

Abstract
Loss of muscle mass and insulin sensitivity are common phenotypic traits of immobilisation and increased inflammatory burden. The suppression of muscle protein synthesis is the primary driver of muscle mass loss in human immobilisation, and includes blunting of post‐prandial increases in muscle protein synthesis. However, the mechanistic drivers of this suppression are unresolved. Immobilisation also induces limb insulin resistance in humans, which appears to be attributable to the reduction in muscle contraction per se. Again mechanistic insight is missing such that we do not know how muscle senses its “inactivity status” or whether the proposed drivers of muscle insulin resistance are simply arising as a consequence of immobilisation. A heightened inflammatory state is associated with major and rapid changes in muscle protein turnover and mass, and dampened insulin‐stimulated glucose disposal and oxidation in both rodents and humans. A limited amount of research has attempted to elucidate molecular regulators of muscle mass loss and insulin resistance during increased inflammatory burden, but rarely concurrently. Nevertheless, there is evidence that Akt (protein kinase B) signalling and FOXO transcription factors form part of a common signalling pathway in this scenario, such that molecular cross‐talk between atrophy and insulin signalling during heightened inflammation is believed to be possible. To conclude, whilst muscle mass loss and insulin resistance are common end‐points of immobilisation and increased inflammatory burden, a lack of understanding of the mechanisms responsible for these traits exists such that a substantial gap in understanding of the pathophysiology in humans endures.
Keywords: carbohydrate metabolism, muscle fuel selection, inactivity, bed-rest, muscle atrophy, muscle protein synthesis, muscle protein breakdown
Introduction
Loss of muscle mass and insulin sensitivity are common phenotypic traits of immobilisation (e.g. bed‐rest or limb casting) as well as being associated with ageing, inflammation and trauma, and chronic non‐communicable disease. In particular, sepsis is associated with major metabolic alterations, including significant losses of muscle mass (Hasselgren et al. 2005) and hyperglycaemia (Mizock, 2001), dysregulation of fat and carbohydrate utilisation (Saeed et al. 1999; Chambrier et al. 2000) and hyperlactataemia (Vary, 1999) that is consistent with the impairment of muscle carbohydrate oxidation and insulin resistance. However, our mechanistic understanding of the aetiology of such metabolic perturbations in immobilisation, acute trauma, sepsis and chronic non‐communicable disease is currently poor, particularly in humans, as is detailed insight of whether these negative traits can be at least partly rescued by interventions such as exercise and/or targeted drug administration in humans. Clarity of understanding towards these gaps in our understanding and new insight regarding the trajectories of change and the molecular drivers of muscle mass loss and insulin resistance is vital if in‐roads are to be made in preserving muscle mass and metabolic health in these scenarios. We therefore focus attention here on the impact of immobilisation and inflammation on the loss of muscle mass and insulin sensitivity in humans.
Immobilisation
Immobilisation induced loss of muscle mass
The maintenance of muscle mass is dependent on the balance between rates of muscle protein synthesis and muscle protein breakdown, where a chronic imbalance results in either the loss or gain of muscle mass. In vivo animal studies utilising stable isotope tracer methodologies to quantify muscle protein turnover in an acute setting conclude that muscle protein synthesis is less (Booth & Seider, 1979) and muscle protein breakdown is greater (Kobayashi et al. 2006) following both 6 and 24 h of cast immobilisation compared to the basal non‐immobilised state. Furthermore, evidence from research involving administration of the proteasome inhibitor velcade to rodents during 3 days of limb immobilisation resulted in an ∼50% sparing of muscle weight, leading to the suggestion that muscle protein breakdown predominates in the rodent during immobilisation (Krawiec et al. 2005). In contrast to rodent studies, it is thought that suppression of muscle protein synthesis is the primary driver of muscle mass loss in the immobilised state in humans (Phillips et al. 2009; Murton & Greenhaff, 2010). For example, De Boer and colleagues detected a 50% decline in the rate of post‐absorptive myofibrillar protein synthesis measured over several hours following 10 days of limb suspension in healthy, young volunteers when compared to baseline. The authors concluded that the decline in myofibrillar protein synthesis, even in these fasted state conditions, was of sufficient magnitude to fully account for the decline in muscle cross‐sectional area recorded in the same volunteers, i.e. the contribution from muscle protein breakdown to total muscle mass loss was paltry (De Boer et al. 2007). Reasons for these inconsistencies probably reside in the undoubted metabolic differences between humans and rodents, most notably the relatively greater rates of muscle protein synthesis and basal metabolic rate in rodents that have limited capacity to maintain metabolic homeostasis (Demetrius, 2005). Furthermore, most rodent studies invariably involve young animals that are still very much in their growth phase compared to adult humans, whilst the magnitude of immobilisation induced stress is likely to be far greater for rodents than for consenting human volunteers. The influence these compounding issues may have is exemplified by the degree of muscle mass loss observed between species, where 3 days of hindlimb cast immobilisation of a rodent has been shown to result in an ∼19% lower muscle mass compared to time‐matched controls (Krawiec et al. 2005), vs. an ∼5% decline in human quadriceps mass after 2 weeks of full‐limb cast immobilisation (Jones et al. 2004). However, it is important to recognise that the body of evidence published to date does not preclude a role for muscle protein breakdown during disuse atrophy in humans. Indeed, increased amounts of ubiquitin protein conjugates (Glover et al. 2008) and increased 3‐methylhistidine (Tesch et al. 2008) release in the first few days of muscle disuse point to an early and possibly transient contribution of muscle protein breakdown to the aetiology of human disuse atrophy. However, whilst any contribution is likely to be comparatively small, the current lack of firm evidence based on the application of tracer methodologies over the time course of muscle immobilisation in humans makes the exact contribution of muscle protein breakdown to the aetiology of disuse‐induced muscle loss in humans speculative. Of note, recent evidence reports a 6‐fold increase in a small number of NCAM (also known as CD56; see Table 1 for definitions of abbreviations) positive muscle fibres following 3 days of immobilisation in healthy male volunteers, pointing to an early denervation process which would presumably ultimately involve changes in both muscle protein synthesis and degradation in these fibres (Demangel et al. 2017).
Table 1.
Abbreviations
| 4E‐BP1 | Eukaryotic translation initiation factor 4E binding protein 1 |
| Akt | Serine/threonine‐specific protein kinase |
| CRP | C‐reactive protein |
| E2 | E2 ubiquitin‐conjugating enzyme |
| eEF2 | Eukaryotic elongation factor‐2 |
| eIF‐4E | Eukaryotic translation initiation factor 4E |
| FAT/CD36 | Fatty acid translocase/Cluster of differentiation 36 |
| FOXO | Forkhead box O |
| GLUT4 | Glucose transporter 4 |
| GSK3α and β | Glycogen synthase kinase 3 α and β |
| IκB | Inhibitor of NF‐κB |
| IL‐1 and ‐6 | Interleukin 1 and 6 |
| IMCL | Intramyocellular lipid |
| IRS‐1 | Insulin receptor signalling protein 1 |
| MAFbx | Muscle atrophy F‐box |
| mTOR | Mammalian target of rapamycin |
| MuRF1 | Muscle ring finger‐1 |
| MYD88 | Myeloid differentiation primary response protein |
| NCAM | Neural cell adhesion molecule |
| NCAM/CD56 | Neural cell adhesion molecule/Cluster of differentiation 56 |
| NF‐κB | Nuclear Factor Kappa Beta |
| p70S6K | Ribosomal protein S6 kinase β‐1 |
| PDC | Pyruvate dehydrogenase complex |
| PDK | Pyruvate dehydrogenase kinase |
| Pi | Inorganic phosphate |
| PIP | phosphatidylinositol‐1,4,5‐trisphosphate |
| PI3K | Phosphoinositide 3‐kinase |
| PTEN | Phosphatase and tensin homolog |
| Smad2,3 | Homologies to the Caenorhabditis elegans (SMA) and Drosophila (AD) family of genes for receptors of the transforming growth factor beta (TGF‐B) superfamily |
| TAG | Triglyceride |
| TLR4 | Toll‐like receptor 4 |
| TNFR1 | Tumor necrosis factor receptor |
| TRIF | TIR‐domain‐containing adapter‐inducing interferon‐β |
| Ub | Ubiquitin |
| TNFα | Tumour necrosis factor α |
Cellular and molecular mechanisms allied to immobilisation induced muscle mass loss
Research has highlighted protein translation initiation, where the ribosomal structure is formed and the associated mRNA transcript becomes bound in response to increased intramuscular amino acid availability and/or muscle contraction, as a key point of regulation of muscle protein synthesis, including in a number of conditions where a decline in the rate of muscle protein synthesis is observed (Vary & Kimball, 1992, 2000; Vary et al. 1994). The Akt/mTOR/p70S6K signalling cascade has been assigned a central role in this nutrient and/or contraction mediated activation of protein translation initiation, and is founded on experiments demonstrating that high frequency electrical stimulation of rodent muscle occurs in parallel with increased phosphorylation of these signalling proteins (Atherton et al. 2005) and muscle specific over‐expression of Akt in transgenic mice results in muscle hypertrophy (Bodine et al. 2001). However, the established notion that increased phosphorylation of Akt/mTOR/p70S6K signalling proteins is a central regulator of muscle protein synthesis in human muscle is debatable given that a disassociation between signalling protein phosphorylation and muscle protein turnover has been demonstrated. For example, stepwise increases in serum insulin concentration at a known and fixed amino acid infusion rate failed to modulate leg protein synthesis any further than amino acid administration alone (with insulin maintained at the post‐absorptive concentration), despite markedly increasing muscle Akt and p70S6K phosphorylation (Greenhaff et al. 2008). Similarly, Wilkinson et al. (2008) demonstrated that acute resistance exercise, but not endurance exercise, was able to increase post‐exercise myofibrillar protein synthesis despite signalling protein phosphorylation being increased to the same extent in both exercise protocols. From the perspective of human immobilisation, evidence suggests that the Akt/mTOR/p70S6K signalling cascade has no obvious role in the decline in muscle protein synthesis given that neither the phosphorylation state nor content of Akt, p70S6K, 4E‐BP1 or eIF‐4E were altered in the post‐absorptive state following 10 or 21 days of limb suspension (de Boer et al. 2007). Furthermore, although immobilisation blunted the increase in muscle protein synthesis in response to amino acid infusion in healthy volunteers when compared to the non‐immobilised contralateral limb (even under conditions of high amino acid provision), this anabolic blunting occurred in the face of similar changes in the phosphorylation state of the Akt/mTOR/p70S6K signalling pathway in both limbs (Glover et al. 2008), highlighting that this pathway cannot be regulating the deficits in post‐absorptive or post‐prandial muscle protein synthesis observed during immobilisation. On balance, it would seem that the precise mechanisms responsible for the decline in muscle mass observed during immobilisation in humans are not at all clear, and available data cast doubt on the measure of protein phosphorylation being a robust proxy of Akt/mTOR/p70S6K signalling pathway flux.
Immobilisation induced muscle insulin resistance
Bed‐rest induces whole body insulin resistance in human volunteers, suggested by the increase in post‐absorptive blood glucose and serum insulin concentrations, and more definitively by reductions in blood glucose clearance following an oral glucose challenge and whole body glucose clearance during an hyperinsulinaemic euglycaemic insulin clamp (Mikines et al. 1991; Sonne et al. 2010, 2011). These adaptations occur within just 3–5 days of immobilisation (Stuart et al. 1988; Smorawinski et al. 2000), and because they are also evident at a local limb level are interpreted as being representative of skeletal muscle insulin resistance. Additionally, insulin resistance develops after 3–14 days of reduced ambulatory activity (Olsen et al. 2008; Krogh‐Madsen et al. 2010). Collectively these studies suggest the reduction in muscle contraction per se drives the deficits in post‐prandial glucose disposal, which may also explain the association between age and insulin resistance reported in the literature. However, the mechanisms underpinning this physiological response are unclear and are considered further below.
Cellular and molecular mechanisms associated with immobilisation induced muscle insulin resistance
Under normal physiological conditions, skeletal muscle glucose transport is a rate limiting step in blood glucose disposal and is thought to occur as a result of blunted insulin signalling and/or GLUT 4 translocation to the plasma membrane (Zierath et al. 1996; Garvey et al. 1998). Maintenance of insulin signalling via the IRS‐1/Akt pathway seems to be essential for insulin mediated muscle glucose uptake because of its involvement in GLUT4 translocation and its dysregulation in type 2 diabetes (Björnholm et al. 1997; Kim et al. 1999; Morino et al. 2005). Moreover in the context of this review, rodent hindlimb immobilisation has been shown to reduce IRS‐1 protein expression and Akt activity (Hirose et al. 2000), whilst bed‐rest has been shown to blunt insulin stimulated Akt phosphorylation in humans (Kiilerich et al. 2011; Mortensen et al. 2013). Furthermore, 7–19 days bed‐rest (Tabata et al. 1999; Op ‘t Eijnde et al. 2001; Biensø et al. 2012) has been shown to reduce muscle GLUT4 protein content. In keeping with a decline in insulin mediated glucose uptake, reduced muscle hexokinase activity and/or expression has also been observed following immobilisation (Ringholm et al. 2011; Biensø et al. 2012), alongside a decline in muscle glycogen synthase activity (Biensø et al. 2012). Whether any of these responses are drivers of immobilisation induced muscle insulin resistance or occur as a consequence of it is unknown. Indeed, we do not yet know how muscle senses its “inactivity status” or the true source of immobilisation induced muscle insulin resistance.
Another important candidate for immobilisation induced impairment of muscle glucose disposal is IMCL accumulation. A positive correlation exists between post‐absorptive plasma free fatty acid concentrations and whole body insulin resistance, and the relationship between IMCL content and insulin resistance is even stronger (Pan et al. 1997 a; Krssak et al. 1999). Furthermore, IMCL content is positively correlated with insulin resistance in healthy volunteers and in first degree relatives of patients with type 2 diabetes (Krssak et al. 1999; Perseghin et al. 1999). It seems logical therefore that IMCL content may play a central role in the aetiology of skeletal muscle insulin resistance during immobilisation. Indeed, an increase in IMCL content has been observed following 28 days of unilateral lower limb suspension and bed‐rest (Manini et al. 2007; Cree et al. 2010), which was less obvious after 7 days bed‐rest (Dirks et al. 2016). An increase in IMCL content during immobilisation could originate from excessive free fatty acid supply secondary to positive energy intake and/or reduced physical activity. Accordingly, the use of acipimox to reduce plasma free fatty acid concentration and IMCL content resulted in a marked improvement in insulin sensitivity in non‐immobilised volunteers (Bajaj et al. 2004). From a mechanistic standpoint, the accumulation of IMCL will increase intracellular fatty acid metabolites, such as long chain fatty acyl CoAs, long chain acylcarnitines, diacylglycerols and ceramides, which collectively will blunt insulin signalling and/or inhibit pyruvate dehydrogenase flux also reducing muscle glucose utilisation. What is the mechanism behind the immobilisation induced increase in IMCL? Physical inactivity reduces lipid oxidation (Ritz et al. 1998; Bergouignan et al. 2006), which has been attributed to a reduction in metabolic rate in this situation (Blanc et al. 2000). Accordingly, the decline in lipid oxidation observed after 7 days bed‐rest was paralleled by the development of whole body insulin resistance (Blanc et al. 2000). One interpretation of these data therefore is that an increase in IMCL content results directly from an immobilisation induced reduction in the rate of lipid oxidation in tandem with a reduction in muscle metabolic rate and a parallel increase in plasma free fatty acid availability. What the time course of muscle IMCL accumulation is during immobilisation and how this relates to the temporal change in muscle insulin sensitivity is unknown. Indeed, whether muscle IMCL accumulation is a driver of immobilisation induced muscle insulin resistance or a consequence of it is unknown.
Finally, a decrease in mitochondrial content and/or mitochondrial function during immobilisation has also been proposed as a potential driver of altered fuel metabolism under these conditions. In support of this, microarray analysis revealed altered mRNA expression of genes involved in mitochondrial bioenergetics and carbohydrate metabolism after 2 and 14 days of immobilisation in healthy volunteers (Abadi et al. 2009). Furthermore, 14 days of immobilisation reduced quadriceps muscle mitochondrial respiratory capacity and protein content, and to the same extent in young and old volunteers (Gram et al. 2014). Importantly, this response was shown to be a function of a decrease in mitochondrial content that accompanies immobilisation rather than a change in intrinsic mitochondrial function per se. This is based on the evidence that the immobilisation induced decline in mitochondrial respiratory capacity disappeared when respiration was normalised to citrate synthase activity (a marker of mitochondrial content). In keeping with this, exercise training following immobilisation restored mitochondrial capacity and citrate synthase activity (Gram et al. 2014). Nevertheless, the same group of authors were able to subsequently show increased mitochondrial reactive oxygen species production in the face of no change in anti‐oxidant capacity, following immobilisation, which was accompanied by decreased mitochondrial ATP generating respiration. However, the consequences of this with respect to altered fuel metabolism under these conditions were not investigated. Exercise training following immobilisation did, however, restore both (Gram et al. 2015).
Inflammation
Inflammation induced loss of muscle mass
Several pro‐inflammatory cytokines (e.g. CRP, IL‐1, IL‐6 and TNFα) have been repeatedly implicated in altered protein homeostatic signalling and atrophy in muscle (Garcia‐Martinez et al. 1993; Haddad et al. 2005). For example, inhibition of TNFα was shown to attenuate skeletal muscle proteolysis during sepsis in rodents (Zamir et al. 1992; Combaret et al. 2002). Moreover, TNFα infusion was shown to elicit MAFbx and MuRF1 upregulation in rodents (Frost et al. 2007). Administration of both IL‐6 and IL‐1 to rats results in increased myofibrillar protein breakdown (Zamir et al. 1991; Goodman, 1994). Sepsis in particular is a complex and potentially fatal condition that results from an uncontrolled, systemic inflammatory response to an infection, and is a major cause of morbidity and mortality in intensive care units (ICUs) worldwide (reviewed in Bone et al. 1992). Muscle wasting occurs rapidly (Puthucheary et al. 2013), the magnitude of which is an independent predictor of mortality in critically ill patients (Ali et al. 2008). Given that muscle wasting is the primary driver of subsequent physical disability in critical illness survivors (Herridge et al. 2003), that continues after discharge (Pfoh et al. 2016) with an associated mortality (Dinglas et al. 2017), it is appropriate that National Institute for Health and Clinical Excellence guidelines highlight physical disability and specifically its driver (muscle wasting) as a public health issue (NICE, 1990). A hallmark for the pathophysiology of sepsis is a large, uncontrolled systemic inflammatory host response to circulating microbial antigens (reviewed in Cohen, 2002), with the most common sites of infection being the lungs, urinary tract and abdominal cavity (Angus et al. 2001). Sepsis‐induced muscle wasting and weakness can clearly have a damaging impact on recovery and survival.
Of further interest, CRP is an acute‐phase protein of hepatic origin that increases following IL‐6 secretion by macrophages and T cells. Its physiological role is to bind to dead or dying cells (and some types of bacteria) in order to activate the complement system, and is therefore a useful marker of systemic inflammation. Circulating CRP concentration can be associated independently with loss of lean body mass (Schaap et al. 2006; Dutra et al. 2017) and age related loss of muscle mass (Schaap et al. 2006). Furthermore, in large observational studies CRP was not associated with age per se, suggesting a separate influence on muscle mass (Puzianowska‐Kuźnicka et al. 2016). In differentiated human myotubes, CRP has been shown to negatively affect cell size directly by decreasing muscle protein synthesis and phosphorylation of Akt and ribosomal protein S6 (Puzianowska‐Kuźnicka et al. 2016).
Cellular and molecular mechanisms allied to inflammation induced muscle mass loss
A limited number of mechanistic experimental research studies focused on muscle mass loss in the context of inflammation has been conducted in patient volunteers, not least because baseline data are often difficult to acquire. This practical problem has been overcome by researchers undertaking relatively short duration endotoxin administration studies in healthy volunteers, which have reported acute decreases in both muscle protein synthesis and muscle protein breakdown, and efflux of muscle amino acids, such that muscle protein balance was unaffected (Vesali et al. 2009). How these observations relate to the accepted accelerated loss of muscle mass in patients under inflammatory conditions (Debigare et al. 2001; Puthucheary et al. 2013) is difficult to rationalise. One possibility is that these acute studies were performed in the post‐absorptive state when muscle protein synthesis will be reduced and muscle protein breakdown increased. However, the limited data available concerning the impact of endotoxin infusion on muscle protein balance in the post‐prandial state in humans is equally baffling as it suggests that whilst amino acid administration can rescue muscle protein loss compared to the post‐absorptive state, calculated muscle protein synthesis and breakdown rates did not differ significantly between interventions (Rittig et al. 2016). Clearly further research is necessary, including muscle tracer incorporation studies. In critically ill patients where stable isotope tracers were used to quantify temporal changes in muscle protein synthesis and leg protein breakdown 24 h after admission to an ICU (Puthucheary et al. 2013), muscle protein synthesis was found to be depressed in patients on day 1 compared with healthy control volunteers in the post‐absorptive state, but by day 7 had increased independent of nutritional status to rates similar to the post‐prandial state in control volunteers. Leg protein breakdown remained elevated throughout the study in the patients. In keeping with these observations, the authors demonstrated that muscle wasting (ultrasound determined muscle cross‐sectional area and the muscle protein‐to‐DNA ratio) occurred early and rapidly during the first week of critical illness, and was more severe among patients with multi‐organ failure compared with single organ failure. These observations of negative muscle protein balance and atrophy occurring early following ICU admittance are in keeping with data depicting elevation of muscle cytokine mRNA and widespread dephosphorylation (inactivation) of proteins regulating translation initiation factor activation and protein synthesis (Akt1, GSK3α and β, mTOR, p70S6K and 4E‐BP1) in patients within 6–8 h of admission to the ICU compared with healthy age‐ and sex‐matched control volunteers (Constantin et al. 2011). In accordance with the observation of elevated leg protein breakdown in such conditions (Puthucheary et al. 2013), this suppression of the Akt/mTOR/p70S6K signalling cascade occurred in tandem with increased muscle‐specific E3‐ligase (MAFbx and MuRF1) and 20S proteasome mRNA and protein expression levels in patients relative to controls (Constantin et al. 2011). The Akt/FOXO signalling pathway has been implicated in the development of muscle atrophy during catabolic conditions, potentially through activation of the ubiquitin/proteasome pathway (Crossland et al. 2008). More specifically, the upregulation of MAFbx and MuRF1 mRNA expression in skeletal muscle in animal models occurs within hours after the onset of endotoxaemia (Dehoux et al. 2003; Wray et al. 2003), and appears to be mediated by dampened Akt phosphorylation activating FOXO transcription factors and increased expression of downstream FOXO gene targets, including MAFbx and MuRF1 (Crossland et al. 2008). This series of events supports the concept that the ubiquitin/proteasome system is an important early driver of muscle protein breakdown, although not the only catabolic driver under such conditions (Deval et al. 2001; Smith et al. 2008).
Overall it would appear there are comprehensive acute alterations in muscle cross‐sectional area, muscle protein turnover and the molecular events considered to regulate muscle protein synthesis and breakdown in patients early following admittance to ICU. Although muscle mass loss is common to both immobilisation and the ICU, we do not know the relative contribution of immobilisation and inflammation to muscle mass loss in this situation. It is clear, however, that the drivers of muscle mass loss are very different between the two scenarios in humans. Furthermore, although pro‐inflammatory cytokines appear to be important in the aetiology of muscle mass loss, it is important to recognise that multiple mediators and pathways contribute to the heightened morbidity and mortality associated with infection and inflammation (reviewed in Jacobi, 2002). Indeed, anti‐cytokine therapies were developed to dampen the systemic inflammatory response, including antibodies against TNFα, soluble TNFα receptors and IL‐1β receptor antagonists. Blockade of TNFα was found to have beneficial effects on survival in animal models of shock (Beutler et al. 1985; Tracey et al. 1987); however, subsequent clinical trials did not show benefit in human sepsis (Reinhart & Karzai, 2001). Furthermore, IL‐1β inhibition using a recombinant receptor antagonist reduced mortality in animal models of shock (Ohlsson et al. 1990), but, again, human trials showed no beneficial effects (Opal et al. 1997).
Inflammation induced muscle insulin resistance
Various catabolic conditions that stimulate muscle atrophy are often associated with insulin resistance, including heightened muscle inflammation (Hasselgren et al. 1987; Dardevet et al. 1994; Wang et al. 2006). Insulin‐stimulated whole body glucose disposal, endogenous glucose production and glucose oxidation have been shown to be impaired in patients with severe sepsis (Chambrier et al. 2000), which is associated with increased mortality (Falciglia et al. 2009).
Cellular and molecular mechanisms associated with inflammation induced muscle insulin resistance
Although current practice is to maintain normoglycaemia in intensive care, detailed mechanistic insight regarding the aetiology of this patient phenotype is missing and current insight has been gained in the main from animal based investigation. Hyperlactataemia is also a frequent manifestation of sepsis, which, based on preclinical research, may be at least partially due to impaired PDC activity and flux limiting mitochondrial pyruvate utilisation as a result of sepsis induced PDK upregulation (Vary, 1999; Alamdari et al. 2008). Given the proposed role of TNFα in insulin resistance (Plomgaard et al. 2005), possibly through suppression of insulin signalling at the level of IRS‐1 (Hotamisligil et al. 1996; del Aguila et al. 1999), it is more than likely that elevated cytokines have some role in the observed changes in carbohydrate metabolism in inflammatory states, and at least partly through dysregulation of Akt signalling (Crossland et al. 2008), not least because divergent in vivo pharmacological approaches that reduce muscle inflammation blunt the dysregulation of muscle carbohydrate metabolism (Crossland et al. 2010, 2017).
It is also of note that a link between circulating CRP and insulin resistance has been established, with associations seen with measures of fat mass, fasting insulin and a number of metabolic disorders (Festa et al. 2000). Just as with protein homeostasis, circulating CRP may not be simply a biomarker of inflammation but may play a causal role in the dysregulation of fuel metabolism. CRP binds and aggregates low density lipoproteins and very low density lipoproteins (de Beer et al. 1982). Additionally, CRP impacts on macrophage phenotype (Dong & Wright, 1996), which purportedly impacts on insulin resistance. However, a direct link between CRP and insulin resistance remains elusive, and causal links have not been established with type 2 diabetes (Brunner et al. 2008). CRP is regulated by other pro‐inflammatory cytokines such as TNFα and IL‐6 for which more robust direct causative links have been established (see above), and as such circulating CRP may be an upstream representative of their activity (Ridker, 2016).
Molecular cross‐talk between muscle atrophy and insulin signalling in inflammation
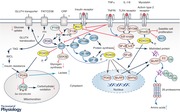
Few studies to date have investigated muscle protein and fuel metabolism concurrently to elucidate potential mutual signalling events that may explain dysregulation of muscle protein and carbohydrate metabolism in inflammation. As previously discussed, Akt signalling appears to be crucial in hypertrophy and atrophy signalling, as well as insulin signalling, and it is increasingly evident that there is cross‐talk between insulin and atrophy signalling processes during heightened inflammation in animal models (Crossland et al. 2008) and patients (Constantin et al. 2011). Specifically, elevated cytokines may result in the impairment (reduced phosphorylation) of Akt1 via inhibition of IRS‐1 (del Aguila et al. 1999), subsequently leading to the dephosphorylation (activation) of FOXO, and, in turn, transcriptional upregulation of FOXO target genes MAFbx, MuRF1 and PDK, as well as decreased phosphorylation (activation) of muscle anabolic signalling proteins (Crossland et al. 2008; Constantin et al. 2011). In support of this, genetically insulin‐resistant, db/db, mice were found to have increased rates of muscle proteolysis, which correlated with altered Akt/FOXO signalling (Wang et al. 2006). Furthermore, strategies aimed at blunting the muscle cytokine response to inflammation in vivo in an animal model of clinical sepsis have been shown to dampen the dysregulation of Akt/FOXO signalling and the abundance of downstream mRNA targets, whilst concomitantly preventing muscle protein loss and the impairment of pyruvate dehydrogenase complex activation and carbohydrate oxidation (Crossland et al. 2010, 2017). Collectively, these findings, along with other reports of dysregulation of Akt signalling in the catabolic, insulin resistant state (Wang et al. 2006), and the proposed role of FOXO in both atrophy and insulin signalling pathways (Kwon et al. 2004; Sandri et al. 2004), suggest that Akt and FOXO form part of a common signalling pathway influencing muscle protein breakdown, protein synthesis and the induction of insulin resistance during inflammation (Abstract figure).
Conclusion
This review highlights that whilst muscle mass loss and insulin resistance are common end‐points of immobilisation and increased inflammatory burden, there is a lack of understanding of the mechanisms responsible for these common events such that a substantial gap in understanding of the pathophysiology exists. It does seem unlikely, however, that the same mechanisms are involved. For example, Akt signalling appears to play a central role in the dysregulation of protein metabolism in conditions of increased inflammatory burden, but not in immobilisation. It is important therefore for future research to determine the mechanisms responsible for the loss of muscle mass and insulin sensitivity during immobilisation and whether the impacts of combined immobilisation and increased inflammatory burden are additive in situations such as intensive care, which could have important clinical ramifications. Importantly, these studies need to be conducted in humans, and will include multiple time‐point measurements over the course of intervention, dynamic measurements of muscle protein turnover and glucose uptake in the post‐prandial state, and will be combined with sensitive measures of muscle composition, intermediary metabolism and modern molecular biology. Such studies will produce major new mechanistic insights into human pathophysiology, with potential application to new therapeutic approaches and opportunities for “reverse translation” to more basic research.
Additional information
Competing interests
None declared.
Author contributions
P.L.G. wrote the first draft of the review manuscript, which all other authors added to and edited. D.C.‐T. created the Abstract figure, which P.L.G. edited. All authors approved the final version of the article. All persons designated as authors qualify for authorship, and all those who are eligible for authorship are listed.
Funding
This work was supported by the Medical Research Council [grant number MR/K00414X/1]; and Arthritis Research UK [grant number 19891, 21595] Centre for Musculoskeletal Ageing Research, the Arthritis Research UK Centre for Sport, Exercise and Osteoarthritis, the Biotechnology and Biological Sciences Research Council and the National Institute for Health Research Nottingham Biomedical Research Centre by contributions to the infrastructure that facilitated generation of this review article.
Acknowledgements
The authors would like to acknowledge the contribution of many colleagues, including not least Professor Michael J. Rennie, to scientific ideas and reflections contained in this manuscript.
Biography
Paul Greenhaff is deputy director of the Medical Research Council (MRC)/Arthritis Research UK (ARUK) Centre for Musculoskeletal Ageing Research, and an active member of the ARUK Centre for Sport, Exercise and Osteoarthritis. Paul's research interests are centred on the loss of muscle mass and the dysregulation of metabolism in immobilisation, inflammation, ageing and disease, and strategies (including exercise, nutrition and pharmacological interventions) to offset these effects.

Edited by: Ole Petersen & Karyn Hamilton
References
- Abadi A, Glover EI, Isfort RJ, Raha S, Safdar A, Yasuda N, Kaczor JJ, Melov S, Hubbard A, Qu X, Phillips SM & Tarnopolsky M (2009). Limb immobilization induces a coordinate down‐regulation of mitochondrial and other metabolic pathways in men and women. PLoS One 4, e6518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamdari N, Constantin‐Teodosiu D, Murton AJ, Gardiner SM, Bennett T, Layfield R & Greenhaff PL (2008). Temporal changes in the involvement of pyruvate dehydrogenase complex in muscle lactate accumulation during lipopolysaccharide infusion in rats. J Physiol 586, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali NA, O'Brien JM Jr, Hoffmann SP, Phillips G, Garland A, Finley JC, Almoosa K, Hejal R, Wolf KM, Lemeshow S, Connors AF Jr & Marsh CB; Midwest Critical Care Consortium (2008). Acquired weakness, handgrip strength, and mortality in critically ill patients. Am J Respir Crit Care Med 178, 261–268. [DOI] [PubMed] [Google Scholar]
- Angus DC, Linde‐Zwirble WT, Lidicker J, Clermont G, Carcillo J & Pinsky MR (2001). Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 29, 1303–1310. [DOI] [PubMed] [Google Scholar]
- Atherton PJ, Babraj J, Smith K, Singh J, Rennie MJ & Wackerhage H (2005). Selective activation of AMPK‐PGC‐1α or PKB‐TSC2‐mTOR signaling can explain specific adaptive responses to endurance or resistance training‐like electrical muscle stimulation. FASEB J 19, 786–788. [DOI] [PubMed] [Google Scholar]
- Bajaj M, Suraamornkul S, Kashyap S, Cusi K, Mandarino L & DeFronzo RA (2004). Sustained reduction in plasma free fatty acid concentration improves insulin action without altering plasma adipocytokine levels in subjects with strong family history of type 2 diabetes. J Clin Endocrinol Metab 89, 4649–4655. [DOI] [PubMed] [Google Scholar]
- Bergouignan A, Schoeller DA, Normand S, Gauquelin‐Koch G, Laville M, Shriver T, Desage M, Le Maho Y, Ohshima H & Gharib C (2006). Effect of physical inactivity on the oxidation of saturated and monounsaturated dietary fatty acids: results of a randomized trial. PLoS Clin Trials 1, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B, Milsark IW & Cerami AC (1985). Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin. Science 229, 869–871. [DOI] [PubMed] [Google Scholar]
- Biensø RS, Ringholm S, Kiilerich K, Aachmann‐Andersen NJ, Krogh‐Madsen R, Guerra B, Plomgaard P, Van Hall G, Treebak JT, Saltin B, Lundby C, Calbet JAL, Pilegaard H & Wojtaszewski JFP (2012). GLUT4 and glycogen synthase are key players in bed rest‐induced insulin resistance. Diabetes 61, 1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnholm M, Kawano Y, Lehtihet M & Zierath JR (1997). Insulin receptor substrate‐1 phosphorylation and phosphatidylinositol 3‐kinase activity in skeletal muscle from NIDDM subjects after in vivo insulin stimulation. Diabetes 46, 524–527. [DOI] [PubMed] [Google Scholar]
- Blanc S, Normand S, Pachiaudi C, Fortrat J‐O, Laville M & Gharib C (2000). Fuel homeostasis during physical inactivity induced by bed rest. J Clin Endocrinol Metab 85, 2223–2233. [DOI] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ & Yancopoulos GD (2001). Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3, 1014–1019. [DOI] [PubMed] [Google Scholar]
- Bone RC, Balk RA, Cerra FB, Dellinger RP, Fein AM, Knaus WA, Schein RM & Sibbald WJ (1992). Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 101, 1644–1655. [DOI] [PubMed] [Google Scholar]
- Booth FW & Seider MJ (1979). Early change in skeletal muscle protein synthesis after limb immobilization of rats. J Appl Physiol Respir Environ Exerc Physiol 47, 974–977. [DOI] [PubMed] [Google Scholar]
- Brunner EJ, Kivimäki M, Witte DR, Lawlor DA, Davey Smith G, Cooper JA, Miller M, Lowe GD, Rumley A, Casas JP, Shah T, Humphries SE, Hingorani AD, Marmot MG, Timpson NJ & Kumari M (2008). Inflammation, insulin resistance, and diabetes – Mendelian randomization using CRP haplotypes points upstream. PLoS Med 5, e155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambrier C, Laville M, Rhzioual Berrada K, Odeon M, Bouletreau P & Beylot M (2000). Insulin sensitivity of glucose and fat metabolism in severe sepsis. Clin Sci (Lond) 99, 321–328. [PubMed] [Google Scholar]
- Cohen J (2002). The immunopathogenesis of sepsis. Nature 420, 885–891. [DOI] [PubMed] [Google Scholar]
- Combaret L, Tilignac T, Claustre A, Voisin L, Taillandier D, Obled C, Tanaka K & Attaix D (2002). Torbafylline (HWA 448) inhibits enhanced skeletal muscle ubiquitin‐proteasome‐dependent proteolysis in cancer and septic rats. Biochem J 361, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantin D, McCullough J, Mahajan RP & Greenhaff PL (2011). Novel events in the molecular regulation of muscle mass in critically ill patients. J Physiol 589, 3883–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cree MG, Paddon‐Jones D, Newcomer BR, Ronsen O, Aarsland A, Wolfe RR & Ferrando A (2010). Twenty‐eight‐day bed rest with hypercortisolemia induces peripheral insulin resistance and increases intramuscular triglycerides. Metabolism 59, 703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossland H, Constantin‐Teodosiu D, Gardiner SM, Constantin D & Greenhaff PL (2008). A potential role for Akt/FOXO signalling in both protein loss and the impairment of muscle carbohydrate oxidation during sepsis in rodent skeletal muscle. J Physiol 586, 5589–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossland H, Constantin‐Teodosiu D, Greenhaff PL & Gardiner SM (2010). Low‐dose dexamethasone prevents endotoxaemia‐induced muscle protein loss and impairment of carbohydrate oxidation in rat skeletal muscle. J Physiol 588, 1333–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossland H, Constantin‐Teodosiu D, Gardiner SM & Greenhaff PL (2017). Peroxisome proliferator‐activated receptor γ agonism attenuates endotoxaemia‐induced muscle protein loss and lactate accumulation in rats. Clin Sci (Lond) 131, 1437–1447. [DOI] [PubMed] [Google Scholar]
- Dardevet D, Sornet C, Attaix D, Baracos VE & Grizard J (1994). Insulin‐like growth factor‐1 and insulin resistance in skeletal muscles of adult and old rats. Endocrinology 134, 1475–1484. [DOI] [PubMed] [Google Scholar]
- de Beer FC, Soutar AK, Baltz ML, Trayner IM, Feinstein A & Pepys MB (1982). Low density lipoprotein and very low density lipoprotein are selectively bound by aggregated C‐reactive protein. J Exp Med 156, 230–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debigare R, Cote CH & Maltais F (2001). Peripheral muscle wasting in chronic obstructive pulmonary disease. Clinical relevance and mechanisms. Am J Respir Crit Care Med 164, 1712–1717. [DOI] [PubMed] [Google Scholar]
- de Boer MD, Selby A, Atherton P, Smith K, Seynnes OR, Maganaris CN, Maffulli N, Movin T, Narici MV & Rennie MJ (2007). The temporal responses of protein synthesis, gene expression and cell signalling in human quadriceps muscle and patellar tendon to disuse. J Physiol 585, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehoux MJ, van Beneden RP, Fernandez‐Celemin L, Lause PL & Thissen JP (2003). Induction of MafBx and Murf ubiquitin ligase mRNAs in rat skeletal muscle after LPS injection. FEBS Lett 544, 214–217. [DOI] [PubMed] [Google Scholar]
- del Aguila LF, Claffey KP & Kirwan JP (1999). TNF‐α impairs insulin signaling and insulin stimulation of glucose uptake in C2C12 muscle cells. Am J Physiol 276, E849–E855. [DOI] [PubMed] [Google Scholar]
- Demangel R, Treffel L, Py G, Brioche T, Pagano AF, Bareille MP, Beck A, Pessemesse L, Candau R, Gharib C, Chopard A & Millet C (2017). Early structural and functional signature of 3‐day human skeletal muscle disuse using the dry immersion model. J Physiol 595, 4301–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetrius L (2005). Of mice and men – When it comes to studying ageing and the means to slow it down, mice are not just small humans. EMBO Rep 6, S39–S44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval C, Mordier S, Obled C, Bechet D, Combaret L, Attaix D & Ferrara M (2001). Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J 360, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinglas VD, Aronson Friedman L, Colantuoni E, Mendez‐Tellez PA, Shanholtz CB, Ciesla ND, Pronovost PJ & Needham DM (2017). Muscle weakness and 5‐year survival in acute respiratory distress syndrome survivors. Crit Care Med 45, 446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirks ML, Wall BT, van de Valk B, Holloway TM, Holloway GP, Chabowski A, Goossens GH & van Loon LJ (2016). One week of bed rest leads to substantial muscle atrophy and induces whole‐body insulin resistance in the absence of skeletal muscle lipid accumulation. Diabetes 65, 2862–2875. [DOI] [PubMed] [Google Scholar]
- Dong Q & Wright JR (1996). Expression of C‐reactive protein by alveolar macrophages. J Immunol 156, 4815–4820. [PubMed] [Google Scholar]
- Dutra MT, Avelar BP, Souza VC, Bottaro M, Oliveira RJ, Nobrega OT & Moreno Lima R (2017). Relationship between sarcopenic obesity‐related phenotypes and inflammatory markers in postmenopausal women. Clin Physiol Funct Imaging 37, 205–210. [DOI] [PubMed] [Google Scholar]
- Falciglia M, Freyberg RW, Almenoff PL, D'Alessio DA & Render ML (2009). Hyperglycemia‐related mortality in critically ill patients varies with admission diagnosis. Crit Care Med 37, 3001–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festa A, D'Agostino R Jr, Howard G, Mykkanen L, Tracy RP & Haffner SM (2000). Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 102, 42–47. [DOI] [PubMed] [Google Scholar]
- Frost RA, Nystrom GJ, Jefferson LS & Lang CH (2007). Hormone, cytokine, and nutritional regulation of sepsis‐induced increases in atrogin‐1 and MuRF1 in skeletal muscle. Am J Physiol Endocrinol Metab 292, E501–E512. [DOI] [PubMed] [Google Scholar]
- Garcia‐Martinez C, Lopez‐Soriano FJ & Argiles JM (1993). Acute treatment with tumour necrosis factor‐α induces changes in protein metabolism in rat skeletal muscle. Mol Cell Biochem 125, 11–18. [DOI] [PubMed] [Google Scholar]
- Garvey WT, Maianu L, Zhu J‐H, Brechtel‐Hook G, Wallace P & Baron AD (1998). Evidence for defects in the trafficking and translocation of GLUT4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest 101, 2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover EI, Phillips SM, Oates BR, Tang JE, Tarnopolsky MA, Selby A, Smith K & Rennie MJ (2008). Immobilization induces anabolic resistance in human myofibrillar protein synthesis with low and high dose amino acid infusion. J Physiol 586, 6049–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman MN (1994). Interleukin‐6 induces skeletal muscle protein breakdown in rats. Proc Soc Exp Biol Med 205, 182–185. [DOI] [PubMed] [Google Scholar]
- Gram M, Vigelsø A, Yokota T, Hansen CN, Helge JW, Hey‐Mogensen M & Dela F (2014). Two weeks of one‐leg immobilization decreases skeletal muscle respiratory capacity equally in young and elderly men. Exp Gerontol 58, 269–278. [DOI] [PubMed] [Google Scholar]
- Gram M, Vigelsø A, Yokota T, Helge JW, Dela F & Hey‐Mogensen M (2015). Skeletal muscle mitochondrial H2O2 emission increases with immobilization and decreases after aerobic training in young and older men. J Physiol 593, 4011–4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhaff PL, Karagounis L, Peirce N, Simpson EJ, Hazell M, Layfield R, Wackerhage H, Smith K, Atherton P, Selby A & Rennie MJ (2008). Disassociation between the effects of amino acids and insulin on signalling, ubiquitin‐ligases and protein turnover in human muscle. Am J Physiol Endocrinol Metab 295, E595–E604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad F, Zaldivar F, Cooper DM & Adams GR (2005). IL‐6‐induced skeletal muscle atrophy. J Appl Physiol (1985) 98, 911–917. [DOI] [PubMed] [Google Scholar]
- Hasselgren PO, Menconi MJ, Fareed MU, Yang H, Wei W & Evenson A (2005). Novel aspects on the regulation of muscle wasting in sepsis. Int J Biochem Cell Biol 37, 2156–2168. [DOI] [PubMed] [Google Scholar]
- Hasselgren PO, Warner BW, James JH, Takehara H & Fischer JE (1987). Effect of insulin on amino acid uptake and protein turnover in skeletal muscle from septic rats. Evidence for insulin resistance of protein breakdown. Arch Surg 122, 228–233. [DOI] [PubMed] [Google Scholar]
- Herridge MS, Cheung AM, Tansey CM, Matte‐Martyn A, Diaz‐Granados N, Al‐Saidi F, Cooper AB, Guest CB, Mazer CD, Mehta S, Stewart TE, Barr A, Cook D & Slutsky AS; Canadian Critical Care Trials Group (2003). One‐year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med 348, 683–693. [DOI] [PubMed] [Google Scholar]
- Hirose M, Kaneki M, Sugita H, Yasuhara S & Martyn JA (2000). Immobilization depresses insulin signaling in skeletal muscle. Am J Physiol Endocrinol Metab 279, E1235–E1241. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF & Spiegelman BM (1996). IRS‐1‐mediated inhibition of insulin receptor tyrosine kinase activity in TNF‐α‐ and obesity‐induced insulin resistance. Science 271, 665–668. [DOI] [PubMed] [Google Scholar]
- Jacobi J (2002). Pathophysiology of sepsis. Am J Health Syst Pharm 59 (Suppl. 1), S3–8. [DOI] [PubMed] [Google Scholar]
- Jones SW, Hill RJ, Krasney PA, O'Conner B, Peirce N & Greenhaff PL (2004). Disuse atrophy and exercise rehabilitation in humans profoundly affects the expression of genes associated with the regulation of skeletal muscle mass. FASEB J 18, 1025–1027. [DOI] [PubMed] [Google Scholar]
- Kiilerich K, Ringholm S, Bienso RS, Fisher JP, Iversen N, van Hall G, Wojtaszewski JF, Saltin B, Lundby C, Calbet JA & Pilegaard H (2011). Exercise‐induced pyruvate dehydrogenase activation is not affected by 7 days of bed rest. J Appl Physiol (1985) 111, 751–757. [DOI] [PubMed] [Google Scholar]
- Kim Y‐B, Nikoulina SE, Ciaraldi TP, Henry RR & Kahn BB (1999). Normal insulin‐dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3‐kinase, in muscle in type 2 diabetes. J Clin Invest 104, 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H, Kato H, Hirabayashi Y, Murakami H & Suzuki H (2006). Modulations of muscle protein metabolism by branched‐chain amino acids in normal and muscle‐atrophying rats. J Nutr 136, 234S–236S. [DOI] [PubMed] [Google Scholar]
- Krawiec BJ, Frost RA, Vary TC, Jefferson LS & Lang CH (2005). Hindlimb casting decreases muscle mass in part by proteasome‐dependent proteolysis but independent of protein synthesis. Am J Physiol Endocrinol Metab 289, E969–E980. [DOI] [PubMed] [Google Scholar]
- Krogh‐Madsen R, Thyfault JP, Broholm C, Mortensen OH, Olsen RH, Mounier R, Plomgaard P, van Hall G, Booth FW & Pedersen BK (2010). A 2‐wk reduction of ambulatory activity attenuates peripheral insulin sensitivity. J Appl Physiol (1985) 108, 1034–1040. [DOI] [PubMed] [Google Scholar]
- Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M & Shulman GI (1999). Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia 42, 113–116. [DOI] [PubMed] [Google Scholar]
- Kwon HS, Huang B, Unterman TG & Harris RA (2004). Protein kinase B‐α inhibits human pyruvate dehydrogenase kinase‐4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes 53, 899–910. [DOI] [PubMed] [Google Scholar]
- Manini TM, Clark BC, Nalls MA, Goodpaster BH, Ploutz‐Snyder LL & Harris TB (2007). Reduced physical activity increases intermuscular adipose tissue in healthy young adults. Am J Clin Nutr 85, 377–384. [DOI] [PubMed] [Google Scholar]
- Mikines KJ, Richter EA, Dela F & Galbo H (1991). Seven days of bed rest decrease insulin action on glucose uptake in leg and whole body. J Appl Physiol (1985) 70, 1245–1254. [DOI] [PubMed] [Google Scholar]
- Mizock BA (2001). Alterations in fuel metabolism in critical illness: hyperglycaemia. Best Pract Res Clin Endocrinol Metab 15, 533–551. [DOI] [PubMed] [Google Scholar]
- Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N, Neschen S, White MF, Bilz S & Sono S (2005). Reduced mitochondrial density and increased IRS‐1 serine phosphorylation in muscle of insulin‐resistant offspring of type 2 diabetic parents. J Clin Invest 115, 3587–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen B, Friedrichsen M, Andersen NR, Alibegovic AC, Hojbjerre L, Sonne MP, Stallknecht B, Dela F, Wojtaszewski JF & Vaag A (2013). Physical inactivity affects skeletal muscle insulin signaling in a birth weight‐dependent manner. J Diabetes Complications 28, 71–78. [DOI] [PubMed] [Google Scholar]
- Murton AJ & Greenhaff PL (2010). Physiological control of muscle mass in humans during resistance exercise, disuse and rehabilitation. Curr Opin Clin Nutr Metab Care 13, 249–254. [DOI] [PubMed] [Google Scholar]
- NICE (2009). NICE clinical guideline 83. Rehabilitation after critical illness. https://www.nice.org.uk/guidance/cg83/evidence/full-guideline-pdf-242292349.
- Ohlsson K, Bjork P, Bergenfeldt M, Hageman R & Thompson RC (1990). Interleukin‐1 receptor antagonist reduces mortality from endotoxin shock. Nature 348, 550–552. [DOI] [PubMed] [Google Scholar]
- Olsen RH, Krogh‐Madsen R, Thomsen C, Booth FW & Pedersen BK (2008). Metabolic responses to reduced daily steps in healthy nonexercising men. JAMA 299, 1261–1263. [DOI] [PubMed] [Google Scholar]
- Opal SM, Fisher CJ Jr, Dhainaut JF, Vincent JL, Brase R, Lowry SF, Sadoff JC, Slotman GJ, Levy H, Balk RA, Shelly MP, Pribble JP, LaBrecque JF, Lookabaugh J, Donovan H, Dubin H, Baughman R, Norman J, DeMaria E, Matzel K, Abraham E & Seneff M (1997). Confirmatory interleukin‐1 receptor antagonist trial in severe sepsis: a phase III, randomized, double‐blind, placebo‐controlled, multicenter trial. The Interleukin‐1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med 25, 1115–1124. [DOI] [PubMed] [Google Scholar]
- Op ‘t Eijnde B, Ursø B, Richter EA, Greenhaff PL & Hespel P (2001). Effect of oral creatine supplementation on human muscle GLUT4 protein content after immobilization. Diabetes 50, 18–23. [DOI] [PubMed] [Google Scholar]
- Pan D, Lillioja S, Kriketos A, Milner M, Baur L, Bogardus C, Jenkins A & Storlien L (1997). Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes 46, 983–988. [DOI] [PubMed] [Google Scholar]
- Perseghin G, Scifo P, De Cobelli F, Pagliato E, Battezzati A, Arcelloni C, Vanzulli A, Testolin G, Pozza G & Del Maschio A (1999). Intramyocellular triglyceride content is a determinant of in vivo insulin resistance in humans: a 1H‐13C nuclear magnetic resonance spectroscopy assessment in offspring of type 2 diabetic parents. Diabetes 48, 1600–1606. [DOI] [PubMed] [Google Scholar]
- Pfoh ER, Wozniak AW, Colantuoni E, Dinglas VD, Mendez‐Tellez PA, Shanholtz C, Ciesla ND, Pronovost PJ & Needham DM (2016). Physical declines occurring after hospital discharge in ARDS survivors: a 5‐year longitudinal study. Intensive Care Med 42, 1557–1566. [DOI] [PubMed] [Google Scholar]
- Phillips SM, Glover EI & Rennie MJ. Alterations of protein turnover underlying disuse atrophy in human skeletal muscle (2009). J Appl Physiol (1985) 107, 645–654. [DOI] [PubMed] [Google Scholar]
- Plomgaard P, Bouzakri K, Krogh‐Madsen R, Mittendorfer B, Zierath JR & Pedersen BK (2005). Tumor necrosis factor‐α induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54, 2939–2945. [DOI] [PubMed] [Google Scholar]
- Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, Hopkinson NS, Phadke R, Dew T, Sidhu PS, Velloso C, Seymour J, Agley CC, Selby A, Limb M, Edwards LM, Smith K, Rowlerson A, Rennie MJ, Moxham J, Harridge SD, Hart N & Montgomery HE (2013). Acute skeletal muscle wasting in critical illness. JAMA 310, 1591–1600. [DOI] [PubMed] [Google Scholar]
- Puzianowska‐Kuźnicka M, Owczarz M, Wieczorowska‐Tobis K, Nadrowski P, Chudek J, Slusarczyk P, Skalska A, Jonas M, Franek E & Mossakowska M (2016). Interleukin‐6 and C‐reactive protein, successful aging, and mortality: the PolSenior study. Immun Ageing 13, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhart K & Karzai W (2001). Anti‐tumor necrosis factor therapy in sepsis: update on clinical trials and lessons learned. Crit Care Med 29, S121–125. [DOI] [PubMed] [Google Scholar]
- Ridker PM (2016). C‐reactive protein to interleukin‐6 to interleukin‐1: moving upstream to identify novel targets for atheroprotection. Circ Res 118, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringholm S, Biensø RS, Kiilerich K, Guadalupe‐Grau A, Aachmann‐Andersen NJ, Saltin B, Plomgaard P, Lundby C, Wojtaszewski JF & Calbet JA (2011). Bed rest reduces metabolic protein content and abolishes exercise‐induced mRNA responses in human skeletal muscle. Am J Physiol Endocrinol Metab 301, E649–E658. [DOI] [PubMed] [Google Scholar]
- Rittig N, Bach E, Thomsen HH, Johannsen M, Jørgensen JO, Richelsen B, Jessen N & Møller N (2016). Amino acid supplementation is anabolic during the acute phase of endotoxin‐induced inflammation: A human randomized crossover trial. Clin Nutr 35, 322–330. [DOI] [PubMed] [Google Scholar]
- Ritz P, Acheson K, Gachon P, Vico L, Bernard J, Alexandre C & Beaufrere B (1998). Energy and substrate metabolism during a 42‐day bed‐rest in a head‐down tilt position in humans. Eur J Appl Physiol 78, 308–314. [DOI] [PubMed] [Google Scholar]
- Saeed M, Carlson GL, Little RA & Irving MH (1999). Selective impairment of glucose storage in human sepsis. Br J Surg 86, 813–821. [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH & Goldberg AL (2004). Foxo transcription factors induce the atrophy‐related ubiquitin ligase atrogin‐1 and cause skeletal muscle atrophy. Cell 117, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaap LA, Pluijm SM, Deeg DJ & Visser M (2006). Inflammatory markers and loss of muscle mass (sarcopenia) and strength. Am J Med 119, 526.e9–526.e17. [DOI] [PubMed] [Google Scholar]
- Smith IJ, Lecker SH & Hasselgren PO (2008). Calpain activity and muscle wasting in sepsis. Am J Physiol Endocrinol Metab 295, E762–E771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smorawinski J, Kaciuba‐Uscilko H, Nazar K, Kubala P, Kaminska E, Ziemba AW, Adrian J & Greenleaf JE (2000). Effects of three‐day bed rest on metabolic, hormonal and circulatory responses to an oral glucose load in endurance or strength trained athletes and untrained subjects. J Physiol Pharmacol 51, 279–289. [PubMed] [Google Scholar]
- Sonne MP, Alibegovic AC, Hojbjerre L, Vaag A, Stallknecht B & Dela F (2010). Effect of 10 days of bedrest on metabolic and vascular insulin action: a study in individuals at risk for type 2 diabetes. J Appl Physiol (1985) 108, 830–837. [DOI] [PubMed] [Google Scholar]
- Sonne MP, Hojbjerre L, Alibegovic AC, Nielsen LB, Stallknecht B, Vaag AA & Dela F (2011). Endothelial function after 10 days of bed rest in individuals at risk for type 2 diabetes and cardiovascular disease. Exp Physiol 96, 1000–1009. [DOI] [PubMed] [Google Scholar]
- Stuart CA, Shangraw RE, Prince MJ, Peters EJ & Wolfe RR (1988). Bed‐rest‐induced insulin resistance occurs primarily in muscle. Metabolism 37, 802–806. [DOI] [PubMed] [Google Scholar]
- Tabata I, Suzuki Y, Fukunaga T, Yokozeki T, Akima H & Funato K (1999). Resistance training affects GLUT‐4 content in skeletal muscle of humans after 19 days of head‐down bed rest. J Appl Physiol (1985) 86, 909–914. [DOI] [PubMed] [Google Scholar]
- Tesch PA, von Walden F, Gustafsson T, Linnehan RM & Trappe TA (2008). Skeletal muscle proteolysis in response to short‐term unloading in humans. J Appl Physiol (1985) 105, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF & Cerami A (1987). Anti‐cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia. Nature 330, 662–664. [DOI] [PubMed] [Google Scholar]
- Vary TC (1999). Down regulation of pyruvate dehydrogenase complex in skeletal muscle during sepsis: implications for sepsis‐induced hyperlactatemia. Sepsis 2, 303–312. [Google Scholar]
- Vary TC, Jurasinski CV, Karinch AM & Kimball SR (1994). Regulation of eukaryotic initiation factor‐II expression during sepsis. Am J Physiol 266, E193–E201. [DOI] [PubMed] [Google Scholar]
- Vary TC & Kimball SR (1992). Sepsis‐induced changes in protein synthesis: differential effects on fast‐ and slow‐twitch muscles. Am J Physiol 262, C1513–C1519. [DOI] [PubMed] [Google Scholar]
- Vary TC & Kimball SR (2000). Effect of sepsis on eIE4E availability in skeletal muscle. Am J Physiol Endocrinol Metab 279, E1178–E1184. [DOI] [PubMed] [Google Scholar]
- Vesali RF, Cibicek N, Jakobsson T, Klaude M, Wernerman J & Rooyackers O (2009). Protein metabolism in leg muscle following an endotoxin injection in healthy volunteers. Clin Sci (Lond) 118, 421–427. [DOI] [PubMed] [Google Scholar]
- Wang X, Hu Z, Hu J, Du J & Mitch WE (2006). Insulin resistance accelerates muscle protein degradation: Activation of the ubiquitin‐proteasome pathway by defects in muscle cell signaling. Endocrinology 147, 4160–4168. [DOI] [PubMed] [Google Scholar]
- Wilkinson SB, Phillips SM, Atherton PJ, Patel R, Yarasheski KE, Tarnopolsky MA & Rennie MJ (2008). Differential effects of resistance and endurance exercise in the fed state on signalling molecule phosphorylation and protein synthesis in human muscle. J Physiol 586, 3701–3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray CJ, Mammen JM, Hershko DD & Hasselgren PO (2003). Sepsis upregulates the gene expression of multiple ubiquitin ligases in skeletal muscle. Int J Biochem Cell Biol 35, 698–705. [DOI] [PubMed] [Google Scholar]
- Zamir O, Hasselgren PO, Kunkel SL, Frederick J, Higashiguchi T & Fischer JE (1992). Evidence that tumor necrosis factor participates in the regulation of muscle proteolysis during sepsis. Arch Surg 127, 170–174. [DOI] [PubMed] [Google Scholar]
- Zamir O, Hasselgren PO, von Allmen D & Fischer JE (1991). The effect of interleukin‐1α and the glucocorticoid receptor blocker RU 38486 on total and myofibrillar protein breakdown in skeletal muscle. J Surg Res 50, 579–583. [DOI] [PubMed] [Google Scholar]
- Zierath JR, He L, Guma A, Odegoard Wahlstrom E, Klip A & Wallberg‐Henriksson H (1996). Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia 39, 1180–1189. [DOI] [PubMed] [Google Scholar]