

Abstract
IgG4-related disease is a fibro-inflammatory condition that can affect nearly any organ system. Common presentations include major salivary and lacrimal gland enlargement, orbital disease, autoimmune pancreatitis, retroperitoneal fibrosis and tubulointerstitial nephritis. This review focuses on the hematologic manifestations of IgG4-related disease, including lymphadenopathy, eosinophilia, and polyclonal hypergammaglobulinemia. The disease can easily be missed by unsuspecting hematologists, as patients may present with clinical problems that mimic disorders such as multicentric Castleman disease, lymphoma, plasma cell neoplasms and hypereosinophilic syndromes. When IgG4-related disease is suspected, serum protein electrophoresis and IgG subclasses are helpful as initial tests but a firm histological diagnosis is essential both to confirm the diagnosis and to rule out mimickers. The central histopathological features are a dense, polyclonal, lymphoplasmacytic infiltrate enriched with IgG4-positive plasma cells (with an IgG4/IgG ratio >40%), storiform fibrosis, and obliterative phlebitis. Importantly for hematologists, the latter two features are seen in all tissues except bone marrow and lymph nodes, making these two sites suboptimal for histological confirmation. Many patients follow an indolent course and respond well to treatment, but a significant proportion may have highly morbid or fatal complications such as periaortitis, severe retroperitoneal fibrosis or pachymeningitis. Corticosteroids are effective but cause new or worsening diabetes in about 40% of patients. Initial response rates to rituximab are high but durable remissions are rare. More intensive lymphoma chemotherapy regimens may be required in rare cases of severe, refractory disease, and targeted therapy against plasmablasts, IgE and other disease biomarkers warrant further exploration.
Example case
An 80-year old Korean man was referred for evaluation of chronic lymphadenopathy, eosinophilia and polyclonal hypergammaglobulinemia. He had had abdominal pain since the 1970s and was initially thought to have Crohn disease, subsequently complicated by idiopathic common bile duct narrowing. In the 1990s, he was found to have a kidney mass on computed tomography which was suspected to be lymphoma, but after the mass was resected, the histology was in keeping with multifocal fibrosclerosis. At the time of referral, his physical examination revealed a low, firm, slightly enlarged, left submandibular gland, no lacrimal gland swelling, and multiple 2 cm or smaller soft inguinal lymph nodes bilaterally. His white blood cell count was 8.3×109/L, eosinophil count 2.0×109/L (normal values <0.7×109/L), creatinine concentration 140 μmol/L, total protein 87 g/L (normal values <82 g/L), with a polyclonal increase in gamma globulins on serum protein electrophoresis of 20.5 g/L (normal values <14 g/L), and total IgG of 28.9 g/L (normal values <18.5 g/dL).
Introduction
Immunoglobulin G4-related disease (IgG4-RD) is a chronic immune-mediated disease that may present with tumefactive lesions, fibrosis, and a polyclonal IgG4-positive (IgG4+) plasma cell-enriched infiltrate in nearly any anatomic site. In many centers, systemic therapy is guided by rheumatologists, yet nearly every medical, surgical and pathology specialty must be aware of this entity and its protean manifestations. Involvement of blood-forming and lymphoid organs, manifesting as lymphadenopathy, eosinophilia, and polyclonal hypergammaglobulinemia, is common and IgG4-RD often mimics other hematologic conditions such as multicentric Castleman disease, lymphoma, plasma cell neoplasms, and hypereosinophilic syndromes (HES). This review provides an overview of IgG4-RD with a focus on aspects most relevant to clinical hematology practice.
In the early 2000s, while searching for non-invasive bio-markers to distinguish sclerosing (autoimmune) pancreatitis from pancreatic cancer, Japanese investigators noticed a fast-moving band in the beta-gamma region of the serum protein electrophoresis of patients with sclerosing pancreatitis. This band represented markedly elevated serum IgG4.1 Subsequently, abundant polyclonal IgG4+ plasma cells were found within a lymphoplasmacytic infiltrate in tissue samples from patients with autoimmune pancreatitis and in surrounding tissues including the liver and gallbladder.2 Once this entity was recognized as a distinct disease with characteristic histological features, many historically “idiopathic” and eponymous disorders such as multifocal fibrosclerosis (mediastinal and retroperitoneal fibrosis), Kuttner tumor (chronic sclerosing sialadenitis) and Reidel thyroiditis (woody infiltration of the thyroid) were found to be part of the IgG4-RD spectrum.3 Numerous names were proposed in the early days of its discovery, including “IgG4-related sclerosing disease”, “IgG4-related systemic disease”, “IgG4-related multi-organ lymphoproliferative syndrome” and “systemic IgG4-related plasmacytic syndrome”. An international group of investigators, primarily from Japan, the USA and Europe met in Boston in 2011 and agreed upon uniform nomenclature and diagnostic criteria.3 The accepted name “IgG4-related disease” reflects the universality of the IgG4+ plasma cell infiltration in involved organs as well as the frequency of elevation in serum IgG4 rather than a pathogenic role for IgG4 per se.4 The wide variety of disease manifestations stems not only from multi-organ involvement, but also from the fact that different organs may be involved in a metachronous fashion. Common presentations are major salivary (parotid and submandibular) and lacrimal gland enlargement (Mickulicz disease), lymphadenopathy, orbital pseudotumor, pancreatitis, sclerosing cholangitis, retroperitoneal fibrosis and tubulointerstitial nephritis.5
Epidemiology and pathophysiology
Underrecognition has hindered accurate estimates of the epidemiological burden of this disease, but a starting point is the prevalence of autoimmune pancreatitis in Japan, which increased from 2.2/100 000 in 2007 to 4.6/100 000 in 2011. The increase is almost certainly due to increased recognition, and given that pancreatic involvement is present in about 20-25% of IgG4-RD cases, the true prevalence of the disease is likely much higher. There is a 2:1 male preponderance and the median age of affected patients at diagnosis is in the sixth to seventh decade of life. Aside from one case report of identical twins with IgG4-RD,6 evidence of genetic susceptibility is sparse. Pediatric cases are rare, but a recent review identified 25 cases in children, of whom 11 had orbital disease and three had autoimmune pancreatitis.7
At first glance, the presence of IgG4 in serum and IgG4+ plasma cells in tissues, increased IgG4+ plasmablasts in serum, and responsiveness to rituximab suggest that B-cell activation drives the disease.8 However, the IgG4 antibody itself is not thought to be pathogenic since it does not bind complement, does not traditionally form immune complexes, and patients with other conditions with markedly elevated serum IgG4, such as IgG4 myeloma, do not develop features of IgG4-RD.9,10 Recent studies have demonstrated that an unconventional population of CD4+SLAMF7+ cytotoxic T lymphocytes is central to the pathogenesis of the disease.11 Histopathologically, polyclonal B cells are found in clusters near these CD4+ T cells, the latter of which are among the most abundant cells within affected tissues. Oligoclonal expansion of these CD4+ cytotoxic T lymphocytes in peripheral blood may explain the high rates of T-cell clonality positivity determined by polymerase chain reaction analysis.12 These CD4+ T cells produce profibrotic cytokines such as interleukin-1, transforming growth factor-beta and interferon-gamma, as well as cytolytic molecules such as granzyme A and B and perforin.13 These cytotoxic T lymphocytes are likely sustained by continuous antigen presentation by B cells and plasmablasts. The responsible autoantigen(s) have not been definitively identified, but annexin A11 and galactin-3 have both recently been implicated in IgG4-RD.14,15 The autoantibody response to galactin-3 is primarily of the IgG4 and IgE isotypes, which correlates with the typical immunoglobulin responses seen in IgG4-RD.
Clinical presentation
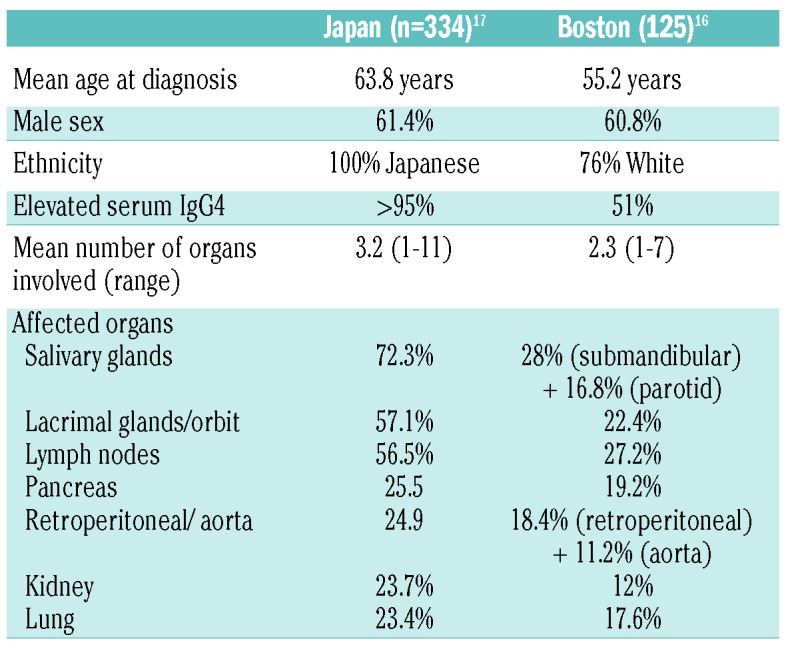
IgG4-RD can affect nearly any organ except synovial tissue. This “fibro-inflammatory” disease presents with tumefactive (puffy) inflammatory infiltrates and fibrosis with a predilection for glandular tissue. Figure 1 illustrates the manifestations of IgG4-RD by organ system. The most common organ manifestations from two large cohorts, one Japanese and one American, are shown in Table 1.16,17 In patients presenting with the better-known features of IgG4-RD, such as autoimmune pancreatitis, orbital disease and major salivary gland involvement, the disease tends to be recognized and histologically confirmed earlier, but patients referred to hematologists may present with less obvious features of IgG4-RD, and a high index of suspicion is needed to arrive at an accurate diagnosis. Common reasons for referral to a hematologist include lymphadenopathy, eosinophilia, and polyclonal hypergammaglobulinemia.
Figure 1.

Manifestations of IgG4-related disease by organ system. The most common primary disease features are indicated in bold.
Table 1.
Clinical characteristics of patients from two large published cohorts.
Lymphadenopathy
IgG4-related lymphadenopathy is one of the three most common manifestations of IgG4-RD, affecting 30-60% of patients with IgG4-RD in most large cohorts (Table 1).5,16,17 IgG4-lymphadenompathy may be generalized or localized (the latter typically contiguous with involved organs such as pancreas and lungs). Parallel enlargement of salivary, lacrimal and parotid glands is common. Five morphological subtypes, all of which display increased IgG4+ plasma cells, have been described (see Figure 2 for examples):18–20
Multicentric Castleman disease-like: preserved nodal architecture with patent sinusoids and hyperplastic follicles; abundant mature plasma cells in interfollicular areas with some eosinophils, similar to lymphadenopathy in multicentric Castleman disease or autoimmune disease.
Reactive follicular hyperplasia: increased IgG4+ plasma cells in germinal centers and often in the interfollicular zones with some eosinophils present.
Interfollicular expansion pattern: expanded interfollicular zones with small lymphocytes, plasmablasts and mature plasma cells and eosinophils which may resemble lymphoma (e.g. angioimmunoblastic lymphoma). Examples are given in Figure 2A–C.
Progressive transformation of germinal center-like: scattered larger or transformed follicles containing plasma cells. An example is given in Figure 2D,E.
Inflammatory pseudotumor-like: lymph node partially effaced by a fibro-inflammatory infiltrate and storiform fibrosis; this subtype is considered most specific for IgG4-RD in lymph nodes. An example is given in Figure 2F.
Figure 2.

Lymph nodes in IgG4-related disease. (A,B) An example of the interfollicular pattern of IgG4-related lymphadenopathy, with mature plasma cells, many expressing IgG4, distributed between benign follicles. (A) Hematoxylin and eosin stain. (B) IgG4 immunohistochemistry. (C) A needle core lymph node biopsy from a different case with the interfollicular pattern (hematoxylin and eosin stain). (D,E) A case of IgG4-lymphadenoapthy with a progressive transformation of the follicular center pattern, with the plasma cells within the follicle proper. (D) Hematoxylin and eosin stain. (E) IgG4 immunohistochemistry. (F) An example of a mass-like lesion (inflammatory pseudotumor) with dense fibrosis and associated follicular hyperplasia in a case of IgG4-lymphadenoapthy (hematoxylin and eosin).
IgG4-related lymphadenopathy has aptly been called both “an underdiagnosed and overdiagnosed entity”:19 underdiagnosed because if it is not included in the differential diagnosis, IgG4 and IgG stains may not be done and the disease may be missed, and overdiagnosed because increased IgG4+ plasma cells may be seen in conditions ranging from Rosai-Dorfman-Destombes disease to inflammatory vasculitis.21,22 Although not the optimal tissue for making the histological diagnosis of IgG4-RD, in a patient with typical clinical features such as autoimmune pancreatitis or retroperitoneal fibrosis, a lymph node biopsy may be sufficient for diagnosis if biopsy of other affected organs is not feasible. Given the low specificity of increased IgG4+ plasma cells in lymph node and the variable histological patterns, the greatest utility of lymph node biopsy is perhaps excluding other diagnoses, such as lymphoma and HHV8-associated Castleman disease. The role of lymph node biopsy is further discussed in the section on “Diagnosis and staging”.
Eosinophilia
Approximately 40% of patients with IgG4-RD have peripheral blood eosinophilia, often accompanied by asthma and atopy.23 Thus, IgG4-RD is an important and underappreciated cause of reactive or secondary eosinophilia.12 HES and IgG4-RD commonly involve the skin, lungs, gas trointestinal tract, and lymph nodes.12 Idiopathic HES and hypereosinophilia of unknown significance are diagnoses of exclusion, and account for a substantial proportion (30-50%) of diagnoses of patients evaluated for eosinophilia.24–26 Evaluating these patients for IgG4-RD is an important and underappreciated aspect of their care. In fact, we previously published a case report with a diagnostic label of idiopathic HES, reviewed by several world experts in eosinophilia who concurred with the diagnosis, which was subsequently found to be IgG4-RD.27,28 Findings of a myeloid clonal disorder such as increased blast cells, abnormal karyotype, mutations in PDGFR-alpha/beta, FGFR-1 and PCM1-JAK2 are not seen in IgG4-RD. However, differentiating lymphocytic-variant HES from IgG4-RD can be more challenging. The aberrant T-cell phenotypes found in lymphocytic-variant HES, including increased CD4+CD3−, CD3+/CD4−/CD8− and CD4+/CD7− T cells, with or without T-cell clonality as determined by polymerase chain reaction analysis, have all been reported in IgG4-RD.12,29 Increased IgG4 deposits have been found in tissue samples from adult and pediatric patients with eosinophilic esophagitis.30–33 In contrast to HES and chronic eosinophilic leukemia, the eosinophilia secondary to IgG4-RD is typically mild to moderate, rarely exceeding 5×109/L and typically quite evanescent, being ablated by steroids or rituximab therapy.
Polyclonal hypergammaglobulinemia
As for eosinophilia, IgG4-RD represents an important new diagnostic consideration in patients with hypergammaglobulinemia. An elevated serum IgG4 level, often accompanied by an increase in IgG1, causes polyclonal hypergammaglobulinemia. Rarely, this elevation can be sufficient to result in polyclonal hyperviscosity syndrome.27,28,34 It is not known what causes the exuberant production of IgG4 immunoglobulins, currently considered an epiphenomenon rather than a contributor to the pathogenesis of the disease.8 Not surprisingly, the serum free light chains tend to be abnormally elevated.35 IgE is often markedly increased, particularly in patients with eosinophilia and atopy, whereas IgA and IgM levels are normal or modestly elevated. Serum IgG4 typically runs in the fast gamma or beta-gamma region on the serum protein electrophoresis, and thus the typical electrophoretic profile of a patient with IgG4-RD demonstrates polyclonal hypergammaglobulinemia with beta-gamma bridging. This sometimes prominent pattern is dependent on IgG4 concentration and is highlighted in Figure 3. The hypergammaglobulinemia can be mistaken for a polyclonal increase in IgA, monoclonal gammopathy of undetermined significance or “biclonal” IgG kappa and lambda gammopathy, as laboratory physicians may not be familiar with the dense bands of IgG-lambda and kappa which in fact represent polyclonal IgG4.36,37 Some patients have even been treated for myeloma before subsequently being found to have IgG4-RD as the cause of their protein abnormalities, plasmacytosis and renal disease.9,38 Laboratory physicians must, therefore, consider the differential diagnosis of beta-gamma bridging and order or suggest additional investigations to clarify clonality and heavy chain composition where necessary.39,40
Figure 3.

Serum protein electrophoresis showing electrophoretic patterns for four patients with mild to gross elevations in IgG4 concentration in between two for patients with low IgG4 concentration. The physicochemical properties of the IgG4 heavy chain result in a relative anodal position (shift toward albumin) of the gamma globulins when the IgG4 becomes the predominant gamma globulin. Apart from IgG4, IgA immunoglobulins are frequently observed in the boundary between the beta and gamma regions. Monoclonal bands may also migrate in this region as shown in the gel (the monoclonal gammopathy in this case is an IgG1 monoclonal band that has physicochemical properties that are atypical for IgG1 immunoglobulins, which are normally found in a more cathodal position). NC: normal control, MG: monoclonal gammopathy.
Prior to the recognition of IgG4-RD, a large case series of 130 patients with polyclonal hypergammaglobulinemia >30 g/L on serum protein electrophoresis showed that the most common single diagnoses were liver disease (79/130, 66%), connective tissue disease (28/130, 22%), chronic infection (8/130, 6%) and hematologic disorders (7/130, 5%).41 In a recent, single-center study of 70 patients with polyclonal increases in IgG ≥20 g/L it was found that 14 (20%) had IgG4-RD as the cause of their hypergammaglobulinemia, indicating that a substantial proportion of patients with hypergammaglobulinemia have IgG4-RD as the underlying cause.42 The discovery of IgG4-RD has also led to increased recognition of other IgG subclass elevations with specific diseases, such as hepatitis C and mono - clonal gammopathy of undetermined significance with IgG1, hypothyroidism and irritable bowel syndrome with IgG2, rheumatoid arthritis with increased IgG3 and IgG1, and celiac disease with IgG4.43
IgG4 myeloma has been described but is rare; one large case series found that 6/158 bone marrow biopsies in myeloma patients expressed IgG4, in keeping with the relatively small fraction of overall circulating gamma globulins normally made up by the IgG4 subtype.10 One case report of IgG4 subtype POEMS has been reported.44 The bone marrow morphology may mimic myeloma with florid plasmacytosis,9,45 although in our experience, bone marrow examination is very insensitive for the diagnosis of IgG4-RD, with many cases showing no increase in plasma cells or lymphocytes despite florid hypergammaglobulinemia.
Important mimickers of IgG4-related disease
The diagnostic challenge of IgG4-RD for hematologists is heightened by overlap of clinical and laboratory features with those of a number of other hematologic diseases including lymphoma, plasma cell neoplasms, and histiocyte disorders (Table 2). In addition to the diseases discussed in this section and presented in Table 2, there are numerous non-hematologic mimickers reviewed in detail elsewhere.46 Careful review of histological specimens and correlation with clinical, laboratory and radiological findings are crucial for solidifying the correct diagnosis. Multicentric Castleman disease and IgG4-RD show considerable overlap given the high frequency of lymphadenopathy, IgG4+ plasma cell-enriched tissue infiltrates and elevated serum IgG4 levels seen in both diseases.18 However, IgG4-RD typically affects older patients, rarely exhibits the “hyper-interleukin-6” systemic inflammatory features of multicentric Castleman disease such as fever and elevated C-reactive protein, and the histological features are distinct. The histiocytic disorders Rosai-Dorfman-Destombes disease and Erdheim-Chester disease both cause inflammatory mass lesions that can mimic IgG4-RD. Histopathological evaluation of Rosai-Dorfman-Destombes disease can show enrichment of IgG4+ plasma cells,22,47 but typically in the context of CD68+ S100+ histiocytes, often associated with emperipolesis. The most recent classification of the histiocyte disorders recommends evaluating suspected cases of Rosai-Dorfman-Destombes disease for increased IgG4+ plasma cells,48 but in the absence of other evidence for a common pathophysiological link, Rosai-Dorfman-Destombes disease is not considered part of the spectrum of IgG4-RD or vice versa.49 One third of patients with Erdheim-Chester disease have retroperitoneal fibrosis, which may raise the suspicion of IgG4-RD; however, more than 95% of patients with Erdheim-Chester disease have skeletal involvement, which, aside from rare cases of IgG4-related angiocentric eosinophilic fibrosis (midline destructive lesions of the head and neck), is generally not seen in IgG4-RD.50 The histology of Erdheim-Chester disease shows a CD68+S100−CD1a− histiocyte infiltration often with “foamy histiocytes”.51 Extra-pulmonary sarcoidosis may share clinical features similar to those of IgG4-RD, including polyclonal hypergammaglobulinemia, lymphadenopathy, pulmonary nodules, sclerosing mesenteritis and pachymeningitis. The association between IgG4-RD and malignant lymphoma has been studied extensively. In Asian patients, mucosal-associated lymphoid tissue (MALT) lymphoma, particularly of ophthalmological tissues has been described, whereas in western populations a variety of histologies (diffuse large B-cell, follicular, lymphoplasmacytic, and MALT) have been reported.52,53 There are also case reports of IgG4-RD concomitant with autoimmune lymphoproliferative syndrome.54,55
Table 2.
Diseases that mimic hematologic manifestations of IgG4-RD (lymphadenopathy, eosinophilia and polyclonal hypergammaglobulinemia).

Case continued
The patient was suspected to have IgG4-RD, so serum IgG subclasses were analyzed. His serum IgG4 level was markedly elevated at 11.6 g/L (normal values <1.35). The tissue blocks from his previous nephrectomy were retrieved and pathology review showed a lymphoplasmacytic infiltrate, moderate tissue eosinophilia and interstitial fibrosis and atrophy. Staining for IgG4 and IgG revealed abundant IgG4+ plasma cells with >40 IgG4+ plasma cells per high power field and an IgG4/IgG ratio >40%. Computed tomography scans of the neck, chest, abdomen and pelvis revealed multiple pulmonary nodules, carinal lymphadenopathy and a soft tissue density encasing the main coronary arteries, previous right nephrectomy and pancreatic atrophy. His IgG4-RD Responder Index activity score was 12.
Diagnosis and staging
A careful history and thorough physical examination, attending to clues such as a history of waxing and waning glandular swelling, sicca symptoms and unexplained pancreatitis or jaundice must be accompanied by serum protein studies. Histological confirmation of the disease is essential, and once a diagnosis has been established, investigations to assess for symptomatic and subclinical organ involvement, such as early retroperitoneal fibrosis and albuminuria, are important for management planning. The IgG4-RD Responder Index is a standardized, validated tool for the evaluation of disease activity at initial evaluation and subsequent follow up.56,57 Suggested laboratory and imaging tests are summarized in Table 3.
Table 3.
Diagnostic and staging tests in IgG4-RD.

Serum protein studies
Approximately 70% of patients with IgG4-RD have an elevated serum IgG4 level. Serum IgG subclasses should be investigated in conjunction with serum protein electrophoresis to exclude a monoclonal paraprotein (Figure 3). Serum IgG4 level has a diagnostic sensitivity ranging from 83-97% and specificity from 60-85% with a general cut-off of “above the upper limit of normal”.58–60 Typically, 1.35 g/L is used as the biomarker cut-off for IgG4-RD (which corresponds to the upper limit of normal for one common commercial method but not another) without specifying the methodology. While mildly elevated serum IgG4 can be seen in many conditions, markedly elevated serum IgG4 (>5 g/L) is approximately 90% specific for IgG4-RD. Aside from methodological differences, serum IgG4 levels in IgG4-RD can vary greatly depending on ethnicity and degree of organ involvement. In the Boston cohort of patients (76% White), only 53 of 103 patients had elevated serum IgG4 levels.16 In contrast, in a cohort of 334 Japanese patients, more than 95% had elevated serum IgG4.17 In our multi-ethnic cohort, we found that Asians have a higher serum IgG4 than non-Asians (median 11.2 g/L versus 2.9 g/L, P=0.0094), and elevated serum IgG4 had a sensitivity of 96% in Asians compared to 67% in non-Asians.61 Patients with multi-organ involvement or of Asian ethnicity typically have elevated serum IgG4, sometimes markedly so, such as the patient in this illustrative case. The serum IgG4/IgG ratio is typically >0.2 in patients with IgG4-RD, although the ratio does not increase the diagnostic specificity of serum IgG4 alone. Flow cytometric detection of plasmablasts may offer a more sensitive modality for diagnosing IgG4-RD, with a reported sensitivity of 95% and specificity of 82% using a cut-off of 900/mL.62 However, the flow cytometry method used to detect plasmablasts is not widely available.
Most centers use immunonephelometry to measure IgG subclasses, which can cause some challenges with interpretation. The two most common immunonephelometric methods (Siemens and Binding Site) correlate well with regard to IgG4, but the absolute IgG4 values differ by approximately 50% at the upper limit of normal.63 IgG4 levels may also be markedly under-reported in cases of extreme IgG4 elevations due to the hook effect. The hook effect, or prozone phenomenon, occurs when an excessive amount of analyte prevents binding of the capture antibody in a sandwich assay, yielding a falsely low or normal result. Erroneously low measurements of serum IgG4 reported in the literature reflect this error.64 Furthermore, IgG4 itself interferes with the nephelometric measurement of IgG1 and IgG2, in particular, which can obscure the immunoglobulin profile that would otherwise highlight the disproportionate elevation of serum IgG4.65 Because of the traditional errors in immunonephelometry, some have mistakenly reported increased serum IgG2 levels as a marker of IgG4-RD.66–68 Our group has recently demonstrated that mass spectrometry is an alternative that eliminates these analytical errors and is more cost-effective than immunonephelometry.65
Histopathology
A firm diagnosis of IgG4-related disease requires histopathological confirmation, except in the case of autoimmune pancreatitis, in which radiological features (diffuse “sausage-like” enlargement of the pancreas with featureless borders and delayed enhancement with or without a capsule-like rim or “halo”) may be sufficiently specific to exclude requirement for tissue biopsy.3,69 As in sarcoidosis, in which non-caseating granulomas may be seen in any of the organs affected by the disease, IgG4-RD demonstrates common histology in most of the multitude of organs that may be affected.
The three major histological features of IgG4-RD in tissue are: (i) a dense, polyclonal lymphoplasmacytic infiltrate enriched with IgG4+ plasma cells; (ii) fibrosis; and (iii) obliterative phlebitis. With regards to the lymphoplasmacytic infiltrate, the number of IgG4+ plasma cells per high-power field (hpf) considered diagnostic varies according to tissue site, from >10/hpf in meninges to >100/hpf in skin. Regardless of the site, the ratio of IgG4+/IgG+ plasma cells is >40% in IgG4-RD. Fibrosis is a histological requirement for the diagnosis of IgG4-RD and should be arranged at least focally in a storiform pattern. Storiform fibrosis is a swirling, “cartwheel” pattern of fibrosis which may have a patchy distribution and can, therefore, be missed with small biopsies. In the obliterative phlebitis of IgG4-RD, venous channels are obliterated by an inflammatory lymphoplasmacytic infiltrate. Expert pathologists recommend looking for arteries/arterioles where the accompanying venous vessel is not readily apparent and may in fact have been replaced by an inflammatory infiltrate; elastin stains may be helpful in identifying completely obliterated vessels.
Other histopathological features include phlebitis without obliteration of the lumen and increased number of eosinophils. As in the illustrative case, archival specimens may be used to confirm a diagnosis, and many patients will have previous biopsies available due to their chronic disease course. As long as a tissue block is still available, IgG4 and IgG stains can be done. When a new biopsy is required, excisional specimens are preferred over core needle biopsies to allow for proper assessment. There are currently no established criteria for the cytological diagnosis of IgG4-RD from a fine needle aspirate.
Increased numbers of IgG4+ plasma cells are seen in all tissues affected by IgG4-RD, but this is not, in itself, a specific finding. Many chronic inflammatory conditions such as vasculitis, inflammatory bowel disease and lymphoma may exhibit increased numbers of IgG4+ plasma cells but do not share the other histological features of storiform fibrosis, obliterative phlebitis and absence of granulomatous inflammation.19,21 Unfortunately, for the purposes of hematologists, bone marrow involvement is uncommon in IgG4-RD and obliterative phlebitis and storiform fibrosis (not to be confused with myelofibrosis) are not typically seen in bone marrow and lymph nodes.70 Furthermore, even when involved, lymph nodes and bone marrow may not show robust elevation in IgG4-expressing plasma cells as compared to the total IgG population, or the findings may be only focal. Patterns of IgG4-RD have not been well established in the bone marrow, but the presence of mature plasma cells would be supportive of marrow involvement, once plasma cell neoplasms are excluded. Some examples of marrow involvement in IgG4-RD are shown in Figure 4.
Figure 4.

Bone marrow specimens involved by IgG4-related disease. Both cases show mature plasma cells distributed throughout the marrow. Ancillary studies established that these plasma cells were polyclonal, excluding a plasma cell neoplasm. (A,C) Hematoxylin and eosin stains. (B,D) IgG4 immunohistochemistry.
In general, only organs with clinical or radiological evidence of involvement are likely to show diagnostic features on biopsy, and thus biopsy should be directed at affected organ(s). Patients with proteinuria or renal lesions on imaging may require kidney biopsy, and renal involvement may demonstrate two distinct histological patterns: hypocomplementic tubulointerstitial nephritis in 80% of cases, and membranoproliferative glomerulonephritis in 20% of cases.70 In patients in whom affected organs are not amenable to biopsy, minor salivary gland (lip) biopsy should be considered. Even without clinical evidence of major salivary gland swelling or sicca symptoms, minor salivary gland biopsy can be a minimally invasive way to reach a histological diagnosis in some patients. One study of 66 patients with suspected IgG4-RD reported a sensitivity of 55% and specificity of 100% for labial salivary gland biopsy.71 From a pragmatic perspective, patients who have classic clinical, laboratory and radiological manifestations of IgG4-RD but are too frail to undergo an attempted biopsy attempt, or in whom small biopsies yield insufficient diagnostic material,72 can be given a working diagnosis of “suspected IgG4-RD” and treated as such provided reasonable efforts have been made to exclude mimickers of IgG4-RD.
Splenic involvement in IgG4-RD remains an enigma. Overt splenomegaly and splenic lesions are rare in confirmed cases of IgG4-RD. A rare entity known as scleros ing angiomatoid transformation of the spleen (SANT) is known to be enriched with IgG4+ plasma cells, but whether this condition is part of the spectrum of IgG4-RD remains unclear, as many patients with sclerosing angiomatoid transformation of the spleen do not exhibit other features of IgG4-RD.73 The inability to biopsy splenic tissue short of full splenectomy hinders the histological characterization of IgG4-RD in this organ.
Imaging and staging
Once histopathological confirmation of the diagnosis has been obtained, the disease can be staged by computed tomography of the chest, abdomen and pelvis.74 Although the orbit is a commonly involved organ in IgG4-RD, the absence of dacryoadenitis is suggestive that there is no underlying orbital pseudotumor and therefore dedicated head or orbital computed tomography is not always necessary.75 Positron emission tomography/computed tomography showed greater sensitivity for the detection of disease in arteries, salivary glands and lymph nodes in a study of 21 patients but further evaluation is needed to clarify which patients benefit from the combined tomographic method over conventional imaging.76 It is important to check the urinary albumin/creatinine ratio and serum C3/C4 levels to assess for renal involvement. IgG4-RD often behaves indolently, but accurate diagnosis and staging are crucial because some patients have asymptomatic but potentially organ- or life-threatening disease such as retroperitoneal fibrosis, periaortitis or coronary arteritis, the last of which may develop suddenly. Moreover, fibrotic disease is typically irreversible, so early treatment is important.
Case continued
Given the patient’s multi-organ involvement, particularly of the coronary arteries, he was treated with two doses of rituximab 1 g administered intravenously 2 weeks apart. His IgG4 level improved from 11.6 g/L to 5.84 g/L after 6 months. A repeat computed tomography scan of the chest and abdomen showed improvement of his coronary artery vasculitis, pulmonary nodules and lymphadenopathy and his post-treatment IgG4-RD Responder Index activity score was 3.
Treatment
Steroids are the first-line therapy for most patients, with an overall response rate of 93% and complete response rate of 66% in a phase 2 trial of 44 patients with IgG4-RD from Japan.77 The regimen used in this trial was prednisone 0.6 mg/kg/day initially with a gradual decrease of 5 mg every 2 weeks.77 Patients with higher eosinophil count, lacrimal gland involvement, five or more organs involved, and higher IgG4-RD Responder Index scores were at higher risk of glucocorticoid failure in a Chinese cohort of 215 patients.78 Some suggest a maintenance dosage of prednisone 5 mg daily for autoimmune pancreatitis specifically, which, as one would expect, decreases relapse rates compared to placebo.79 However, the toxicity associated with steroids is well known, with 40% of patients experiencing new or worsening diabetes. Blood glucose levels should be checked regularly in all patients receiving steroid therapy and many of our patients are also followed by a diabetes specialist. Disease-modifying anti-rheumatic drugs are not very effective for induction of remission but may have a role in maintenance of remission for some patients.80–82 In the absence of prospective clinical trials, an international consensus guideline on IgG4-RD treatment had only a 46% expert agreement on whether disease-modifying anti-rheumatic drugs should be started from the outset of treatment or not.83
Rituximab has been shown to be highly effective for IgG4-RD, with a response rate of 97% (29 out of 30 patients) in one prospective trial.74 The majority of patients did not require concomitant corticosteroids from the time of enrollment. However, rituximab is difficult to access due to cost, particularly outside of the USA, and remissions are often short-lived. In a French database study of 33 patients who received rituximab, a clinical response was seen in 29/31 (93.5%) patients, but 13/31 (41.9%) responders relapsed during a mean follow up of 24.8 months (with the relapses occurring at a mean of 19 months after rituximab therapy).84 The severe infection rate was estimated to be 12.1/100 patient years and three patients had hypogammaglobulinemia <5 g/L. In our practice, we often keep patients on a low dose of maintenance prednisone to maintain remission after rituximab treatment.
A number of emerging therapeutic options show promise. An open label, phase 2 clinical trial of a humanized anti-CD19 antibody (Xmab®5871) showed that this treatment decreased the IgG-RD Responder Index score by ≥2 points in 12/12 patients who completed the study.85 Successful use of lymphoma chemoimmunotherapy regimens such as fludarabine and rituximab and bendamustine and rituximab have been reported for two steroid- and rituximab-refractory cases.28 Whether these patients needed T-cell directed therapy or simply more potent immunosuppression than can be achieved by steroids or rituximab alone requires further exploration. Elotuzumab is an enticing agent because of the expression of SLAMF7 both on circulating plasmablasts and on the CD4+ cytotoxic T lymphocytes thought to drive the disease process. Anti-IgE therapy with omalizumab is also a potential target for those with severe atopic disease or asthma and elevated IgE levels.
Conclusions
IgG4-RD is an important condition for hematologists to recognize, both because of its common hematologic manifestations of lymphadenopathy, eosinophilia and polyclonal hypergammaglobulinemia, as well as its overlap with other hematologic inflammatory and neoplastic diseases. When IgG4-RD is suspected, measurement of serum IgG subclasses is a simple, non-invasive screening test; levels >5 g/L (normal <1.35 g/L) are highly suspicious for IgG4-RD. Regardless of serum IgG4 level, the definitive diagnosis requires histology, preferably in an affected organ other than lymph node or bone marrow, to confirm IgG4-RD and to exclude its many mimics. Early recognition and treatment with steroids, rituximab, or other immunosuppressive therapies, is essential to prevent complications such as fibrosis, periaortitis, and renal failure.
Key points
IgG4-RD is an important cause of eosinophilia, lymphadenopathy and polyclonal hypergammaglobulinemia. Hematologists should include IgG4-RD in the differential diagnosis of these abnormalities.
Other common manifestations of IgG4-RD include autoimmune pancreatitis, obstructive jaundice, orbital pseudotumor, lacrimal and salivary gland swelling, retroperitoneal fibrosis, and tubulointerstitial nephritis.
Serum protein electrophoresis and IgG subclass evaluation should be performed in patients with suspected IgG4-RD. Serum IgG4 levels are elevated in approximately 70% of cases. Mildly increased serum IgG4 (1.5-5 g/L) is a non-specific finding, but a markedly elevated level (>5 g/L) is 90% specific for IgG4-RD.
A firm diagnosis requires histological confirmation based on the International Consensus Criteria which include a dense lymphoplasmacytic infiltrate, storiform fibrosis and obliterative phlebitis. There must be an increased number of IgG4+ plasma cells with an IgG4:IgG plasma cell ratio >40%.
Early recognition and diagnosis are essential because patients typically respond well to steroids or rituximab in the early stages of the disease, but fibrotic disease and late complications such as chronic pancreatitis are often irreversible.
Supplementary Material
Acknowledgments
The authors thank the clinicians and pathologists of the IgG4 West working group in Vancouver for their invaluable collaboration, and Dr. Erica Peterson for providing critical comments on the draft of this manuscript.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/3/444
References
- 1.Hamano H, Kawa S, Horiuchi A, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344(10):732–738. [DOI] [PubMed] [Google Scholar]
- 2.Kamisawa T, Funata N, Hayashi Y, et al. A new clinicopathological entity of IgG4-related autoimmune disease. J Gastroenterol. 2003;38(10):982–984. [DOI] [PubMed] [Google Scholar]
- 3.Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–1192. [DOI] [PubMed] [Google Scholar]
- 4.Stone JH, Khosroshahi A, Deshpande V, et al. Recommendations for the nomenclature of IgG4-related disease and its individual organ system manifestations. Arthritis Rheum. 2012;64(10):3061–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mahajan VS, Mattoo H, Deshpande V, Pillai SS, Stone JH. IgG4-related disease. Annu Rev Pathol. 2014;9:315–347. [DOI] [PubMed] [Google Scholar]
- 6.Grados A, Vaysse T, Ebbo M, Carbonnel F, Schleinitz N. IgG4-related disease in monozygotic twins: a case report. Ann Intern Med. 2017;166(2):153–155. [DOI] [PubMed] [Google Scholar]
- 7.Karim F, Loeffen J, Bramer W, et al. IgG4-related disease: a systematic review of this unrecognized disease in pediatrics. Pediatr Rheumatol Online J. 2016;14(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–551. [DOI] [PubMed] [Google Scholar]
- 9.Tang SH, Lin MH, Du JS, Liu YC, Hsiao HH, Liu TC. IgG4-related disease with bone marrow involvement mimicking multiple myeloma. Br J Haematol. 2017;177(5):673. [DOI] [PubMed] [Google Scholar]
- 10.Geyer JT, Niesvizky R, Jayabalan DS, et al. IgG4 plasma cell myeloma: new insights into the pathogenesis of IgG4-related disease. Mod Pathol. 2014;27(3):375–381. [DOI] [PubMed] [Google Scholar]
- 11.Mattoo H, Mahajan VS, Maehara T, et al. Clonal expansion of CD4(+) cytotoxic T lymphocytes in patients with IgG4-related disease. J Allergy Clin Immunol. 2016;138(3):825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carruthers MN, Park S, Slack GW, et al. IgG4-related disease and lymphocyte-variant hypereosinophilic syndrome: a comparative case series. Eur J Haematol. 2016;98(4):378–387. [DOI] [PubMed] [Google Scholar]
- 13.Mattoo H, Stone JH, Pillai S. Clonally expanded cytotoxic CD4(+) T cells and the pathogenesis of IgG4-related disease. Autoimmunity. 2017;50(1):19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hubers LM, Vos H, Schuurman AR, et al. Annexin A11 is targeted by IgG4 and IgG1 autoantibodies in IgG4-related disease. Gut. 2018;67(4):728–735. [DOI] [PubMed] [Google Scholar]
- 15.Perugino CA, AlSalem SB, Mattoo H, et al. Identification of galectin-3 as an autoantigen in patients with IgG4-related disease. J Allergy Clin Immunol. 2018. May 29 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace ZS, Deshpande V, Mattoo H, et al. IgG4-related disease: clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015;67(9): 2466–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamada K, Yamamoto M, Saeki T, et al. New clues to the nature of immunoglobulin G4-related disease: a retrospective Japanese multicenter study of baseline clinical features of 334 cases. Arthritis Res Ther. 2017;19(1):262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato Y, Kojima M, Takata K, et al. Systemic IgG4-related lymphadenopathy: a clinical and pathologic comparison to multicentric Castleman’s disease. Mod Pathol. 2009;22(4):589–599. [DOI] [PubMed] [Google Scholar]
- 19.Cheuk W, Chan JK. Lymphadenopathy of IgG4-related disease: an underdiagnosed and overdiagnosed entity. Semin Diagn Pathol. 2012;29(4):226–234. [DOI] [PubMed] [Google Scholar]
- 20.Wick MR, O’Malley DP. Lymphadenopathy associated with IgG4-related disease: diagnosis & differential diagnosis. Semin Diagn Pathol. 2017;35(1):61–66. [DOI] [PubMed] [Google Scholar]
- 21.Chang SY, Keogh KA, Lewis JE, et al. IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener’s): a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases. Hum Pathol. 2013;44(11): 2432–2437. [DOI] [PubMed] [Google Scholar]
- 22.Menon MP, Evbuomwan MO, Rosai J, Jaffe ES, Pittaluga S. A subset of Rosai-Dorfman disease cases show increased IgG4-positive plasma cells: another red herring or a true association with IgG4-related disease? Histopathology. 2014;64(3):455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy. 2014;69(2): 269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gotlib J. World Health Organization-defined eosinophilic disorders: 2017 update on diagnosis, risk stratification, and management. Am J Hematol. 2017;92(11):1243–1259. [DOI] [PubMed] [Google Scholar]
- 25.Chen YY, Khoury P, Ware JM, et al. Marked and persistent eosinophilia in the absence of clinical manifestations. J Allergy Clin Immunol. 2014;133(4):1195–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu Z, Boddu PC, Loghavi S, et al. A multi-modality work-up of patients with hypereosinophilia. Am J Hematol. 2018;93(11): 1337–1346. [DOI] [PubMed] [Google Scholar]
- 27.Chen LY, Lai EJ, Collins DR, Ostrow DN, Sreenivasan GM. A young woman with episodic angioedema, papilledema, and eosinophilia. Am J Hematol. 2010;85(2):124–127. [DOI] [PubMed] [Google Scholar]
- 28.Chen LY, Wong PC, Noda S, Collins DR, Sreenivasan GM, Coupland R. Polyclonal hyperviscosity syndrome in IgG4-related disease and associated conditions. Clin Case Rep. 2015;3(4):217–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Khoury P. Lymphocytic-variant hypereosinophilic syndromes. The Hematologist. 2017;14(6):6. [Google Scholar]
- 30.Mohammad N, Avinashi V, Chan E, Vallance BA, Portales-Casamar E, Bush JW. Pediatric eosinophilic esophagitis is associated with increased lamina propria immunoglobulin G4-positive plasma cells. J Pediatr Gastroenterol Nutr. 2018;67(2):204–209. [DOI] [PubMed] [Google Scholar]
- 31.Chen C, Chen K, Huang X, Wang K, Qian S. Concurrent eosinophilia and IgG4-related disease in a child: a case report and review of the literature. Exp Ther Med. 2018;15(3): 2739–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Clayton F, Fang JC, Gleich GJ, et al. Eosinophilic esophagitis in adults is associated with IgG4 and not mediated by IgE. Gastroenterology. 2014;147(3):602–609. [DOI] [PubMed] [Google Scholar]
- 33.Zukerberg L, Mahadevan K, Selig M, Deshpande V. Esophageal intrasquamous IgG4 deposits: an adjunctive marker to distinguish eosinophilic esophagitis from reflux esophagitis. Histopathology. 2016;68(7): 968–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong PC, Fung AT, Gerrie AS, et al. IgG4-related disease with hypergammaglobulinemic hyperviscosity and retinopathy. Eur J Haematol. 2013;90(3):250–256. [DOI] [PubMed] [Google Scholar]
- 35.Grados A, Ebbo M, Boucraut J, et al. Serum immunoglobulin free light chain assessment in IgG4-related disease. Int J Rheumatol. 2013;2013:426759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jawad MD, Go RS, Witzig TE, Mikhael JR, Ravindran A, Murrray DL. Pseudo-monoclonal gammopathy: a report of four cases. Haematologica. 2017;102(11):e466–e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobs JF, van der Molen RG, Keren DF. Relatively restricted migration of polyclonal IgG4 may mimic a monoclonal gammopathy in IgG4-related disease. Am J Clin Pathol. 2014;142(1):76–81. [DOI] [PubMed] [Google Scholar]
- 38.Costa MS, Silva A, Costa L, Rodrigues TB, Lemos S, Garrido J. Immunoglobulin G4-related disease mimicking multiple myeloma. Port J Nephrol Hypert. 2018;32(3):217–222. [Google Scholar]
- 39.Finn WG, Gulbranson R, Fisher S, et al. Detection of polyclonal increases in immunoglobulin G4 subclass by distinct patterns on capillary serum protein electrophoresis: diagnostic pitfalls and clinical observations in a study of 303 cases. Am J Clin Pathol. 2016;146(3):303–311. [DOI] [PubMed] [Google Scholar]
- 40.McCudden CR, Jacobs JFM, Keren D, Caillon H, Dejoie T, Andersen K. Recognition and management of common, rare, and novel serum protein electrophoresis and immunofixation interferences. Clin Biochem. 2017;51:72–79. [DOI] [PubMed] [Google Scholar]
- 41.Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc. 2001;76(5):476–487. [DOI] [PubMed] [Google Scholar]
- 42.Zhao EJ, Carruthers MN, Li CH, Mattman A, Chen LYC. Prevalence of IgG4-Related Disease in Patients with Hypergammaglobulinemia. XXXVII World Congress of the International Society of Hematology (ISH 2018); Sept 14, 2018, 2018; Vancouver, British Columbia, Canada. [Google Scholar]
- 43.Engelhart S, Glynn RJ, Schur PH. Disease associations with isolated elevations of each of the four IgG subclasses. Semin Arthritis Rheum. 2017;47(2):276–280. [DOI] [PubMed] [Google Scholar]
- 44.Zheng M, Zhou P, Zheng K, et al. A special subtype of POEMS syndrome: IgG4 subtype. Am J Transl Res. 2016;8(2):588–596. [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu J, Wu B. Immunoglobulin G4-positive plasma cell myeloma. Blood. 2015;126(19): 2254. [DOI] [PubMed] [Google Scholar]
- 46.Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2014;385(9976): 1460–1471. [DOI] [PubMed] [Google Scholar]
- 47.Zhang X, Hyjek E, Vardiman J. A subset of Rosai-Dorfman disease exhibits features of IgG4-related disease. Am J Clin Pathol. 2013;139(5):622–632. [DOI] [PubMed] [Google Scholar]
- 48.Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131(26):2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deshpande V, Khosroshahi A, Nielsen GP, Hamilos DL, Stone JH. Eosinophilic angiocentric fibrosis is a form of IgG4-related systemic disease. Am J Surg Pathol. 2011;35(5):701–706. [DOI] [PubMed] [Google Scholar]
- 51.Estrada-Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017;1(6):357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sato Y, Ohshima K, Ichimura K, et al. Ocular adnexal IgG4-related disease has uniform clinicopathology. Pathol Int. 2008;58(8):465–470. [DOI] [PubMed] [Google Scholar]
- 53.Bledsoe JR, Wallace ZS, Stone JH, Deshpande V, Ferry JA. Lymphomas in IgG4-related disease: clinicopathologic features in a Western population. Virchows Arch. 2017;472(5):839–852. [DOI] [PubMed] [Google Scholar]
- 54.van de Ven A, Seidl M, Drendel V, et al. IgG4-related disease in autoimmune lymphoproliferative syndrome. Clin Immunol. 2017;180:97–99. [DOI] [PubMed] [Google Scholar]
- 55.Langan RC, Gill F, Raiji MT, et al. Autoimmune pancreatitis in the autoimmune lymphoproliferative syndrome (ALPS): a sheep in wolves’ clothing? Pancreas. 2013;42(2):363–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carruthers MN, Stone JH, Deshpande V, Khosroshahi A. Development of an IgG4-RD Responder Index. Int J Rheumatol. 2012;2012:259408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wallace ZS, Khosroshahi A, Carruthers MD, et al. An international, multi-specialty validation study of the IgG4-related disease responder index. Arthritis Care Res (Hoboken). 2018;70(11):1671–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH. The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2015;74(1):14–18. [DOI] [PubMed] [Google Scholar]
- 59.Culver EL, Sadler R, Simpson D, et al. Elevated serum IgG4 levels in diagnosis, treatment response, organ involvement, and relapse in a prospective IgG4-related disease UK cohort. Am J Gastroenterol. 2016;111(5):733–743. [DOI] [PubMed] [Google Scholar]
- 60.Masaki Y, Kurose N, Yamamoto M, et al. Cutoff values of serum IgG4 and histopathological IgG4+ plasma cells for diagnosis of patients with IgG4-related disease. Int J Rheumatol. 2012;2012:580814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qi R, Chen LYC, Park S, et al. Utility of serum IgG4 levels in a multiethnic population. Am J Med Sci. 2018;355(1):61–66. [DOI] [PubMed] [Google Scholar]
- 62.Wallace ZS, Mattoo H, Carruthers M, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Parker AR, Carr-Smith HD. Calibration differences and the prozone phenomenon in IgG4-related disease: comment on the article by Khosroshahi et al. Arthritis Rheumatol. 2014;66(7):1964–1965. [DOI] [PubMed] [Google Scholar]
- 64.Khosroshahi A, Cheryk LA, Carruthers MN, Edwards JA, Bloch DB, Stone JH. Brief Report: spuriously low serum IgG4 concentrations caused by the prozone phenomenon in patients with IgG4-related disease. Arthritis Rheumatol. 2014;66(1):213–217. [DOI] [PubMed] [Google Scholar]
- 65.van der Gugten G, DeMarco ML, Chen LYC, et al. Resolution of spurious immunonephelometric IgG subclass measurement discrepancies by LC-MS/MS. Clin Chem. 2018;64(4):735–742. [DOI] [PubMed] [Google Scholar]
- 66.Chan ASY, Mudhar H, Shen SY, et al. Serum IgG2 and tissue IgG2 plasma cell elevation in orbital IgG4-related disease (IgG4-RD): potential use in IgG4-RD assessment. Br J Ophthalmol. 2017;101(11):1576–1582. [DOI] [PubMed] [Google Scholar]
- 67.Dunkley L, Mudhar HS. IgG4-related disease presenting with raised serum IgG2-real timeline of IgG4-RD? Rheumatology. 2017;57(1):197–199. [DOI] [PubMed] [Google Scholar]
- 68.Mattman A, Chen LYC, van der Gugten G, et al. Comment on: IgG4-related disease presenting with raised serum IgG2-real timeline of IgG4-RD? Rheumatology. 2018;57(6):1126–1127. [DOI] [PubMed] [Google Scholar]
- 69.Chari ST, Takahashi N, Levy MJ, et al. A diagnostic strategy to distinguish autoimmune pancreatitis from pancreatic cancer. Clin Gastroenterol Hepatol. 2009;7(10): 1097–1103. [DOI] [PubMed] [Google Scholar]
- 70.Mann S, Seidman MA, Barbour SJ, Levin A, Carruthers M, Chen LY. Recognizing IgG4-related tubulointerstitial nephritis. Can J Kidney Health Dis. 2016;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moriyama M, Ohta M, Furukawa S, et al. The diagnostic utility of labial salivary gland biopsy in IgG4-related disease. Mod Rheumatol. 2016;26(5):725–729. [DOI] [PubMed] [Google Scholar]
- 72.Arora K, Rivera M, Ting DT, Deshpande V. The histologic diagnosis of IgG4-related disease on small biopsies: challenges and pitfalls. Histopathology. 2018. November 8 [Epub ahed of print] [DOI] [PubMed] [Google Scholar]
- 73.Kuo TT, Chen TC, Lee LY. Sclerosing angiomatoid nodular transformation of the spleen (SANT): clinicopathological study of 10 cases with or without abdominal disseminated calcifying fibrous tumors, and the presence of a significant number of IgG4+ plasma cells. Pathol Int. 2009;59(12):844–850. [DOI] [PubMed] [Google Scholar]
- 74.Carruthers MN, Topazian MD, Khosroshahi A, et al. Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis. 2015;74(6):1171–1177. [DOI] [PubMed] [Google Scholar]
- 75.Wallace ZS, Khosroshahi A, Jakobiec FA, et al. IgG4-related systemic disease as a cause of “idiopathic” orbital inflammation, including orbital myositis, and trigeminal nerve involvement. Surv Ophthalmol. 2012;57(1): 26–33. [DOI] [PubMed] [Google Scholar]
- 76.Ebbo M, Grados A, Guedj E, et al. Usefulness of 2-[18F]-fluoro-2-deoxy-D-glucose-positron emission tomography/computed tomography for staging and evaluation of treatment response in IgG4-related disease: a retrospective multicenter study. Arthritis Care Res (Hoboken). 2014;66(1):86–96. [DOI] [PubMed] [Google Scholar]
- 77.Masaki Y, Matsui S, Saeki T, et al. A multi-center phase II prospective clinical trial of glucocorticoid for patients with untreated IgG4-related disease. Mod Rheumatol. 2017;27(5):849–854. [DOI] [PubMed] [Google Scholar]
- 78.Wang L, Zhang P, Wang M, et al. Failure of remission induction by glucocorticoids alone or in combination with immunosuppressive agents in IgG4-related disease: a prospective study of 215 patients. Arthritis Res Ther. 2018;20(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Masamune A, Nishimori I, Kikuta K, et al. Randomised controlled trial of long-term maintenance corticosteroid therapy in patients with autoimmune pancreatitis. Gut. 2017;66(3):487–494. [DOI] [PubMed] [Google Scholar]
- 80.Hart PA, Topazian MD, Witzig TE, et al. Treatment of relapsing autoimmune pancreatitis with immunomodulators and rituximab: the Mayo Clinic experience. Gut. 2013;62(11):1607–1615. [DOI] [PubMed] [Google Scholar]
- 81.Yunyun F, Yu C, Panpan Z, et al. Efficacy of cyclophosphamide treatment for immunoglobulin G4-related disease with addition of glucocorticoids. Sci Rep. 2017;7(1):6195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yunyun F, Yu P, Panpan Z, et al. Efficacy and safety of low dose mycophenolate mofetil treatment for immunoglobulin G4-related disease. Rheumatology (Oxford, England). 2018. August 14 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 83.Khosroshahi A, Wallace ZS, Crowe JL, et al. International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol. 2015;67(7):1688–1699. [DOI] [PubMed] [Google Scholar]
- 84.Ebbo M, Grados A, Samson M, et al. Long-term efficacy and safety of rituximab in IgG4-related disease: data from a French nationwide study of thirty-three patients. PLoS One. 2017;12(9):e0183844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stone JH, Wallace ZS, Perugino CA, et al. A Trial of XmAb (R) 5871, a reversible inhibitor of CD19+ cells, in IgG4-related disease. Arthritis Rheumatol. 2016;68(Suppl 10). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.