

Abstract
Key points
In humans, the vasodilatory response to skeletal muscle contraction is mediated in part by activation of inwardly rectifying potassium (KIR) channels. Evidence from animal models suggest that KIR channels serve as electrical amplifiers of endothelium‐dependent hyperpolarization (EDH).
We found that skeletal muscle contraction amplifies vasodilatation to the endothelium‐dependent agonist ACh, whereas there was no change in the vasodilatory response to sodium nitroprusside, an endothelium‐independent nitric oxide donor.
Blockade of KIR channels reduced the exercise‐induced amplification of ACh‐mediated vasodilatation. Conversely, pharmacological activation of KIR channels in quiescent muscle via intra‐arterial infusion of KCl independently amplified the vasodilatory response to ACh.
This study is the first in humans to demonstrate that specific endothelium‐dependent vasodilatory signalling is amplified in the vasculature of contracting skeletal muscle and that KIR channels may serve as amplifiers of EDH‐like vasodilatory signalling in humans.
Abstract
The local vasodilatory response to muscle contraction is due in part to the activation of inwardly rectifying potassium (KIR) channels. Evidence from animal models suggest that KIR channels function as ‘amplifiers’ of endothelium‐dependent vasodilators. We tested the hypothesis that contracting muscle selectively amplifies endothelium‐dependent vasodilatation via activation of KIR channels. We measured forearm blood flow (Doppler ultrasound) and calculated changes in vascular conductance (FVC) to local intra‐arterial infusion of ACh (endothelium‐dependent dilator) during resting conditions, handgrip exercise (5% maximum voluntary contraction) or sodium nitroprusside (SNP; endothelium‐independent dilator) which served as a high‐flow control condition (n = 7, young healthy men and women). Trials were performed before and after blockade of KIR channels via infusion of barium chloride. Exercise augmented peak ACh‐mediated vasodilatation (ΔFVC saline: 117 ± 14; exercise: 236 ± 21 ml min−1 (100 mmHg)−1; P < 0.05), whereas SNP did not impact ACh‐mediated vasodilatation. Blockade of KIR channels attenuated the exercise‐induced augmentation of ACh. In eight additional subjects, SNP was administered as the experimental dilator. In contrast to ACh, exercise did not alter SNP‐mediated vasodilatation (ΔFVC saline: 158 ± 35; exercise: 121 ± 22 ml min−1 (100 mmHg)−1; n.s.). Finally, in a subset of six subjects, direct pharmacological activation of KIR channels in quiescent muscle via infusion of KCl amplified peak ACh‐mediated vasodilatation (ΔFVC saline: 97 ± 15, KCl: 142 ± 16 ml min−1 (100 mmHg)−1; respectively; P < 0.05). These findings indicate that skeletal muscle contractions selectively amplify endothelium‐dependent vasodilatory signalling via activation of KIR channels, and this may be an important mechanism contributing to the normal vasodilatory response to exercise in humans.
Keywords: Inwardly rectifying potassium channels, exercise hyperaemia, endothelium‐dependent hyperpolarization
Key points
In humans, the vasodilatory response to skeletal muscle contraction is mediated in part by activation of inwardly rectifying potassium (KIR) channels. Evidence from animal models suggest that KIR channels serve as electrical amplifiers of endothelium‐dependent hyperpolarization (EDH).
We found that skeletal muscle contraction amplifies vasodilatation to the endothelium‐dependent agonist ACh, whereas there was no change in the vasodilatory response to sodium nitroprusside, an endothelium‐independent nitric oxide donor.
Blockade of KIR channels reduced the exercise‐induced amplification of ACh‐mediated vasodilatation. Conversely, pharmacological activation of KIR channels in quiescent muscle via intra‐arterial infusion of KCl independently amplified the vasodilatory response to ACh.
This study is the first in humans to demonstrate that specific endothelium‐dependent vasodilatory signalling is amplified in the vasculature of contracting skeletal muscle and that KIR channels may serve as amplifiers of EDH‐like vasodilatory signalling in humans.
Introduction
During skeletal muscle contraction a number of local metabolic, endothelial, mechanical and humoral signals interact to elicit vasodilatation and increase blood flow and oxygen delivery to support tissue metabolic demand (Saltin, 2007). Exercise‐induced vasodilatation is achieved through activation of signalling pathways associated with the endothelium and vascular smooth muscle and ascends from the site of metabolic demand to upstream feed arteries, redirecting blood to metabolically active skeletal muscle (Segal, 2005). Evidence from animal models point to endothelium‐dependent signalling as the critical pathway for coordinating ascending vasodilatation (Emerson & Segal, 2000; Duza & Sarelius, 2004; Murrant et al. 2004; Wolfle et al. 2007). In particular, studies have identified endothelial‐derived hyperpolarization (EDH) as the key mediator of ascending vasodilatation in response to skeletal muscle contraction (Segal & Jacobs, 2001), and disruption of the endothelium and/or EDH signalling prevents the spread of vasodilatation along the vessel and significantly reduces the hyperaemic response to muscle contraction (Milkau et al. 2010; Sinkler & Segal, 2017).
EDH occurs in response to a rise in intracellular Ca2+ concentration ([Ca2+]) due to release of internal stores from the endoplasmic reticulum, as well as activation of discrete Ca2+‐permeable ion channels (e.g. transient receptor potential channels) located within myoendothelial gap junctions (MEGJs) (Kerr et al. 2015; Senadheera et al. 2012). Rising intracellular [Ca2+] in turn activates small‐ and intermediate‐conductance Ca2+‐activated K+ (KCa) channels resulting in efflux of K+ from the endothelial cell and subsequent membrane hyperpolarization (Behringer & Segal, 2012). Hyperpolarization of membrane potential can spread between endothelial cells along the length of the blood vessel through homocellular gap junctions, as well as directly to vascular smooth muscle through MEGJs (Kerr et al. 2012). The resulting vascular smooth muscle cell hyperpolarization closes voltage‐gated Ca2+ channels, lowers intracellular Ca2+ and induces vasodilatation. Importantly, recent studies have identified a role for inwardly rectifying potassium channels (KIR) acting as electrical ‘amplifiers’, or ‘boosters’, of EDH (Smith et al. 2008; Sonkusare et al. 2016). In this context, KIR channels demonstrate negative slope conductance over physiological membrane potentials, where membrane hyperpolarization facilitates greater KIR channel activity which further increases K+ efflux thus amplifying the initial hyperpolarizing signal (Jackson, 2017).
With respect to exercise hyperaemia, our laboratory and others have identified a significant contribution of KIR channel activity to the vasodilatory response to skeletal muscle contraction. In humans, KIR channels are activated immediately in response to a single muscle contraction (Crecelius et al. 2013 a), and contribute to the vasodilatation observed throughout exercise onset as well as during steady‐state exercise (Crecelius et al. 2014, 2015 b; Racine et al. 2018). Evidence from animal models support K+ efflux from contracting skeletal muscle as a primary stimulus for vascular KIR channel activation (Mohrman & Sparks, 1974; Armstrong et al. 2007). In this context, skeletal muscle K+ efflux serves as a feedforward mechanism coupling skeletal muscle contraction and the initiation of vasodilatation. Additionally, other stimuli that increase during exercise, such as intravascular ATP, may activate KIR channels and participate in vasodilatation (Crecelius et al. 2012). Thus, it is possible that once activated, KIR channels serve to amplify endothelium‐dependent vasodilatation, further contributing to blood flow control in contracting skeletal muscle.
Accordingly, in the present study we tested the primary hypothesis that contracting skeletal muscle selectively amplifies endothelium‐dependent vasodilatation. To do so, we determined the forearm vascular responses to Ach (endothelium‐dependent) and sodium nitroprusside (SNP, endothelium‐independent) at rest, during a high flow control condition, and during mild intensity handgrip exercise in humans. Second, we hypothesized that amplification of endothelium‐dependent vasodilatory signalling is due in part to KIR channel activation in humans. To investigate the role of KIR channels, we determined the responses to ACh in contracting muscle during KIR channel blockade via barium chloride (BaCl2), and in quiescent muscle during pharmacological KIR channel activation via exogenous KCl infusions.
Methods
Ethical approval and human subjects
With Colorado State University Institutional Review Board approval (14‐5392H), and after informed written consent, 15 young healthy subjects (9 men, 6 women; age, 23 ± 1 years; weight, 74 ± 4 kg; height, 170 ± 1 cm; body mass index, 23 ± 1 kg/m2; means ±SEM) participated in this study. All subjects were free from overt cardiovascular disease as assessed from a medical history, were sedentary to moderately active, non‐smokers, non‐obese, normotensive and not taking any medications. Female subjects were studied during the early follicular phase of their menstrual cycle or placebo phase of oral contraceptive use to minimize any potential cardiovascular effects of sex‐specific hormones. All studies were performed in the Human Cardiovascular Physiology Laboratory located at Colorado State University following a 12‐h fast with the subjects in the supine position and in accordance with the Declaration of Helsinki, except for registration in a database.
Body composition and forearm volume
Dual‐energy X‐ray absorptiometry (DEXA; Hologic, Bedford, MA, USA) was used to determine body composition. A regional analysis of the experimental forearm area (proximal to distal radio‐ulnar joint) from the whole body DEXA scan was performed to determine forearm volume (FAV). Drug doses were normalized according to FAV where appropriate.
Arterial catheterization, arterial blood pressure and heart rate
A 20‐gauge, 7.6‐cm catheter was placed in the brachial artery of the non‐dominant arm under aseptic conditions after local anaesthesia (2% lidocaine) for local administration of study drugs and mean arterial pressure (MAP) measurement as described previously (Dinenno & Joyner, 2003; Richards et al. 2014; Hearon et al. 2016). Heart rate (HR) was determined using a three‐lead ECG (Cardiocap/5; Datex‐Ohmeda, Louisville, CO, USA).
Forearm blood flow and vascular conductance
A 12‐MHz linear‐array ultrasound probe (Vivid 7; General Electric, Milwaukee, WI, USA) was used to determine brachial artery mean blood velocity (MBV) and brachial artery diameter. For blood velocity measurements, the probe insonation angle was maintained at 60° and the frequency used was 5 MHz. The Doppler shift frequency spectrum was analysed via a Multigon 500M TCD spectral analyser (Multigon Industries, Mt. Vernon, NY, USA), from which mean velocity was determined as a weighted mean of the spectrum of Doppler shift frequencies. Brachial artery diameter was measured in triplicate at end diastole and between contractions where applicable, at the end of each time‐point as described below. Forearm blood flow (FBF) was then calculated as described previously (Crecelius et al. 2015 b). Forearm vascular conductance (FVC) was calculated as (FBF/MAP) × 100 and expressed as ml min−1 100 mmHg−1. All studies were performed in a cool (20–22°C) temperature‐controlled environment with a fan directed toward the forearm to minimize the contribution of skin blood flow to forearm haemodynamics.
Rhythmic handgrip exercise
Maximal voluntary contraction (MVC; all subjects mean 38 ± 3 kg) was determined for the experimental arm as the average of three maximal squeezes of a handgrip dynamometer (Stoelting, Chicago, IL, USA) that were within 3% of each other. Forearm exercise was performed with weight corresponding to 5% MVC (mean 2.1 ± 0.2 kg) attached to a pulley system and lifted 4–5 cm over the pulley at a duty cycle of 1 s contraction: 2 s relaxation (20 contractions·min−1) using both visual and auditory feedback to ensure the correct timing. We chose this mild intensity rhythmic handgrip exercise to limit the contribution of systemic haemodynamics and reflex activation of the sympathetic nervous system on exercise hyperaemia, and thus our experimental model isolates the effects of muscle contractions on local vascular control mechanisms.
Vasoactive drug administration
All vasoactive drug infusions occurred via the brachial artery catheter to create a local effect in the forearm, and saline was utilized as a control infusate. Specific timing and duration of infusions are provided under the Experimental protocols and presented in Fig. 1. Ach (endothelium‐dependent vasodilator; Michol‐E, Novartis, Basel, Switzerland) and SNP [endothelium‐independent nitric oxide (NO) donor; Hospira, Lake Forest, IL, USA] were initially infused at 2.25 and 3.33 μg (dl FAV)−1 min−1, respectively, and adjusted thereafter to ensure a moderate rise in FBF similar to that observed during mild intensity (5% MVC) exercise. Importantly, with this level of vasodilatation there is a large vasodilatory reserve, providing sufficient resolution to observe amplification of the vasodilatory response to a subsequent dose of ACh and SNP (final average doses: ACh: 2.05 μg (dl FAV)−1 min−1; SNP: 2.88 μg min−1). To activate KIR channels, potassium chloride (KCl; Hospira) was administered at 0.20 mmol min−1, equal to the largest dose of KCl given by our lab previously without subject discomfort (Crecelius et al. 2012). To block KIR channels, barium chloride (BaCl2; 10%, w/v, BDH3238, EMD Chemicals, Gibbstown, NJ, USA) was infused at 0.9 μmol (dl FAV)−1 min−1, with a minimum dose of 8 μmol min−1 to a maximum dose of 10 μmol min−1 for 3 min as a loading dose and continued throughout the experimental trial (an additional 6 min) at the same dose (Crecelius et al. 2015 b; Hearon et al. 2017). Our laboratory and others have demonstrated previously that infusion of BaCl2 inhibits the majority of the dilatory response to exogenous KCl (∼60–70%) (Dawes et al. 2002; Dwivedi et al. 2005), and reduces the vasodilatory response to handgrip exercise (∼30%) (Crecelius et al. 2014, 2015 b), consistent with effective inhibition of KIR channels.
Figure 1. Experimental protocols.
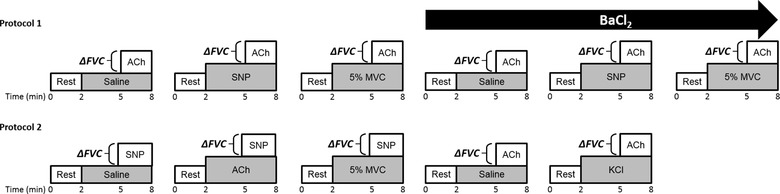
After catheterization of the brachial artery and subject instrumentation, the change in forearm vascular conductance (ΔFVC) in response to ACh or sodium nitroprusside (SNP) was assessed during rest, handgrip exercise at 5% maximal voluntary contraction (MVC) or a high flow control vasodilator condition. Protocol 1: the vasodilatory response to ACh was assessed at rest, during 5% MVC exercise and during a high flow control SNP infusion, before and after infusion of barium chloride (BaCl2) to block inwardly rectifying potassium (KIR) channels. Protocol 2: the vasodilatory response to SNP was assessed at rest, during 5% MVC exercise and during a high flow control ACh infusion. In a subset of subjects, the vasodilatory response to ACh was assessed before and after pharmacological activation of KIR channels via infusion of potassium chloride (KCl).
Experimental protocols
After catheterization and experimental set up, subjects were briefly familiarized with the handgrip exercise modality and FBF was measured to estimate steady‐state FBF responses. Sufficient rest was given to allow stabilization of forearm haemodynamics. Resting haemodynamic data were acquired for 2 min before the start of each experimental condition. Subsequently, the vasodilatory response to a single dose of ACh or SNP was assessed during three experimental conditions: (1) during resting control (saline) conditions, (2) during mild intensity (5% MVC) handgrip exercise, or (3) during a control vasodilatory stimulus to elevate resting flows to match those observed during handgrip exercise (Fig. 1). The initial vasodilator infusion served as a high flow control condition where the rate of the infusion was adjusted until steady‐state forearm blood flow matched what was observed during handgrip exercise, and remained constant for the remainder of the protocol. Steady‐state haemodynamics were achieved within ∼3 min during handgrip exercise and the flow‐matched vasodilator infusion (see below for specific protocol). Thereafter, infusion of a single dose of ACh or SNP was initiated and the vasodilatory response was measured continuously throughout the final 3 min of the experimental condition (representative tracings depicted in Fig. 2). Subjects rested for 15 min after each trial to ensure return to resting baseline conditions and washout of any pharmacological agents.
Figure 2. Representative tracings of exercise‐induced amplification of ACh‐mediated vasodilatation before and after inhibition of inwardly rectifying potassium channels.
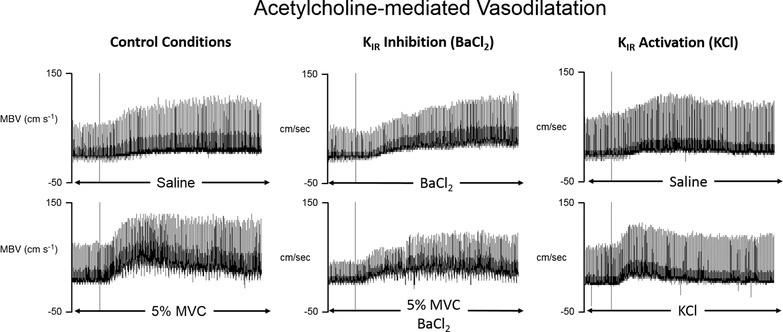
Each panel depicts the mean blood velocity response to an intra‐arterial infusion of Ach (initiated at each vertical line) maintained for 3 min during various conditions (indicated below each panel). Left panels: under control conditions, 5% MVC exercise significantly amplified the peak vasodilatory response to ACh compared to resting skeletal muscle (saline; Fig. 3). This effect was not observed with infusion of the endothelium‐independent nitric oxide donor, sodium nitroprusside (SNP; Fig. 5). Middle panels: surprisingly, inhibition of inwardly rectifying potassium (KIR) channels (BaCl2 infusion) increased the control response to ACh in quiescent skeletal muscle. However, there was no further exercise‐induced amplification of ACh after inhibition of KIR channels (Fig. 3). Right panels: pharmacological activation of KIR channels via infusion of KCl significantly amplified the peak vasodilatory response to ACh, similar to handgrip exercise (Fig. 6).
Protocol 1: ACh‐mediated vasodilatation during exercise
In seven subjects (3 male, 4 female), a saline control trial was performed first to ensure a moderate vasodilatory response to ACh was observed. Subsequently, the same dose of ACh was infused during steady‐state 5% MVC handgrip exercise or during steady‐state infusion of SNP matched to the hyperaemia observed during 5% exercise to serve as the ‘high flow’ control vasodilator condition. Following the first three trials (saline, 5% MVC, SNP), BaCl2 was administered and the same three experimental trials were repeated to investigate the role of KIR channels in amplifying the vasodilatory response to ACh during handgrip exercise. Based on our previous work demonstrating that blockade of KIR channels reduces steady‐state exercise hyperaemia ∼30% (Crecelius et al. 2014, 2015 b), the dose of SNP was reduced to account for the effect of KIR channel blockade on exercise hyperaemia. The order of the 5% MVC exercise and SNP high flow control trials was counterbalanced across subjects, and conducted in the same order before and after blockade of KIR channels (Fig. 1, Protocol 1).
Protocol 2A: SNP‐mediated vasodilatation during exercise
In eight subjects (6 male, 2 female), a saline control trial was performed first to establish the normal vasodilatory response to SNP and to ensure a moderate response was observed. Subsequently, the same dose of SNP was infused during steady‐state 5% MVC exercise or during steady‐state infusion of ACh matched to the hyperaemia observed during 5% exercise to serve as the ‘high flow’ control vasodilator condition. The order of the 5% MVC exercise and ACh high flow control trials was counterbalanced across subjects (Fig. 1, Protocol 2).
Protocol 2B: ACh‐mediated vasodilatation during pharmacological activation of KIR channels
To further investigate a potential role for KIR channel activity in amplifying vasodilatory responses to ACh, the same dose of ACh was infused in a subgroup of six subjects (4 male, 2 female) at rest and during infusion of KCl to pharmacologically activate KIR channels in quiescent muscle. Two subjects were unable to participate in this protocol due to time constraints. These final two trials were randomized and conducted after the initial three SNP trials in protocol 2A (Fig. 1, Protocol 2).
Data acquisition and analysis
Data were collected and stored on computer at 250 Hz and analysed off‐line with signal‐processing software (WinDaq; DATAQ Instruments, Akron, OH, USA). The FBF, MAP and FVC responses were analysed in 3‐s bins for 15 s before, and for 180 s after initiation of the ACh or SNP infusion. If the MBV signal quality obtained during a 3‐s cycle was altered due to operator error, a mathematical average of the MBV in the preceding and subsequent bins was used. This occurred in <2% of all bins analysed. Absolute change in FVC was calculated for each bin as the change in FVC from baseline (i.e. the average FVC for the 15 s prior to the start of the vasodilator infusion). Peak change in FVC was identified as the greatest change in FVC from baseline for any 3‐s bin during the 180 s of vasodilator infusion. The peak vasodilatory response was chosen as the primary outcome in order to avoid the potential confounding influence of metabolic autoregulation during exercise (discussed below). The magnitude of amplification of the vasodilatory response to ACh or SNP during experimental conditions (exercise or high flow control) was calculated as the difference between the peak change in FVC observed during the experimental condition and the resting (saline) control condition (ΔFVC5% exercise/High flow − ΔFVCsaline). HR was determined at rest and over the last minute of each hyperaemic condition.
Statistical analysis
All values are reported as means ± SEM. Comparisons of HR, MAP and forearm haemodynamics were assessed by two‐way (time point × trial) repeated measures ANOVA. For Protocol 1, the peak change in FVC was assessed using a two‐way (condition × blockade) repeated measures ANOVA. For Protocol 2, the peak change in FVC was assessed using a one‐way repeated measures ANOVA. In both protocols Student–Newman–Keuls post hoc pairwise comparisons were made with significance set a priori at P < 0.05. Data sets are not powered to detect differences between men and women, although no trends for bimodal distribution were readily apparent in this investigation.
Results
Protocol 1: ACh‐mediated vasodilatation during exercise (n = 7)
Haemodynamics at rest, immediately prior to ACh infusion (pre‐ACh), and at the absolute peak response to ACh are presented in Table 1. Resting FBF and FVC were similar across all conditions. Prior to ACh infusion (pre‐ACh), there was no difference in steady‐state FVC between 5% MVC exercise and SNP conditions, and both were higher than saline control (FVC Pre‐ACh: saline: 31 ± 3, SNP: 85 ± 9, 5% MVC: 90 ± 11 ml min−1 100 mmHg−1; both P < 0.05 vs. saline). During control saline conditions, ACh‐mediated vasodilatation elicited a peak ΔFVC of 117 ± 14 ml min−1 100 mmHg−1 (Fig. 3 C). The peak vasodilatory response to ACh was not altered during SNP infusion (peak ACh ΔFVC: saline: 117 ± 14, SNP: 130 ± 22 ml min−1 100 mmHg−1; P > 0.05; Fig. 3 C). In contrast, exercise more than doubled the peak vasodilatory response to ACh compared to saline control (peak ACh ΔFVC: 5% MVC: 236 ± 21 ml min−1 100 mmHg−1; P < 0.05 vs. saline and SNP) (Fig. 3 C).
Table 1.
Protocol 1: forearm and systemic haemodynamics at rest, pre‐ACh, and peak ACh‐mediated vasodilatation
| Condition | Trial | Timepoint | Forearm blood flow (ml min−1) | Mean arterial pressure (mmHg)# | Forearm vascular conductance (ml min−1 100 mmHg−1) | Heart rate (beats min−1)§ |
|---|---|---|---|---|---|---|
| Control | Saline | Rest | 26 ± 3 | 91 ± 2 | 29 ± 3 | 57 ± 4 |
| Pre‐ACh | 29 ± 3 | 91 ± 2 | 31 ± 3 | 56 ± 3 | ||
| Peak ACh | 133 ± 12 | 92 ± 2 | 146 ± 13 | – | ||
| SNP | Rest | 31 ± 3 | 94 ± 3 | 33 ± 3 | 54 ± 4 | |
| Pre‐ACh | 79 ± 9* | 92 ± 3 | 85 ± 9* | 58 ± 4 | ||
| Peak ACh | 197 ± 12* | 93 ± 3 | 214 ± 18* | – | ||
| 5% MVC | Rest | 29 ± 3 | 92 ± 3 | 32 ± 3 | 57 ± 4 | |
| Pre‐ACh | 84 ± 11* | 93 ± 3 | 90 ± 11* | 62 ± 4 | ||
| Peak ACh | 295 ± 17*, † | 92 ± 3 | 323 ± 19*, † | – | ||
| BaCl2 | Saline | Rest | 24 ± 4 | 94 ± 3 | 25 ± 3 | 59 ± 4 |
| Pre‐ACh | 22 ± 3 | 97 ± 4 | 22 ± 2 | 57 ± 4 | ||
| Peak ACh | 214 ± 28‡ | 97 ± 3 | 219 ± 24 | – | ||
| SNP | Rest | 28 ± 6 | 99 ± 3 | 28 ± 5 | 59 ± 4 | |
| Pre‐ACh | 66 ± 11*, ‡ | 98 ± 2 | 67 ± 9*, ‡ | 59 ± 4 | ||
| Peak ACh | 266 ± 25* | 95 ± 3 | 280 ± 27* | – | ||
| 5% MVC | Rest | 27 ± 3 | 95 ± 3 | 29 ± 3 | 57 ± 4 | |
| Pre‐ACh | 55 ± 5*, ‡ | 96 ± 2 | 58 ± 5*, ‡ | 62 ± 3 | ||
| Peak ACh | 257 ± 24* | 96 ± 2 | 268 ± 24* | – |
Values are shown as means ± SEM; ACh: acetylcholine; BaCl2: barium chloride; SNP: sodium nitroprusside; MVC: maximum voluntary contraction.
*P < 0.05 vs. saline within condition.
†P < 0.05 vs. SNP within condition.
‡P < 0.05 vs. time point between condition.
#P < 0.05 effect of condition.
§P < 0.05 effect of trial.
Figure 3. Exercise amplifies peak ACh‐mediated vasodilatation.
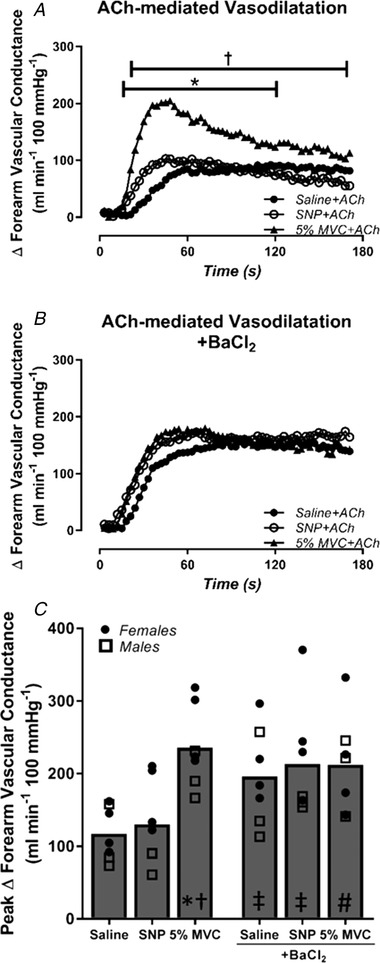
The change in forearm vascular conductance (FVC) in response to ACh during saline control, sodium nitroprusside (SNP) (high flow control) and mild intensity exercise (5% MVC) before and after inhibition of inwardly rectifying potassium (KIR) channels with barium chloride (BaCl2). A, exercise significantly augments peak ACh‐mediated ΔFVC relative to saline and high flow control (SNP) conditions. B and C, inhibition of KIR channels significantly increased the peak vasodilatory response to ACh during saline and high flow (SNP) conditions relative to control conditions, although inhibition of KIR channels prevented the exercise‐induced amplification of ACh‐mediated vasodilatation (P = 0.07). *P < 0.05 vs. saline within condition; † P < 0.05 vs. SNP within condition; ‡ P < 0.05 vs. respective control condition; # P = 0.07 vs. 5% MVC in control conditions.
Infusion of BaCl2 to block KIR channels did not impact resting haemodynamics compared to control conditions (Table 1). Similar to previous reports (Crecelius et al. 2014, 2015 b), BaCl2 reduced the vasodilatory response to 5% MVC exercise by ∼30% (Pre‐ACh FVC: 5% MVC: 90 ± 11, 5% MVC + BaCl2: 58 ± 5 ml min−1 100 mmHg−1; P < 0.05; Table 1), consistent with effective blockade of KIR channels. Prior to ACh infusion (Pre‐ACh), there was no difference in steady‐state FVC between 5% MVC exercise and SNP conditions, and both remained higher than saline control (Pre‐ACh FVC: saline + BaCl2: 22 ± 2, SNP + BaCl2: 67 ± 9, 5% MVC + BaCl2: 58 ± 5 ml min−1 100 mmHg−1; both P < 0.05 vs. saline). Peak change in FVC to ACh during saline + BaCl2 infusion was greater than that observed during control saline conditions (peak ΔFVC: ACh + saline: 117 ± 14, Ach + BaCl2: 196 ± 25 ml min−1 100 mmHg−1; P < 0.05; Fig. 3 C). Similar to control conditions, the peak vasodilatory response to ACh was not altered during SNP + BaCl2 compared with saline + BaCl2 (peak ACh ΔFVC: saline + BaCl2: 196 ± 25 ml min−1 100 mmHg−1, SNP + BaCl2: 213 ± 30 ml min−1 100 mmHg−1; P > 0.05; Fig. 3 C). Importantly, after blockade of KIR channels, there was no further exercise‐induced amplification of ACh vasodilatation (peak ACh ΔFVC: 5% MVC + BaCl2: 212 ± 25 ml min−1 100 mmHg−1; P > 0.05 vs. saline + BaCl and SNP + BaCl) (Fig. 3 C & Fig. 4).
Figure 4. Amplification of ACh‐mediated vasodilatation during exercise is attenuated by blockade of inwardly rectifying potassium channels.
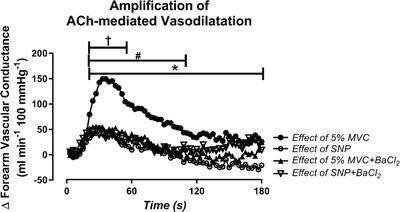
The effect of 5% MVC exercise and sodium nitroprusside (SNP) on ACh‐mediated vasodilatation (amplification of ACh), calculated by subtracting the change in FVC observed during control infusion from the change in FVC in response to ACh during exercise or SNP (ΔFVC − ΔFVC control). A, exercise amplifies ACh‐mediated vasodilatation beyond what is observed during a passive high flow control condition (SNP). B and C, blockade of inwardly rectifying potassium (KIR) channels with barium chloride (BaCl2) attenuated the effect of exercise on ACh‐mediated vasodilatation with no change on the effect of SNP. * P < 0.05, effect of 5% MVC vs. effect of SNP; † P < 0.05, effect of 5% MVC vs. effect of SNP + BaCl2; # P < 0.05, effect of 5% MVC vs. effect of 5% MVC + BaCl2.
Protocol 2A: SNP‐mediated vasodilatation during exercise (n = 8)
Haemodynamics at rest, immediately prior to SNP (Pre‐SNP) and at the absolute peak response to SNP are presented in Table 2. Resting FBF and FVC were similar across all conditions. Prior to SNP infusion (Pre‐SNP), there was no difference in steady‐state FVC between 5% MVC exercise and ACh conditions, and both were higher than saline control (FVC Pre‐SNP: saline: 37 ± 5, ACh: 98 ± 10, 5% MVC: 101 ± 11 ml min−1 100 mmHg−1; both P < 0.05 vs. saline). During control saline conditions, SNP elicited a peak ΔFVC of 158 ± 35 ml min−1 100 mmHg−1 (Fig. 5 B). Infusion of ACh had no impact on subsequent peak ΔSNP‐mediated dilatation (peak SNP ΔFVC: saline: 158 ± 35, ACh: 126 ± 37 ml min−1 100 mmHg−1, P > 0.05 vs. saline) (Fig. 5 B). Similarly, there was no exercise‐induced augmentation of SNP vasodilatation (peak SNP ΔFVC: 5% MVC: 121 ± 22 ml min−1 100 mmHg−1; P > 0.05) (Fig. 5 B)
Table 2.
Protocols 2A and 2B: forearm and Systemic Haemodynamics at rest, Pre‐SNP or ACh, and peak SNP‐ or ACh‐mediated vasodilatation
| Trial | Timepoint | Forearm blood flow (ml min−1) | Mean arterial pressure (mmHg) | Forearm vascular conductance (ml min−1 100 mmHg−1) | Heart rate (beats min−1) |
|---|---|---|---|---|---|
| Saline | Rest | 33 ± 5 | 93 ± 3 | 36 ± 6 | 56 ± 4 |
| Pre‐SNP | 33 ± 4 | 91 ± 4 | 37 ± 5 | 56 ± 4 | |
| Peak SNP | 159 ± 28 | 90 ± 4 | 182 ± 36 | – | |
| ACh | Rest | 32 ± 3 | 96 ± 4 | 34 ± 4 | 55 ± 4 |
| Pre‐SNP | 91 ± 8*, † | 94 ± 3 | 98 ± 10*, † | 56 ± 3 | |
| Peak SNP | 197 ± 34 | 91 ± 5 | 221 ± 39 | – | |
| 5% MVC | Rest | 31 ± 4 | 94 ± 3 | 34 ± 4 | 57 ± 5 |
| Pre‐SNP | 94 ± 10*, † | 94 ± 3 | 101 ± 11*, † | 59 ± 4 | |
| Peak SNP | 195 ± 26 | 90 ± 3 | 222 ± 30 | – | |
| Saline | Rest | 24 ± 4 | 98 ± 5 | 25 ± 4 | 54 ± 3 |
| Pre‐ACh | 27 ± 3 | 99 ± 5 | 28 ± 4 | 55 ± 4 | |
| Peak ACh | 119 ± 11 | 97 ± 4 | 124 ± 14 | – | |
| KCl | Rest | 29 ± 2 | 94 ± 5 | 31 ± 3 | 57 ± 4 |
| Pre‐ACh | 56 ± 5*, † | 98 ± 3 | 58 ± 6*, † | 57 ± 5 | |
| Peak ACh | 192 ± 17‡ | 99 ± 8 | 195 ± 18‡ | – |
Values are shown as means ± SEM; ACh: acetylcholine; SNP: sodium nitroprusside; KCl: potassium chloride; MVC: maximum voluntary contraction.
*P < 0.05 vs. rest within trial.
†P < 0.05 vs. saline baseline.
‡P < 0.05 vs. saline peak ACh.
Figure 5. Exercise does not amplify SNP‐mediated vasodilatation.
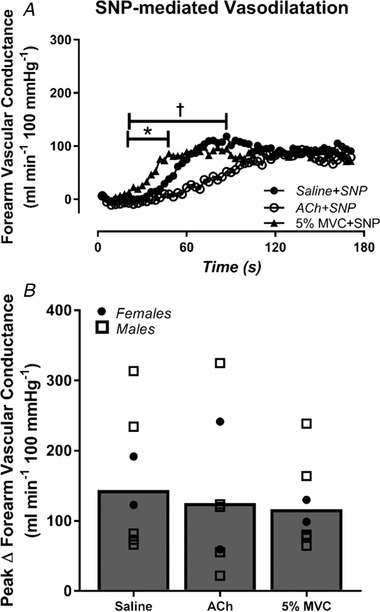
The change in forearm vascular conductance (FVC) in response to sodium nitroprusside (SNP) during saline control, Ach (high flow control) and mild intensity exercise (5% MVC). A, there were no differences in SNP‐mediated vasodilatation quantified as ΔFVC from baseline. B, peak ΔFVC in response to SNP was not different during exercise or ACh (high flow control) compared to control saline conditions. * P < 0.05, 5% MVC vs. saline; † P < 0.05, 5% MVC vs. ACh.
Protocol 2B: ACh‐mediated vasodilatation during pharmacological activation of KIR channels (n = 6)
Haemodynamics at rest, immediately prior to ACh (Pre‐ACh) and the absolute peak response to ACh are presented in Table 2. Resting FBF and FVC were similar across all conditions. As expected, steady‐state FVC was higher during infusion of KCl compared to saline control (Pre‐ACh FVC: saline: 28 ± 4, KCl: 58 ± 6 ml min−1 100 mmHg−1; P < 0.05). During control saline conditions, ACh elicited a peak ΔFVC of 96 ± 12 ml min−1 100 mmHg−1 (Fig. 6 B). Pharmacological activation of KIR channels in quiescent muscle, via infusion of KCl, amplified the peak vasodilatation in response to ACh by ∼50% (peak ΔFVC: 139 ± 14 ml min−1 100 mmHg−1; P < 0.05 vs. saline) (Fig. 6 B).
Figure 6. Infusion of KCl in quiescent skeletal muscle amplifies peak ACh‐mediated vasodilatation.
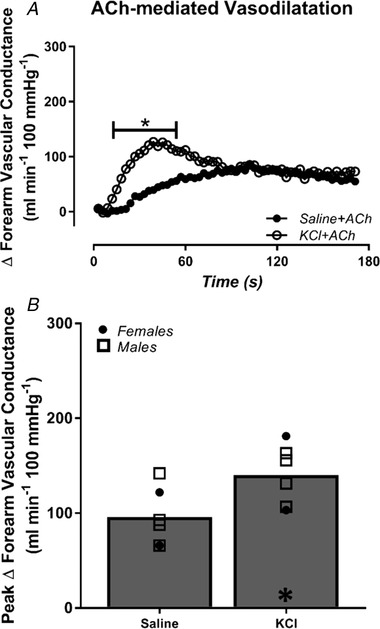
The change in forearm vascular conductance (FVC) in response to ACh is presented during control saline, and infusion of potassium chloride (KCl) to activate inwardly rectifying potassium (KIR) channels in quiescent muscle. Peak ACh‐mediated vasodilatation was amplified during KCl infusion compared to saline control. * P < 0.05 vs. saline.
Discussion
The results from this investigation are the first in humans to demonstrate that contracting skeletal muscle differentially amplifies certain types of vasodilatory signalling. First, we show that the peak response to the endothelium‐dependent vasodilator ACh is significantly amplified during mild intensity handgrip exercise. In contrast, there is no amplification of the endothelium‐independent NO donor SNP, which suggests that the amplification of ACh is independent of NO signalling and probably represents amplification of EDH during exercise. Second, while the control response to ACh was greater in the presence of BaCl2, inhibition of KIR channels prevented any further exercise‐induced amplification of ACh‐mediated vasodilatation. Third, pharmacological activation of KIR channels in quiescent muscle amplifies peak ACh‐mediated vasodilatation similar to exercise. Taken together, these findings demonstrate that endothelium‐dependent vasodilatory signalling is amplified in contracting skeletal muscle and provides initial evidence that KIR channels may act as amplifiers of EDH‐like vasodilatation during exercise in humans.
Endothelium‐dependent vasodilatory signalling during exercise
In animal models, skeletal muscle contraction causes a conducted vasodilatory response that relies on spread of EDH along the endothelium to upstream arterioles (Segal & Jacobs, 2001; Duza & Sarelius, 2004; Murrant et al. 2017). Furthermore, disruption of the endothelium (Segal & Jacobs, 2001; Sinkler & Segal, 2017), or genetic knock out of endothelial SKca channels and gap junction protein connexin 40 (Milkau et al. 2010), significantly attenuates contraction‐induced hyperaemia, highlighting the critical importance of the endothelium for mediating the vasodilatory response to exercise. The present study demonstrates for the first time in humans that the vasculature of contracting skeletal muscle is extraordinarily sensitive to endothelium‐dependent vasodilatory signalling elicited by ACh. Importantly, there was no change in the sensitivity of the vasculature to SNP‐mediated vasodilatation during exercise, confirming that the amplification of ACh in contracting skeletal muscle is not due to a generalized sensitization of the vasculature to all dilatory signals. Additionally, high flow control conditions reveal that the amplification is not explained by changes in baseline vessel tone, blood flow or drug delivery. Rather, the augmentation of ACh represents a distinct phenomenon that is specific to ACh‐mediated, endothelium‐dependent vasodilatory signalling. The fundamental signal underlying ACh vasodilatation is a rise in endothelial cell calcium that will broadly result in the generation of EDH and the production of autacoids, such as NO, that will cause vascular smooth muscle relaxation. It has been demonstrated in animal models that administration of a direct NO donor (SNP) causes vasodilatation independent of a rise in endothelial cell calcium (Tallini et al. 2007). NO can act directly on smooth muscle cell K+ channels, and cGMP‐dependent protein kinases/phosphatases to dephosphorylate myosin light chains and cause smooth muscle relaxation (Koeppen et al. 2004). Considering there was no amplification of NO‐mediated vasodilatation in our experimental approach, it is likely that endothelial cell calcium and initiation of EDH‐like signalling is necessary to observe amplification of ACh (Smith et al. 2008; Sonkusare et al. 2016).
Interestingly, the dramatic and rapid increase in vasodilatation followed by a gradual decline back towards baseline levels observed in response to ACh during exercise and KCl infusion (Figs 4 and 6) mirrors the temporal pattern of changes in intracellular calcium, membrane potential and specifically Kca channel activation in response to ACh in animal models (Murrant et al. 2004; Wölfle et al. 2009; Behringer & Segal, 2015). The similarity of these temporal patterns supports the idea that the amplification of ACh during exercise reflects EDH‐like signalling mediated through changes in intracellular calcium or membrane potential. The gradual decline in forearm vascular conductance after the initial peak may also represent metabolic autoregulation whereby over‐perfusion of a metabolically active tissue can washout vasoactive metabolites, reduce vasodilatory signalling and thus decrease blood flow in an attempt to ‘correct’ the mismatch between oxygen delivery and oxygen demand. In either case, it is possible that augmentation of endothelium‐dependent signalling may serve as a feedforward mechanism that acts to initiate immediate alterations in vascular tone that are sustained or fine‐tuned by alternative feedback dilatory pathways that were not activated during these pharmacological manipulations.
Role of KIR channels as electrical amplifiers
KIR channels are thought to be a primary mechanism by which the vasodilatory response to EDH is amplified and conducted within the resistance vasculature (Jackson, 2017). KIR channel activity is in part regulated by voltage‐dependent Mg2+ inhibition such that hyperpolarization of the vasculature results in removal of the Mg2+ inhibition and thus greater extrusion of K+ outside of the cell membrane. This unique voltage‐dependent property of KIR channels allows them to operate as ‘electrical amplifiers’ whereby hyperpolarization of the endothelium results in greater hyperpolarization via activation of KIR channels, in essence amplifying the original stimulus (Jantzi et al. 2006; Sonkusare et al. 2016). Animal models have convincingly demonstrated that KIR channels serve to both amplify and conduct EDH initiated by ACh and other endothelium‐dependent vasodilatory substances (Rivers et al. 2001; Smith et al. 2008). Our laboratory has previously identified a primary role for KIR channels in mediating the net vasodilatory response to skeletal muscle contraction (Crecelius et al. 2013 a, 2014), although it was not clear whether KIR channels serve as amplifiers of endothelium‐dependent vasodilatory signalling at rest or during exercise in humans.
To test this hypothesis, we assessed the amplification of ACh‐mediated vasodilatation during exercise before and after administration of BaCl2 to inhibit KIR channels. Unexpectedly, infusion of BaCl2 increased the peak vasodilatory response to ACh under resting conditions (Fig. 3 C). The unexpected augmentation of ACh during BaCl2 infusion could lead to the interpretation that KIR channels act to restrain endothelium‐dependent vasodilation under resting conditions. However, this scenario is unlikely as KIR channel activation in the endothelium or smooth muscle causes vasodilatation, and the activation of Gq/11 coupled receptors and subsequent initiation of EDH and production of NO (as would be expected during infusion of ACh) have all been linked to activation, not inhibition, of KIR channels (Schubert et al. 2004; Jackson, 2017). The more likely interpretation is that a non‐specific interaction with BaCl2 augmented the vasodilatory response to ACh in this experimental preparation, thus changing the ‘normal control’ response to ACh (discussed in ‘Experimental considerations’). Regardless, when compared to the ACh dilatory response in the presence of BaCl2, the exercise‐induced amplification of ACh was no longer present after inhibition of KIR channels (Fig. 4). It is important to note that the lack of augmentation cannot be explained simply by diminished vasodilatory reserve remaining to observe amplification of the dilatory response, as our laboratory and others have shown greater vasodilatory capacity during both pharmacological and physiological (e.g. exercise) stimuli (Kirby et al. 2012; Crecelius et al. 2013 b). Even when the peak ACh response during 5% MVC exercise + BaCl2 is compared to the control 5% MVC exercise condition, the peak ACh‐mediated change in FVC tended to be lower after inhibition of KIR channels, but not statistically different (P = 0.07).
Therefore, to further test the hypothesis that KIR channels can amplify endothelium‐dependent vasodilatation in humans, infusion of KCl was used to pharmacologically activate KIR channels in the absence of changes in metabolic demand. During pharmacological activation of KIR channels, the peak response to ACh was amplified in a manner that was characteristically similar to handgrip exercise (Figs 2 and 6). In both cases, the most dramatic effect on ACh‐mediated vasodilatation occurred within the first 20 s after initiation of the vasodilatory response, followed by a gradual decline in vascular conductance back to control ACh levels (Figs 3 A, 4 and 6 A). These are the first data in humans to demonstrate that KCl can amplify endothelium‐dependent vasodilatation, and are largely supportive of our findings that activation of KIR channels amplifies Ach‐mediated vasodilatation in contracting muscle. In addition to greater KIR channel conductance directly amplifying the vasodilatory response to EDH, an indirect effect of changes in membrane potential associated with exercise or KCl infusion may independently influence the response to ACh via alterations in calcium signalling. In this context, Behringer & Segal ( 2015) demonstrated that the rise in intracellular calcium in response to ACh becomes progressively greater as membrane potential becomes more hyperpolarized, consistent with the greater electrochemical driving force of calcium. In the present set of studies, both handgrip exercise and KCl infusion would be expected to hyperpolarize the vasculature (Segal & Jacobs, 2001; Burns et al. 2004) resulting in a greater calcium influx in response to the same dose of ACh. Given the experimental limitations of in vivo investigations in humans, it is currently not possible to determine if exercise or KCl augment ACh‐mediated dilatation due to direct KIR‐mediated amplification of EDH, or secondary to greater activation of SK/IKca channels due to elevated calcium influx. In either case, it appears that KIR channels serve as a mechanism through which endothelium‐dependent dilatory signals are amplified in humans.
Integration of KIR channel activity in human skeletal muscle vasculature
Collective data from our laboratory and others (Dawes et al. 2002; Dwivedi et al. 2005) indicate that at rest, the vasculature of human skeletal muscle may operate somewhere near the ‘activation potential’ for KIR channels. Our laboratory has typically shown a modest reduction in vascular conductance after administration of BaCl2 to resting forearm skeletal muscle, suggesting some contribution of KIR channels to resting vascular tone (Crecelius et al. 2012, 2013 b, 2014; Hearon et al. 2017). Although slightly variable across conditions (∼0–20%), the data from the present study are generally consistent with a modest activation of KIR channels under resting conditions in humans (Table 1). From this resting membrane potential, small changes in vascular signalling, in this case via muscle contraction or low doses of KCl, will hyperpolarize membrane potential leading to greater KIR channel conductance. Any additional or subsequent hyperpolarizing signal (e.g. SK/IKca activation) will result in greater KIR channel conductance and augmentation of electrical communication along arterioles. In the context of exercise hyperaemia, K+ released from skeletal muscle fibres may elevate interstitial K+ and directly activate KIR channels immediately upon the initiation of skeletal muscle contraction eliciting a feedforward vasodilatory response (Mohrman & Sparks, 1974; Armstrong et al. 2007). Indeed, KIR channels have been shown to contribute to the rapid‐onset vasodilatory response observed after a single brief muscle contraction, suggesting that activation of KIR channels is among the first signalling events during exercise (Armstrong et al. 2007; Crecelius et al. 2013 a). We propose a working hypothesis (Fig. 7) that the presence of increased interstitial K+ and increased KIR channel activity immediately at the onset of muscle contractions will elicit feedforward vasodilatation and increase the activity of KIR channels such that any subsequent local and/or metabolic EDH‐like signalling initiated by other substances within the vasculature of contracting muscle (Clifford & Hellsten, 2004; Casey & Joyner, 2011) will be amplified via greater conductance through KIR channels. The initial actions of extracellular K+ resulting in rapid vasodilatation and amplification of EDH‐like signals will facilitate robust and rapid conduction of vasodilatory signalling to upstream arterioles and feed arteries and ensure adequate blood flow and oxygen delivery to active skeletal muscle.
Figure 7. Working hypothesis on KIR channel‐mediated regulation of skeletal muscle blood flow.
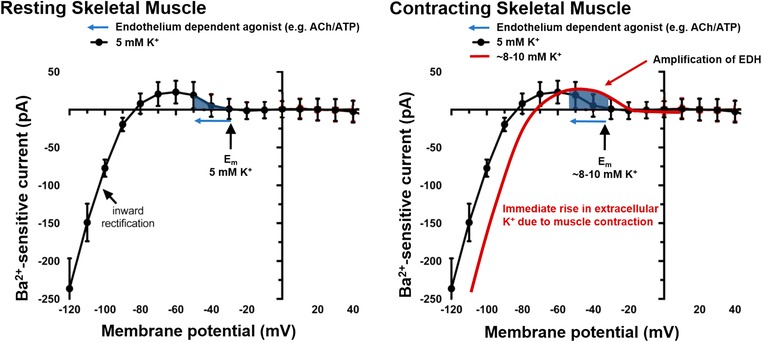
In resting skeletal muscle (left panel), the prevailing extracellular potassium levels (∼4–5 mM) and membrane potential (E m) are near the activation potential of KIR channels, resulting in mild activation of KIR channels and a modest contribution to resting vascular tone. Application of certain endothelium‐dependent agonists may hyperpolarize membrane potential (leftward arrow) and recruit current through KIR channels to elicit vasodilatation. Immediately upon initiation of exercise (right panel), efflux of K+ from skeletal muscle fibres will increase extracellular [K+] (∼8–10 mM) shifting the I–V relationship (solid line) rightward, resulting in activation of KIR channels and feedforward vasodilatation. Subsequently, any increase in endothelium‐dependent signalling associated with muscle contractions will be amplified via greater conductance through KIR channels. Thus, for the same EDH stimulus (e.g. ACh or ATP) during muscle contractions, there will be greater recruitment of KIR channel conductance and amplification of the original stimulus, facilitating greater upstream conduction of vasodilator signalling (figure adapted from Jackson, 2017). [Color figure can be viewed at wileyonlinelibrary.com]
Experimental considerations
For this initial investigation, we chose to use ACh as the endothelium‐dependent vasodilatory stimulus for a few key reasons. First, the endothelium dependence and downstream signalling mechanisms of ACh are well characterized (Behringer & Segal, 2012). Second, our laboratory recently established that the vasodilatation initiated by ACh within contracting skeletal muscle appears to be independent of the production of the endothelial autacoids NO and prostaglandins (Hearon et al. 2016). The resulting vasodilatation is thought to rely heavily on EDH (Hoepfl et al. 2002; Dabisch et al. 2004; Hilgers et al. 2006; De Wit, 2010) and demonstrates vasomotor properties that are unique to EDH‐like signalling, including the ability to attenuate sympathetic vasoconstriction (Kurjiaka & Segal, 1995; Hearon et al. 2016). Third, the vasodilatory response to ACh in humans has previously been shown to have little contribution from KIR channels under resting conditions (Dwivedi et al. 2005; Crecelius et al. 2012), which was confirmed in this set of experiments.
The exaggerated peak vasodilatation to ACh in the presence of BaCl2 observed in this investigation is similar to previous observations of exaggerated vasoconstrictor responses to the α1‐adrenergic agonist phenylephrine in the presence of BaCl2 (Crecelius et al. 2015 b; Hearon et al. 2017). Both phenylephrine and ACh are Gq/11 protein coupled receptors that rely on calcium entry into the endothelium or vascular smooth muscle, respectively. It is possible that small changes in membrane potential or calcium entry in the presence of elevated extracellular BaCl2 alter the downstream response to Gq/11 protein coupled receptor activation. The shared signalling mechanisms could explain why BaCl2 has been observed to augment both vasodilatory and vasoconstrictor responses under resting conditions.
Finally, future studies will be required to determine if responses of other candidate vasodilators believed to be involved in the regulation of blood flow are amplified in contracting skeletal muscle. Along these lines, intravascular (circulating) ATP increases during exercise and elicits vasodilatation and is thought to be an important contributor to exercise hyperaemia in humans (Gonzalez‐Alonso et al. 2002; Kirby et al. 2012, 2013; Crecelius et al. 2015 a). Interestingly, both ATP and ACh cause vasodilatation by binding to Gq/11 protein coupled receptors (P2y2 and M3, respectively) and initiating EDH (Garland et al. 2011). Activated KIR channels are the primary downstream effectors of ATP mediated vasodilatation in humans (Crecelius et al. 2012; Hearon et al. 2017), and therefore may be a physiological contributor to exercise‐induced vasodilatory signalling (Hearon & Dinenno, 2017), and perhaps contribute to the amplification of other vasodilatory substances (Kwan et al. 2003; Liu et al. 2004; Fujii et al. 2015). Future studies should address whether ATP‐mediated signalling is amplified during exercise, and whether ATP itself is capable of amplifying or altering the vasoactive properties of other vasodilators in a similar manner to exercise.
Conclusions
The results of the present investigation demonstrate for the first time that endothelium‐dependent ACh‐mediated vasodilatation is amplified in contracting skeletal muscle of humans. In contrast, the endothelium‐independent vasodilator SNP is not augmented, demonstrating that specific dilatory pathways may be amplified during skeletal muscle contraction. Furthermore, the amplification of ACh may be due in part to activation of KIR channels, which is consistent with animal studies demonstrating that KIR channels may act as electrical amplifiers of endothelium‐dependent vasodilatory signalling. We hypothesize that K+ efflux from skeletal muscle may be a key feedforward vasodilatory signal that activates vascular KIR channels to cause vasodilatation and amplify EDH‐mediated vasodilatory signalling in humans.
Additional information
Competing interests
None.
Author contributions
All experiments were performed in The Human Cardiovascular Physiology Laboratory, Colorado State University, Fort Collins, CO, USA. C.M.H. and F.A.D. contributed to conception and design of the experiments, collection, analysis and interpretation of the data, and writing the manuscript. J.C.R. and M.L.R. contributed to the experimental design, interpretation of data, and provided critical revision of the manuscript. G.J.L and D.G.L. contributed to the experimental design, provided invasive methodology for data collection, and critical revision of the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by the National Institutes of Health award NIH R01HL119337 (F.A.D.)
Biography
Christopher Hearon Jr received his undergraduate degree from Texas Tech University (2010), his MS from the University of Colorado Boulder (2012) and PhD under Dr Frank Dinenno in the Human Cardiovascular Physiology Laboratory at Colorado State University (2016). He is currently the recipient of a Post‐Doctoral Ruth L. Kirschstein National Research Service Award mentored by Dr Benjamin Levine at the University of Texas Southwestern Medical Center. His research focuses on the signalling mechanisms regulating peripheral vascular tone with a particular interest in the role of the sympathetic nervous system in ageing, pathophysiological conditions and environmental extremes.

Edited by: Harold Schultz & Fernando Santana
Linked articles: This article is highlighted in a Perspectives article by Welsh. To read this article, visit https://doi.org/10.1113/JP277513.
References
- Armstrong ML, Dua AK & Murrant CL (2007). Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2012). Spreading the signal for vasodilatation: implications for skeletal muscle blood flow control and the effects of ageing. J Physiol 590, 6277–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behringer EJ & Segal SS (2015). Membrane potential governs calcium influx into microvascular endothelium: integral role for muscarinic receptor activation. J Physiol 593, 4531–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns WR, Cohen KD & Jackson WF (2004). K+‐induced dilation of hamster cremasteric arterioles involves both the Na+/K+‐ATPase and inward‐rectifier K+ channels. Microcirculation (New York, NY: 1994) 11, 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey DP & Joyner MJ (2011). Local control of skeletal muscle blood flow during exercise: influence of available oxygen. J Appl Physiol (1985) 111, 1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford PS & Hellsten Y (2004). Vasodilatory mechanisms in contracting skeletal muscle. J Appl Physiol (1985) 97, 393–403. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS & Dinenno FA (2015. a). Intravascular ATP and the regulation of blood flow and oxygen delivery in humans. Exerc Sport Sci Rev 43, 5–13. [DOI] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Hearon CM, Luckasen GJ, Larson DG & Dinenno FA (2015. b). Contracting human skeletal muscle maintains the ability to blunt α1‐adrenergic vasoconstriction during KIR channel and Na+/K+‐ATPase inhibition. J Physiol 593, 2735–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Luckasen GJ, Larson DG & Dinenno FA (2012). ATP‐mediated vasodilatation occurs via activation of inwardly rectifying potassium channels in humans. J Physiol 590, 5349–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Kirby BS, Luckasen GJ, Larson DG & Dinenno FA (2013. a). Mechanisms of rapid vasodilation after a brief contraction in human skeletal muscle. Am J Physiol Heart Circ Physiol 305, H29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Luckasen GJ, Larson DG & Dinenno FA (2014). KIR channel activation contributes to onset and steady‐state exercise hyperemia in humans. Am J Physiol Heart Circ Physiol 307, H782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crecelius AR, Richards JC, Luckasen GJ, Larson DG & Dinenno FA (2013. b). Reactive hyperemia occurs via activation of inwardly rectifying potassium channels and Na+/K+‐ATPase in humans. Circ Res 113, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabisch PA, Liles JT, Taylor JT, Sears BW, Saenz R & Kadowitz PJ (2004). Role of potassium channels in the nitric oxide‐independent vasodilator response to acetylcholine. Pharmacol Res 49, 207–215. [DOI] [PubMed] [Google Scholar]
- Dawes M, Sieniawska C, Delves T, Dwivedi R, Chowienczyk PJ & Ritter JM (2002). Barium reduces resting blood flow and inhibits potassium‐induced vasodilation in the human forearm. Circulation 105, 1323–1328. [DOI] [PubMed] [Google Scholar]
- De Wit C (2010). Different pathways with distinct properties conduct dilations in the microcirculation in vivo. Cardiovasc Res 85, 604–613. [DOI] [PubMed] [Google Scholar]
- Dinenno FA & Joyner MJ (2003). Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory? J Physiol 553, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duza T & Sarelius IH (2004). Increase in endothelial cell Ca2+ in response to mouse cremaster muscle contraction. J Physiol 555, 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi R, Saha S, Chowienczyk PJ & Ritter JM (2005). Block of inward rectifying K+ channels (KIR) inhibits bradykinin‐induced vasodilatation in human forearm resistance vasculature. Arterioscler Thromb Vasc Biol 25, e7–9. [DOI] [PubMed] [Google Scholar]
- Emerson GG & Segal SS (2000). Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 86, 94–100. [DOI] [PubMed] [Google Scholar]
- Fujii N, Halili L, Singh MS, Meade RD & Kenny GP (2015). Intradermal administration of ATP augments methacholine‐induced cutaneous vasodilation but not sweating in young males and females. Am J Physiol Regul Integr Comp Physiol 309, R912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora Ka (2011). EDHF: Spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Alonso J, Olsen DB & Saltin B (2002). Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res 91, 1046–1055. [DOI] [PubMed] [Google Scholar]
- Hearon CM, Jr & Dinenno FA (2017). KIR channels mediate vasodilation but not sympatholysis. Channels (Austin) 11, 495–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearon CM, Kirby BS, Luckasen G, Larson D & Dinenno FA (2016). Endothelium‐dependent vasodilatory signalling modulates α1‐adrenergic vasoconstriction in contracting skeletal muscle of humans. J Physiol 594, 7435–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearon CM, Richards JC, Racine ML, Luckasen GJ, Larson DG, Joyner MJ & Dinenno FA (2017). Sympatholytic effect of intravascular ATP is independent of nitric oxide, prostaglandins, Na+/K+‐ATPase and K IR channels in humans. J Physiol 595, 5175–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgers RHP, Todd J & Webb RC (2006). Regional heterogeneity in acetylcholine‐induced relaxation in rat vascular bed: role of calcium‐activated K+ channels. Am J Physiol Heart Circ Physiol 291, H216–222. [DOI] [PubMed] [Google Scholar]
- Hoepfl B, Rodenwaldt B, Pohl U & De Wit C (2002). EDHF, but not NO or prostaglandins, is critical to evoke a conducted dilation upon ACh in hamster arterioles. Am J Physiol Heart Circ Physiol 283, H996–1004. [DOI] [PubMed] [Google Scholar]
- Jackson WF (2017). Boosting the signal: endothelial inward rectifier K+ channels. Microcirculation 24, e12319–e12319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jantzi MC, Brett SE, Jackson WF, Corteling R, Vigmond EJ & Welsh DG (2006). Inward rectifying potassium channels facilitate cell‐to‐cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol 291, H1319–1328. [DOI] [PubMed] [Google Scholar]
- Kerr PM, Tam R, Ondrusova K, Mittal R, Narang D, Tran CHT, Welsh DG & Plane F (2012). Endothelial feedback and the myoendothelial projection. Microcirculation 19, 416–422. [DOI] [PubMed] [Google Scholar]
- Kerr PM, Wei R, Tam R, Sandow SL, Murphy TV, Ondrusova K, Lunn SE, Tran CHT, Welsh DG & Plane F (2015). Activation of endothelial IKCa channels underlies NO‐dependent myoendothelial feedback. Vascular pharmacology 74, 130–138. [DOI] [PubMed] [Google Scholar]
- Kirby BS, Crecelius AR, Richards JC & Dinenno FA (2013). Sources of intravascular ATP during exercise in humans: critical role for skeletal muscle perfusion. Exp Physiol 98, 988–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby BS, Crecelius AR, Voyles WF & Dinenno FA (2012). Impaired skeletal muscle blood flow control with advancing age in humans: attenuated ATP release and local vasodilation during erythrocyte deoxygenation. Circ Res 111, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeppen M, Feil R, Siegl D, Feil S, Hofmann F, Pohl U & de Wit C (2004). cGMP‐dependent protein kinase mediates NO‐ but not acetylcholine‐induced dilations in resistance vessels in vivo. Hypertension 44, 952–955. [DOI] [PubMed] [Google Scholar]
- Kurjiaka DT & Segal SS (1995). Interaction between conducted vasodilation and sympathetic nerve activation in arterioles of hamster striated muscle. Circ Res 76, 885–891. [DOI] [PubMed] [Google Scholar]
- Kwan HY, Leung PC, Huang Y & Yao X (2003). Depletion of intracellular Ca2+ stores sensitizes the flow‐induced Ca2+ influx in rat endothelial cells. Circ Res 92, 286–292. [DOI] [PubMed] [Google Scholar]
- Liu C, Mather S, Huang Y, Garland CJ & Yao X (2004). Extracellular ATP facilitates flow‐induced vasodilatation in rat small mesenteric arteries. Am J Physiol Heart Circ Physiol 286, H1688–1695. [DOI] [PubMed] [Google Scholar]
- Milkau M, Köhler R & de Wit C (2010). Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J 24, 3572–3579. [DOI] [PubMed] [Google Scholar]
- Mohrman DE & Sparks HV (1974). Role of potassium ions in the vascular response to a brief tetanus. Circ Res 35, 384–390. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Duza T, Kim MB, Cohen KD & Sarelius IH (2004). Arteriolar dilations induced by contraction of hamster cremaster muscle are dependent on changes in endothelial cell calcium. Acta Physiol Scand 180, 231–238. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Lamb IR & Novielli NM (2017). Capillary endothelial cells as coordinators of skeletal muscle blood flow during active hyperemia. Microcirculation 24. [DOI] [PubMed] [Google Scholar]
- Racine ML, Crecelius AR, Luckasen GJ, Larson DG & Dinenno FA (2018). Inhibition of Na+/K+‐ATPase and KIR channels abolishes hypoxic hyperaemia in resting but not contracting skeletal muscle of humans. J Physiol 596, 3371–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards JC, Luckasen GJ, Larson DG & Dinenno FA (2014). Role of α‐adrenergic vasoconstriction in regulating skeletal muscle blood flow and vascular conductance during forearm exercise in ageing humans. J Physiol 592, 4775–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivers RJ, Hein TW, Zhang C & Kuo L (2001). Activation of barium‐sensitive inward rectifier potassium channels mediates remote dilation of coronary arterioles. Circulation 104, 1749–1753. [DOI] [PubMed] [Google Scholar]
- Saltin B (2007). Exercise hyperaemia: magnitude and aspects on regulation in humans. J Physiol 583, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert R, Krien U, Wulfsen I, Schiemann D, Lehmann G, Ulfig N, Veh RW, Schwarz JR & Gago H (2004). Nitric oxide donor sodium nitroprusside dilates rat small arteries by activation of inward rectifier potassium channels. Hypertension 43, 891–896. [DOI] [PubMed] [Google Scholar]
- Segal SS & Jacobs TL (2001). Role for endothelial cell conduction in ascending vasodilatation and exercise hyperaemia in hamster skeletal muscle. J Physiol 536, 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal SS (2005). Regulation of blood flow in the microcirculation. Microcirculation 12, 33–45. [DOI] [PubMed] [Google Scholar]
- Senadheera S, Kim Y, Grayson, TM , Toemoe S, Kochukov MY, Abramowitz J, Housley GD, Bertrand RL, Chadha PS, Bertrand PP, Murphy TV, Tare M, Birnbaumer L, Marrelli SP & Sandow SL (2012). Transient receptor potential canonical type 3 channels facilitate endothelium‐derived hyperpolarization‐mediated resistance artery vasodilator activity. Cardiovascular Research 95, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkler SY & Segal SS (2017). Rapid versus slow ascending vasodilatation: intercellular conduction versus flow‐mediated signalling with tetanic versus rhythmic muscle contractions. J Physiol 595, 7149–7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ & Welsh DG (2008). KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol 586, 1147–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Dalsgaard T, Bonev AD & Nelson MT (2016). Inward rectifier potassium (Kir2.1) channels as end‐stage boosters of endothelium‐dependent vasodilators. J Physiol 594, 3271–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallini YN, Brekke JF, Shui B, Doran R, Hwang S‐m, Nakai J, Salama G, Segal SS & Kotlikoff MI (2007). Propagated endothelial Ca2+ waves and arteriolar dilation in vivo: measurements in Cx40BAC GCaMP2 transgenic mice. Circ Res 101, 1300–1309. [DOI] [PubMed] [Google Scholar]
- Wolfle SE, Schmidt VJ, Hoepfl B, Gebert A, Alcolea S, Gros D & de Wit C (2007). Connexin45 cannot replace the function of connexin40 in conducting endothelium‐dependent dilations along arterioles. Circ Res 101, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Wölfle SE, Schmidt VJ, Hoyer J, Köhler R & De Wit C (2009). Prominent role of KCa3.1 in endothelium‐derived hyperpolarizing factor‐type dilations and conducted responses in the microcirculation in vivo. Cardiovasc Res 82, 476–483. [DOI] [PubMed] [Google Scholar]