleaved Spz5. Quantification of cleaved Spz5-HA to total Spz5-HA indicates Fur2 is (L) necessary and (N) sufficient for Spz5-HA processing. (P) Quantification of cleaved Spz5-HA to total Spz5-HA indicates neuronal apoptosis drives Spz5 processing. (R) Quantification of cleaved Spz5-HA to total Spz5-HA indicates blocking neuronal apoptosis decreases Spz5-HA processing. Error bars are SEM. n is located at bottom of each column. n.s., not significantly different. **, P<0.01; ***, P<0.001. Values for mean and SEM are located in Table S6.
We assayed whether neuronal Furin knockdown increases Dcp-1 debris. Reducing neuronal Fur1 expression via two RNAi lines results in modest increases in Dcp-1 (Figure 6 G). We find that reducing Fur2 via three RNAi lines leads to more pronounced increases in apoptotic debris of 74%, 53%, and 42% (Figure 6 E-G). Hence, while knockdown of either Furin results in increased persistent neuronal corpses, loss of Fur2 has more severe consequences. Together, these data are consistent with the idea that neuronal Spz5 activation is regulated by Furin proteolysis.
If Spz5 signaling is controlled at the level of proteolysis, Furin overexpression might drive elevated release of the mature, cleaved ligand and decrease Dcp-1 debris similarly to glial Toll-6 overexpression. While overexpression of Fur1 has no effect (Figure 6 H), Fur2 overexpression results in a 25% decrease in neuronal debris (Figure 6 H). These findings suggest that neuronal Fur2 overexpression accelerates corpse clearance by a Toll-6-mediated cortex glial pathway. If so, neuronal Fur2 overexpression might also result in elevated Drpr levels in cortex glia. Indeed, overexpression of Fur2 in neurons drives a 30% increase in cortex glial Drpr (Figure 6 I). These data imply that neuronal Fur2, likely via Spz5 processing, tunes cortex glial phagocytic capabilities to promote the timely clearance of apoptotic debris.
We next undertook a series of biochemical experiments to investigate a possible link between Fur2 activity, Spz5 processing, and phagocytosis of dying neurons. We assessed whether Fur2 is necessary for Spz5 cleavage. On immunoblot, we observe both full length (~50 kDa) and mature Spz5 (~19 kDa) in CNS lysates from animals expressing Spz5-HA in all neurons (Figure 6 K). Demonstrating that Fur2 is necessary to generate mature Spz5, neuron-specific loss of Fur2 results in a 53% reduction in mature Spz5 relative to total Spz5 (Figure 6 K-L; Figure S5 C). On the other hand, overexpression of neuronal Fur2 drives a 3.8-fold increase in Spz5 cleavage (Figure 6 M-N; Figure S5 D). Thus, Fur2 is necessary and sufficient for Spz5 processing in vivo.
Our genetic data suggest that Fur2-dependent cleavage of Spz5 in dying neurons prompts cortex glia to upregulate the phagocytic program. Could Spz5 proteolysis be specifically triggered in dying neurons? To test this idea, we assayed if acutely inducing neuronal apoptosis drives increased levels of mature Spz5. We overexpressed the pro-apoptotic gene Head-involution defective (Hid) in HS-Hid transgenic animals via a 75 min heat shock and investigated whether the amount of cleaved Spz5 is altered. We observe elevated Dcp-1 labeling in these brains (Figure S5 F-G), demonstrating increased apoptosis. Moreover, in line with our hypothesis, we find a 2.1-fold increase in the fraction of mature Spz5 relative to total Spz5 in these experiments (Figure 6 O-P) without a change in total protein levels (Figure S5 E). As an alternative approach, we utilized CHX to acutely drive apoptosis in the L3 CNS. We treated animals expressing Spz5-HA in all neurons with 50 mM CHX and asked whether the fraction of cleaved Spz5 relative to total Spz5 is altered. Similar to Hid over-expression, CHX treatment increases the fraction of cleaved Spz5 by 2.3-fold (Figure S5 H-J). Thus, acute induction of apoptosis leads to increased levels of cleaved Spz5. Arguing that apoptosis upregulates Fur2 activity post-transcriptionally, we do not observe changes in Fur2 transcription in the CNS in acute apoptosis paradigms (Figure S5 K).
If neuronal apoptosis drives Spz5 processing, we predicted that blocking apoptosis would inhibit Spz5 cleavage. To approach this question, we overexpressed P35 in all neurons, which is sufficient to decrease Dcp-1 accumulation (Figure S5 M-O). Strikingly, Spz5 cleavage is decreased 40% compared to total Spz5 when neuronal apoptosis is inhibited (Figure 6 Q-R; Figure S5 L). Taken together, our data indicate that neuronal apoptosis triggers Furin-mediated Spz5 processing to drive activation of glial Toll-6 signaling and phagocytosis.
Impaired cortex glial Toll-6 signaling causes early-onset neurodegeneration
Are there long-term consequences of impaired Toll-6-FoxO signaling in cortex glia? We began by assessing whether Toll-6 pathway mutants exhibit increased Dcp-1 debris in adults. We first quantified apoptotic debris in Toll-6 pathway mutants on the day of eclosion and find that Toll-6 and foxO nulls display roughly double the amount of apoptotic debris as wild type (Figure 7 A-D). To establish the specific contribution of cortex glial Toll-6 signaling, we quantified apoptotic debris in cortex glial knockdown brains. Selective loss of Toll-6, dSARM, or foxO in cortex glia causes an average 87% increase in apoptotic debris in the adult brain (Figure 7 E, S6 A-D). In contrast, neuronal knockdown does not result in an adult Dcp-1 phenotype (Figure 7 F). These data demonstrate that cortex glial Toll-6 signaling limits the accumulation of apoptotic debris in adult, as well as in developing, brains
Figure 7. Impaired cortex glial Toll-6 signaling causes early-onset neurodegeneration.

(A-C) Representative z-projections of adult brains of indicated genotypes labeled with anti-Dcp-1. Scale bar is 100 μm (DPE = days post eclosion). (D-F) Quantification indicates that Toll-6 signaling in cortex glia limits Dcp-1 debris. (G-J) Representative 5 μm paraffin sections stained with hematoxylin and eosin of indicated genotypes. (K) Quantification of adult degeneration indicates that cortex glial Toll-6 signaling prevents degeneration. (L) Schematic representation of data presented in this paper. n is noted at the bottom of each column. n.s., not significantly different; **, P<0.01; ***, P<0.001. Values for mean and SEM are located in Table S7.
Since we observe elevated apoptotic debris in adult brains, we asked if these mutants display neurodegeneration. Defects in phagocytosis play a role in multiple degenerative contexts (Etchegaray et al., 2016; Kleinberger et al., 2014; Sokolowski and Mandell, 2011). In flies, pan-glial drpr knockdown leads to excess neurodegeneration in adults (Draper et al., 2014; Etchegaray et al., 2016). Degeneration is marked by the accumulation of neuropathological vacuolar lesions (Cao et al., 2013; Fergestad et al., 2006). Since Toll-6, dSARM, and foxO mutants have impaired phagocytosis, we asked if loss of pathway function enhanced neurodegeneration.
We adapted a neurodegeneration index for phenotypic quantification (Figure S6 E) (Cao et al., 2013). Briefly, we assigned brains a neurodegeneration score based the amount of tissue that contained lesions, taking into account the number and size of the vacuoles. For example, a brain with one large lesion (Figure S6 E1) would receive a lower score than a brain with many smaller holes (Figure S6 E3 & 4). We initially tried to query null mutants for vacuolization, but dSARM nulls do not survive the pupal stage, and foxO nulls have severely impaired lifespan. Thus, we turned to cortex glial RNAi-mediated knockdown of Toll-6, dSARM, or foxO, which do not exhibit lifespan alterations (Figure S6 F). In controls, severe vacuolization does not begin until 35 days post-eclosion (DPE; Figure 7 K). In contrast, severe degeneration can be detected at 10 DPE when Toll-6, dSARM, or FoxO are lost in cortex glia (Figure 7 G-K). The finding that mutant brains show significant neurodegeneration 25 days before controls indicates that impaired cortex glial Toll-6 signaling results in early-onset neurodegeneration. Together, our findings argue that a Toll-6-mediated innate immune pathway directs cortex glial clearance of apoptotic debris during development and prevents early-onset neurodegeneration in the adult.
Discussion
Neurons and glia are intimately associated. This close relationship allows glia to detect and rapidly respond to changes in neuronal health. Given the highly interdependent nature of neuronal and glial functions across development, surprisingly little is understood concerning the molecular pathways coordinating their activities. Here we define a signaling interaction between dying neurons and glia in the developing brain that prepares glia for phagocytosis. We find that apoptosis induces proteolytic activation of the Toll-6 ligand, Spz5, in dying neurons. Neuronal activation of the ligand triggers the Toll-6 pathway in cortex glia, which then drives expression of the Drpr engulfment receptor to permit efficient phagocytosis of neuronal corpses (Figure 7 L). Given that this cue is upstream of the “eat-me” cue and signals neuronal distress, we refer to it as a priming cue—a type of “find-me” signal. This priming signal is poised to increase both specificity and speed of corpse clearance.
Toll-6 as a glial sensor for damaged/dying neurons
Accurate recognition and efficient clearance of cellular corpses involves a tightly-regulated sequence of molecular signals. Apoptotic cells can secrete soluble “find-me” signals which serve as chemo-attractants to attract the phagocyte. Next, engulfment is initiated when “eat-me” signals on the dying cell bind receptors on the phagocyte (Elliott et al., 2009; Flannagan et al., 2012; Ravichandran, 2010; Segawa and Nagata, 2015). Given that cortex glia provide trophic support through sustained, close physical contact with neurons, a chemo-attractive “find-me” cue is superfluous. However, recent evidence suggests that the function of “find-me” signals extends beyond physical recruitment of the phagocyte (Elliott and Ravichandran, 2016). Namely, macrophages are attracted to dying cells by the “find-me” cue sphingosine 1-phosphate (S1P). Importantly, S1P signaling also promotes upregulation of phagocytosis receptors on macrophages such as MerTK and MGF-E8 and is required for efficient phagocytosis (Kolb and Martinez, 2016; Luo et al., 2016). Thus, the S1P “find-me” signal regulates both recruitment and priming of macrophages, indicating a similarity between the functions of Spz5 and S1P. Given that Spz5 promotes upregulation of Drpr and readies cortex glia for engulfment, we propose that it prepares, or primes, glia for phagocytosis. We propose that Spz5 is a “find-me” cue in the Drosophila CNS, since its priming function is consistent with an established function of mammalian “find-me” cues.
We became interested in the Spz ligand family after uncovering a glial requirement for Toll-6 in clearing neuronal corpses, which raised the exciting possibility that the receptor might be activated in response to a neuronal signal. We screened the six endogenous Toll ligands and found that only Spz5 fulfilled for the requirements for such a cue. Multiple lines of evidence argue that Spz5 serves as a distress signal from dying neurons that activates a glial Toll-6 pathway required to promote phagocytosis. First, we do not detect defects in neuronal or glial number in these backgrounds indicative of a trophic functions. Additionally, we do not find defects in gross morphology of cortex glia suggestive of a more generic defect in neuron-glia communication. Second, Spz5 and Toll-6 display cleanly separable cellular requirements. Spz5 is required specifically in neurons, while Toll-6 is required only in glia. Loss of either neuronal Spz5 or glial Toll-6 results in indistinguishable phenotypes—reduced expression of the Drpr engulfment receptor and increased persistent apoptotic debris. Third, Drpr overexpression fully rescues increased Dcp-1 accumulation in pathway mutants. Since Drpr is the primary engulfment receptor in the CNS (Hilu-Dadia et al., 2018; Tasdemir-Yilmaz and Freeman, 2014), these results demonstrate that phagocytosis is specifically impaired in the mutants. Fourth, pathway over-activation results in reduced apoptotic debris, consistent with the hypothesis that this “priming” pathway normally accelerates debris clearance. Specifically, neuronal overexpression of Furin2 or glial overexpression of Toll-6 both lead to reduced accumulation of Dcp-1 in the CNS. Fifth, acute induction of apoptosis leads to increased levels of the mature Spz5, suggesting that apoptosis induces Spz5 signaling by triggering Fur2-mediated processing. And sixth, blocking neuronal apoptosis inhibits Spz5 cleavage. Together, these results argue that Spz5 is an instructive cue released by dying neurons that signals to glia to activate their phagocytic program.
Our findings indicate that Toll-6 activates FoxO to regulate Drpr at the transcriptional level since Drpr transcript levels are down 71% in FoxO mutant brains. While we have not tested whether FoxO directly regulates Drpr transcription, we identified a number of FoxO binding sites at the Drpr locus. Specifically, we found three putative FoxO binding sites [5’-TTGTTTAT/C-3’] upstream of the Drpr transcriptional start site (−1,735, −3,367, and −6,281 bp) and three sites downstream (3,632, 5,638, and 8,341 bp). It is also possible that FoxO regulates Drpr transcription via another transcription factor(s). Regardless, our findings indicate that Drpr expression is dynamically controlled during development to tune glial competence for phagocytosis. Drpr is similarly known to be induced following injury (Logan et al., 2012). Interestingly, the dAP1 transcriptional complex and Stat92E transcription factor regulate glial Drpr transcription following axon injury (Doherty et al., 2014; MacDonald et al., 2013; Musashe et al., 2016). Thus, Drpr levels are tightly controlled during development and in response to injury.
Our data indicate that Toll-6 is likely not, itself, a phagocytic receptor but instead acts upstream of key engulfment machinery. This is in keeping with TLR functions in immunity, where TLR activation induces expression of a phagocytic gene program in macrophages (Doyle et al., 2004). Moreover, a seminal paper found that TLR2/4 promotes macrophage-mediated phagocytosis of microbial pathogens (Blander and Medzhitov, 2004). Mammalian TLRs have also been shown to promote phagocytosis of axonal debris following injury (Church et al., 2017; Rajbhandari et al., 2014). Likewise, TLRs in neurodegeneration and disease promote microglial activation and initiate signaling cascades necessary to clear toxic proteins (Burguillos et al., 2015; ReedGeaghan et al., 2009 ). Our work suggests exciting parallels between the molecular mechanisms governing glial phagocytosis in development and disease/degeneration. In the future, it will be critical to determine whether Toll-6 also activates the phagocytic machinery in response to injury.
Together, these findings offer insight into neuron-glia interactions in the developing CNS. We propose that an immune signaling mechanism functions as a priming cue to activate glial phagocytosis of apoptotic neurons. We propose that this signal results in a valence change in the neuron-glia relationship, facilitating a switch in the glial state from supportive to degenerative. Thus, this study underscores the importance of local cues in tuning glial responsiveness to changes in neuronal homeostasis.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for information, protocols, reagents, or stocks should be directed to and will be fulfilled by the lead contact, Heather T. Broihier (heather.broihier@case.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
D. melanogaster stocks
Drosophila melanogaster stocks were raised on standard molasses formulation food at 25°C (most crosses) or 29°C (adult degeneration experiments). Both male and female flies were included in all analyses. Sex was not considered since phenotypes were generally evenly distributed, suggesting no detectable sex contribution.
The following lines were used: OregonR (OR; wild type), UAS-FoxOF19−5 (Hwangbo et al., 2004), UAS-dSARM (Osterloh et al., 2012), UAS-Toll-6 (Yagi et al., 2010), UAS-Toll-6TIRdead (BDSC 64076), UAS-Draper-I (BDSC 67035), UAS-Spz5-HA (FlyORF F003080), UAS-Fur1R (BDSC 63077), UAS-Fur1X (BDSC 63078), UAS-Fur2 (BDSC 63081), FoxOΔ94 (BDSC 42220), dSARM4621 (Osterloh et al., 2012), dSARM4705 (Osterloh et al., 2012), Toll-6EX13 (Yagi et al., 2010), MyD88C03881 (Ward et al., 2015), DraperΔ5 (MacDonald et al., 2006), Toll-7g1−1 (Yagi et al., 2010), Toll-7AW1 (Ward et al., 2015), Df(2R)BSC279 (BDSC 23664), Df(2R)BSC280 (BDSC 23665), Mi{MIC}foxoMI02660 (BDSC 37585), Mi{MIC}Toll-6MI02127 (BDSC 34467), TI{TI}Rab7EYFP (BDSC 62545), QUAS-Nuc-LacZ (BDSC 30007), UAS-mCD8::GFP (BDSC 32185, 32186, 32194), UAS-GFP.NLS (BDSC 4775 or 4776), UAS-mCherry.NLS (BDSC 38425), Wrapper932i-Gal4DBD, Nrv2-VP16AD (Ctxglia-split; Coutinho-Budd et al., 2017), Wrapper932i-Gal4DBD, Nrv2-VP16AD, UAS-mCD8::GFP (Coutinho-Budd et al., 2017), Wrapper932i-Gal4DBD, Nrv2-VP16AD, UAS-mCD8::Cherry (Coutinho-Budd et al., 2017), Wrapper932i-QF2 (Coutinho-Budd et al., 2017), ElavC155Gal4 (BDSC 458), RepoGal4 (Lee and Jones, 2005), Inx2Gal4 (BDSC 12834), NP577Gal4 (Awasaki et al., 2008; Hayashi et al., 2002), SpgGal4 (Stork et al., 2008), Nrv2Gal4 (Sun et al., 1999), NP6293Gal4 (Awasaki et al., 2008; Hayashi et al., 2002), AlrmGal4 (BDSC 67032), Mz0709Gal4 (Ito et al., 1995), dSARMGal4 (Kyoto DGRC 105471), UAS-Toll-6 RNAiv928 (VDRC 928), UAS-Toll-6 RNAi BDSC (BDSC 56048), UAS-FoxO RNAi v107786 (VDRC 107786), UAS-FoxO RNAi BDSC (BDSC 32427), UAS-dSARM RNAi v102044 (VDRC 102044), UAS-dSARM RNAi v105369 (VDRC 105369), UAS-Dl RNAi B27650 (BDSC 27650), UAS-Dl RNAi B34938 (BDSC 34938), UAS-Drpr RNAi3B (MacDonald et al., 2006), UAS-Drpr RNAi BDSC (VDSC 36732), UAS-Drpr-I RNAi (Logan et al., 2012), UAS-Spz RNAi B58499 (BDSC 58499), UAS-Spz RNAiv105017 (VDRC 105017), UAS-Spz RNAiB28538 (BDSC 28538), UAS-Spz2 RNAi v108894 (VDRC 108894), UAS-Spz2 RNAi v110495 (VDRC 110495), UAS-Spz3 RNAi BDSC (BDSC 6958), UAS-Spz3 RNAi v102871 (VDRC 102871), UAS-Spz3 RNAi v18949 (VDRC 18949), UAS-Spz4 RNAi BDSC (BDSC 60044), UAS-Spz5 RNAi v102389 (VDRC 102389), UAS-Spz5 RNAi v41295 (VDRC 41295), UAS-Spz6 RNAi BDSC (BDSC 57510), UAS-Fur1 RNAi 41914 (BDSC 41914), UAS-Fur1 RNAi 42481(BDSC 42481), UAS-Fur2 RNAi 51743 (BDSC 51743), UAS-Fur2 RNAi 42577 (BDSC 42577), UAS-Fur2 RNAi 25959 (BDSC 25959), and P{hs-hid} (BDSC 7757).
METHOD DETAILS
Immunofluorescence and antibodies
Third instar larval (L3) or adult (at 0 DPE) CNS were rapidly dissected in PBS and fixed in 4% paraformaldehyde in PBS or PTX (1X PBS; 0.1% Triton X-100). Fixed brains were washed and permeabilized in PBT (1X PBS, 0.1% Triton X-100, 1% BSA) and incubated in primary antibody overnight at 4°C. After incubation in primary antibody, samples were washed 3× 10 min in PBT and incubated in secondary antibody for 2 hr at room temperature (RT) while nutating. Following incubation with secondary antibody, samples were washed 3× 10 min in PTX and mounted in either 60% glycerol (larval CNS) or Vectashield (Vector labs; adult brains). The following primary antibodies were used: rabbit anti-cleaved Drosophila Dcp-1 (Asp216) (Cell Signaling Technology; at 1:300), chicken anti-GFP (Abcam; at 1:600), rat anti-mCherry (Thermo Fisher; at 1:500), mouse anti-repo (DSHB; at 1:10), rabbit anti-beta Galactosidase antibody (ICL lab; at 1:1000), guinea pig anti-FoxO (Nechipurenko and Broihier, 2012; at 1:20), guinea pig anti-Dbx (gift from J. Skeath; at 1:1000), guinea pig anti-Hb9 (Broihier and Skeath, 2002; at 1:1000), rabbit anti-Even skipped (gift from J. Skeath; at 1:1000), and mouse anti-Draper (5D14; DSHB; at 1:300). Alexa-conjugated species-specific secondary antibodies were obtained from Thermo Fisher and used at a 1:300.
Image acquisition
Fluorescent 16-bit images were acquired on either an inverted Leica TCS SP8 (Leica Biosystems) using Application Suite X software (Leica Biosystems) or an upright Zeiss LSM 800 (Carl Zeiss) using Zen software (Carl Zeiss). The following objectives were used on the Zeiss LSM 800 unless otherwise noted: 20x Plan-Apochromat (0.8 NA) air objective, 20x HC PL APO (0.75 NA) air objective (used on Leica SP8), 40x Fluar (1.3 NA) oil immersion objective, and 63x W N-Achroplan (0.9 NA) dipping objective. Adult degeneration was imaged using an Axioplan 2 upright microscope (Carl Zeiss) using a 20x objective. Brains imaged on the Axioplan 2 were acquired at RT using AxioVision microscopy software. 3D volume renderings were generated from raw confocal images using Imaris image analysis software (Bitplane, Switzerland).
Dcp-1 quantification
Dcp-1 positive particles were quantified using the cell counter function of Fiji (National Institutes of Health). We quantified the total number of Dcp-1 particles within both lobes of the L3 brain and all segments of the L3 VNC.
Draper quantification
Control and mutant genotypes were uniformly processed and imaged using identical acquisition settings. Complete z-stacks were acquired with optimized confocal settings to ensure that oversaturation did not occur. Average intensity was quantified from 3 μm thick z-projections that began with the first appearance of the neuropil in the brain using Fiji (National Institutes of Health). For loss-of-function and cortex glia knockdown experiments, an ROI was defined by GFP labeling cortex glia expressing UAS-mCD8::GFP. For analysis of neuronal knockdown of Spz ligands and furins, we only analyzed Draper intensity around the neuropil that in an area where the staining resembled cortex glial processes. ROI area did not seem to differ in experimental and control groups. No modifications to any images were made before quantification. When possible, quantification was performed blinded to genotype.
qRT-PCR
Between 25–50 L3 CNS were dissected, tissue was homogenized in TRIzol (Thermo Fisher Scientific), and RNA was isolated using a chloroform extraction. cDNA was prepared using SuperScript III First Strand Synthesis SuperMix (Thermo Fisher Scientific), and qPCR was performed for 40 cycles with the StepOne Plus Real Time PCR system (Thermo Fisher Scientific). All TaqMan probes (Dm02134593_g1, Dm01832226_g1, and Dm01832224_g1) and reagents were purchased from the Life Technologies database. CT values of experimental genes were normalized to CT values for the housekeeping gene RpII140. Relative gene expression was determined using the ΔΔCT method. Statistical analyses were performed on ΔCT values. We performed 4–7 biological replicates per target gene and 2–3 technical replicates per biological replicate. TURBO DNA-free kit (Thermo Fisher Scientific) was used to eliminate genomic DNA contamination from samples.
Annexin V labeling & quantification
L3 expressing UAS-mCD8::GFP in cortex glia were rapidly dissected and pinned into fillets to expose the CNS on individual Sil-Guard plates at RT. Larvae were then washed 3× 3 min in 1X PBS and then incubated in Annexin V conjugated to Alexa-594 (Thermo Fisher Scientific) for 30 min at 25°C. Annexin V was used at a 1:4 dilution in 1X Annexin Binding Buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2; pH 7.4). Following incubation, larvae were washed 3× 5’ in 1X PBS and live-imaging was performed in 1X PBS using an upright Zeiss LSM 800 (Carl Zeiss).
Z-stacks were acquired for all experiments and the total number of Annexin V particles/5 μm was calculated by quantifying the total number of Annexin V particles within one brain lobe in 10 consecutive, in focus, sections.
The proportion of engulfed Annexin V particles was calculated by counting the number of Annexin V particles within cortex glial membranes and dividing that number by the total number of Annexin V particles within the cortex region. Since some of the animals moved during imaging, we quantified engulfed Annexin V particles in one central, in focus z section that displayed bright Annexin V and GFP labeling. Quantification was performed blinded to genotype.
Dextran labeling
2.5 mM 10 kDa Dextran-568 (Thermo Fisher Scientific) was diluted 1:100 and applied to dissected L3 filets for 30 min at 25°C. Brain lobes were imaged following 3× 5 min washes in PBS.
Rab7-YFP Quantification
Z stacks were acquired and the following metrics were measured using Fiji (National Insititutes of Heath). The proportion of Rab7-YFP donuts within cortex glial membranes (labeled with Ctxglia-split>mCD8::Cherry), and the proportion of Rab7-YFP donuts surrounding either Elav, Dcp-1, or Repo was quantified in serial z sections that encompassed 6 μm total. The total number of Rab7-YFP donuts within cortex glial membranes of each genotype was quantified in serial z sections encompassing 7.5 μm. Quantification was performed blinded to genotype.
Cycloheximide treatment & quantification
L3s were rapidly dissected into fillets and washed in PBS as described above. Dissected larvae were treated with 10 mM (for Annexin V experiments) or 50 mM (for Spz5 cleavage experiments) cycloheximide (CHX) to induce apoptosis. CHX was diluted in 1X PBS and larvae were incubated in CHX for 1 hr at 25°C. For Annexin V labeling: Larvae were washed 3× 5 min in 1X PBS following CHX treatment and subsequently labeled with Annexin V and imaged as described above. The number of Annexin V positive particles was quantified using methods described above. Control larvae were incubated in PBS using an identical treatment paradigm.
Furin cleavage site prediction
Potential Furin cleavage sites in Spz5 were identified using the ProP 1.0 prediction tool (Duckert et al., 2004).
Drosophila protein extracts and immunoblots
CNS lysates were prepared from ~12 wandering L3 larvae by extracting and immediately homogenizing the CNS in 2x Laemmli sample buffer (Bio-Rad Laboratories). Lysates were subsequently heated for 5 min at 95°C. Approximately 12 CNS equivalents/lane were loaded onto 4–20% SD-PAGE gels (Bio-Rad Laboratories). Protein was transferred to IR-compatible PVDF membranes. Immunoblots were probed with REVERT total protein stain (LI-COR Biosciences) and imaged using the Odyssey imaging system (LI-COR Biosciences). Membranes were blocked in 1:1 TBS Odyssey blocking buffer: TBS (LI-COR Biosciences) for 1 hour at RT and incubated overnight at 4°C in primary antibody. Membranes were washed in 4 × 5 min in TBST and incubated in 1:5000 of the appropriate IR dye conjugated secondary antibody in blocking buffer for 1–1.5 hours at RT. Membranes were imaged using the Odyssey imaging system (LI-COR Biosciences).
Spz5-HA was quantified using ImageStudio Lite Software (LI-COR Biosciences). Furin-cleaved Spz5-HA (~20 kDa) was measured and normalized to total HA protein (all other Spz5-HA bands). Total HA protein was measured by determining the sum of the values of the detectable Spz5-HA bands. Some cleavage bands (those between 25–30 kDa) were weakly and inconsistently detectable, but when detected were quantified within total Spz5-HA measurements.
For HS-Hid experiments, L3 were placed in a vial of fresh molasses food and heat shocked at 37°C for 75 minutes in a water bath. Con trols were kept at 25°C.
Lifespan analysis
Between 10–11 vials of 15–30 flies per genotype were collected at the day of eclosion and kept at 25°C. Flies were transferred to fresh vials every 2–3 days. The number of surviving flies was recorded daily.
Histology of adult brains
Histology on adult brains was performed similar to Cao et al., 2013. In brief, flies were collected upon eclosion and aged at 29°C. Fly heads were severed and placed into a metal collar for sectioning. Heads were fixed in Carnoy’s fixative (60% ethanol, 30% chloroform, 10% acetic acid) overnight at 4°C. Following fixation, heads were washed and placed in 70% ethanol 2× 10 min followed by 100% ethanol 2× 10 min, and then processed into paraffin using standard histological procedures. Embedded heads were sectioned at 5 μm and stained with a hematoxylin and eosin (H & E) stain.
Scoring of neurodegeneration
Sequential sections of adult brains labeled with H & E were imaged and quantified. Neurodegeneration is indicated by vacuolar lesions in the adult brain and five levels of neurodegeneration (0, 1, 2, 3, and 4) were defined for quantification (Fig. S6 E). For most genotypes, 9–16 brains were scored. Scoring of brains was performed blinded to genotype.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were preformed and graphs were generated using Prism 6 (GraphPad Software). In all bar graphs and scatter dot plots, error bars are presented as mean ± SEM. In all box and whisker plots, the whiskers represent minimum and maximum data points, and the line within the box depicts median values. All pairwise sample comparisons were performed using a Mann-Whitney test. A Kruskal-Wallis test was used to compare each sample with other samples in a group of three or more, and a Dunn’s multiple comparison test was subsequently performed. A two-way ANOVA followed by a Dunnetts’s multiple comparisons test was performed on adult degeneration data only (Figure 7 K). In all figures, p-values for statistical tests are as follows: n.s., not significant; *, P< 0.05; **, P<0.01; ***, P<0.001. n values can be found on each graph. Statistical details can be found within Tables S1, S3–7.
Supplementary Material
Highlights.
Dying neurons signal to phagocytic glia via a Toll receptor pathway during development
Dying neurons activate Furin-dependent processing of the Toll-6 ligand, Spz5
Cortex glia sense cleaved Spz5 via Toll-6-dSARM signaling
Toll-6 signaling drives transcriptional upregulation of the Draper engulfment receptor
Acknowledgements
We thank Marc Freeman, Mary Logan, and Jim Skeath for reagents. We thank Kelsey Herrmann for experimental assistance and for comments on the manuscript. We are grateful to Tiffany Kearse for sectioning and imaging adult brains. We thank Rich Lee, Jay Myers and Alex Huang for use of Imaris software and assistance with 3D volume renderings. We thank Nan Liu and Jim Sears for technical assistance. We appreciate Pola Philippidou’s comments on the manuscript. We also appreciate helpful discussions with members of the Broihier and Landreth labs. We thank the Vienna Drosophila Resource Center, Bloomington Drosophila Stock Center, and the Kyoto DGRC for fly stocks. We thank Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at the University of Iowa for antibodies. Adult brains were sectioned and processed using the Tissue Resources Core Facility of the Comprehensive Cancer Center of CWRU and University Hospitals of Cleveland (P30 CA43703). The Leica TCS SP8 was obtained via NIH Office of Research Infrastructure Shared Instrumentation Grant (S10OD016164). This work was supported by National Institutes of Health grants R21NS090369 and R01NS095895 to H.T. Broihier.
Footnotes
Declaration of Interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akagawa H, Hara Y, Togane Y, Iwabuchi K, Hiraoka T, Tsujimura H, 2015. The role of the effector caspases drICE and dcp-1 for cell death and corpse clearance in the developing optic lobe in Drosophila. Dev. Biol 404, 61–75. [DOI] [PubMed] [Google Scholar]
- Awasaki T, Lai S-L, Ito K, Lee T, 2008. Organization and Postembryonic Development of Glial Cells in the Adult Central Brain of Drosophila. J. Neurosci 28, 13742–13753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R, 2004. Regulation of phagosome maturation bu signals from toll-like receptors. Science (80-. ). 304, 1014–1018. [DOI] [PubMed] [Google Scholar]
- Bolkan BJ, Booker R, Goldberg ML, Barbash DA, 2007. Developmental and cell cycle progression defects in Drosophila hybrid males. Genetics 177, 2233–2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguillos MA, Svensson M, Schulte T, Boza-Serrano A, Garcia-Quintanilla A, Kavanagh E, Santiago M, Viceconte N, Oliva-Martin MJ, Osman AM, et al. , 2015. Microglia-secreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 10, 1626–1638. [DOI] [PubMed] [Google Scholar]
- Cao Y, Chtarbanova S, Petersen AJ, Ganetzky B, 2013. Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc. Natl. Acad. Sci. 110, E1752–E1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, et al. , 2013. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church JS, Milich LM, Lerch JK, Popovich PG, McTigue DM, 2017. E6020, a synthetic TLR4 agonist, accelerates myelin debris clearance, Schwann cell infiltration, and remyelination in the rat spinal cord. Glia 65, 883–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho-Budd JC, Sheehan AE, Freeman MR, 2017. The secreted neurotrophin spätzle 3 promotes glial morphogenesis and supports neuronal survival and function. Genes Dev. 31, 2023–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Sheehan AE, Bradshaw R, Fox AN, Lu TY, Freeman MR, 2014. PI3K Signaling and Stat92E Converge to Modulate Glial Responsiveness to Axonal Injury. PLoS Biol. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SE, O’Connell RM, Miranda GA, Vaidya SA, Chow EK, Liu PT, Suzuki S, Suzuki N, Modlin RL, Yeh W-C, et al. , 2004. Toll-like Receptors Induce a Phagocytic Gene Program through p38. J. Exp. Med 199, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper I, Mahoney LJ, Mitsuhashi S, Pacak CA, Salomon RN, Kangy PB, 2014. Silencing of DRPR leads to muscle and brain degeneration in adult drosophila. Am. J. Pathol 184, 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckert P, Brunak S, Blom N, 2004. Prediction of proprotein convertase cleavage sites. Protein Eng. Des. Sel 17, 107–112. [DOI] [PubMed] [Google Scholar]
- Dunst S, Kazimiers T, von Zadow F, Jambor H, Sagner A, Brankatschk B, Mahmoud A, Spannl S, Tomancak P, Eaton S, Brankatschk M, 2015. Endogenously Tagged Rab Proteins: A Resource to Study Membrane Trafficking in Drosophila. Dev. Cell 33, 351–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, et al. , 2009. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Ravichandran KS, 2016. The Dynamics of Apoptotic Cell Clearance. Dev. Cell 38, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etchegaray JI, Elguero EJ, Tran JA, Sinatra V, Feany MB, McCall K, 2016. Defective Phagocytic Corpse Processing Results in Neurodegeneration and Can Be Rescued by TORC1 Activation. J. Neurosci 36, 3170–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fergestad T, Ganetzky B, Palladino MJ, 2006. Neuropathology in Drosophila membrane excitability mutants. Genetics 172, 1031–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannagan RS, Jaumouillé V, Grinstein S, 2012. The Cell Biology of Phagocytosis. Annu. Rev. Pathol. Mech. Dis 7, 61–98. [DOI] [PubMed] [Google Scholar]
- Foldi I, Anthoney N, Harrison N, Gangloff M, Verstak B, Nallasivan MP, AlAhmed S, Zhu B, Phizacklea M, Losada-Perez M, et al. , 2017. Three-tier regulation of cell number plasticity by neurotrophins and Tolls in Drosophila. J. Cell Biol 216, 1421–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fourgeaud L, Traves PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, Callaway P, Zagorska A, Rothlin CV, Nimmerjahn A, Lemke G, 2016. TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, 2015. Drosophila central nervous system glia. Cold Spring Harb. Perspect. Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR, Delrow J, Kim J, Johnson E, Doe CQ, 2003. Unwrapping glial biology: Gcm target genes regulating glial development, diversification, and function. Neuron 38, 567–580. [DOI] [PubMed] [Google Scholar]
- Gay NJ, Gangloff M, 2007. Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem 76Gay, N., 141–165. [DOI] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Sasaki Y, Diantonio A, Milbrandt J, 2013. Sarm1Mediated Axon Degeneration Requires Both SAM and TIR Interactions. J. Neurosci 33, 13569–13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke ML, Kielian T, 2011. Toll-like receptors in health and disease in the brain: mechanisms and therapeutic potential. Clin. Sci 121, 367–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay BA, Wolff T, Rubin GM, 1994. Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121–2129. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Ito K, Sado Y, Taniguchi M, Akimoto A, Takeuchi H, Aigaki T, Matsuzaki F, Nakagoshi H, Tanimura T, et al. , 2002. GETDB, a database compiling expression patterns and molecular locations of a collection of gal4 enhancer traps. Genesis 34, 58–61. [DOI] [PubMed] [Google Scholar]
- Hilu-Dadia R, Hakim-Mishnaevski K, Levy-Adam F, Kurant E, 2018. Draper-mediated JNK signaling is required for glial phagocytosis of apoptotic neurons during Drosophila metamorphosis. Glia 1–13. [DOI] [PubMed] [Google Scholar]
- Horng T, Medzhitov R, 2001. Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 98, 12654–12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwangbo DS, Gersham B, Tu M, Palmer M, 2004. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body 429, 562–567. [DOI] [PubMed] [Google Scholar]
- Ito K, Urban J, Technau GM, 1995. Distribution, classification, and development of Drosophila glial cells in the late embryonic and early larval ventral nerve cord. Roux’s Arch. Dev. Biol 204, 284–307. [DOI] [PubMed] [Google Scholar]
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, et al. , 2014. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci. Transl. Med 6. [DOI] [PubMed] [Google Scholar]
- Kochan T, Singla A, Tosi J, Kumar A, 2012. Toll-like receptor 2 ligand pretreatment attenuates retinal microglial inflammatory response but enhances phagocytic activity toward Staphylococcus aureus. Infect. Immun 80, 2076–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb JP, Martinez J, 2016. Bon EPOtit! S1P-Mediated EPO Signaling Whets a Macrophage’s Appetite for Apoptotic Cells. Immunity 44, 209–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurant E, 2011. Keeping the CNS clear: Glial phagocytic functions in Drosophila. Glia 59, 1304–1311. [DOI] [PubMed] [Google Scholar]
- Kurant E, Axelrod S, Leaman D, Gaul U, 2008. Six-Microns-Under Acts Upstream of Draper in the Glial Phagocytosis of Apoptotic Neurons. Cell 133, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannan E, Vandergaast R, Friesen PD, 2007. Baculovirus Caspase Inhibitors P49 and P35 Block Virus-Induced Apoptosis Downstream of Effector Caspase DrICE Activation in Drosophila melanogaster Cells. J. Virol 81, 9319–9330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BP, Jones BW, 2005. Transcriptional regulation of the Drosophila glial gene repo. Mech. Dev. 122, 849–862. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, et al. , 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay SA, Wasserman SA, 2014. Conventional and non-conventional Drosophila Toll signaling. Dev. Comp. Immunol 42, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan MA, 2017. Glial contributions to neuronal health and disease: new insights from Drosophila. Curr. Opin. Neurobiol 47, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan MA, Hackett R, Doherty J, Sheehan A, Speese SD, Freeman MR, 2012. Negative regulation of glial engulfment activity by Draper terminates glial responses to axon injury. Nat. Neurosci. 15, 722–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo B, Gan W, Liu Z, Shen Z, Wang J, Shi R, Liu Y, Liu Y, Jiang M, Zhang Z, Wu Y, 2016. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 44, 287–302. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, Freeman MR, 2006. The Drosophila Cell Corpse Engulfment Receptor Draper Mediates Glial Clearance of Severed Axons. Neuron 50, 869–881. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Doherty J, Hackett R, Freeman MR, 2013. The c-Jun kinase signaling cascade promotes glial engulfment activity through activation of draper and phagocytic function. Cell Death Differ 20, 1140–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIlroy G, Foldi I, Aurikko J, Wentzell JS, Lim MA, Fenton JC, Gay NJ, Hidalgo A, 2013. Toll-6 and Toll-7 function as neurotrophin receptors in the Drosophila melanogaster CNS. Nat. Neurosci 16, 1248–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin CN, Broihier HT, 2018. Keeping Neurons Young and Foxy: FoxOs Promote Neuronal Plasticity. Trends Genet 34, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin CN, Nechipurenko IV, Liu N, Broihier HT, 2016. A Toll receptor-FoxO pathway represses Pavarotti/MKLP1 to promote microtubule dynamics in motoneurons. J. Cell Biol 214, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musashe DT, Purice MD, Speese SD, Doherty J, Logan MA, 2016. Insulin-like Signaling Promotes Glial Phagocytic Clearance of Degenerating Axons through Regulation of Draper. Cell Rep 16, 1838–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechipurenko IV, Broihier HT, 2012. FoxO limits microtubule stability and is itself negatively regulated by microtubule disruption. J. Cell Biol 196, 345–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Kotter MR, Franklin RJM, 2009. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 132, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, Bowie AG, 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol 7, 353–364. [DOI] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox N, Adalbert R, Powell EH, Sheehan AE, Avery M, Hackett R, Logan M, et al. , 2012. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337, 481–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Holmes C, 2014. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol 10, 217–224. [DOI] [PubMed] [Google Scholar]
- Potter CJ, Tasic B, Russler EV, Liang L, Luo L, 2010. The Q system: A repressible binary system for transgene expression, lineage tracing, and mosaic analysis. Cell 141, 536–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajbhandari L, Tegenge MA, Shrestha S, Ganesh Kumar N, Malik A, Mithal A, Hosmane S, Venkatesan A, 2014. Toll-like receptor 4 deficiency impairs microglial phagocytosis of degenerating axons. Glia 62, 1982–1991. [DOI] [PubMed] [Google Scholar]
- Ravichandran KS, 2010. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med 207, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, Landreth GE, 2009. CD14 and Toll-Like Receptors 2 and 4 Are Required for Fibrillar A -Stimulated Microglial Activation. J. Neurosci 29, 11982–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes S, Ebert S, Regen T, Agarwal A, Tauber SC, Czesnik D, Spreer A, Bunkowski S, Eiffert H, Hanisch UK, Hammerschmidt S, Nau R, 2010. Toll-like receptor stimulation enhances phagocytosis and intracellular killing of nonencapsulated and encapsulated Streptococcus pneumoniae by murine microglia. Infect. Immun 78, 865–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogulja-Ortmann A, Luer K, Seibert J, Rickert C, Technau GM, 2007. Programmed cell death in the embryonic central nervous system of Drosophila melanogaster. Development 134, 105–116. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B, 2012. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K, Nagata S, 2015. An Apoptotic “Eat Me” Signal: Phosphatidylserine Exposure. Trends Cell Biol. 25, 639–650. [DOI] [PubMed] [Google Scholar]
- Shklover J, Mishnaevski K, Levy-Adam F, Kurant E, 2015. JNK pathway activation is able to synchronize neuronal death and glial phagocytosis in Drosophila. Cell Death Dis 6, e1649–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shklyar B, Sellman Y, Shklover J, Mishnaevski K, Levy-Adam F, Kurant E, 2014. Developmental regulation of glial cell phagocytic function during Drosophila embryogenesis. Dev. Biol 393, 255–269. [DOI] [PubMed] [Google Scholar]
- Sierra A, Abiega O, Shahraz A, Neumann H, 2013. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front. Cell. Neurosci 7, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowski JD, Mandell JW, 2011. Phagocytic clearance in neurodegeneration. Am. J. Pathol 178, 1416–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stork T, Engelen D, Krudewig A, Silies M, Bainton RJ, Klambt C, 2008. Organization and Function of the Blood Brain Barrier in Drosophila. J. Neurosci. 28, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Xu P, Salvaterra PM, 1999. Dynamic visualization of nervous system in live Drosophila. Proc. Natl. Acad. Sci. U. S. A. 96, 10438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdemir-Yilmaz OE, Freeman MR, 2014. Astrocytes engage unique molecular programs to engulf pruned neuronal debris from distinct subsets of neurons. Genes Dev. 28, 20–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauszig-Delamasure S, Bilak H, Capovilla M, Hoffmann J. a, Imler J-L, 2002. Drosophila MyD88 is required for the response to fungal and Gram-positive bacterial infections. Nat. Immunol 3, 91–97. [DOI] [PubMed] [Google Scholar]
- Tung TT, Nagaosa K, Fujita Y, Kita A, Mori H, Okada R, Nonaka S, Nakanishi Y, 2013. Phosphatidylserine recognition and induction of apoptotic cell clearance by Drosophila engulfment receptor Draper. J. Biochem 153, 483–491. [DOI] [PubMed] [Google Scholar]
- Valanne S, Wang J-H, Rämet M, 2011. The Drosophila Toll signaling pathway. J. Immunol 186, 649–656. [DOI] [PubMed] [Google Scholar]
- Venken KJT, Simpson JH, Bellen HJ, 2011. Genetic manipulation of genes and cells in the nervous system of the fruit fly. Neuron 72, 202–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C, 1995. A novel assay for apoptosis Flow cytometric detection of phosphatidylserine early apoptotic cells using fluorescein labelled expression on Annexin V. ournal Immunol. Methods 184, 39–51. [DOI] [PubMed] [Google Scholar]
- Ward A, Hong W, Favaloro V, Luo L, 2015. Article Toll Receptors Instruct Axon and Dendrite Targeting and Participate in Synaptic Partner Matching in a Drosophila Olfactory Circuit. Neuron 85, 1013–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B, 2015. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 36, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi Y, Nishida Y, Ip YT, 2010. Functional analysis of Toll-related genes in Drosophila. Dev. Growth Differ. 52, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo W, Gautier J, 2004. Early neural cell death: Dying to become neurons. Dev Biol 274, 233–244. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, Piña-Crespo JC, Zhang M,Z, et al. , 2018. TREM2 Is a Receptor for β-Amyloid that Mediates Microglial Function. Neuron 97, 1023–1031.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.