

Abstract
Rationale:
Voltage-gated Na+ channel (INa) function is critical for normal cardiac excitability. However, the Na+ channel “late” component (INa,L) is directly associated with potentially fatal forms of congenital and acquired human arrhythmia. Ca2+/calmodulin-dependent kinase II (CaMKII) enhances INa,L in response to increased adrenergic tone. However, the pathways that negatively regulate the CaMKII/Nav1.5 axis are unknown and essential for the design of new therapies to regulate the pathogenic INa,L.
Objective:
To define phosphatase pathways that regulate INa,L in vivo.
Methods and Results:
A mouse model lacking a key regulatory subunit (B56α) of the protein phosphatase 2A (PP2A) holoenzyme displayed aberrant action potentials following adrenergic stimulation. Unbiased computational modeling of B56α knockout (KO) mouse myocyte action potentials revealed an unexpected role of PP2A in INa,L regulation that was confirmed by direct INa,L recordings from B56α KO myocytes. Further, B56α KO myocytes display decreased sensitivity to isoproterenol-induced induction of arrhythmogenic INa,L, and reduced CaMKII-dependent phosphorylation of Nav1.5. At the molecular level, PP2A/B56α complex was found to localize and co-immunoprecipitate with the primary cardiac Nav channel, Nav1.5.
Conclusions:
PP2A regulates Nav1.5 activity in mouse cardiomyocytes. This regulation is critical for pathogenic Nav1.5 “late” current and requires PP2A-B56α. Our study supports B56α as a novel target for the treatment of arrhythmia.
Subject Terms: Animal Models of Human Disease, Arrhythmia, Basic Science Research, Calcium Cycling/Excitation-Contraction Coupling, Cell Signaling/Signal Transduction
Keywords: PP2A, ankyrin, CaMKII, Nav1.5, arrhythmia (mechanisms), physiology/function, phosphorylation, phosphatase
INTRODUCTION
Voltage-gated Na+ channels (Nav) play a critical role in regulation of myocyte membrane excitability and cardiac function. Nav channel current (INa) is a large amplitude, short duration inward current that is regulated by rapid channel activation and immediate inactivation.1, 2 However, a small “late” component of INa (INa,L) is present at baseline. This INa,L increases in response to heightened adrenergic conditions and has been directly associated with potentially fatal forms of congenital and acquired human arrhythmia.3–6 Thus, the mechanisms underlying INa,L regulation have been a critical area of translational research both in academics and industry.7–10
Ca2+/calmodulin-dependent kinase II (CaMKII)-dependent phosphorylation of Nav1.5 is directly linked with increased INa,L. Increased CaMKII-dependent phosphorylation Nav1.5 (pS571; Nav1.5 phosphorylation at amino acid 571) is now a signature of adrenergic imbalance in human heart disease and animal models of cardiovascular disease.4, 11–16 Despite the clear impact of the Nav1.5/CaMKII regulatory axis in human disease, we are unaware of any in vitro or in vivo data on the molecular mechanisms that negatively regulate the CaMKII/Nav1.5 axis. We expect these data to advance the design of new therapies aimed at regulating arrhythmogenic INa,L.
There are three major protein phosphatase (PP) families: tyrosine, serine-threonine, and dual specificity phosphatases, with serine-threonine phosphatases (e.g. PP1, PP2A and PP2B) responsible for approximately 90% of de-phosphorylation events in the heart. While both PP1 and PP2B have been tightly linked with cardiac signaling and disease, PP2A function has been relatively less explored. Unlike many phosphatase enzymes, the PP2A holoenzyme is formed from three subunits. The core enzyme has constitutive activity and is comprised of a catalytic (C) and scaffolding (A) subunit, each of which are encoded by two genes.17 Unique to the PP2A class of phosphatases is the third, regulatory (B) subunit family with thirteen members each encoded by a separate gene to regulate PP2A tissue- and cell-expression, activity, and subcellular distribution.18
Here, we utilized an unbiased approach using the cardiac action potential of B56α deficient mice to define novel roles of PP2A in cardiac excitability. Our data implicate an unexpected and key role of PP2A, and more specifically PP2A/B56α, in the regulation of Nav1.5 phosphorylation and INa,L. Specifically, action potentials (APs) recorded from B56α knockout (KO) mice (show increased myocyte PP2A activity) displayed decreased sensitivity to APD prolongation in response to isoproterenol treatment. Computational modeling supported a potential role for INa,L in AP phenotypes and direct recording of INa,L from B56α KO myocytes illustrated that B56α KO mice are insensitive to isoproterenol-induced arrhythmogenic INa,L. Mechanistically, we identify that PP2A/B56α complex is localized at the intercalated disc with the primary cardiac Nav channel, Nav1.5. We illustrate that the B56α-associated PP2A complex is critical for Nav channel activity as Nav1.5 pS571 is reduced in mice lacking B56α. Finally, we illustrate that the B56α/PP2A is a bona fide regulatory molecule for Nav1.5/INa,L in myocytes, as B56α KO myocytes are insensitive to isoproterenol-induced augmentation of INa,L. In summary, our findings define a pathway that regulates CaMKII-dependent phosphorylation in the heart, and identify a potential novel target to suppress arrhythmogenic activity of INa in heart.
METHODS
The data that support the findings of this study are available from the corresponding author on reasonable request.
Animals.
All animal procedures were approved and in accordance with institutional guidelines (Institutional Animal Care and Use Committee; The Ohio State University). All mice used were age-matched (male and female littermates) and were housed in the same facility (temperature and humidity), consumed the same diet, provided water ad libitum, and kept on identical 12-h light/dark cycles. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. B56α KO mice (backcrossed >7 generations for this study) were created as described.19
Detailed descriptions on the materials and methods used in this study are provided in the Online Supplement.
RESULTS
B56α knockout mice display altered cardiomyocyte excitability.
Our group previously identified the B56α class of PP2A regulatory subunits as key regulatory proteins in heart. Work from our group and others implicate this pathway as essential for regulation of RyR2 phosphorylation and cardiac calcium signaling.19 Finally, B56α KO mice illustrate that this subunit behaves as an auto-inhibitor of PP2A activity in heart and B56α KO hearts display increased PP2A activity and reduced spontaneous calcium release in response to increased sympathetic activity.19
To test the impact of B56α deficiency on cardiomyocyte excitability, APs were recorded from isolated cardiac myocytes of WT mice or mice lacking B56α (B56α KO) at 0.5 and 1.0 Hz. B56α KO myocytes displayed significantly shortened AP duration (APD) at 50%, 75% and 95% repolarization (APD50, APD75, and APD95) compared with WT myocytes. Specifically, APD50, APD75, and APD95 were reduced 53%, 46%, and 32% respectively, in B56α KO ventricular myocytes paced at 1-Hz (Figure 1A–B; p<0.05). Action potential amplitude (APA) and maximum upstroke velocity (dv/dtmax) demonstrated a downward trend in B56α KO myocytes, but did not achieve statistical significance (Figure 1C–D). While we previously implicated B56α and PP2A with the regulation of RyR2 in myocytes19, these APD data support potential new roles of PP2A-dependent regulation in myocyte excitability.
Figure 1. B56α KO mice display aberrant cardiomyocyte excitability.

(A-B) Representative action potential (APs, 1.0 Hz pacing) and summary of APD at 50%, 75% and 95% repolarization for 0.5 and 1.0 Hz pacing in WT and B56α KO myocytes. (C-D) AP amplitudes (APA) and maximum upstroke velocity (dv/dtmax) in WT and B56α KO myocytes. Results are shown for 0.5 and 1.0 Hz pacing frequencies (for B-D, WT, N=3; n= 9 and B56α KO, N=3; n=8; *p<0.05).
B56α KO mice display decreased sensitivity to adrenergic stimulation.
Based on the role of PP2A in autonomic regulation of the heart, we tested the impact of beta-adrenergic stimulation on myocyte excitability in WT and B56α KO myocytes. When exposed to 100nM isoproterenol (Iso), WT myocytes exhibited a 42% and 47% prolongation of APD95 at 0.5 Hz and 1.0 Hz pacing respectively (Figure 2A,C; 1 Hz). In contrast, 100nM Iso did not alter APD95 in B56α KO myocytes at either pacing frequency (Figure 2B, D, 1Hz). Most notably, B56α KO myocytes displayed nearly identical repolarization profiles late in the AP. No significant changes in APA or dv/dtmax were identified between WT or B56α KO myocytes following Iso treatment (Figure 2E–H). Together, these new data support key roles of PP2A in regulation of cardiac excitability at baseline and following adrenergic stimulation. Importantly, these findings support new roles of PP2A in the regulation of late phases of the cardiac action potential.
Figure 2. B56α KO ventricular myocytes display decreased sensitivity to isoproterenol-induced APD prolongation.

(A-D) Representative APs (1.0 Hz pacing) and summary of APD at 50%, 75% and 95% repolarization at 0.5 and 1.0 Hz pacing in WT and B56α KO myocytes ±100nM Iso. (E-H) Action potential amplitudes (APA) and maximum upstroke velocity (dv/dtmax) in WT and B56α KO myocytes ±Iso. Results are shown for 0.5 and 1.0 Hz pacing frequencies (WT, N=3; n=9 and B56α KO, N=3; n=8 *p<0.05).
Identification of Nav1.5 as PP2A target in heart.
To define potential new targets of the PP2A/B56α complex in heart, computational modeling was performed. Briefly, partial least-squares regression analysis was performed using the Hund-Rudy AP model to identify sets of parameters that produced the best-fit to experimental data from WT and B56α KO AP measurements (APD, APA, and dv/dtmax; representative regression coefficients are shown for APD in Figure 3A).20–23 This unbiased approach supported work from our group and others linking PP2A with cardiac ion channels and transporters important for intracellular calcium handling (Figure 3A; Cav1.2, NCX, SERCA2a).19, 24 However, this analysis predicted a new link between PP2A/B56α and INa regulation. While not the focus of this manuscript, the findings also suggest a potential role in ICa,L regulation. Specifically, the simulations predicted an important role for altered INa,L, but not INa,peak, in abnormal cell membrane excitability observed in the setting of B56α deficiency. In fact, the unbiased computational analysis identified a likely 60–70% decrease in INa,L as a mechanistic determinant of altered repolarization in B56α KO myocytes (Figure 3B–C).
Figure 3. Identification of Nav1.5 as PP2A target.

Computer simulations to predict the mechanism underlying changes in cell excitability in the setting of B56α-deficiency. (A) Regression coefficients showing relative impact of changes in ion channel conductance/transport rates on action potential duration at 90% repolarization (APD) in the Hund-Rudy dynamic cell model. Abbreviations are as follows: inwardly rectifying K current (gK1), rapid delayed rectifier K+ current (gKr), slow delayed rectifier K+ current (gKs), L-type Ca2+ current (gCa(L)), fast inward Na+ current (gNa), subspace and bulk Na+/Ca2+ exchanger (gNaCa(ss) and gNaCa(bulk), respectively), Na+/K+ ATPase (gNak), late Na+ current (gNa,L), Ca2+ release from SR (grel), transient outward K+ current (gto), Ca2+ translocation from network to junctional SR (gtr) and Ca2+ uptake into SR (gup). (B) APD90 (expressed relative to WT) in experiment (B56α KO), and in the following computational models: 1) model with parameter values identified in the regression analysis with smallest sum-of-squared error compared to experiment (Mutant), and 2) model with parameter values selected as average of 10 best solutions (Mutant10). (C) Corresponding model parameters (expressed relative to WT) for Mutant and Mutant10 computational models.
Identification of Nav1.5 as PP2A target in heart.
Based on computational predictions, we tested the impact of B56α deficiency on INa properties in isolated mouse cardiomyocytes. As shown in Figure 4A–C, we observed no difference in the current-voltage relationship or peak INa at baseline between WT and B56α KO myocytes (p=N.S.). Further, we observed no difference in INa voltage-dependent activation, steady-state voltage-dependent inactivation, or time-dependent recovery between WT and B56α KO myocytes (Figure 4D–E; p=N.S.). Detailed analysis of these properties by gender did not identify a significant difference in whole cell INa properties or cell capacitance between male or female mice (Online Figure I; p=N.S.). However, direct recording of INa,L illustrated that integration of INa from 50 to 100 msec was ~40 % lower at baseline in B56α KO myocytes compared to WT myocytes (Figure 4F–G; WT 9.65 ±1.87 vs. 5.82±1.06 nA.msec in B56α KO; p<0.05; no statistical difference in cell capacitance, Figure 4H). In summary, INa,L is reduced in B56α KO mouse cardiomyocytes at baseline compared with WT myocytes.
Figure 4. B56α KO myocytes display decreased INa,L.
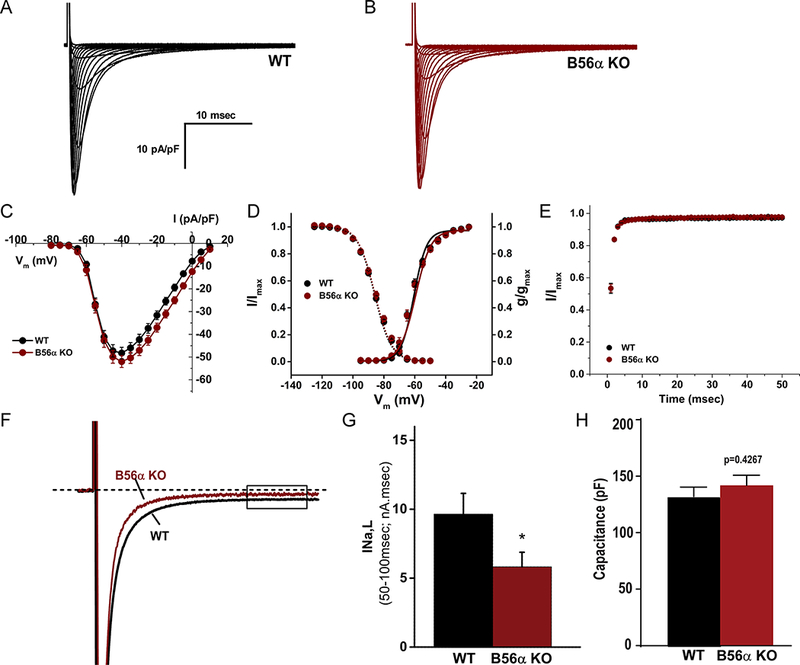
(A-B) Representative recordings of whole cell INa from WT and B56α KO ventricular myocytes. (C) Current-voltage relationship, (D) voltage-dependent activation and voltage-dependent inactivation curves, and (E) time-dependent recovery of INa in WT and B56α KO ventricular myocytes. No significant difference was observed in peak INa at experimental voltages ranging from −60 to −15mV (p=N.S.), in V½ as determined by Boltzmann fits of the steady-state voltage-dependent inactivation (p=N.S.) and time-dependent recovery (p=N.S.; N=5,5, n=22,21 combined genders). (F) Representative INa,L traces from WT and B56α KO ventricular myocytes. (G) Quantification of INa,L from WT and B56α KO ventricular myocytes at baseline (WT, N=4; n=10 and B56α KO, N=4; n=9; *p<0.05). (H) No difference in cell capacitance was observed between WT and B56α KO myocytes (p=N.S.; shown for INa,L and INa experiments).
B56α/PP2A complex is localized with CaMKII at the myocyte intercalated disc.
As prior work has focused on the link between PP2A and transverse-tubule/sarcoplasmic reticulum calcium handling machinery19, 25–27, we next investigated the predicted association of PP2A with the primary cardiac Nav channel, Nav1.5 (Online Figure IIA–J). While present at multiple membrane domains28,29, Nav1.5 is enriched at the cardiac intercalated disc (Online Figure IIA).30, 31 In support of a regulatory role of B56α/PP2A for INa,L regulation, populations of B56α as well as PP2A catalytic and scaffolding subunits were also localized to the intercalated disc of WT myocytes (Online Figure IIC, E,F,G,). In the heart, CaMKIIδ phosphorylation of Nav1.5 is critical for regulation of INa availability and INa,L.13, 32 As previously described13, and in support of a role for dual INa adrenergic regulation by CaMKII/PP2A, we observed localization of CaMKIIδ at the intercalated disc (Online Figure IID). Finally, the adapter protein ankyrin-G, localized primarily to the intercalated disc (Online Figure IIB) is critical for both the targeting and regulation of Nav1.5.33,31, 34 Specifically, ankyrin-G directly interacts with Nav1.5 and βIV-spectrin, a CaMKII-scaffolding protein at the intercalated disc (Online Figure IIH).13 In our studies, we observed significant expression of CaMKIIδ and ankyrin-G at sites co-localized in triple-labeling experiments with PP2A subunits at the intercalated disc (Online Figure III–J). In summary, B56α and PP2A core enzyme subunits are localized with the CaMKII regulatory complex. However, it is important to note that while enriched at the intercalated disc, these components, as well as Nav1.5 populations, are present at other membrane domains as previously demonstrated by multiple groups28,29. As expected, the localization of ankyrin-G, CaMKIIδ, and PP2A-C were independent of the B56α expression (Online Figure III).
PP2A/B56α regulatory complex associates with membrane complex.
Based on our new data, we performed co-immunoprecipitation experiments using detergent-soluble lysates of mouse left ventricle to determine whether the PP2A holoenzyme associates with the Nav1.5 regulatory complex. We observed association of ankyrin-G and CaMKIIδ with Nav1.5 (Figure 5A–B). Further, we demonstrated association of the PP2A catalytic subunit (PP2A-C, Figure 5C), the PP2A B56α regulatory subunit (Figure 5D), and the PP2A scaffolding subunit (PP2A-A) with Nav1.5 by co-immunoprecipitation (Figure 5E). Ankyrin-G also associated with PP2A-C by co-immunoprecipitation (Figure 5F). Finally, we confirmed our findings in human heart, through co-immunoprecipitation of Nav1.5 with PP2A subunits from detergent-soluble lysates of non-failing human left ventricle (Figure 5G). In summary, the PP2A holoenzyme exists in complex with the Nav1.5 complex in heart.
Figure 5. PP2A/B56α complex associates with Nav1.5 regulatory complex.

(A-E) Co-immunoprecipitation experiments were performed from detergent-soluble lysates of adult mouse hearts using beads conjugated to Nav1.5 Ig or control Ig. Bound protein was eluted and immunoblotted with ankyrin-G (A), CaMKIIδ (B), PP2A-C (C), PP2A-B56α (D) or PP2A-A (E). Data are representative of experiments repeated three times. (F) Co-immunoprecipitation experiments were performed from detergent-soluble lysates of adult mouse hearts using beads conjugated to PP2A-C Ig or control Ig. Bound protein was immunoblotted with ankyrin-G. (G) Co-immunoprecipitation experiments were performed from human left ventricular lysates (non-failing hearts) using beads conjugated to Nav1.5Ig or control Ig. Bound protein was immunoblotted with PP2A-C. Experiments were repeated three times. (H-K) Pull-down experiments were performed using detergent-soluble extracts of WT mouse hearts using glutathione S-transferase (GST) or GST-AnkG. Bound protein was analyzed using antibodies specific for PP2A-B56α (H), PP2A-C (I), PP2A-A (J) or PP2A-B56γ (L). Data are representative of experiments repeated three times.
We previously identified an interaction of ankyrin-B (unique gene product from ankyrin-G) with B56α in heart.35 Ankyrin-B directly associates with the C-terminus of B56α and is necessary for the targeting of B56α to the cardiac dyad to modulate CaMKII-dependent regulation of RyR2.19, 25, 36, 37 Based on our new findings, we tested a potential interaction of ankyrin-G with B56α. Purified GST-AnkG, but not GST- alone associated with B56α from detergent-soluble lysates from mouse heart (Figure 5H). Further GST-ankyrin-G, but not GST associated with PP2A catalytic and scaffolding subunits, supporting the interaction of ankyrin-G with the full holoenzyme (Figure 5I–J). This interaction was specific, as we did not observe association of ankyrin-G with the structurally similar (but lacks the ankyrin-binding motif) PP2A B56α regulatory subunit (Figure 5K). Thus, ankyrin-G (and thus Nav1.5) associates with PP2A via the B56α regulatory subunit.
B56α KO mice display aberrant CaMKII-dependent Nav1.5 phosphorylation.
Nav1.5 pS571 phosphorylation is increased in both ischemic and non-ischemic human heart disease, as well as in canine and mouse models of disease.4 Finally, mice harboring Nav1.5 phosphomimetic S571E display increased INa,L, altered myocyte electrical regulation and arrhythmia phenotypes.12 To test the impact of B56α-associated PP2A on cardiac Nav1.5, we evaluated Nav1.5 phosphorylation using affinity-purified Ig directed against pS571 (Figure 6A–G). As expected, we did not observe a significant increase in total Nav1.5 protein levels between WT and B56α KO mice following isoproterenol treatment (Figure 6A,E). Additionally, we observed no difference in total CaMKIIδ between WT and B56α KO mice (Figure 6A, D). However, compared with WT mouse hearts, we observed a significant decrease in cardiac Nav1.5 pS571 levels in hearts of B56α KO mice (Figure 6A,F). This change was significant when normalizing for total Nav1.5 protein expression levels (Figure 6F). Notably, total PP2A-C levels were unchanged between WT and B56α KO mice (Figure 6A, C). In summary, our findings support a role of B56α-linked PP2A in regulation of Nav1.5 phosphorylation.
Figure 6. B56α KO mice display decreased Nav1.5 Ser571 phosphorylation.

(A-G) Immunoblots and quantitative analysis of PP2A-B56α (WT, N=5; B56α KO, N=7; p<0.05), PP2A-C (WT, N=7; B56α KO, N=7; p=N.S.), CaMKIIδ (WT, N=7; B56α KO, N=7; p=N.S.) Nav1.5 (WT, N=11; B56α KO, N=11; p=N.S.), Nav1.5 pSer571 (WT, N=11; B56α KO, N=10; p<0.05) and GAPDH expression in WT and B56α KO mouse hearts following isoproterenol treatment. B56α KO mice showed a significant decrease in both B56α expression and Nav1.5 Ser571 phosphorylation, as well as the ratio of Nav1.5 Ser571 phosphorylation to Nav1.5 expression (WT, N=11; B56α KO, N=10; p<0.05) following isoproterenol treatment. For Nav1.5pS571/total Nav1.5ratio, data is expressed based on normalized protein expression versus GAPDH as shown in E and F. We observed no difference in GAPDH expression between genotypes (WT, N=11; B56α KO, N=10; p=N.S.).
B56α is required for INa,L regulation.
To test the role of B56α for INa and INa,L regulation, we evaluated peak and late INa in WTand B56α KO myocytes ± isoproterenol treatment to mimic increased adrenergic tone. As expected, isoproterenol treatment induced an increase in both peak and INa,L in WT myocytes (Figure 7A, C-D; p<0.05). In contrast, consistent with a critical role of B56α in INa,L regulation, we observed no significant difference of peak INa or INa,L in B56α KO myocytes±isoproterenol treatment (Figure 7B–D; p=N.S.; no significant difference in cell capacitance between genotypes). In fact, at baseline, we observed a reduction in INa,L between WT and B56α KO myocytes likely representing the basal increase in PP2A-dependent phosphatase activity in the B56α KO heart.19, 38, 39,40 In summary, myocytes lacking B56α have reduced sensitivity to isoproterenol-mediated increases in INa,L and importantly arrhythmogenic INa,L.
Figure 7. B56α KO myocytes are insensitive to isoproterenol-induced increases of INa,L.

(A-B) Representative voltage-gated Na+ current (INa) traces from WT and B56α KO ventricular myocytes ± isoproterenol (100nM). (C-D) Summary data (mean ± SEM) for peak INa (at −30 mV), and INa,L in response to 100nM Iso (*p<0.05). (E) Cell capacitance was not different between groups (p=N.S., WT, WT Iso N=5, 4; n=10, 9 and B56α KO, B56α KO Iso N=4,4; n= 9,7).
DISCUSSION
In vertebrates, cellular activity is tightly governed by highly evolved protein signaling platforms. Key to this regulation is the control of protein function via reversible protein phosphorylation. In the heart, protein phosphorylation is a central signaling axis with implications for excitation-contraction coupling, transcriptional regulation and metabolism.41, 42 Alterations in protein phosphorylation are present in both congenital and acquired forms of heart disease including fatal arrhythmias and heart failure.43–45 While the protein kinase axis has received appropriate attention and has been vital in specific cardiovascular therapies, the removal of phosphate groups by phosphatases is relatively less studied, although mounting data support important roles for PP1 (protein phosphatase type 1) and PP2A in cardiovascular disease.46–49 For example, we and others have illustrated that B56α is critical for targeting key populations of PP2A to the cardiac T-tubule/SR through the interaction of B56α with ankyrin-B.35 Mice lacking ankyrin-B display a loss of T-tubule/SR B56α targeting and alterations in CaMKII/PP2A signaling, contributing to increased adrenergic tone and substrate for the development of catecholamine-based arrhythmias.35, 36 Further, PP2A activity has been linked to a number of key ion channel activities including the L-type calcium-channel (Cav1.2)24, ryanodine receptor 2 (RyR2)19, Na+/Ca+ exchanger (NCX)50, and Na+/K+ ATPase.35, 51
Work from our group and others has defined a critical role of CaMKII in Nav1.5 regulation for normal cardiac electrical function and in disease. Specifically, CaMKII-dependent phosphorylation of Nav1.5 at sites including serine 571 have been highly associated with decreased Nav1.5 channel availability and increased INa,L.11 In fact, mice harboring a phosphomimetic S/E residue at Ser571 display increased INa,L and increased arrhythmia susceptibility, whereas ablation of the serine site by S/A substitution abrogates effects of isoproterenol on INa,L and burden of arrhythmia.12 Notably, CaMKII is tightly linked with its effector molecule Nav1.5 via interaction with the βIV-spectrin/ankyrin-G scaffold.13 While other Nav channel subunits are present in heart52, 53,54 we did not observe interaction of either Nav1.6 or Nav β subunits with the PP2A complex in co-immunoprecipitation assays (Online Figure IVA–B). However, future experiments will be necessary to definitively define the role of PP2A for Nav1.5-independent INa in heart.
Our new data expand the ankyrin-G/Nav1.5/CaMKIIδ/βIV-spectrin protein complex and define the specific signaling pathway underlying negative/inhibitory modulation of Nav1.5 and INa,L, as well as other potential membrane and submembrane effectors at the cardiac intercalated disc (Figure 8). Specifically, our new data implicate PP2A via interaction with B56α, as critical for AP duration, both at baseline and following adrenergic stimulation. Our new findings illustrate key potential downstream membrane complexes as targets for PP2A/B56α including INa,L. We demonstrate that B56α is localized with Nav1.5 and CaMKII, as well as ankyrin-G and βIV-spectrin at the intercalated disc, and biochemical experiments show both co-immunoprecipitation of the full PP2A holoenzyme (catalytic, regulatory, scaffolding subunits) and interaction of B56α with ankyrin-G, and Nav1.5. Notably, in line with prior findings illustrating elevated Nav1.5 pSer571 phosphorylation and elevated INa,L in the presence of heightened adrenergic state, B56α KO hearts displayed reduced Nav1.5 pSer571 phosphorylation and INa,L in response to isoproterenol treatment. In summary, our new findings define a key nodal regulatory complex in heart, as well as a new molecular target for the regulation of Nav1.5, and specifically the pathogenic INa,L.
Figure 8. Intercalated disc Nav1.5 macromolecular signaling complex.

Via direct interaction of Nav1.5 with ankyrin-G and βIV spectrin, CaMKIIδ resides in close proximity with its target molecule, Nav1.5. Our new findings implicate ankyrin-G in targeting the PP2A complex via B56α to the Nav1.5 intercalated disc complex to balance CaMKII-dependent phosphorylation. Together, CaMKIIδ and PP2A regulate INa,L in heart.
This is the first study evaluating the role of PP2A in the negative regulation of CaMKII/NaV1.5; thus there are limitations. First, it is important to note that although our data support PP2A as a central regulatory molecule for Nav1.5 and INa,L, our study does not preclude secondary mechanisms. Certainly, INa regulation is complex and relies on multiple phosphorylation and secondary post-translational modifications.55–57 Second, PP1 is a dominant phosphatase, particularly in heart. Thus, we cannot rule out key roles of PP1 and even other non-traditional cardiac protein kinases and phosphatases for Nav1.5 regulation.58, 59 Third, while our studies were performed in vivo, it will be critical for expansion of these studies into large animal and even human models. Fourth, while B56α KO mice have served as an excellent first-generation model to understand PP2A signaling in vivo, secondary small molecule inhibitors will be the next logical step to ensure appropriate and selective acute or chronic B56α inhibition depending on the pathology. Nonetheless, these studies provide compelling new evidence supporting a critical role of local signaling complexes for ion channel regulation in health and disease. Finally, while prior data supports that reduced basal INa,L in B56α KO myocytes is a function of increased basal phosphatase activity in B56α hearts, future experiments will be necessary to define the direct and indirect (compensatory) changes that result in altered INa,L in the chronic absence of B56α.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Cardiac function depends on the coordinated activity of voltage-gated Na+ channels (Nav). Nav channel current (INa) is a short-duration inward current caused by rapid channel activation and immediate inactivation.
Na+ channel “late” component (INa,L) is present at baseline and increases in response to adrenergic stimulation. This late component is directly associated with potentially fatal forms of congenital and acquired human arrhythmia.
Ca2+/calmodulin-dependent kinase II (CaMKII) enhances INa,L in response to increased adrenergic tone. CaMKII-dependent phosphorylation Nav1.5 ( Nav1.5 phosphorylation at amino acid 571) is a hallmark of adrenergic imbalance in human heart disease and animal models of cardiovascular disease.
What New Information Does This Article Contribute?
We define a pathway that negatively regulates CaMKII-dependent phosphorylation in the heart.
The PP2A holoenzyme exists in complex with the Nav1.5 complex in heart. These findings support a regulatory role for PP2A-B56α in Nav1.5 phosphorylation.
Although critical for normal cardiac excitability, defects in the activity of the primary voltage-gated Nav channel, Nav1.5 are linked with both congenital and acquired forms of cardiac arrhythmia. More specifically, the Na+ channel “late” component (INa,L) is directly associated with potentially fatal forms of arrhythmia. Ca2+/calmodulin-dependent kinase II (CaMKII) enhances INa,L in response to elevated adrenergic tone and this current may support arrhythmia. Nevertheless, the molecular mechanisms that negatively regulate the CaMKII/Nav1.5 pathway are not well known. Our findings delineate a role of PP2A/B56α in the modulation of INa,L. This work illustrates that PP2A-based pathways regulate CaMKII-dependent phosphorylation in the heart and support a potential target to suppress the arrhythmogenic activity of Nav1.5.
Acknowledgments
SOURCES OF FUNDING
The authors are supported by NIH grants HL135754, HL134824, HL139348, HL135096, and HL114383 (PJ Mohler), HL135096, HL134824, HL114893 (TJ Hund), and HL089598, HL091947, and HL117641 (X Wehrens). Work is supported by the Ohio State JB Project.
Nonstandard Abbreviations and Acronyms:
- Aps
Action potentials
- APA
Action potential amplitude
- APD
Action potential duration
- CaMKII
Ca2+/calmodulin-dependent kinase II
- dv/dtmax
Maximum upstroke velocity
- INa
Nav channel current
- INa,L
late component of INa
- KO
Knockout
- Nav1.5
voltage-gated Na+ channel 1.5
- PP2A
Protein phosphatase 2A
- PP1
Protein phosphatase 1
- RyR2
Ryanodine receptor 2
Footnotes
In December 2018, the average time from submission to first decision for all original research papers submitted to Circulation Research was 14.99 days.
DISCLOSURES
XHTW is a founding partner of Elex Biotech, a start-up company that developed drug molecules that target ryanodine receptors for the treatment of cardiac arrhythmia disorders. Other authors have no conflicts.
REFERENCES
- 1.Chen-Izu Y, Shaw RM, Pitt GS, Yarov-Yarovoy V, Sack JT, Abriel H, Aldrich RW, Belardinelli L, Cannell MB, Catterall WA, Chazin WJ, Chiamvimonvat N, Deschenes I, Grandi E, Hund TJ, Izu LT, Maier LS, Maltsev VA, Marionneau C, Mohler PJ, Rajamani S, Rasmusson RL, Sobie EA, Clancy CE and Bers DM. Na+ channel function, regulation, structure, trafficking and sequestration. J Physiol. 2015;593:1347–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abriel H Cardiac sodium channel Na(v)1.5 and interacting proteins: Physiology and pathophysiology. J Mol Cell Cardiol. 2009. [DOI] [PubMed] [Google Scholar]
- 3.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M and Undrovinas AI. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–52. [DOI] [PubMed] [Google Scholar]
- 4.Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, Sag CM, Hund TJ, Mohler PJ, Belardinelli L, Hasenfuss G, Maier LS and Sossalla S. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J Mol Cell Cardiol. 2013;61:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ and Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–83. [DOI] [PubMed] [Google Scholar]
- 6.Belardinelli L, Giles WR, Rajamani S, Karagueuzian HS and Shryock JC. Cardiac late Na(+) current: proarrhythmic effects, roles in long QT syndromes, and pathological relationship to CaMKII and oxidative stress. Heart Rhythm. 2015;12:440–8. [DOI] [PubMed] [Google Scholar]
- 7.Frommeyer G, Milberg P, Maier LS and Eckardt L. Late sodium channel inhibition: The most promising antiarrhythmic principle in the near future? Curr Med Chem. 2013. [DOI] [PubMed] [Google Scholar]
- 8.Hund TJ and Mohler PJ. Nav channel complex heterogeneity: new targets for the treatment of arrhythmia? Circulation. 2014;130:132–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E and Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation. 2013;127:575–84. [DOI] [PubMed] [Google Scholar]
- 10.Burashnikov A and Antzelevitch C. Role of late sodium channel current block in the management of atrial fibrillation. Cardiovascular drugs and therapy / sponsored by the International Society of Cardiovascular Pharmacotherapy. 2013;27:79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, Leymaster ND, Dun W, Wright PJ, Cardona N, Qian L, Mitchell CC, Boyden PA, Binkley PF, Li C, Anderson ME, Mohler PJ and Hund TJ. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation. 2012;126:2084–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ and Hund TJ. Voltage-Gated Sodium Channel Phosphorylation at Ser571 Regulates Late Current, Arrhythmia, and Cardiac Function In Vivo. Circulation. 2015;132:567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, Gudmundsson H, Kline CF, Davidson NP, Cardona N, Rasband MN, Anderson ME and Mohler PJ. A beta(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O’Rourke B, Akar FG and Tomaselli GF. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res. 2010;85:454–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herren AW, Weber DM, Rigor RR, Margulies KB, Phinney BS and Bers DM. CaMKII Phosphorylation of Na(V)1.5: Novel in Vitro Sites Identified by Mass Spectrometry and Reduced S516 Phosphorylation in Human Heart Failure. J Proteome Res. 2015;14:2298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marionneau C, Lichti CF, Lindenbaum P, Charpentier F, Nerbonne JM, Townsend RR and Merot J. Mass spectrometry-based identification of native cardiac Nav1.5 channel alpha subunit phosphorylation sites. J Proteome Res. 2012;11:5994–6007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janssens V and Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. The Biochemical journal. 2001;353:417–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sents W, Ivanova E, Lambrecht C, Haesen D and Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J. 2013;280:644–61. [DOI] [PubMed] [Google Scholar]
- 19.Little SC, Curran J, Makara MA, Kline CF, Ho HT, Xu Z, Wu X, Polina I, Musa H, Meadows AM, Carnes CA, Biesiadecki BJ, Davis JP, Weisleder N, Gyorke S, Wehrens XH, Hund TJ and Mohler PJ. Protein phosphatase 2A regulatory subunit B56alpha limits phosphatase activity in the heart. Sci Signal. 2015;8:ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Onal B, Gratz D and Hund T. LongQt: A cardiac electrophysiology simulation platform. MethodsX. 2016;3:589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hund TJ, Decker KF, Kanter E, Mohler PJ, Boyden PA, Schuessler RB, Yamada KA and Rudy Y. Role of activated CaMKII in abnormal calcium homeostasis and I(Na) remodeling after myocardial infarction: insights from mathematical modeling. J Mol Cell Cardiol. 2008;45:420–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onal B, Gratz D and Hund TJ. Ca(2+)/calmodulin-dependent kinase II-dependent regulation of atrial myocyte late Na(+) current, Ca(2+) cycling, and excitability: a mathematical modeling study. Am J Physiol Heart Circ Physiol. 2017;313:H1227–H1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glynn P, Unudurthi SD and Hund TJ. Mathematical modeling of physiological systems: an essential tool for discovery. Life Sci. 2014;111:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall DD, Feekes JA, Arachchige Don AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC and Hell JW. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochemistry. 2006;45:3448–59. [DOI] [PubMed] [Google Scholar]
- 25.Camors E, Mohler PJ, Bers DM and Despa S. Ankyrin-B reduction enhances Ca spark-mediated SR Ca release promoting cardiac myocyte arrhythmic activity. J Mol Cell Cardiol. 2012;52:1240–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Terentyev D, Belevych AE, Terentyeva R, Martin MM, Malana GE, Kuhn DE, Abdellatif M, Feldman DS, Elton TS and Gyorke S. miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104:514–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiken S, Gaburjakova M, Guatimosim S, Gomez AM, D’Armiento J, Burkhoff D, Wang J, Vassort G, Lederer WJ and Marks AR. Protein kinase A phosphorylation of the cardiac calcium release channel (ryanodine receptor) in normal and failing hearts. Role of phosphatases and response to isoproterenol. J Biol Chem. 2003;278:444–53. [DOI] [PubMed] [Google Scholar]
- 28.Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, Fishman GI and Delmar M. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011;8:1923–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shy D, Gillet L and Abriel H. Cardiac sodium channel NaV1.5 distribution in myocytes via interacting proteins: the multiple pool model. Biochim Biophys Acta. 2013;1833:886–94. [DOI] [PubMed] [Google Scholar]
- 30.Lowe JS, Palygin O, Bhasin N, Hund TJ, Boyden PA, Shibata E, Anderson ME and Mohler PJ. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008;180:173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Makara MA, Curran J, Little SC, Musa H, Polina I, Smith SA, Wright PJ, Unudurthi SD, Snyder J, Bennett V, Hund TJ and Mohler PJ. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res. 2014;115:929–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM and Maier LS. Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J Clin Invest. 2006;116:3127–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunha SR and Mohler PJ. Cardiac ankyrins: Essential components for development and maintenance of excitable membrane domains in heart. Cardiovasc Res. 2006;71:22–9. [DOI] [PubMed] [Google Scholar]
- 34.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG and Bennett V. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1.5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhasin N, Cunha SR, Mudannayake M, Gigena MS, Rogers TB and Mohler PJ. Molecular basis for PP2A regulatory subunit B56alpha targeting in cardiomyocytes. Am J Physiol Heart Circ Physiol. 2007;293:H109–19. [DOI] [PubMed] [Google Scholar]
- 36.DeGrande S, Nixon D, Koval O, Curran JW, Wright P, Wang Q, Kashef F, Chiang D, Li N, Wehrens XH, Anderson ME, Hund TJ and Mohler PJ. CaMKII inhibition rescues proarrhythmic phenotypes in the model of human ankyrin-B syndrome. Heart Rhythm. 2012;9:2034–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, Murphy N, Kilic A, Higgins R, Binkley PF, Boyden PA, Carnes CA, Anderson ME, Hund TJ and Mohler PJ. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem. 2013;288:1032–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho US and Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445:53–7. [DOI] [PubMed] [Google Scholar]
- 39.Gergs U, Boknik P, Buchwalow I, Fabritz L, Matus M, Justus I, Hanske G, Schmitz W and Neumann J. Overexpression of the catalytic subunit of protein phosphatase 2A impairs cardiac function. J Biol Chem. 2004;279:40827–34. [DOI] [PubMed] [Google Scholar]
- 40.Brewis N, Ohst K, Fields K, Rapacciuolo A, Chou D, Bloor C, Dillmann W, Rockman H and Walter G. Dilated cardiomyopathy in transgenic mice expressing a mutant A subunit of protein phosphatase 2A. Am J Physiol Heart Circ Physiol. 2000;279:H1307–18. [DOI] [PubMed] [Google Scholar]
- 41.Shan J, Kushnir A, Betzenhauser MJ, Reiken S, Li J, Lehnart SE, Lindegger N, Mongillo M, Mohler PJ and Marks AR. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J Clin Invest. 2010;120:4388–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bers DM. Ca(2)-calmodulin-dependent protein kinase II regulation of cardiac excitation-transcription coupling. Heart rhythm : the official journal of the Heart Rhythm Society. 2011;8:1101–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiang DY, Lebesgue N, Beavers DL, Alsina KM, Damen JM, Voigt N, Dobrev D, Wehrens XH and Scholten A. Alterations in the interactome of serine/threonine protein phosphatase type-1 in atrial fibrillation patients. J Am Coll Cardiol. 2015;65:163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiang DY, Alsina KM, Corradini E, Fitzpatrick M, Ni L, Lahiri SK, Reynolds J, Pan X, Scott L Jr., Heck AJR and Wehrens XH. Rearrangement of the Protein Phosphatase 1 Interactome During Heart Failure Progression. Circulation. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heijman J, Ghezelbash S, Wehrens XH and Dobrev D. Serine/Threonine Phosphatases in Atrial Fibrillation. J Mol Cell Cardiol. 2017;103:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Federico M, Portiansky EL, Sommese L, Alvarado FJ, Blanco PG, Zanuzzi CN, Dedman J, Kaetzel M, Wehrens XHT, Mattiazzi A and Palomeque J. Calcium-calmodulin-dependent protein kinase mediates the intracellular signalling pathways of cardiac apoptosis in mice with impaired glucose tolerance. J Physiol. 2017;595:4089–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uchinoumi H, Yang Y, Oda T, Li N, Alsina KM, Puglisi JL, Chen-Izu Y, Cornea RL, Wehrens XHT and Bers DM. CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J Mol Cell Cardiol. 2016;98:62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang X, Wang T, Lin X, Yue X, Wang Q, Wang G, Fu Q, Ai X, Chiang DY, Miyake CY, Wehrens XHT and Chang J. Genetic deletion of Rnd3/RhoE results in mouse heart calcium leakage through upregulation of protein kinase A signaling. Circ Res. 2015;116:e1–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116:1413–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulze DH, Muqhal M, Lederer WJ and Ruknudin AM. Sodium/calcium exchanger (NCX1) macromolecular complex. J Biol Chem. 2003;278:28849–55. [DOI] [PubMed] [Google Scholar]
- 51.Kimura T, Han W, Pagel P, Nairn AC and Caplan MJ. Protein phosphatase 2A interacts with the Na,K-ATPase and modulates its trafficking by inhibition of its association with arrestin. PLoS ONE. 2011;6:e29269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dhar Malhotra J, Chen C, Rivolta I, Abriel H, Malhotra R, Mattei LN, Brosius FC, Kass RS and Isom LL. Characterization of sodium channel alpha- and beta-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103:1303–10. [DOI] [PubMed] [Google Scholar]
- 53.O’Malley HA and Isom LL. Sodium Channel beta Subunits: Emerging Targets in Channelopathies. Annu Rev Physiol. 2015;77:481–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T and Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng J, Valdivia CR, Vaidyanathan R, Balijepalli RC, Ackerman MJ and Makielski JC. Caveolin-3 suppresses late sodium current by inhibiting nNOS-dependent S-nitrosylation of SCN5A. J Mol Cell Cardiol. 2013;61:102–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ and Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R and Dudley SC Jr. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahern CA, Zhang JF, Wookalis MJ and Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res. 2005;96:991–8. [DOI] [PubMed] [Google Scholar]
- 59.Jespersen T, Gavillet B, van Bemmelen MX, Cordonier S, Thomas MA, Staub O and Abriel H. Cardiac sodium channel Na(v)1.5 interacts with and is regulated by the protein tyrosine phosphatase PTPH1. Biochem Biophys Res Commun. 2006;348:1455–62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.