

Abstract
Non-nucleoside inhibitors of HIV-1 reverse transcriptase (NNRTIs), which bind to an allosteric site 10 −15Å from the polymerase active site, play a central role in anti-HIV chemotherapy. Though NNRTIs have been known for 30 years, the pathways by which they bind and unbind from HIV-RT have not been characterized. In crystal structures for complexes, three channels are found to extend from the NNRTI binding site to the exterior of the protein, while added mystery comes from the fact that the binding site is collapsed in the unliganded protein. To address this issue, metadynamics simulations have been performed to elucidate the unbinding of four NNRTIs from HIV-RT. A general and transferable collective variable (CV) defined by the distance between the center-of-mass (COM) of the binding pocket and COM of the ligand is used to follow the dynamics while minimizing the bias. The metadynamics also allows computation of the barriers to unbinding, which are compared with the observed potencies of the compounds in an antiviral assay.
Graphical Abstract
INTRODUCTION
Non-nucleoside reverse transcriptase inhibitors (NNRTI) are an integral part of current anti-HIV therapies, and they are typically used in combination with nucleoside reverse transcriptase inhibitors (NRTIs) to treat HIV infections.1,2 NNRTIs inhibit the HIV-1 reverse transcriptase (RT) enzyme by binding to the non-nucleoside inhibitor binding pocket (NNIBP), which is situated 10 – 15 Å from the polymerase active site.3 As with other classes of anti-HIV compounds, mutations of the target proteins arise readily and there is a need for the continued development of new drugs.3–5 The optimization of new inhibitors usually focuses on performance in enzymatic or cell assays, though there is increasing interest in maximizing the amount of time the drug spends bound to the target by minimizing the unbinding rate constant (koff). Recent studies have shown that drug efficacy sometimes correlates better with binding kinetics than with affinity alone.6 In particular, results for HIV-1 protease have shown that inhibition constant values (Ki) correlate well with koff, and resistance to mutations is enhanced by higher unbinding rates.7 To better understand variations in koff for NNRTIs, it is of fundamental importance to know the unbinding pathways from the NNIBP. Though there have been several reports where MD simulations were used to help understand the dynamics of HIV-RT with and without a bound NNRTI and how NNRTI binding inhibits polymerase activity,8–11 details on how NNRTIs enter or exit the binding pocket are still lacking.
Thus, the present work was undertaken to elucidate the exit pathways taken by NNRTIs and to better understand the factors affecting the kinetics of NNRTI unbinding. The inhibitor that was initially studied, JLJ135 (Figure 1), was discovered in our laboratory.12 It is a potent NNRTI with an EC50 of 2 nM in a standard assay for cytopathic protection of infected human T-cells. Subsequently, we reported a 1.95-Å X-ray crystal structure for it in complex with wild-type HIV-1 RT, as illustrated in Figure 2.13 Notably, the dimethylallyl group of the inhibitor is positioned in the center of the cluster of aromatic residues, Tyr181, Tyr188, Phe227, and Phe229; the cyano group extends into the open space between Phe227, Pro226, and Tyr318; there are hydrogen bonds between the aminopyrimidine fragment and the backbone of Lys101; and the C5-C6 edge of the pyrimidine ring is directed below the Lys101-Glu138 salt-bridge. Much rearrangement occurs on binding NNRTIs, as the binding site is collapsed in the apo protein with Tyr181 and Tyr188 in a down orientation filling the binding site.3 Though there is no direct evidence, the region projecting forward in Figure 2 between Lys101, Lys103, Glu138, and Val179 has been referred to as the “entrance” to the binding site,14,15 and we did report analogs of JLJ135 with solubilizing substituents such as morpholinylpropoxy extending into that region.13,15 There are two additional channels leading from the binding site to the bulk solvent, the “tunnel” and “groove” or “eastern channel”.15 The former leads past Tyr188 and Phe227 towards the polymerase active site, while the latter leads downward in Figure 2 past Pro226 and Pro236.
Figure 1.
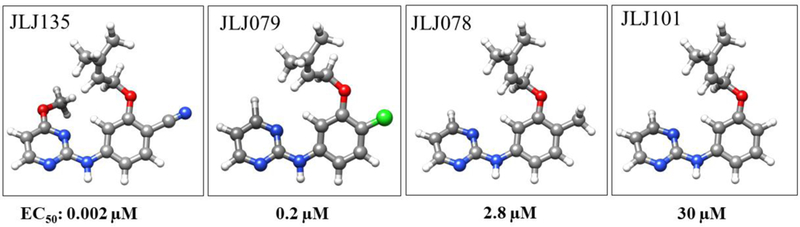
NNRTIs considered in this study and their experimental EC50 values for inhibition of viral replication in infected human T-cells.
Figure 2.
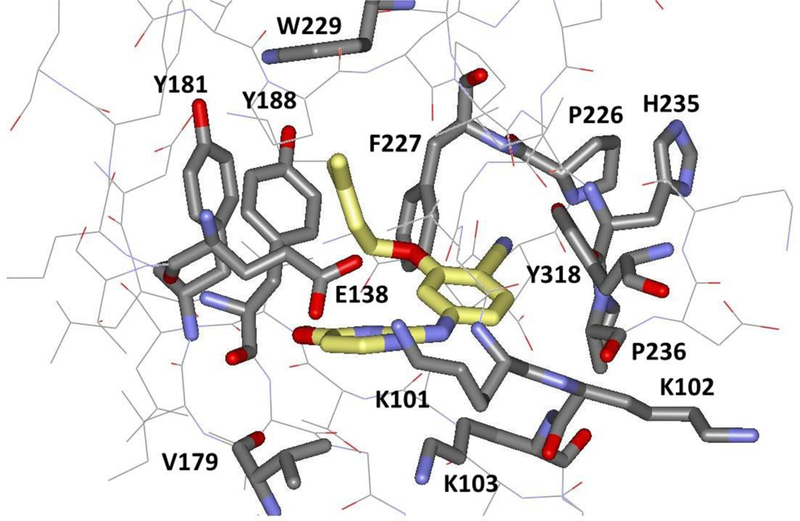
Illustration of the complex of JLJ135 and HIV-1 RT with key residues in the binding site highlighted (PDB ID: 4KO0).13 Carbon atoms of the inhibitor are in yellow.
It is unclear a priori which channel is preferred by an NNRTI for entrance or exit. Shen et al. have used steered molecular dynamics (MD) simulations to study exit of α-APA through the entrance channel, but this study does not address the intrinsic preference as the NNRTI was forced to leave via the entrance channel.14 Recent work by Bellucci et al.16 and the bifunctional RT inhibitors reported by Bailey et al. 17 have shown that NNRTIs can occupy the tunnel region, while other NNRTIs extend far in the groove.18 Thus, the preferred entrance/exit pathway remains unclear in the absence of a directional bias. A variety of methods featuring molecular dynamics with enhanced sampling and/or biasing potentials are now popular for modeling protein-ligand binding and unbinding kinetics.19,20 The present choice was to use metadynamics because it can be applied without introducing a directional bias for unbinding.21
Specifically, the unbinding pathways of JLJ135 and three close analogs (Figure 1) have been studied. The metadynamics was applied using a reaction coordinate or “collective variable” (CV), which was defined as the distance between the center of mass (COM) of Cα atoms of entrance, tunnel, and groove residues and the COM of the ligand (Figure 3). This choice promotes the departure of the ligand from the binding pocket without adding any directional bias. Metadynamics facilitates the rapid exploration of the chosen CV by periodically adding a small, repulsive Gaussian bias potential at the current CV value to discourage the system from revisiting an already explored region.21 Furthermore, once the system samples sufficiently the expanse of the CV, the deposited bias potentials can be used to estimate the underlying free energy surface or potential of mean force (PMF). Metadynamics has been applied successfully to study protein-ligand binding/unbinding kinetics, and reviews of the method and applications are available.19–24
Figure 3.
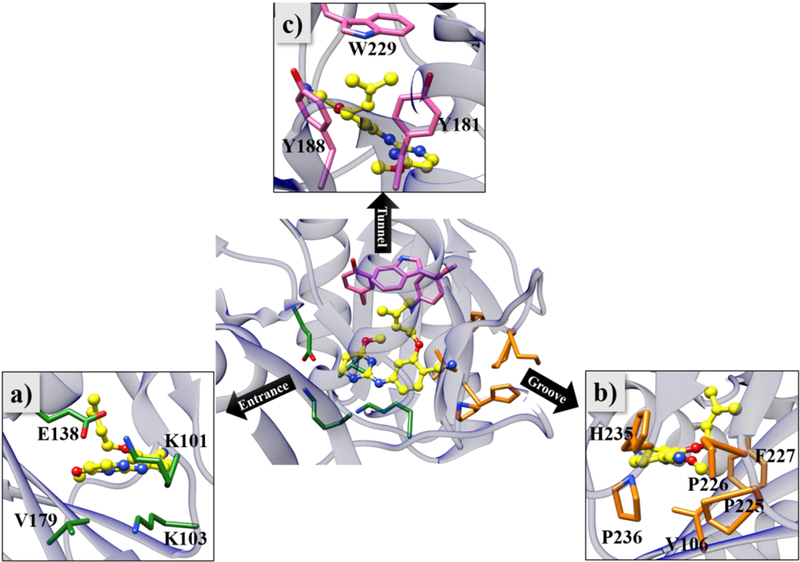
HIV-RT in complex with JLJ135 (PDB ID: 4KO0). Residues lining the three different exit channels, namely (a) entrance, (b) groove and (c) tunnel, are shown in orange, green, and pink respectively, and JLJ135 is shown with carbon atoms colored yellow.
In a recent report, Copeland proposed that in vivo cellular activity is better correlated with drug residence time than with the equilibrium dissociation constant (Kd).6 Metadynamics simulations were used previously to estimate drug residence times; however, these calculations employed a system-specific multi-dimensional CV and often were computationally taxing.25 Here, the average unbinding PMFs are calculated using a small number of metadynamics simulations with a transferable CV. The resulting unbinding barriers from the PMFs also offer a metric for ranking the compounds, which can be compared with observed activity data. For this purpose, three analogs of JLJ135 were added for study that feature small differences in structure, but significant changes in EC50 values from the infected T-cell assay using wild-type HIV-1 (Figure 1).12,26 JLJ101 is the parent compound with an EC50 of 30 µM. JLJ078 just adds a methyl group at the 4-position of the phenyl ring, which is directed towards the groove, but this improves the EC50 to 2.8 µM. Replacement of the methyl group with a chlorine in JLJ079 provides another 10-fold enhancement in potency to 0.2 µM. This change is attributed to enhancement of the acidity of the amino group, which is hydrogen-bonded to the carbonyl group of Lys101, and to improved electrostatic interactions for the C-Cl dipole with nearby residues including His235. A dramatic 100-fold increase in potency to 0.002 µM then occurs in progressing to JLJ135 stemming from replacement of the chlorine by the more polar cyano group and from addition of the methoxy group on the pyrimidine ring, which makes favorable van der Waals contacts in the vicinity of Tyr181. Thus, there is progressive filling of the binding site and improvement of non-bonded interactions along this series. Though the size difference between JLJ078 and JLJ079 is slight, the series should provide a reasonable test of the ability of PMFs calculated from unbinding metadynamics to discriminate the highly potent JLJ135 from compounds with moderate or weak activity. This assumes the expectation that kon for such a congeneric series is relatively constant compared to koff.6,23,25
SIMULATION METHODOLOGY
The MD simulations were initiated using the 1.95-Å crystal structure of HIV-RT complexed with JLJ135 (PDB ID: 4KO0)13 with missing residues added using the Modeller Plugin in UCSF Chimera.27,28 The initial complexes for JLJ101, JLJ079, and JLJ078 were generated by making the small modifications to JLJ135. Each protein-ligand complex was prepared using the solvate and the autoionize plugins in the VMD package,29 and solvated in a TIP3P30 water box with a padding of 12 Å, and neutralized by adding chloride ions. In all, the system consisted of 981 protein residues, 39,572 water molecules, 8 chloride ions, and the ligand in a ca. 134 × 118 × 88-Å periodic cell. Force-field parameters and topologies of the ligands were obtained from the LigParGen server.31 The OPLS-AA/M32 and OPLS-AA/1.14*CM1A-LBCC33,34 force fields were used for representing the protein and ligands, respectively. All MD simulations were carried out with the NAMD 2.11 package.35 Non-bonding interactions were truncated with a residue-based cutoff of 10 Å, and a switching function was applied between 8 and 10 Å. The Particle Mesh Ewald (PME) method was used for correcting long-range electrostatics, and the SHAKE algorithm was used for constraining the lengths of covalent bonds with hydrogen atoms. Before the metadynamics was invoked, each protein-ligand complex was energy-minimized for 5000 steps, equilibrated for 2 nanoseconds (ns) with constraints of 10 kcal/(mol Å2) on all protein heavy atoms, and was further equilibrated without any constraints for 5 ns. These MD calculations used a 2 femtosecond (fs) time-step with a Langevin thermostat and barostat to maintain temperature and pressure at 298 K and 1 atm.
The resultant final structures provided the starting point for the metadynamics simulations. For each of the four protein-ligand complexes, six independent metadynamics simulations were performed using the Colvars module (version 2015-09-16)36 of NAMD 2.11 in the NVT ensemble at 298 K, with a 2-fs time-step. The distance between the COM of the ligand, and the COM of Cα atoms of residues in the tunnel, entrance, and groove was used as the CV (Figure 3). Different Gaussian parameters and deposition rates were explored yielding choices of 0.2 kcal/mol for the height, 0.14 Å for the width, and deposition every 1000 time-steps. Larger heights and more frequent deposition increased lead to greater noise in the computed PMFs. Each run was terminated when the CV reached 25 Å. The VMD and MDTraj37 software packages were used for analyzing trajectories.
RESULTS AND DISCUSSION
Unbinding of JLJ135 and Analogs.
The 24 individual PMFs and their averages obtained from the six metadynamics simulations for the four ligands are illustrated in Figure S1 of the Supplementary Information. In all cases, the NNRTIs were found to exit via the entrance channel in the vicinity of the salt-bridge between Lys101 and Glu138 (Figures S2–S6). Since all the simulations showed that unbinding of the inhibitors occurs via the entrance channel, detailed analyses focused on the most potent compound, JLJ135. The unbinding PMF from the first metadynamics run for JLJ135 is representative and highlights the existence of three stages in the binding, as shown in Figure 4. The stages reflect exploration of the native complex, a pre-complex, and the unbound state with CV values of ca. 0–5, 7–14, and >15 Å, and with a significant barrier separating the native and pre-complex regions (Figure 4a). In this trajectory, the native complex is explored for about 4 ns, followed by 2 ns in the pre-complex region (Figure 4b).
Figure 4.
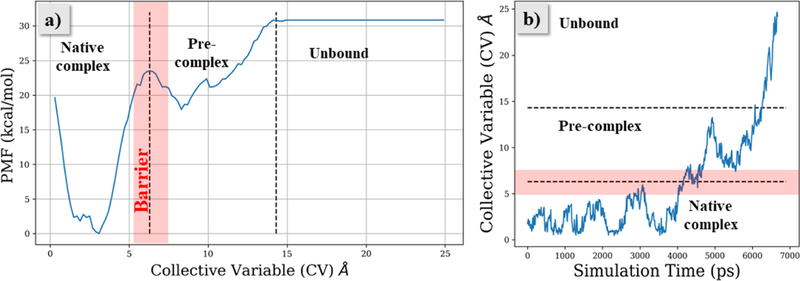
(a) Potential of mean force (PMF) profile for JLJ135 unbinding from HIV-RT. (b) Progression of the CV for the metadynamics trajectory with time through three stages of unbinding. The barrier region separating native and pre-complex states is shown in red.
Snapshots representative of the different stages of unbinding are shown in Figure 5. In the beginning of the metadynamics simulation, JLJ135 is in the native-complex region (Figure 5a), stabilized by the numerous interactions identified above (Figure 1) including contacts made by the O-dimethylally (ODMA) and methoxy groups with Tyr181, Tyr188, and Trp229, and the hydrogen bonds with Lys101. The binding pocket is further formed by the Lys101-Glu138 salt bridge, and the network of hydrogen bonds around the charged Lys101, Lys103, and Glu138 residues.
Figure 5.
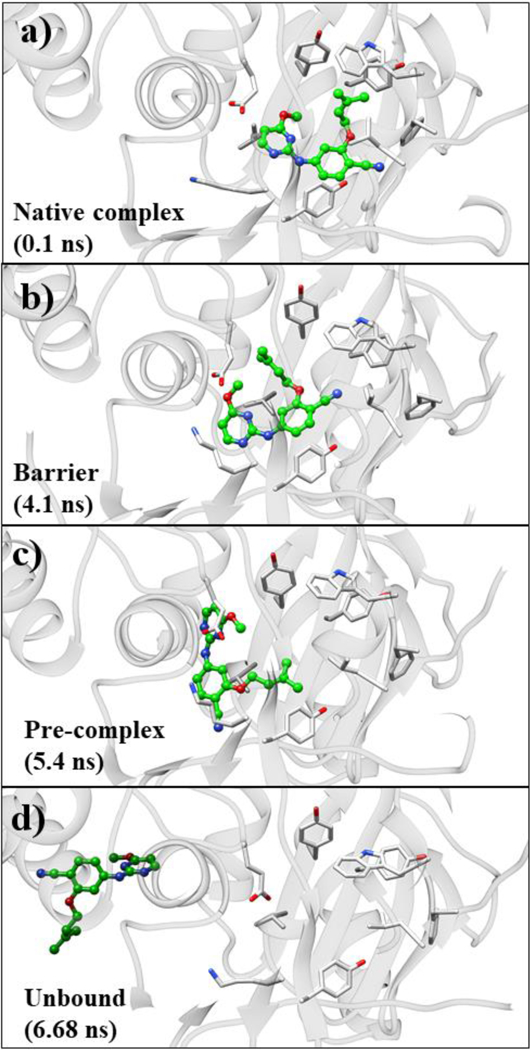
Snapshots of conformations sampled during unbinding of JLJ135. From the top: (a) native complex, (b) barrier between native complex and pre-complex, (c) pre-complex, and (d) unbound state.
As the unbinding progresses, JLJ135 moves towards the entrance channel near t = 3 ns after disruption of the salt-bridge between Lys101 and Glu138; however, the ODMA group is still out-of-plane from the diarylamino core. These variations are documented in Figures 6a–b and 7, respectively. In Figure 6, the COM-COM distance based on all side-chain atoms is plotted vs. simulation time for Lys101-Glu138 and Leu100-Val179, while Figure 7 shows the variations for a dihedral angle Φ that reflects the planarity of the inhibitor. Owing to resulting clashes with entrance channel residues, the inhibitor goes back to the bound-state region. At around t = 4.1 ns (Figure 4b), the ODMA group starts to sample conformations that are more in-plane with the core (Figure 7). The flatter structure is able to squeeze out of the binding site and the inhibitor moves into the barrier region without disruption of the salt-bridge between Lys101 and Glu138 (Figures 5, 6a, 7). The time in the barrier region is short since it is a free-energy maximum (Figure 4a) with reduced attractive non-bonded interactions, especially with residues Tyr181, Tyr188, Trp229, and Lys101, and with the ligand’s conformation more planar than in the bound or unbound states (Figure 7b).
Figure 6.
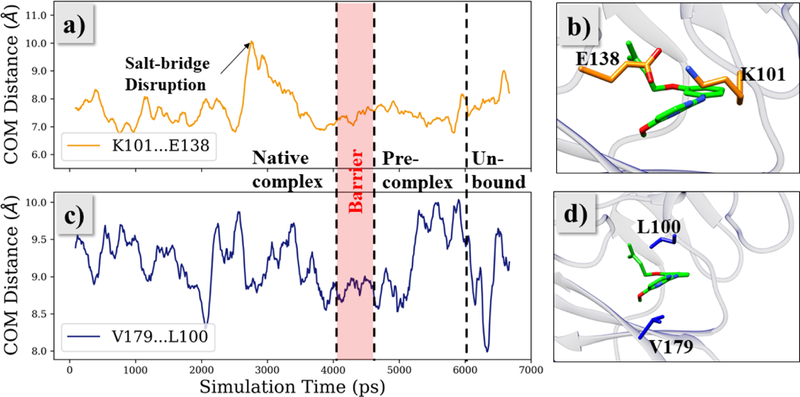
Rearrangement of binding pocket residues during the unbinding of JLJ135, as seen from the side-chain COM distances between Lys101-Glu138 (a, b) and Val179-Leu100 (c, d).
Figure 7.
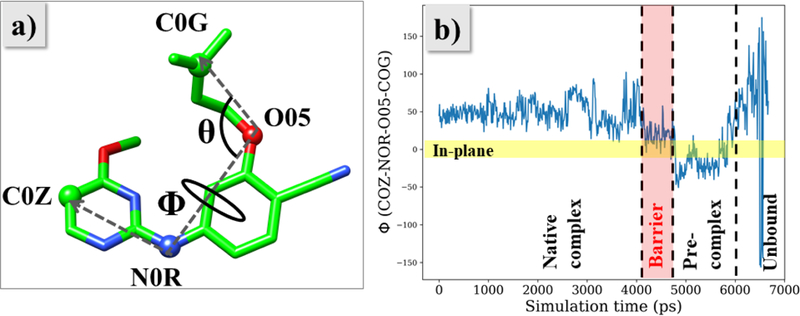
(a) The angles Φ and θ describe the orientation of the ODMA group relative to the diarylamino core and compactness of the ligand. (b) Evolution of Φ during the unbinding of JLJ135.
Once JLJ135 crosses the barrier, the pre-complex state is reached, as illustrated at t = 5.4 ns in Figure 5c and in Figure 8. At this stage, the front-edge of the ligand is solvent exposed, and there are hydrogen bonds with water for the aminopyrimidine fragment as well as a hydrogen bond between the cyano group and the ammonium group of Lys103. The inhibitor has flattened and slipped under the Lys101-Glu138 salt bridge, and there are substantial contacts between the ODMA group and Leu100 and between the phenyl ring of the inhibitor and Val179; there are no longer contacts with Tyr181, Tyr188, and Trp229. To allow this positioning, the distance between Leu100 and Val179 increases by about 1 Å, then it declines once the ligand is expelled (Figure 6c). At t = 6.3 ns, the CV reaches 15 Å and JLJ135 is fully unbound. In this trajectory, the inhibitor then reorients into a more extended geometry with the pyrimidine ring closer to the entrance channel (Figure 5d).
Figure 8.
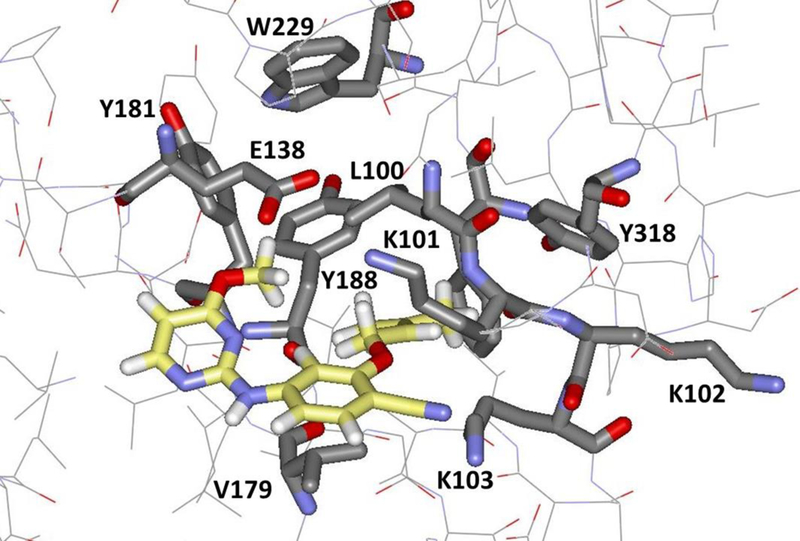
Illustration of the pre-complex of JLJ135 and HIV-1 RT with key residues in the binding site highlighted. Carbon atoms of the inhibitor are in yellow.
In reversing the trajectory to gain insight on the binding process, the structure of the pre-complex is very interesting. It indicates that the ODMA group is inserted first into the binding site with Tyr181 and Tyr188 in their “up” conformations. The ODMA group is co-planar with the attached phenyl ring during the insertion and it passes over Val179, which can be viewed as a gatekeeper. The important steric role of Val179 was also noted in the steered MD study of the unbinding of the NNRTI α-APA (Figure 9).14 From a conformational search with the OPLS/1.14*CM1A force field, the lowest-energy conformer for JLJ135 is very similar to the bound one illustrated in Figure 2, while the fourth conformer, which is 1.1 kcal/mol higher in energy, is similar to the conformation of the pre-complex in Figure 8. Thus, the conformational penalty to populate the co-planar form is not large. After insertion, the conformation of the ODMA group evolves to the lower-energy non-planar one, which allows good contacts to be established with Tyr181, Tyr188, and Trp229. Simultaneously, the cyanophenyl fragment rotates counterclockwise in Figure 8 and enters the binding site, followed finally by the 4-methoxypyrimidine fragment. A similar sequence can be expected to occur for other relatively small NNRTIs such that the substructure that makes the important stabilizing contacts with the Tyr181-Tyr-188-Trp229 cluster is inserted first (or leaves last). The steered MD study did show this sequence for α-APA with the 2-acetyl-5-methylphenyl substituent leaving last;13 however, the pathways are unclear for larger NNRTIs, for example, delavirdine or JLJ636 (Figure 9), which feature substantial extension into the groove region. This is illustrated for JLJ636 in Figure 10.38
Figure 9.
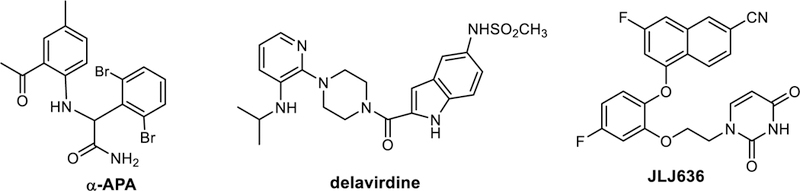
Structures of additional NNRTIs.
Figure 10.
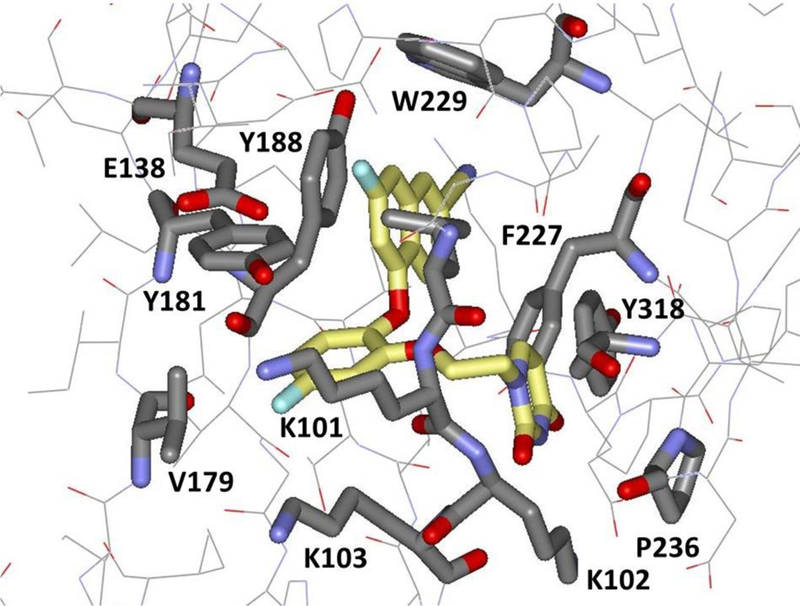
Illustration from the crystal structure (PDB ID: 5TW3) of JLJ636 bound to wild-type HIV-RT. Extension into the groove region near Pro236 is noted.
Metadynamics for Ranking Compounds.
Because of the general nature of the CV in this study, it can be used to examine the unbinding of any NNRTI and can be applied for identifying molecules with substantial unbinding barriers, i.e., low kd values. Such compounds are expected to show high efficacy in assays for anti-viral activity. For the four NNRTIs considered here (Figure 1), the PMFs that were obtained from averaging over the results of the six metadynamics runs for each compound are illustrated in Figure 11. There are significant quantitative uncertainties as indicated by the standard deviation bars in Figure 11, though the order of barrier heights separating the native and pre-complex is expected to reflect the difficulty in extracting the NNRTIs from the binding site. In order to reduce the uncertainties in the barrier heights, multiple transitions between the bound complex and unbound states need to be sampled. However, with the metadynamics methodology, the simulations were terminated once the ligand is unbound and equilibrium between bound and unbound states is not considered.
Figure 11.
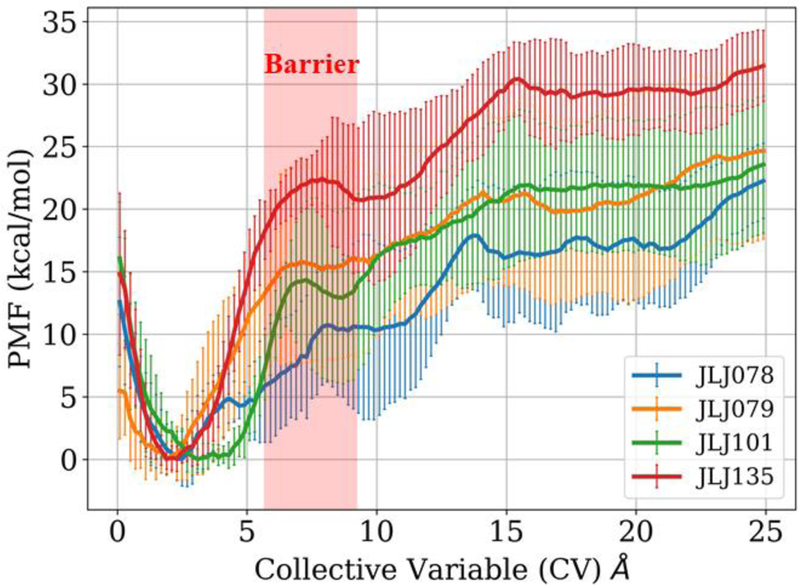
Unbinding potential of mean force (PMF) profiles of the four NNRTIs considered in this study. The barrier region separating native and pre-complex states is highlighted in red.
With these caveats, it is clear in Figure 11 that the most potent compound, JLJ135, does yield the highest barrier for exiting the binding site. Furthermore, the next most potent compound, JLJ079, gives the second highest barrier height, and the shape of its PMF for CV = 3 – 5 Å is similar to that for JLJ135. The PMFs for the two weakest NNRTIs, JLJ078 and JLJ101, are softer at low CV values indicating that they are more mobile in the binding site, though the order of their barrier heights is inconsistent with their relative potencies. The greater mobility is reasonable as the smaller inhibitors, JLJ078 and JLJ101, are making fewer contacts in the binding site. In addition, JLJ101, which lacks a buttressing substituent adjacent to the ODMA group has a greater preference for co-planar structures. A conformational search for JLJ101, like the one above for JLJ135, now yields a nearly planar structure as the second conformer, only 0.2 kcal/mol above the lowest-energy conformer, while conformers like that for bound JLJ135 are now 2–3 kcal/mol higher in energy. Thus, JLJ101 is less well preorganized for binding and it has a greater preference for planar conformers that facilitate transit out of the binding site. Overall, results for a much greater number of diverse NNRTIs are needed to be confident in general patterns; however, it does appear that the barrier heights and PMF shapes in Figure 11 do distinguish the two most potent NNRTIs from the two weakest inhibitors.
CONCLUSIONS
Three channels emanate from the binding site for NNRTIs, namely, the entrance, groove and tunnel. In this study, metadynamics simulations have been performed to establish the preferred unbinding pathways of the NNRTI JLJ135 and three analogs. The simulations were performed using a collective variable (CV) that was defined as the distance between the COM of the inhibitor and the COM of Cα atoms of residues at the entrances to the three channels. All six unbinding metadynamics runs for each of the four inhibitors showed that they prefer to unbind via the entrance channel. The resultant potentials of mean force (PMFs) were consistent with a three-state model for the unbinding starting with the bound state, passing over a barrier to a pre-complex, and then further dissociation to the fully unbound state. The inhibitors were found to adopt a relatively planar geometry as they slip out of the binding site under Lys101 and Glu138, and squeeze between Leu100 and Val179 (Figure 8). The average PMFs from the 24 runs also showed indications of ability to rank potency of the inhibitors; the two most potent inhibitors yielded higher barriers to achieve the pre-complex state and narrower free-energy wells for the bound state than the two least potent inhibitors. Further application of metadynamics to diverse NNRTIs is desirable to better assess the utility of the method for ranking the potency of compounds and to ascertain if other exit channels are used for NNRTIs that, for example, fill the groove region more than the inhibitors studied here.
Supplementary Material
ACKNOWLEDGEMENTS
Gratitude is expressed for support to the National Institutes of Health (GM32136 and AI44616) and to the facilities and staff of the Yale University Faculty of Arts and Sciences High Performance Computing Center. Gratitude is also expressed to Mr. Vladimiras Oleinikovas for discussions.
Footnotes
SUPPORTING INFORMATION
Six Figures provide details on the PMFs and trajectories from the 24 metadynamics runs for the four inhibitors. This information is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
REFERENCES
- 1.De Clercq E The nucleoside reverse transcriptase inhibitors, nonnucleoside reverse transcriptase inhibitors, and protease inhibitors in the treatment of HIV infections (AIDS). Adv. Pharmacol 2013, 67, 317–358. [DOI] [PubMed] [Google Scholar]
- 2.Gubernick SI; Félix N; Lee D; Xu JJ; Hamad B The HIV therapy market. Nature Rev. Drug Desc 2016, 15, 451–452. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds C; de Koning CB; Pelly SC; van Otterlo WAL; Bode ML In search of a treatment for HIV – current therapies and the role of non-nucleoside reverse transcriptase inhibitors (NNRTIs). Chem. Soc. Rev 2012, 41, 4657–4670. [DOI] [PubMed] [Google Scholar]
- 4.Iyidogan P; Anderson KS Current perspectives on HIV-1 antiretroviral drug resistance. Viruses 2014, 6, 4095–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jorgensen WL Computer-aided discovery of anti-HIV Agents. Bioorg. Med. Chem 2016, 24, 4768–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Copeland RA The drug–target residence time model: a 10-year retrospective. Nat. Rev. Drug Discovery 2016, 15, 87–95. [DOI] [PubMed] [Google Scholar]
- 7.Maschera B; Darby G; Palú G; Wright LL; Tisdale M; Myers R; Blair ED; Furfine ES Human immunodeficiency virus. Mutations in the viral protease that confer resistance to saquinavir increase the dissociation rate constant of the protease-saquinavir complex. J. Biol. Chem 1996, 271, 33231–33235. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Z; Madrid M; Evanseck JD; Madura JD Effect of a bound non-nucleoside RT inhibitor on the dynamics of wild-type and mutant HIV-1 reverse transcriptase. J. Am. Chem. Soc 2005, 127, 17253–17260. [DOI] [PubMed] [Google Scholar]
- 9.Ivetac A; McCammon JA Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. J. Mol. Biol 2009, 388, 644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Madrid M; Lukin JA; Madura JD; Ding J; Arnold E Molecular dynamics of HIV-1 reverse transcriptase indicates increased flexibility upon DNA binding. Proteins: Struct., Funct., Bioinf 2001, 45, 176–182. [DOI] [PubMed] [Google Scholar]
- 11.Vijayan RS; Arnold E; Das K Molecular dynamics study of HIV-1 RT-DNA-nevirapine complexes explains NNRTI inhibition and resistance by connection mutations. Proteins: Struct., Funct., Bioinf 2014, 82, 815–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruiz-Caro J; Basavapathruni A; Kim JT; Bailey CM; Wang L; Anderson KS; Hamilton AD; Jorgensen WL Optimization of diarylamines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett 2006, 16, 668–671. [DOI] [PubMed] [Google Scholar]
- 13.Bollini M; Frey KM; Cisneros JA; Spasov KA; Das K; Bauman JD; Arnold E; Anderson KS; Jorgensen WL Extension into the entrance channel of HIV-1 reverse transcriptase--crystallography and enhanced solubility. Bioorg. Med. Chem. Lett 2013, 23, 5209–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen L; Shen J; Luo X; Cheng F; Xu Y; Chen K; Arnold E; Ding J; Jiang H Steered molecular dynamics simulation on the binding of NNRTI to HIV-1 RT. Biophys. J 2003, 84, 3547–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ekkati AR; Bollini M; Domaoal RA; Spasov KA; Anderson KS; Jorgensen WL Discovery of dimeric inhibitors by extension into the entrance channel of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett 2012, 22, 1565–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bellucci L; Angeli L; Tafi A; Radi M; Botta M Unconventional plasticity of HIV-1 reverse transcriptase: how inhibitors could open a connection “gate” between allosteric and catalytic sites. J. Chem. Inf. Model 2013, 53, 3117–3122. [DOI] [PubMed] [Google Scholar]
- 17.Bailey CM; Sullivan TJ; Iyidogan P; Tirado-Rives J; Chung R; Ruiz-Caro J; Mohamed E; Jorgensen WL; Hunter R; Anderson KS Bifunctional inhibition of human immunodeficiency virus type 1 reverse transcriptase: mechanism and proof-of-concept as a novel therapeutic design strategy. J. Med. Chem 2013, 56, 3959–3968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leung CS; Zeevaart JG; Domaoal RA; Bollini M; Thakur VV; Spasov KA; Anderson KS; Jorgensen WL Eastern extension of azoles as non-nucleoside inhibitors of HIV-1 reverse transcriptase; cyano group alternatives. Bioorg. Med. Chem. Lett 2010, 20, 2485–2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Vivo M; Masetti M; Bottegoni G; Cavalli A Role of molecular dynamics and related methods in drug discovery. J. Med. Chem 2016, 59, 4035–4061. [DOI] [PubMed] [Google Scholar]
- 20.Bruce NJ; Ganotra GK; Kokh DB; Sadiq SK; Wade RC New approaches for computing ligand-receptor binding kinetics. Curr. Opin. Struct. Biol 2018, 49, 1–10. [DOI] [PubMed] [Google Scholar]
- 21.Laio A; Parrinello M Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiwary P; Limongelli V; Salvalaglio M; Parrinello M Kinetics of protein-ligand unbinding: Predicting pathways, rates, and rate-limiting steps. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E386–E391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cavalli A; Spitaleri A; Saladino G; Gervasio FL Investigating drug-target association and dissociation mechanisms using metadynamics-based algorithms. Acc. Chem. Res 2015, 48, 277–285. [DOI] [PubMed] [Google Scholar]
- 24.Casasnovas R; Limongelli V; Tiwary P; Carloni P; Parrinello M, Unbinding kinetics of a p38 MAP kinase type II inhibitor from metadynamics simulations. J. Am. Chem. Soc 2017, 139, 4780–4788. [DOI] [PubMed] [Google Scholar]
- 25.Callegari D; Lodola A; Pala D; Rivara S; Mor M; Rizzi A; Capelli AM Metadynamics simulations distinguish short- and long-residence-time inhibitors of cyclin-dependent kinase 8. J. Chem. Inf. Model 2017, 57, 159–169. [DOI] [PubMed] [Google Scholar]
- 26.Thakur VV; Kim JT; Hamilton AD; Bailey CM; Domaoal RA; Wang L; Anderson KS; Jorgensen WL Optimization of pyrimidinyl- and triazinyl-amines as non-nucleoside inhibitors of HIV-1 reverse transcriptase. Bioorg. Med. Chem. Lett 2006, 16, 5664–5667. [DOI] [PubMed] [Google Scholar]
- 27.Sali A; Blundell TL Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol 1993, 234, 779–815. [DOI] [PubMed] [Google Scholar]
- 28.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 29.Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- 30.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- 31.Dodda LS; Cabeza de Vaca I; Tirado-Rives J; Jorgensen WL LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res 2017, 45, W331–W336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robertson MJ; Tirado-Rives J; Jorgensen WL Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput 2015, 11, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen WL; Tirado-Rives J Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 6665–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dodda LS; Vilseck JZ; Tirado-Rives J; Jorgensen WL 1.14*CM1A-LBCC: Localized bond-charge corrected CM1A charges for condensed-phase simulations. J. Phys. Chem. B 2017, 121, 3864–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kalé L; Schulten K Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiorin G; Klein ML; Hénin J Using collective variables to drive molecular dynamics simulations. Mol. Phys 2013, 111, 3345–3362. [Google Scholar]
- 37.McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernández CX; Schwantes CR; Wang LP; Lane TJ; Pande VS MDTraj: A modern open library for the analysis of molecular dynamics trajectories. Biophys. J 2015, 109, 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee W-G; Frey KM; Gallardo-Macias R; Spasov KA; Bollini M; Anderson KS; Jorgensen WL Picomolar inhibitors of HIV-1 reverse transcriptase: design and crystallography of naphthyl phenyl ethers. ACS Med. Chem. Lett 2014, 5, 1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.